
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Design principles for a nanoconfined enzyme cascade electrode via reaction–diffusion modelling
Bhavin
Siritanaratkul

Stephenson Institute for Renewable Energy and the Department of Chemistry University of Liverpool, Liverpool, L69 7ZF, UK. E-mail: Bhavin.Siritanaratkul@liverpool.ac.uk
First published on 10th March 2023
Abstract
The study of enzymes by direct electrochemistry has been extended to enzyme cascades, with a key development being the ‘electrochemical leaf’: an electroactive enzyme is immobilized within a porous electrode, providing in situ cofactor (NADP(H)) regeneration for a co-immobilized downstream enzyme. This system has been further developed to include multiple downstream enzymes, and it has become an important tool in biocatalysis, however, the local environment within the porous electrode has not been investigated in detail. Here, we constructed a 1D reaction–diffusion model, comprising the porous electrode with 2 kinds of enzymes immobilized, and an enzyme-free electrolyte diffusion layer. The modelling results show that the rate of the downstream enzyme is a key parameter, and that substrate transport within the porous electrode is not a main limiting factor. The insights obtained from this model can guide future rational design and improvement of these electrodes and immobilized enzyme cascade systems.
Introduction
In biological systems, cascades with many steps and components are confined together in small volumes, leading to higher efficiencies due to shorter diffusion distances for reaction intermediates. Inspired by biological cascades, the ‘electrochemical leaf’ is a recently developed platform technology: an electron-driven, nanoconfined enzymatic cascade, immobilized within a porous indium tin oxide (ITO) electrode.1–4 The key enzyme is ferredoxin-NADP+ reductase (FNR), a component of the photosynthesis cascade. When FNR is directly in contact with a suitable electrode surface, it is electroactive, able to quasi-reversibly catalyse the 2e− interconversion of NADP+/NADPH, a recyclable redox cofactor. When another NADP(H)-dependent enzyme is co-immobilized into the same electrode pores, this leads to a significant enhancement in the measured current.The electrochemical leaf has been demonstrated to work with various downstream enzymes, in H2 or light-driven systems,5,6 for large-scale biosynthesis,7 or even extended to a 4 or 5-enzyme cascade.8,9 A key attribute of the system is that all enzyme components are confined within the same porous volume, with pore sizes on the scale of 5–100 nm. However, the local environment within the porous electrode is largely unexplored, and rational design of such a nanoconfined cascade electrode cannot be undertaken without understanding the effects of electrode structure (porosity, thickness, etc.) and enzyme distribution.
Reaction–diffusion modelling is a common tool used to elucidate and understand the relationships between mass transport, the local environment, and the reaction rate for a variety of reactions and geometries. For example, in electrochemical CO2 reduction, the local pH and CO2 concentration at the electrode surface are important parameters, and modelling has demonstrated that they can differ significantly from the bulk values at high currents.10,11 Reaction–diffusion modelling has also been previously used to investigate enzyme-immobilized electrodes,12,13 but these studies mainly focussed on the activity of single enzymes (hydrogenases, formate dehydrogenases) and their interactions with buffering species or the local CO2 supply.
Classical electrochemistry with a coupled homogeneous reaction is well-known (i.e. the EC′, so-called ‘catalytic’ mechanism),14 but the electrochemical leaf which is a porous electrode with all components immobilized (i.e. no bulk homogeneous reaction) presents distinct differences. The current–voltage behaviour of an enzyme in porous electrodes has been investigated by modelling, but only for a single enzyme (with no coupled downstream reaction) where nanoconfinement may have no significance.15 In this paper, we use reaction–diffusion modelling to calculate concentration profiles for a porous electrode with multiple enzymes immobilized, and investigate the effects of various parameters such as the downstream reaction rate and electrode porosity. These modelling results provide guidelines for future design and development of the electrochemical leaf system towards practical-scale current densities.
