
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
High pressure treatment promotes the deteriorating effect of cationic antimicrobial peptides on bacterial membranes†
Simon
Kriegler
a,
Michel W.
Jaworek
a,
Rosario
Oliva
*b and
Roland
Winter
 *a
*a
aDepartment of Chemistry and Chemical Biology, Biophysical Chemistry, TU Dortmund University, Otto-Hahn-Str. 4a, 44227 Dortmund, Germany. E-mail: roland.winter@tu-dortmund.de
bDepartment of Chemical Sciences, University of Naples Federico II, Via Cintia 4, 80126 Naples, Italy. E-mail: rosario.oliva2@unina.it
First published on 29th March 2023
Abstract
The helical structure that cationic antimicrobial peptides (cAMPs) adopt upon interaction with membranes is key to their activity. We show that a high hydrostatic pressure not only increases the propensity of cAMPs to adopt a helical conformation in the presence of bacterial lipid bilayer membranes, but also in bulk solution, and the effect on bacterial membranes persists even up to 10 kbar. Therefore, high-pressure treatment could boost cAMP activity in high-pressure food processing to extend the shelf-life of food.
1. Introduction
Cationic antimicrobial peptides (cAMPs) are a class of short peptides, whose sequence is enriched in hydrophobic as well as basic residues, typically in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 or 1:2 ratio.1,2 cAMPs are well known for their antibacterial activity, but they are also effective against viruses, fungi and even tumour cells.1 Moreover, recently, it was shown that cAMPs can also strongly perturb membraneless organelles, such as the ones formed by the LAF-1 protein and RNA, as well as the fibrillation pathways of proteins involved in neurodegenerative diseases, such as α-synuclein in Alzheimer's.3,4 In general, cAMPs affect the viability of pathogens by interacting with the lipid matrix of their cell membrane and disrupting the integrity and function of the lipid membrane.5 Some cAMPs can also cross the lipid bilayer, targeting intracellular components, such as the protein machinery and nucleic acids.5 The selectivity against pathogens is due to the presence of abundant negatively charged lipids in the bacterial membrane, such as phosphatidylglycerol (PG), phosphatidic acid (PA), and cardiolipin (CL). For example, the cytoplasmic membrane of Escherichia coli contains 20–25% PGs.2 The lack of a specific receptor is thought to be the main reason for the rare development of resistance in bacteria to cAMPs, so that cationic AMPs have come into focus as novel antibiotics. In addition, due to the lack of anionic lipids in the outer leaflet of eukaryotic membranes, cAMPs show low cytotoxic effects on mammalian cells.2 Therefore, cAMPs can be used not only in medical applications but also in other fields such as the cosmetic industry and in food preservation (such as nisin and bacteriocins).6–9 The food industry relies heavily on the use of chemicals (e.g., nitrites, sulphites and benzoates) to extend the shelf-life of foodstuff and destroy disease-causing foodborne bacteria. Unfortunately, the use of these chemicals can alter the quality of the food and can be harmful to human health.7 Moreover, pathogenic and spoilage bacteria have developed resistance mechanisms to them due to their massive use.10 cAMPs can be used as an alternative as they are generally recognized as safe and do not alter the quality (e.g., nutritional) and organoleptic (e.g., color, flavor, and sensory) properties of foods.7
:1 or 1:2 ratio.1,2 cAMPs are well known for their antibacterial activity, but they are also effective against viruses, fungi and even tumour cells.1 Moreover, recently, it was shown that cAMPs can also strongly perturb membraneless organelles, such as the ones formed by the LAF-1 protein and RNA, as well as the fibrillation pathways of proteins involved in neurodegenerative diseases, such as α-synuclein in Alzheimer's.3,4 In general, cAMPs affect the viability of pathogens by interacting with the lipid matrix of their cell membrane and disrupting the integrity and function of the lipid membrane.5 Some cAMPs can also cross the lipid bilayer, targeting intracellular components, such as the protein machinery and nucleic acids.5 The selectivity against pathogens is due to the presence of abundant negatively charged lipids in the bacterial membrane, such as phosphatidylglycerol (PG), phosphatidic acid (PA), and cardiolipin (CL). For example, the cytoplasmic membrane of Escherichia coli contains 20–25% PGs.2 The lack of a specific receptor is thought to be the main reason for the rare development of resistance in bacteria to cAMPs, so that cationic AMPs have come into focus as novel antibiotics. In addition, due to the lack of anionic lipids in the outer leaflet of eukaryotic membranes, cAMPs show low cytotoxic effects on mammalian cells.2 Therefore, cAMPs can be used not only in medical applications but also in other fields such as the cosmetic industry and in food preservation (such as nisin and bacteriocins).6–9 The food industry relies heavily on the use of chemicals (e.g., nitrites, sulphites and benzoates) to extend the shelf-life of foodstuff and destroy disease-causing foodborne bacteria. Unfortunately, the use of these chemicals can alter the quality of the food and can be harmful to human health.7 Moreover, pathogenic and spoilage bacteria have developed resistance mechanisms to them due to their massive use.10 cAMPs can be used as an alternative as they are generally recognized as safe and do not alter the quality (e.g., nutritional) and organoleptic (e.g., color, flavor, and sensory) properties of foods.7