Simulation methods
1. Components and geometry
Here we adopt a 1D geometry with the x-direction normal to the electrode surface, with an electrode layer possessing a given thickness (L) and porosity (ε), and the adjoining layer of electrolyte, referred to as the diffusion layer (with thickness δ). A scheme of the model is shown in Fig. 1. We assume this diffusion layer is stagnant (i.e. no convection), and the composition at the boundary with the well-mixed bulk electrolyte is pinned to the bulk values.16 The thickness of the diffusion layer can vary greatly, depending on the cell and electrode geometry, and flow/stirring conditions, in the range of 100–500 μm for a typical flat electrode in a stirred batch cell. Within the porous layer, two kinds of enzymes are immobilized (i.e. no enzymes are present in the diffusion layer or the bulk solution), uniformly dispersed throughout the electrode. One enzyme is the electroactive enzyme (FNR in the electrochemical leaf, denoted as E1 here for a generic model), and here we consider the reduction reaction (E1 consumes 2 electrons to reduce NADP+ to NADPH) as follows:| NADP+ + 2e− + H+ → NADPH with rate R1 | (Reaction 1) |
| S + NADPH + H+ → P + NADP+ with rate R2 | (Reaction 2) |
 | ||
| Fig. 1 Scheme of the 1D-model of a porous electrode, with two kinds of immobilized enzymes (E1 the electroactive NADP(H)-regeneration enzyme, and E2 the NADP(H)-dependent downstream enzyme). The electrode thickness and diffusion layer thickness are not shown to scale. The substrate is transported from the bulk by diffusion to the electrode layer, where both the S → P and NADP(H) recycling reactions occur. | ||
2. Governing equations and boundary conditions
We consider 5 chemical species within the electrolyte-filled portion of the porous electrode and the diffusion layer: NADP+, NADPH, E2 substrate (S), E2 product (P), and H+. As the reaction is usually conducted with a large concentration of supporting electrolyte, we assume migration to be negligible, and the only the processes affecting the concentration profiles are reaction and diffusion.16




Outside the electrode layer, the rates R1 and R2 are set to zero, since both E1 and E2 are present only within the electrode.
Within the electrode layer, the local electrochemical rate of E1 is assumed to follow the Butler–Volmer equation,16 which at sufficient overpotential reduces to the Tafel equation. The rate is first-order with respect to [NADP+], since the reaction occurs at [NADP+] values well below Km. At each position within the electrode, the current is assumed to apply uniformly to the proportion of the electrode that is occupied by electrolyte,11 therefore the rate R1 is

The rate of E2 (R2) is assumed to follow Michaelis–Menten kinetics for 2 substrates:20

The boundary at the back contact of the electrode (i.e. the deeply-buried end, farthest away from the solution) is set to be the no flux condition. At the boundary of the diffusion layer and bulk electrolyte, all species are set to the same concentration as the bulk.
The simulation was conducted in COMSOL 5.0 using the Coefficient Form PDE interface, and the simulation was run for 20 s, at which the model is considered to have reached steady state (the concentration of each species changed by less than 0.3% in the final time step).
3. Modelling parameters
Table 1 shows the diffusion coefficients of the various species used in the model. The diffusion coefficients for NAD(H) and NADP(H) should be similar, and a range of values has been reported in literature (2.0–6.7 × 10−10 m2 s−1).21–25 Here we use the value obtained from previous electrochemical measurements2 which is close to the midpoint of this range. The diffusion coefficients of organic substrates and products in water can vary greatly (∼3–15 × 10−10 m2 s−1),26 here we have taken a representative value (7.5 × 10−10 m2 s−1). The exact values do not significantly affect the trends in results reported here, with the only key value affected being the mass-transport limited current, which scales linearly with the diffusion coefficient (see Results below for explanation).| Species | Value |
|---|---|
| NADP+ | 4.2 × 10−10 |
| NADPH | 4.2 × 10−10 |
| S | 7.5 × 10−10 |
| P | 7.5 × 10−10 |
| H + | 9.3 × 10−9 |
Transport properties within porous materials can differ from their bulk values, depending on the porosity (defined as the void volume fraction of the electrode) and tortuosity of the porous media (the ratio of an effective path length to the edge length), and this can be understood as diffusion becoming more difficult due to the longer path length and smaller cross-section area for transport.27–29 Here we applied a commonly-used model, the Bruggeman correction, which for random blocking spheres is as follows:

| Parameter | Value | Units |
|---|---|---|
| Electrode layer thickness (L) | 5 | μm |
| Electrode porosity (ε) | 0.5 | — |
| Diffusion layer thickness (δ) | 100 | μm |
| E1 heterogeneous electrochemical rate constant | 1 × 10−4 | m s−1 |
| Bulk E2 substrate (S) concentration | 10 | mM |
| Bulk E2 product (P) concentration | 0 | mM |
| E2 concentration within the electrode | 1 | mM |
| E2 Ki,S | 1 | mM |
| E2 Km,S | 1 | mM |
| E2 Km,NADPH | 1 | mM |
| Temperature (T) | 298 | K |
Other parameters are listed in Table 2. Unless stated otherwise, the electrode thickness was set to 5 μm and the porosity set to 0.5, which are typical for a porous ITO electrode used in previous experimental work. The temperature was set to typical room temperature (298 K), and electrolyte effects were not considered.
Results
First, to demonstrate that the model is a good representation of this electrochemical cascade system, we simulated the current density at steady state (taken to be after 20 s) as a function of applied potential, for a range of heterogeneous rate electrochemical constants (k0) for electron transfer to E1 (Fig. 2). This is equivalent to a linear sweep voltammetry at very slow scan rates, and the current is plotted as negative in this figure for ease of comparison with typical voltammograms that use the convention of reduction current as negative. The coupled enzyme E2 is present only within the electrode layer (at a concentration of 1 mM), and the cofactor NADP+ is present in the bulk at 10 μM, a typical concentration used in previous reports (which means the initial NADP+ concentration throughout the electrode layer and electrolyte diffusion layer is also 10 μM). With a low heterogeneous electrochemical rate constant, a distinct overpotential requirement can be observed, while at higher heterogeneous rate constants the currents quickly reach a plateau, where the rate of NADP+ consumption by E1 (measured as the current) is limited by the maximal rate of NADP+ generation from E2 turnover under these conditions. This modelled current–voltage relationship (a current onset, followed by a coupled reaction-limited plateau) agrees qualitatively with the previous experimental results,2,3 as well as with the voltammogram of an EC′ or ‘catalytic’ mechanism from electrochemical theory.14 In the following results, the applied potential is fixed at −0.3 V vs the formal potential of NADP+/NADPH (E0), which is a sufficiently large overpotential, and the heterogeneous electrochemical rate constant for E1 is fixed at 1 × 10−4 m s−1, since the regime of interest is where the E1 is not the limiting component. | ||
| Fig. 2 (A) Steady state current density as a function of applied potential (equivalent to slow-scan linear sweep voltammetry) with various heterogeneous electrochemical rate constants. (B) Concentration profiles of NADP+, NADPH, and the substrate and product of E2 throughout the simulated region. The electrode layer is 5 μm, on the left side, and the bulk electrolyte is towards the right side. Simulation conditions: porosity 0.5, [NADP+] 10 μM, E2 kcat 1 × 102 s−1. | ||
Fig. 2B shows an example concentration profile of NADP+, NADPH, and the substrate (S) and product (P) of E2 (with the E2 kcat of 1 × 102 s−1). The bulk concentration of NADP+ and the E2 substrate were 10 μM and 10 mM, respectively. Under cathodic operation, the NADPH concentration is maintained at a high level within the pores, since the E1 is reducing NADP+ to NADPH. Under these conditions, although E2 is actively turning over, the E2 substrate is still maintained at a relatively high concentration within the pores, but if the E2 rate is sufficiently increased (see below), E2 substrate supply will become limiting.