There are also other ways to extend the shelf-life of food, for example by thermal treatment (pasteurization) which is widely used. However, thermal treatment can result in the loss of nutrients or the formation of toxic compounds.11 Another option is high-pressure (HP) treatment, developed in the 1990s in Japan, in which foodstuff (e.g., fruit juice, vegetables, seafood, dairy and meat products) is subjected to pressures of 3–7 kbar for several minutes at ambient temperature,11–14 a technology that is recognized as the most successful non-thermal food processing method.14 During HP treatment, the integrity of the cellular membranes of pathogens is destroyed, and denaturation of endogenous proteins can also contribute to pathogen death.14–16 This treatment, as pointed out by the European Food Safety Authority (EFSA),17 is effective in killing pathogens and, at the same time, has only minimal effects on the taste, appearance, and nutritional properties of food. For example, HP treatment is an excellent method for reducing levels of Listeria monocytogenes, the presence of which in food is a serious concern.17
As it is still difficult to achieve complete microbial inactivation (e.g., of bacterial spores) by a single HP treatment in some cases, synergistic or additive effects on microbial inactivation, which could reduce the survival of pressure-resistant subpopulations, are desirable. With the idea of combining the advantages of using cAMPs with those of HP treatment, in this work we investigated the interaction of a short cAMP, named (P)GKY20, derived from the human thrombin, with a prototypical bacterial model membrane composed of 80% DOPE (1,2-oleoyl-sn-glycero-3-phosphoethanolamine) and 20% DOPG (1-palmitoyl-2-oleoyl-sn-glycero-3-phospho-(1′-rac-glycerol)), a membrane composition typical for E. coli cells. The major lipid components of animal cells are phosphatidylcholines (PC), which are rarely present in the bacterial membrane.18
Biological membranes are amongst the most pressure-sensitive cellular components. The structure and phase transitions that a particular lipid bilayer membrane can adopt depend on the lipid chain length and degree of unsaturation, on the headgroup structure and its charge, however.32,36,37 Phospholipid bilayers commonly undergo structural phase transitions, including a gel-to-fluid (liquid-crystalline, Lα) main (chain-melting) transition. The effect of increasing pressure is generally opposite to that of increasing temperature. A slope of about 10–20 °C kbar−1 was observed for the chain-melting transition of phosphatidylcholines.32,36 The application of pressure causes an ordering of the lipid chains, resulting in a decreased cross-sectional area and splay of the lipid hydrocarbon tails. The chain length of fluid phospholipid bilayers increases by ∼1 Å kbar−1 only, whereas the lipid chain cross-sectional area decreases by ∼10 Å2 kbar−1. The density of the gel phase is about 3.5% higher compared to the fluid phase, and the compressibility of the gel phase is about a factor 2–3 smaller than that of the fluid phase. The lateral self-diffusion coefficient in the fluid phase decreases by ∼30% from 1 bar to 300 bar, and as much as 70% at the fluid-to-gel transition, and a significant increase in bending rigidity of fluid vesicles is observed as well upon pressurization, about 30% per 100 bar (ref. 32, 36, 37 and references therein). Interestingly, in more complex and biologically more relevant membranes, depending on the temperature, an overall ordered lipid state is generally reached at 1–3 kbar pressures as well, which is also the pressure range where the function of membrane-associated proteins ceases.36,37
Different biophysical techniques, including high-pressure fluorescence and FT-IR spectroscopy, microscopy and calorimetry, were employed to disentangle the combined effects of (P)GKY20 and HP on liposomes mimicking the bacterial membrane. The peptide (P)GKY20 has a charge of +5 at physiological pH, a low haemolytic activity against eukaryotic cells, and it is active against a wide range of bacterial strains, especially Gram-negative ones.19,20 Previously, we showed that, under ambient conditions (T = 25 °C, p = 1 bar), (P)GKY20 perturbs PC–PG bilayers through the formation of lipid domains which ultimately leads to the solubilization of the bilayer, according to a carpet-like mechanism.20 The results reported here show that (P)GKY20 retains its ability to interact and to perturb bacterial model membranes even under high pressure stress in the kbar regime. Therefore, in principle, it can be used in HP treatment of foods to increase their shelf-life and increase the efficiency of HP treatment.
2. Materials and methods
2.1 Materials
The antimicrobial peptide (P)GKY20 (sequence: (P)GKYGFYTHVFRLKKWIQKVI, molecular weight 2609.16 g mol−1)21,22 in lyophilized powder was purchased from GenScript (Germany), with a purity ≥ 95% (see Fig. S1 and S2, ESI† for the HPLC and the mass spectra). All lipids including 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphoethanolamine (POPE), 1-palmitoyl-2-oleoyl-sn-glycero-3-phospho-(1′-rac-glycerol) sodium salt (POPG), 1,2-dioleoyl-sn-glycero-3-phosphoethanolamine (DOPE) and 1,2-dioleoyl-sn-glycero-3-phospho-(1′-rac-glycerol) sodium salt (DOPG) were purchased from Avanti Polar Lipids (Alabaster, USA). The fluorescent dye used in fluorescence confocal microscopy, N-(lissamine rhodamine B sulfonyl)-1,2-dihexadecanoyl-sn-glycero-3-phosphoethanolaminetriethylammonium salt (N-Rh-DHPE), was obtained from Molecular Probes (Invitrogen, California, USA). Laurdan (6-dodecanoyl-N,N-dimethyl-2-naphthylamine) and DPH (1,6-diphenyl-1,3,5-hexatrien) were purchased from Sigma-Aldrich (St Louis, USA). The Tris–HCl and the sodium phosphate salts for buffer preparation were obtained from Sigma-Aldrich (St Louis, USA). Bidistilled water was used for all buffer preparations.2.2 Liposome preparation
Lipid samples for DSC, FT-IR, fluorescence and circular dichroism spectroscopy were prepared by mixing lipid stock solutions at a concentration of 10 mg mL−1 in chloroform. If necessary, the desired amount of 1 mM Laurdan was added to the solution. After evaporating the solvent with a stream of nitrogen and a freeze dryer under vacuum, 10 mM Tris (Tris(hydroxymethyl)aminomethane) buffer at pH 7.4 was added (only for the CD measurements, 20 mM sodium phosphate buffer was used). DSC and FT-IR measurements required the formation of multilamellar vesicles (MLVs). To prepare the vesicles, the solutions were treated for 20 min in an ultrasonic bath, which was heated above the gel-to-fluid phase transition temperature, Tm. Finally, the samples were frozen and thawed five times using a water bath (above Tm) and liquid nitrogen. For the fluorescence emission and circular dichroism measurements, the formation of unilamellar vesicles was performed via extrusion through a polycarbonate membrane with a size of 100 nm by means of a miniextruder from Avanti Polar Lipids (Alabaster, USA).2.3 Fourier-Transform Infrared (FTIR) spectroscopy