Next, we demonstrate the importance of the downstream enzyme E2 in increasing the achievable current density. When there was no E2 present (Fig. 3A, grey trace), the steady-state current is determined by transport of NADP+ from the bulk. With E2 present in the porous layer (Fig. 3A, dark to light red traces, with darker shades signifying higher kcat of E2, all at a constant E2 concentration of 1 mM within the electrode) the reaction of E2 consumes NADPH and produces NADP+, thus increasing the flux of NADP+ for E1 to reduce, resulting in an enhanced current. (In this model, increasing the E2 rate at a constant E2 loading is equivalent to increasing the loading at a constant E2 rate, as the rate of Reaction (2) is governed by the product of these two parameters). This current enhancement is observable even down to [NADP+] of a few μM, which is consistent with experimental results that achieved a significant coupled current in the presence of a downstream enzyme.1,2 At higher [NADP+], the current reaches a transport-limited plateau (of E2 substrate from the bulk to the electrode outer surface), and at the highest [NADP+] values (∼10 mM, i.e. when the bulk [NADP+] is on the same order as the E2 substrate concentration) this plateau is exceeded due to the addition of the current from reduction of bulk [NADP+].
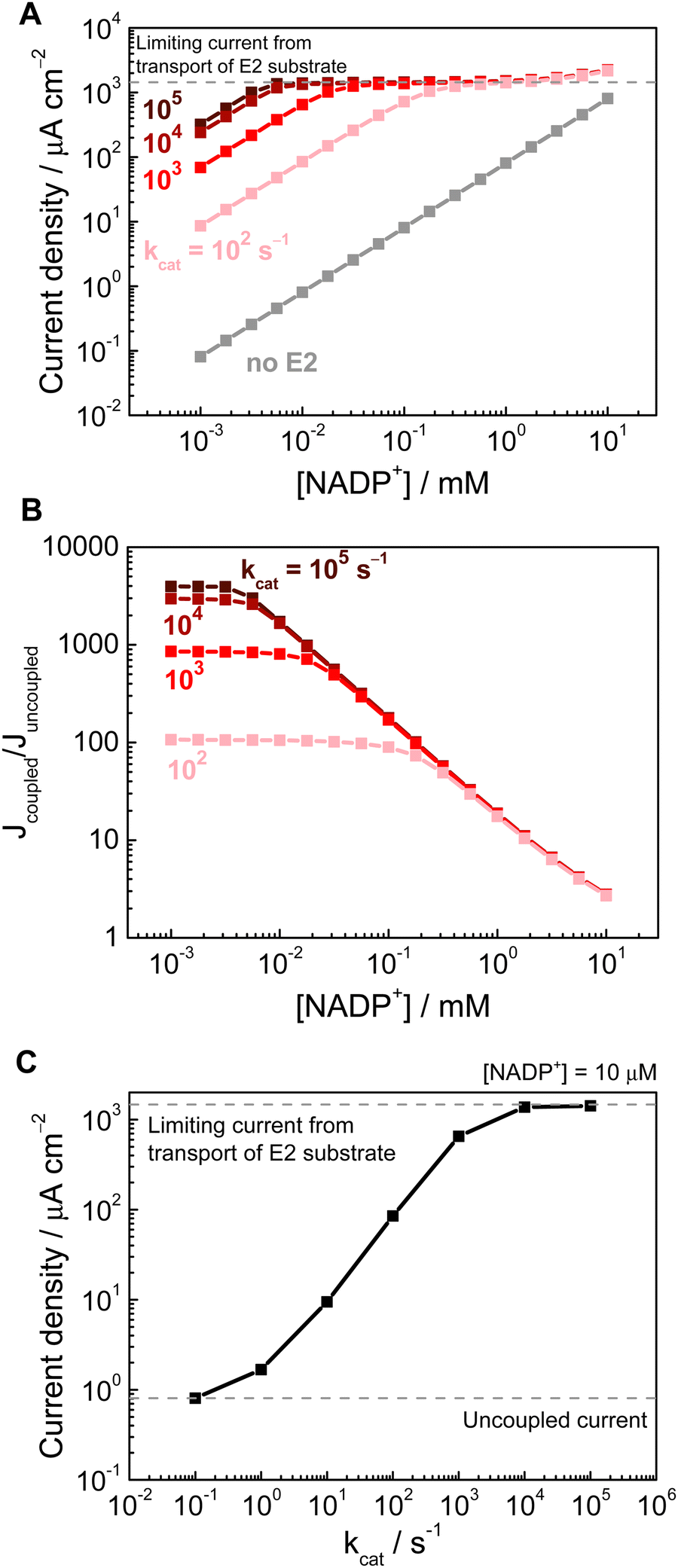 | ||
| Fig. 3 (A) Current density of the porous electrode with only E1 (uncoupled, grey trace), and with E1 and E2 both present (coupled, dark to light red traces), as a function of [NADP+]. (B) The current enhancement factor (the ratio of the coupled and uncoupled currents) as a function of [NADP+]. (C) Current density as a function of E2 rate (kcat) at constant [NADP+] 10 μM. Simulation conditions: electrode thickness 5 μm, porosity 0.5, E2 loading 1 mM, applied potential −0.3 V vs. E0. | ||
Using the concept of a Nernst diffusion layer, the transport-limited current can be estimated from

The increase in current with an E2 present can be quantified as the enhancement factor, i.e. the ratio of the currents with and without the coupled reaction (Fig. 3B). This current enhancement can be compared to a recent experimental work that demonstrated an enhancement factor of ∼25 at [NADP+] ∼50 μM.31 The results here show that the coupled current does increase with higher [NADP+] (by >100–1000 times compared to the uncoupled current, depending on the E2 activity), but at higher [NADP+] (>100 μM) the contribution from the coupled current can become obscured by the bulk supply of [NADP+], resulting in markedly smaller enhancement factors. In the context of a cofactor recycling system, there is a need to balance the requirement for higher cofactor turnover number (i.e. lower cofactor concentration) but still maintain a high rate (i.e. higher current). The actual numbers may vary slightly with different simulation parameters, but trends obtained here demonstrates that modelling can provide a helpful guide for reaction engineering, and that using cofactors at mM scale concentration is inefficient since this offers limited enhancement in rate. Although the present model is a simplified 1D model, with no accounting for pH, temperature, and electrolyte effects, it did capture the current enhancement effect from pore-confined cofactor recycling. Future work will involve extending the model to more detailed 2D and 3D geometries, as well as incorporating additional reaction condition parameters.
Fig. 3C shows the effect of the E2 rate (kcat), at a constant E2 loading, and [NADP+] of 10 μM. The slope of ∼0.9 on a log–log plot, for the intermediate kcat values (between 1 and 1000) indicates that the current scales linearly with the overall rate of E2 reaction. At higher E2 rates, the current plateaus due to E2 substrate transport as explained above. This result underscores the importance of enhancing the coupled enzyme rates, and that increasing the E2 loading is key in achieving high performance. In line with this modelled trend, previous experimental results using an alcohol dehydrogenase as the E2 showed that the steady-state current increased with concentration of E2 used in the loading solution.3
One important question is the distribution of reaction rates within the porous electrode layer. In experimental work, the enzymes are usually loaded onto the porous electrode by dropcasting a concentrated drop of enzyme solution, then the excess enzyme is rinsed off. Therefore, it is reasonable to assume a uniform concentration of enzymes within the electrode. Do deeply-buried downstream enzymes (E2) participate to the same extent as enzymes located near the electrode surface? Fig. 4 shows the local rate of the E2 reaction, as a function of the position within the electrode layer (with 0 being the deeply-buried back contact, and 1 being the outermost surface adjoining the electrolyte diffusion layer). At lower E2 rates, all of the E2 are turning over at a uniform rate, regardless of the position within the electrode. The local concentration of NADPH is maintained at a high level, and a substantial concentration of the E2 substrate remains available throughout the electrode. Only at the highest E2 rate, where the total current density reaches the E2 substrate transport limit, the substrate becomes less available within the deeper pores, leading to lowered E2 rates.