First, an H/D exchange was carried out on the peptide via dialysis against D2O followed by lyophilization. The MLVs as well as the peptide were prepared in 20 mM Tris buffer in D2O at a pH of 7.0. A peptide concentration of 2 wt% (7.6 mM) was measured in the absence and presence of the liposome system (DOPE:DOPG, 80:20, total concentration 190 mM) in a lipid-to-peptide molar ratio of 50:1. FTIR measurements were performed using a Nicolet 6700 (Thermo Fisher Scientific) spectrometer equipped with a liquid-nitrogen cooled MCT-detector (HgCdTe). Per measurement, respectively per spectrum, 128 scans were recorded in the wavenumber range between 4000 and 650 cm−1 at 25 °C. The spectra were processed with Happ–Genzel apodization using the Omnic 7.2 spectral processing software. Temperature control was achieved using an external circulating water thermostat. The sample chamber was continuously purged with CO2-free dry air to achieve a good signal-to-noise ratio. A membrane-driven diamond anvil cell (Diacells VivoDac, Almax easyLab) equipped with type IIa diamonds (Almax easyLab), which was connected to an automated pneumatic pressure controller (Diacells iGM Controller, Almax easyLab), was used to achieve high pressures of up to 12 kbar. Barium sulfate powder was used as an internal pressure calibrant to determine the pressure values.23 The equilibration time for each pressure was chosen to be 5 min before collecting the data. The analysis of the measured spectra was performed using the Thermo Grams 8.0 software package. Each spectrum was buffer subtracted and subsequently smoothed. To determine the relative secondary structural changes, the area of the amide I' band (1700–1600 cm−1) of the spectra was normalized to 1. The number and positions of the sub-bands were determined by using two mathematical operations, Fourier self-deconvolution (FSD) and 2nd derivative spectroscopy. The amide I' band of the peptide possesses six sub-bands, which can be assigned to particular secondary structure elements as described before.24 The relative changes in the population of secondary structure elements were obtained by using mixed Gaussian–Lorentzian line-shape functions in the fitting procedure.25
2.4 Circular dichroism (CD) spectroscopy
Far-UV circular dichroism spectra were acquired by means of a Jasco J-715 spectropolarimeter from Jasco Corporation (Tokyo, Japan). The temperature was set at 25 °C by means of a circulating water bath directly connected to a quartz cuvette (path length of 0.1 cm). The concentration of (P)GKY20 peptide was 50 μM. Instead, the lipid concentration was 2.5 mM so that the spectrum of the peptide in the presence of LUVs was acquired at the lipid-to-peptide ratio of 50:1. The following instrument parameters were used: scan rate of 50 nm min−1, response time of 2 s, and bandwidth of 5 nm. The reported spectra are the results of 5 accumulations. From each spectrum, an appropriate blank (buffer and lipid dispersion in buffer) was subtracted. To avoid interference from Tris buffer, sodium phosphate 20 mM, pH 7.4, was used.
2.5 Differential scanning calorimetry (DSC)
Since DOPE and DOPG vesicles exhibit a transition temperature below 0 °C, for the DSC experiments, liposomes composed of POPE and POPG were used, instead. The suspensions investigated contained a lipid concentration of 5 wt% in 20 μL, and were transferred to Tzero hermetic pans (TA instruments, New Castle, USA). The peptide was added at a lipid-to-peptide molar ratio of 50:1. After 5 min of equilibration at the starting temperature, the measurements were performed with a heating rate of 1 °C min−1 by using a Q20 Differential Scanning Calorimeter from TA instruments (New Castle, DE, USA).
2.6 Fluorescence spectroscopy
A K2 multifrequency phase modulation fluorometer (from ISS Inc., Champaign, IL, USA) was used to record the fluorescence emission spectra of Laurdan in a pressure range from 1 to 2000 bar at the temperature of 25 °C. The vesicle concentration was 3 mM while the concentration of Laurdan was 2.2 μM. The measurements were started 30 min after incubation with the peptide. An excitation wavelength of 390 nm was chosen to collect the Laurdan emission between 410 and 500 nm. The generalized polarization (GP), which is a measure of the lateral lipid order and the fluidity of the membrane, was calculated using the following equation: GP = (I440nm − I490nm)/(I440nm + I490nm).26,27 The different intensity maxima indicate the gel and fluid phase of the membrane. While the intensity at 440 nm represents an ordered gel-like phase, the intensity at 490 nm is indicative of a fluid (liquid-crystalline) phase.2.7 Confocal fluorescence microscopy
The formation of giant unilamellar vesicles (GUVs) was required to visualize vesicles in a confocal fluorescence microscopy experiment. The fluorophore N-Rhodamine DHPE was added to the lipid solution at a concentration of 1 mg mL−1 to obtain a ratio of labeled to unlabeled lipid of 1:500. Then, the PVA-assisted preparation was performed as described previously.28 An objective lens of type CFI Plan Apochromat Lambda 100 × Oil, NA 1.45, WD 0.13 (Nikon, Tokyo, Japan) was used to magnify the GUVs. The fluorophore was excited by a laser combiner Oxxius Simply Light, L4Cc-CSB-130 (Lannion, France) or a mercury-vapor lamp (Hg 100 W, Nikon, Tokyo, Japan) and the fluorescence images recorded on a CCD camera (Andor iXon Ultra, Acal BFi, Germany). A 1 mM peptide stock solution was added after vesicle preparation using an incubation time of 1 h.