 | ||
| Fig. 4 Distribution of E2 rates within the electrode layer, at various E2 rates. Simulation conditions: electrode thickness 5 μm, porosity 0.5, [NADP+] 10 μM, applied potential −0.3 V vs. E0. | ||
Another possibly significant parameter is the porosity of the electrode. Fig. 5 shows the dependence of the current density on the porosity (from 0.4–0.8), at different rates of E2 while holding the E2 concentration constant. (In practice, the porosity is not completely independent of enzyme loading, as higher porosity should provide a higher internal surface area for enzymes to adsorb, but here for ease of comparison the loading is set to be constant). Regardless of the E2 rate, the porosity has a very small effect on the achievable current density (the current increase is <5% when comparing lowest and highest porosity). This is consistent with the results from Fig. 4, which indicated that substrate transport within the electrode is not a main limiting factor.
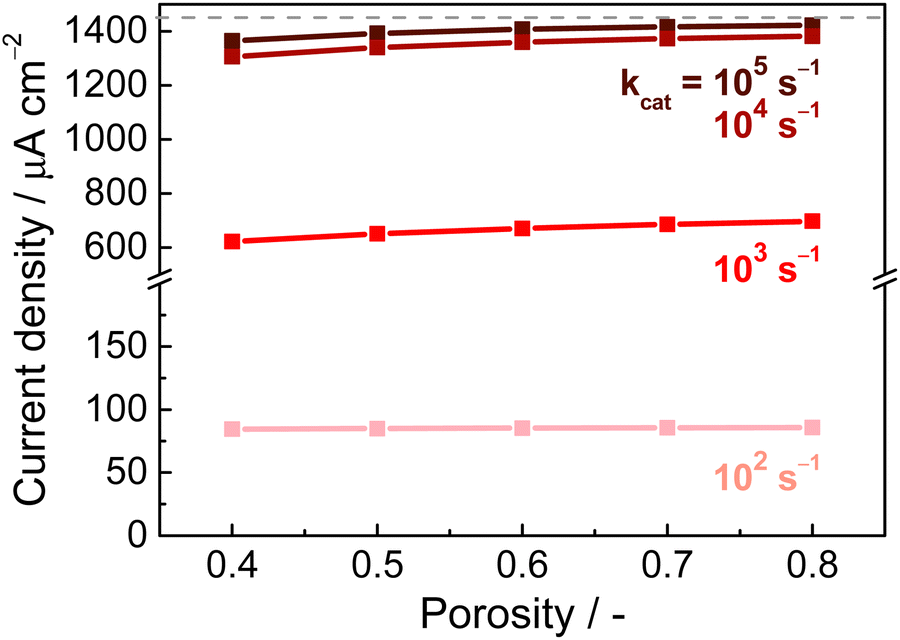 | ||
| Fig. 5 Effect of porosity on current density, at various E2 rates. Simulation conditions: electrode thickness 5 μm, porosity 0.5, [NADP+] 10 μM, applied potential −0.3 V vs. E0. | ||
From our reaction–diffusion model, we can see that the ultimate limiting factor is the transport of the E2 substrate from the bulk to the electrode surface, but experimental results to date are still well below this limit. In this regime, transport within the electrode is not limiting, the E2 substrate concentration is maintained in the pores, and the key limiting parameter is the total E2 rate within the electrode. Based on this insight, we can predict that thicker electrodes (i.e. overall increased enzyme loading, therefore increased total E2 rates) will achieve higher currents. Looking towards the future, when the transport limit is reached (for example by increasing the E2 loading significantly), how do we break this limitation? At that point, it would be valuable to redesign the cell, using a flow system, to achieve a smaller diffusion layer thickness. For example, a CO2 electrolyzer with forced catholyte flow-through with a porous catalyst layer was able to achieve unprecedented current densities (>3 A cm−2), attributed partly to diffusion thicknesses on the scale of 1 μm.32
Conclusions
We have constructed a reaction–diffusion model to investigate a porous electrode containing an immobilized enzyme cascade with cofactor recycling. When operating below the substrate transport-limited regime, the recycling of the cofactor within the pores enhances the achievable current density, with a trade-off between higher cofactor turnover and higher currents. The modelled trends are consistent with previous experimental results, and provides a guide for future improvements of this system. The most important parameter is the total rate of the downstream enzyme, which could be increased by either using E2 with higher intrinsic rates, or increasing the E2 loading. Substrate transport within the pores is not limiting, therefore deeper-buried enzymes still operate at a similar extent as the shallower ones, until the E2 substrate transport-limited regime is reached. The porosity of the electrode has marginal direct effects on the current, but may have indirect effects in increasing enzyme loading. When approaching the substrate transport-limited regime, the diffusion layer thickness is key, and further improvements will depend on cell and electrode design to minimize the diffusion layer thickness.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The author thanks Prof. Fraser A. Armstrong and Dr. Clare F. Megarity for valuable discussions. This work was partly funded by the Early Career Researchers and Returners Fund (University of Liverpool).References
- F. A. Armstrong, B. Cheng, R. A. Herold, C. F. Megarity and B. Siritanaratkul, From Protein Film Electrochemistry to Nanoconfined Enzyme Cascades and the Electrochemical Leaf, Chem. Rev., 2022 DOI:10.1021/acs.chemrev.2c00397.
- B. Siritanaratkul, C. F. Megarity, T. G. Roberts, T. O. M. Samuels, M. Winkler, J. H. Warner, T. Happe and F. A. Armstrong, Transfer of photosynthetic NADP+/NADPH recycling activity to a porous metal oxide for highly specific, electrochemically-driven organic synthesis, Chem. Sci., 2017, 8(6), 4579–4586 RSC.
- C. F. Megarity, B. Siritanaratkul, R. S. Heath, L. Wan, G. Morello, S. R. FitzPatrick, R. L. Booth, A. J. Sills, A. W. Robertson, J. H. Warner, N. J. Turner and F. A. Armstrong, Electrocatalytic Volleyball: Rapid Nanoconfined Nicotinamide Cycling for Organic Synthesis in Electrode Pores, Angew. Chem., Int. Ed., 2019, 58(15), 4948–4952 CrossRef CAS PubMed.
- C. F. Megarity, B. Siritanaratkul, B. Cheng, G. Morello, L. Wan, A. J. Sills, R. S. Heath, N. J. Turner and F. A. Armstrong, Electrified Nanoconfined Biocatalysis with Rapid Cofactor Recycling, ChemCatChem, 2019, 11(23), 5662–5670 CrossRef CAS.
- L. Wan, C. F. Megarity, B. Siritanaratkul and F. A. Armstrong, A hydrogen fuel cell for rapid, enzyme-catalysed organic synthesis with continuous monitoring, Chem. Commun., 2018, 54(8), 972–975 RSC.
- G. Morello, B. Siritanaratkul, C. F. Megarity and F. A. Armstrong, Efficient Electrocatalytic CO2 Fixation by Nanoconfined Enzymes via a C3-to-C4 Reaction That Is Favored over H2 Production, ACS Catal., 2019, 9(12), 11255–11262 CrossRef CAS.