3. Results and discussion
In order to explore the impact of the peptide (P)GKY20 on the stability and lipid chain packing of POPE/POPG (80:20 mol:mol) liposomes, differential scanning calorimetry (DSC) experiments were performed. Fig. 1A shows the DSC heating scans of multilamellar vesicles composed of POPE:POPG before and after the addition of (P)GKY20, at a lipid-to-peptide ratio (L:P) of 50:1 after 1.5 h and 24 h of incubation. Since DOPE:DOPG exhibits a transition temperature below 0 °C only, which is not suitable for DSC experiments, POPE:POPG vesicles were used instead.
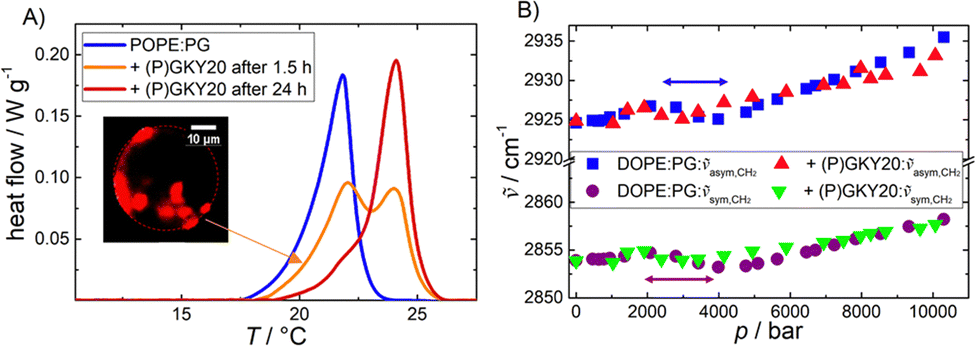 | ||
| Fig. 1 (A) DSC heating scans of POPE:POPG (80:20) multilamellar vesicles in the absence and presence of (P)GKY20 (at L:P = 50:1) after different incubation times. The inset shows a confocal fluorescence microscopy snapshot of a DHPE-rhodamine-labeled μm-sized giant unilamellar vesicle of DOPE:DOPG in the presence of the peptide (the fluorophore is expected to cluster in DOPE-enriched domains (red areas)). (B) Pressure-dependence of the symmetric and antisymmetric CH2 vibrational band peak maxima of DOPE:DOPG (80:20) in the absence and presence of the peptide (L:P = 50:1). | ||
The DSC peak, reporting on the gel-to-liquid phase transition of the lipid mixture, in the absence of peptide, is centred at ∼22 °C with an associated enthalpy change of ΔHm = 20.5 J g−1, which is essentially due to the melting of the lipid hydrocarbon chains (all the thermodynamic parameters are reported in Table 1), in agreement with literature data.29 Upon addition of the peptide, after 1.5 h of incubation, the DSC scan shows peaks at ∼22 °C and ∼24 °C. After 24 h of incubation, the low-temperature peak was less pronounced. These data are clear evidence that (P)GKY20 promotes a redistribution of the lipids within the lipid bilayer, most likely through the preferential interaction with the anionic PGs, which ultimately leads to the formation of lipid domains that are characterized by different transition temperatures. Such domain formation was also previously observed for a PC–PG mixture.20 Moreover, these results indicate a stabilizing effect of the peptide on the gel phase of the bacterial model membrane, suggesting localization of the peptide at the membrane interface. Indeed, the ΔHm value associated with the phase transition (Table 1) is not significantly affected by the presence of the peptide, supporting the notion that the peptide is localized at the water–membrane interface.30
:POPG (80:20) multilamellar vesicles in the absence and presence of (P)GKY20 at a lipid to peptide ratio of 50:1 obtained by means of DSC measurements
To monitor the effect of the peptide on the packing of the lipid chains in the hydrophobic core, high-pressure FT-IR spectroscopic experiments were carried out in the range from 1 to 10000 bar. Fig. 1B depicts the wavenumber shifts associated with the symmetric (∼2854 cm−1 at 1 bar) and antisymmetric (∼2925 cm−1 at 1 bar) CH2 stretching vibrational bands of DOPE–DOPG vesicles in the absence and presence of the peptide. Changes in the CH2 vibrational band positions are indicative of conformational changes of the lipid acyl chains, such as changes in the trans–gauche ratio induced by pressure.31 The position of the bands increases linearly with pressure in the case of a mere elastic compression.31 Changes in the slope between 2 and 4 kbar indicate the pressure-induced phase transition from the fluid phase (Lα) to the more ordered gel-phase (Lβ′). A similar behavior was observed for pure DOPE liposomes (ptransition ≈ 1.5 kbar at 20 °C).32 In the presence of the peptide, the Lα-to-Lβ′ transition shifts to slightly lower pressures, indicating stabilization of the gel phase of the membrane, in accordance with the DSC data, revealing a surface-binding mode of the peptide also under pressure. Insertion of the peptide into the hydrophobic core would lead to a less compact and more disordered membrane. Overall, the data reported so far indicate that the peptide interacts with this bacterial model membrane positioning at the water–membrane interface, and induces lipid domains, but does not penetrate the interior of the hydrophobic core of the bilayer, both at ambient and kbar pressures.
Fluorescence spectroscopy with the Laurdan probe was used to investigate the effects of the peptide on the membrane properties in the interfacial region as a function of pressure. This fluorescence probe localizes in the proximity of the head group region of the lipid bilayer and, thus, reports on changes in the interfacial region of the bilayer.33Fig. 2A shows the general polarization (GP) values of Laurdan embedded in DOPE:DOPG (80:20 mol:mol) liposomes in the absence and in the presence of 1, 2, and 10 μM of the peptide in the pressure range from 1 to 2000 bar at ambient temperature (20 °C). For the pure PE–PG bilayer system, the GP value is ∼0.1 at 1 bar, which indicates that the membrane is in the fluid-like (Lα) phase. Increasing the pressure up to 2 kbar caused an almost linear increase of the GP-value, indicating a higher degree of lipid order. Pressurization leads to a more densely packed lipid bilayer which occupies a smaller partial molar volume.32–38 Upon addition of (P)GKY20, at all concentrations studied, a significant increase of the GP value at 1 bar was observed, revealing that the peptide induces a rigidification and partial dehydration of the membrane surface. Remarkably, this effect is already visible at the lowest peptide concentration used (at L:P = 3000:1), highlighting the strong membrane perturbation ability of the peptide in the whole pressure-range covered.
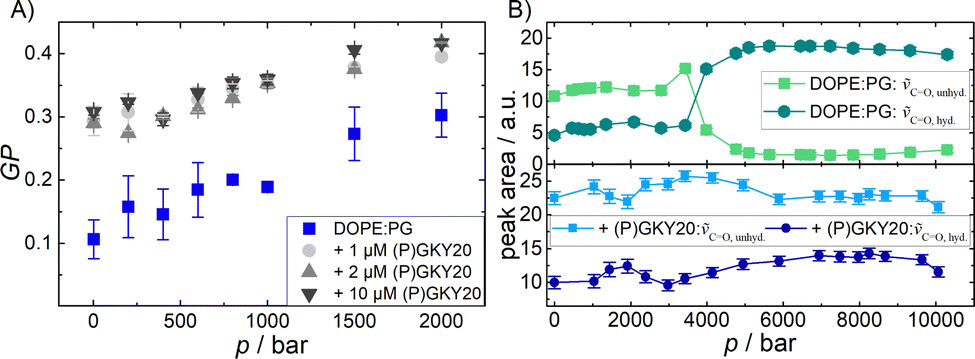 | ||
Fig. 2 (A) Pressure-dependent Laurdan GP-values of DOPE:DOPG (80:20) vesicles at 20 °C with and without (P)GKY20, at the indicated concentrations. (B) Peak area of bound and free C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bands from DOPE:DOPG (80:20) vesicles in the absence (top) and presence (bottom) of (P)GKY20 as a function of pressure. O bands from DOPE:DOPG (80:20) vesicles in the absence (top) and presence (bottom) of (P)GKY20 as a function of pressure. | ||
Additional information regarding the level of hydration at the membrane–water interface can be obtained from analysis of the stretching band region of the carbonyl (CO) group which is centred at ∼1735 cm−1 (Fig. S3, ESI†). The lipid molecules possess two CO groups (one at the sn-1, the other at the sn-2 chain of the lipid molecule), whose wavenumbers differ by about 3–4 cm−1, generally resulting in a rather broad peak.31,39 The CO band position is sensitive to the extent of hydrogen bonding and hence also to the level of hydration, leading to a shift to lower wavenumbers.31,39,40 To quantify the amount of H-bonded vs. non-H-bonded carbonyl groups, the band was fitted with two Gaussian functions. The peak at ∼1724 cm−1 corresponds to CO involved in H-bonds and the peak at ∼1738 cm−1 represents largely free, non-H-bonded CO groups. Fig. 2B depicts the corresponding peak areas of the CO vibrational band of DOPE:DOPG vesicles as a function of pressure in the absence and in the presence of (P)GKY20 at the lipid-to-peptide ratio L:P = 50:1. For pure liposomes, at p = 1 bar, the area of the peak attributed to non H-bonded CO groups is the most prominent one. At 3–4 kbar, the pressure region where also changes in the CH2 stretching vibrations were observed, a phase transition occurred and the whole band shifts to lower wavenumbers (Fig. S3, ESI†). The area of the peak at ∼1738 cm−1 decreased and the area of the peak at ∼1724 cm−1 increased concomitantly, indicating an increase in H-bonding beyond the phase transition. This is a very peculiar behavior since it is expected that increasing pressure favors a more compact and ordered lipid phase, leading to a decrease of the level of hydration and hence H-bonding to CO groups.31 Our observations can be rationalized by invoking changes in intermolecular interactions among PE and PG groups at high pressures. At low pressure, where the bilayer is in the fluid phase, the negatively charged oxygen of the phosphate moiety of PG can establish H-bonds with the positively charged –NH2+ moiety of the PE's headgroup, significantly strengthening inter-lipid contacts and hampering major accumulation of water molecules at the level of CO groups.18,41 At high pressure, where the membrane adopts a densely packed gel phase, the lipid headgroups acquire an orientation that is now most likely perpendicular to the membrane surface. This leads to a breaking of these inter-lipid H-bonds, leading to an increase of the 1724 cm−1 band intensity, allowing some water molecules to gain access to the CO groups again and increase their number of H-bonds. Interestingly, in the presence of (P)GKY20, no such changes in the peaks area were observed in the whole pressure range explored (Fig. 2B). This is clear evidence that the peptide strongly interacts with the bilayer surface at all pressures up to 10 kbar, preventing pressure-induced phase changes of the lipid bilayer system as observed in the peptide-free liposomes.
The data reported above strongly support the idea that the (P)GKY20 peptide interacts with the lipid bilayer at the level of the membrane–water interface, i.e., mainly with the lipid headgroup. In this context, molecular dynamics (MD) simulations would be desirable to yield a more detailed and molecular level understanding of the binding process.42 MD studies for the (P)GKY20 peptide interacting with model biomembranes are not currently available. However, simulations are reported for the interaction of GKY25 with lipid A. The peptide GKY25 is composed by the same sequence of (P)GKY20, with the addition of residues DQFGE at its C-terminus. Holdbrook and colleagues43 reported that GKY25 can interact with the phosphate moiety of lipid A, displacing calcium ions. Interestingly, the region including the residues KYGFY of GKY25 (which are also present in the sequence of (P)GKY20) was found to interact strongly with the headgroups of lipid A. In a similar study,44 it was reported that the residues K2, T7, H8, R11, K13 and K14 of GKY25 interact with the headgroups of lipid A. Instead, residues Y3, F5, Y6 and F10, which are all aromatic, are positioned at the interface between the lipid head and tail parts of lipid A. Since phosphate groups are also present in phospholipids, such as in PEs and PGs in our study, we can assume that (P)GKY20 also interacts effectively at the headgroup level with lipid moieties and, thus, it is localized in this region of the lipid bilayer.
It is well known that most of the linear cAMPs adopt a helical structure upon binding to membranes, which seems fundamental to their biological activity.2 The peptide (P)GKY20 is not an exception, as was shown in a study using PC–PG vesicles.20 In order to verify that (P)GKY20 can adopt an helical structure in the presence of DOPE:DOPG, circular dichroism (CD) spectra were recorded (Fig. S3, ESI†). The acquired spectra clearly show that the peptide is not structured when dissolved in buffer, as evidenced by the presence of a minimum around 200 nm. Upon addition of the anionic lipid vesicles, the CD spectrum indicates that the peptide adopts mainly an α-helical structure. Indeed, two well defined minima at around 210 nm and 225 nm are present. To explore the impact of HP on (P)GKY20, HP FT-IR spectroscopy was employed to determine the changes in the secondary structure elements of (P)GKY20 in the absence and presence of the lipid vesicles. Fig. 3A shows the FT-IR-spectrum of (P)GKY20 in buffer solution at 1 bar and at high pressures up to 10 kbar. Secondary structure analysis (see the Materials and methods section for details) reveals that the conformation of the peptide is mainly composed of β-turns and random-coil structures (Fig. 3B) at 1 bar, in agreement with the CD data. Upon subjecting to pressurization of up to 10 kbar, the shape of amide I′ band changes. The percentage of β-turns and random coils decreases, while the amount of α-helices increases strongly (∼three-fold). This behavior is similar to that observed for membrane-incorporated gramicidin,45 where an increase of the helical content with pressure was reported for the membrane-embedded peptide. Here, an increase of helical content was already observed for the peptide in the absence of liposomes. A lower partial volume for the helical state, compared to the unfolded one, was also observed through molecular dynamics simulations and FT-IR experiments on the model peptide AK16.46,47
 | ||
| Fig. 3 FTIR-analysis of the amide I′ band of (P)GKY20 in the absence (A and B) and presence (C and D) of DOPE:DOPG vesicles at the L:P ratio 50:1. Panels A and C show the pressure dependent changes of the amide-I′ (1600–1700 cm−1) region and panels B and D the relative changes of secondary structure elements of the peptide, i.e., β-turns (black, 1672 cm−1), α-helices (green, 1654 cm−1), β-sheets (red, 1631 and 1672 cm−1), random coils (blue, 1644 cm−1), and side chains (orange, 1616 cm−1). | ||
As can be clearly seen, pressure favors the α-helical structure, which occupies less volume compared to the other secondary structures. This is a remarkable finding since the helical structure is fundamental for the action of cAMPs; in other words, HP is expected to booster the peptide's biological activity. Indeed, a high-pressure induced activation of biological activities of lysozyme and the polycyclic 34-residue peptide nisin against E. coli was already observed in vivo.48 Moreover, a small but significant increase of the antibacterial activities of lactoferrin and its derivatives under pressure was reported in meat samples against Gram-positive bacteria.49 These examples highlight that an in vivo boost in the activity of peptides and proteins can be reached upon pressurization. In the presence of lipid vesicles (Fig. 3C), a significant change in the shape of the amide I′ band is observed. Deconvolution analysis revealed that the percentage of α-helix content increased dramatically and applying HP in the presence of lipid vesicles has only a minor effect on the distribution of secondary structure elements, revealing that HP does not hamper peptide binding and changing the peptide's conformational state imposed by the presence of the lipid bilayer.
4. Conclusions
In conclusion, this work demonstrates that the cAMP (P)GKY20 can interact and affect, through the formation of lipid domains by recruiting anionic lipids, bacterial model membranes composed of PEs and PGs. The interaction with the lipid interface is also associated with a more compact membrane as the peptide screens the repulsion among the lipid head groups, and the formation of a helical conformation which leads to an amphipathic structure, facilitating the interaction with and the perturbation of the membrane (see the schematic in Fig. 4). We showed that the population of this ‘active’ helical conformation is even enhanced in bulk solution upon pressurization which is used in high-pressure food processing technologies. Moreover, the application of HP does not affect the peptide's membrane perturbation propensity in the pressure range commonly used for HP food treatment. Thus, combined processing modes using antimicrobial peptides synergistically with high pressure should be able to open new exciting avenues for their application in high-pressure food processing.14 | ||
| Fig. 4 Proposed action mechanism of (P)GKY20 with bacterial model membranes at low and high pressure: preferential binding to anionic lipids coupled with peptide folding into an α-helical conformation and consequential formation of large domains enriched with anionic lipids. After reaching a threshold peptide concentration, bilayer disruption and lipid extraction occurs.20 High pressure leads to an increase in the population of active α-helical conformers and leaves the peptide-membrane interaction intact even at kbar pressures. | ||
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work has in part been funded by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) under Germany′s Excellence Strategy – EXC-2033 – Projektnummer 390677874. Rosario Oliva is grateful to the Italian MUR for being granted a research associated position (PON R&I 2014-2020, CUP: E65F21003250003).References
- S. Nayab, M. A. Aslam, S. U. Rahman, Z. U. D. Sindhu, S. Sajid, N. Zafar, M. Razaq, R. Kanwar and Amanullah, Int. J. Pept. Res. Ther., 2022, 28, 46 CrossRef CAS.
- V. Teixeira, M. J. Feio and M. Bastos, Prog. Lipid Res., 2012, 51, 149–177 CrossRef CAS PubMed.
- R. Oliva, S. K. Mukherjee, Z. Fetahaj, S. Möbitz and R. Winter, Chem. Commun., 2020, 56, 11577–11580 RSC.
- R. Oliva, S. K. Mukherjee, L. Ostermeier, L. A. Pazurek, S. Kriegler, V. Bader, D. Prumbaum, S. Raunser, K. F. Winklhofer, J. Tatzelt and R. Winter, Chem. – Eur. J., 2021, 27, 11845–11851 CrossRef CAS PubMed.
- Y. Huan, Q. Kong, H. Mou and H. Yi, Front. Microbiol., 2020, 11, 582779 CrossRef PubMed.
- M. Rahnamaeian and A. Vilcinskas, Appl. Microbiol. Biotechnol., 2015, 99, 8847–8855 CrossRef CAS PubMed.
- M. Rai, R. Pandit, S. Gaikwad and G. Kövics, J. Food Sci. Technol., 2016, 53, 3381–3394 CrossRef CAS PubMed.
- Y. Liu, D. E. Sameen, S. Ahmed, J. Dai and W. Qin, Trends Food Sci. Technol., 2021, 112, 471–483 CrossRef CAS.
- Y. L. Vishweshwaraiah, A. Acharya, V. Hegde and B. Prakash, npj Sci. Food, 2021, 5, 26 CrossRef PubMed.
- J. L. Romero, M. J. Grande Burgos, R. Pérez-Pulido, A. Gálvez and R. Lucas, Front. Microbiol., 2017, 8, 1650 CrossRef PubMed.
- M. van Boekel, V. Fogliano, N. Pellegrini, C. Stanton, G. Scholz, S. Lalljie, V. Somoza, D. Knorr, P. R. Jasti and G. Eisenbrand, Mol. Nutr. Food Res., 2010, 54, 1215–1247 CrossRef CAS PubMed.
- N. K. Rastogi, K. S. M. S. Raghavarao, V. M. Balasubramaniam, K. Niranjan and D. Knorr, Crit. Rev. Food Sci. Nutr., 2007, 47, 69–112 CrossRef CAS PubMed.
- Stress Responses of Food Pathogens, ed. T. Ding, X. Liao and J. Feng, Springer Nature, Switzerland, 2022 Search PubMed.
- Q. Xia, A. Liu, G. I. Denoya, C. Yang, F. J. Barba, H. Yu and X. Chen, Front. Nutr., 2022, 9, 878904 CrossRef PubMed.
- P. Putnik, Ž. Kresoja, T. Bosiljkov, A. Režek Jambrak, F. J. Barba, J. M. Lorenzo, S. Roohinejad, D. Granato, I. Žuntar and D. Bursać Kovačević, Food Chem., 2019, 279, 150–161 CrossRef CAS PubMed.
- H. M. Ulmer, H. Herberhold, S. Fahsel, M. G. Gänzle, R. Winter and R. F. Vogel, Appl. Environ. Microbiol., 2002, 68, 1088–1095 CrossRef CAS PubMed.
- K. Koutsoumanis, et al. , EFSA J., 2022, 20, e07128 CAS.
- K. Murzyn, T. Róg and M. Pasenkiewicz-Gierula, Biophys. J., 2005, 88, 1091–1103 CrossRef CAS PubMed.
- G. Kasetty, P. Papareddy, M. Kalle, V. Rydengård, M. Mörgelin, B. Albiger, M. Malmsten and A. Schmidtchen, Antimicrob. Agents Chemother., 2011, 55, 2880–2890 CrossRef CAS PubMed.
- R. Oliva, P. Del Vecchio, A. Grimaldi, E. Notomista, V. Cafaro, K. Pane, V. Schuabb, R. Winter and L. Petraccone, Phys. Chem. Chem. Phys., 2019, 21, 3989–3998 RSC.
- R. Oliva, M. Campanile, P. del Vecchio, F. Pizzo, A. Bosso, R. Winter and L. Petraccone, Phys. Chem. Chem. Phys., 2022, 24, 7994–8002 RSC.
- K. Pane, L. Durante, E. Pizzo, M. Varcamonti, A. Zanfardino, V. Sgambati, A. Di Maro, A. Carpentieri, V. Izzo, A. Di Donato, V. Cafaro and E. Notomista, PLoS One, 2016, 11, e0146552 CrossRef PubMed.
- P. Wong and D. Moffat, Appl. Spectrosc., 1989, 43, 1279–1281 CrossRef CAS.
- A. Adochitei and G. Drochioiu, Rev. Roum. Chim., 2011, 56, 783–791 CAS.
- G. Panick and R. Winter, Biochemistry, 2000, 39, 1862–1869 CrossRef CAS PubMed.
- J. R. Lakowicz, Principles of Fluorescence Spectroscopy, Springer, New York, 3rd edn, 2006 Search PubMed.
- L. A. Bagatolli, Springer Ser. Fluoresc., 2013, 13, 3–36 CAS.
- A. Weinberger, F.-C. Tsai, G. H. Koenderink, T. F. Schmidt, R. Itri, W. Meier, T. Schmatko, A. Schröder and C. Marques, Biophys. J., 2013, 105, 154–164 CrossRef CAS PubMed.
- B. Pozo Navas, K. Lohner, G. Deutsch, E. Sevcsik, K. A. Riske, R. Dimova, P. Garidel and G. Pabst, Biochim. Biophys. Acta, Biomembr., 2005, 1716, 40–48 CrossRef CAS PubMed.
- O. Cañadas and C. Casals, in Lipid-Protein Interactions, ed. J. H. Kleinschmidt, Springer New York, New York, 2019, vol. 2003, pp. 91–106 Search PubMed.
- O. Reis, R. Winter and T. W. Zerda, Biochim. Biophys. Acta, Biomembr., 1996, 1279, 5–16 CrossRef PubMed.
- R. Winter and C. Jeworrek, Soft Matter, 2009, 5, 3157–3173 RSC.
- T. Parasassi and E. Gratton, J. Fluoresc., 1995, 5, 59–69 CrossRef CAS PubMed.
- C. Bernsdorff, A. Wolf, R. Winter and E. Gratton, Biophys. J., 1997, 72, 1264–1277 CrossRef CAS PubMed.
- K. Shin, H. Maeda, T. Fujiwara and H. Akutsu, Biochim. Biophys. Acta, Biomembr., 1995, 1238, 42–48 CrossRef PubMed.
- R. Winter, Annu. Rev. Biophys., 2019, 48, 441–461 CrossRef CAS PubMed.
- J.-M. Knop, S. Mukherjee, M. W. Jaworek, S. Kriegler, M. Manisegaran, Z. Fetahaj, L. Ostermeier, R. Oliva, S. Gault, C. S. Cockell and R. Winter, Chem. Rev., 2023, 123, 73–104 CrossRef CAS PubMed.
- N. L. C. McCarthy, O. Ces, R. V. Law, J. M. Seddon and N. J. Brooks, Chem. Commun., 2015, 51, 8675–8678 RSC.
- R. N. Lewis, R. N. McElhaney, W. Pohle and H. H. Mantsch, Biophys. J., 1994, 67, 2367–2375 CrossRef CAS PubMed.
- A. Blume, W. Huebner and G. Messner, Biochemistry, 1988, 27, 8239–8249 CrossRef CAS PubMed.
- P. Garidel and A. Blume, Eur. Biophys. J., 2000, 28, 629–638 CrossRef CAS PubMed.
- R. G. Huber, T. S. Carpenter, N. Dube, D. A. Holdbrook, H. I. Ingólfsson, W. A. Irvine, J. K. Marzinek, F. Samsudin, J. R. Allison, S. Khalid and P. J. Bond, in, Lipid-Protein Interactions, ed. J. H. Kleinschmidt, Springer, New York, 2019, vol. 2003, pp. 1–30 Search PubMed.
- D. A. Holdbrook, S. Singh, Y. K. Choong, J. Petrlova, M. Malmsten, P. J. Bond, N. K. Verma, A. Schmidtchen and R. Saravanan, Biochim. Biophys. Acta, Biomembr., 2018, 1860, 2374–2384 CrossRef CAS PubMed.
- R. Saravanan, D. A. Holdbrook, J. Petrlova, S. Singh, N. A. Berglund, Y. K. Choong, S. Kjellström, P. J. Bond, M. Malmsten and A. Schmidtchen, Nat. Commun., 2018, 9, 2762 CrossRef PubMed.
- J. Eisenblätter, M. Zein and R. Winter, Prog. Biotechnol., 2002, 19, 131–138 Search PubMed.
- Y. Mori and H. Okumura, Proteins: Struct., Funct., Bioinf., 2014, 82, 2970–2981 CrossRef CAS PubMed.
- T. Takekiyo, T. Imai, M. Kato and Y. Taniguchi, Biochim. Biophys. Acta., 2006, 1764, 355–363 CrossRef CAS PubMed.
- C. Garcia-Graells, B. Masschalck and C. W. Michiels, J. Food Prot., 1999, 62, 1248–1254 CrossRef CAS PubMed.
- A. Del Olmo, J. Calzada and M. Nunez, Meat Sci., 2011, 90, 71–76 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Materials and used methods, sample preparation, and further results. See DOI: https://doi.org/10.1039/d3cp00560g |
| This journal is © the Owner Societies 2023 |