- B. Cheng, L. Wan and F. A. Armstrong, Progress in Scaling up and Streamlining a Nanoconfined, Enzyme-Catalyzed Electrochemical Nicotinamide Recycling System for Biocatalytic Synthesis, ChemElectroChem, 2020, 7(22), 4672–4678 CrossRef CAS PubMed.
- C. F. Megarity, T. R. I. Weald, R. S. Heath, N. J. Turner and F. A. Armstrong, A Nanoconfined Four-Enzyme Cascade Simultaneously Driven by Electrical and Chemical Energy, with Built-in Rapid, Confocal Recycling of NADP(H) and ATP, ACS Catal., 2022, 12(15), 8811–8821 CrossRef CAS PubMed.
- G. Morello, C. F. Megarity and F. A. Armstrong, The power of electrified nanoconfinement for energising, controlling and observing long enzyme cascades, Nat. Commun., 2021, 12(1), 340 CrossRef CAS PubMed.
- L.-C. Weng, A. T. Bell and A. Z. Weber, Modeling gas-diffusion electrodes for CO2 reduction, Phys. Chem. Chem. Phys., 2018, 20(25), 16973–16984 RSC.
- T. Burdyny and W. A. Smith, CO2 reduction on gas-diffusion electrodes and why catalytic performance must be assessed at commercially-relevant conditions, Energy Environ. Sci., 2019, 12(5), 1442–1453 RSC.
- E. Edwardes Moore, J. Cobb Samuel, M. Coito Ana, R. Oliveira Ana, A. C. Pereira Inês and E. Reisner, Understanding the local chemical environment of bioelectrocatalysis, Proc. Natl. Acad. Sci. U. S. A., 2022, 119(4), e2114097119 CrossRef PubMed.
- S. J. Cobb, V. M. Badiani, A. M. Dharani, A. Wagner, S. Zacarias, A. R. Oliveira, I. A. C. Pereira and E. Reisner, Fast CO2 hydration kinetics impair heterogeneous but improve enzymatic CO2 reduction catalysis, Nat. Chem., 2022, 14(4), 417–424 CrossRef CAS PubMed.
- A. J. Bard and L. R. Faulkner, Electrochemical Methods, Wiley, 2nd edn, 2001 Search PubMed.
- H. Le and R. G. Compton, Electrochemical processes mediated via adsorbed Enzymes: Flat and porous electrodes Compared. Understanding Nano-confinement, J. Electroanal. Chem., 2021, 895, 115448 CrossRef CAS.
- R. G. Compton, E. Kätelhön, K. R. Ward and E. Laborda, Understanding voltammetry: simulation of electrode processes, World Scientific, 2014 Search PubMed.
- K. Nakamura, R. Yamanaka, T. Matsuda and T. Harada, Recent developments in asymmetric reduction of ketones with biocatalysts, Tetrahedron: Asymmetry, 2003, 14(18), 2659–2681 CrossRef CAS.
- C. W. Bradshaw, W. Hummel and C. H. Wong, Lactobacillus kefir alcohol dehydrogenase: a useful catalyst for synthesis, J. Org. Chem., 1992, 57(5), 1532–1536 CrossRef CAS.
- J. An, Y. Nie and Y. Xu, Structural insights into alcohol dehydrogenases catalyzing asymmetric reductions, Crit. Rev. Biotechnol., 2019, 39(3), 366–379 CrossRef CAS PubMed.
- A. Cornish-Bowden, Fundamentals of enzyme kinetics, John Wiley & Sons, 2013 Search PubMed.
- J. Moiroux and P. J. Elving, Mechanistic aspects of the electrochemical oxidation of dihydronicotinamide adenine dinucleotide (NADH), J. Am. Chem. Soc., 1980, 102(21), 6533–6538 CrossRef CAS.
- A. Domenech, E. Garcia-Espana, J. A. Ramirez, B. Celda, M. Carmen Martinez, D. Monleon, R. Tejero, A. Bencini and A. Bianchi, A thermodynamic, electrochemical and molecular dynamics study on NAD and NADP recognition by 1,4,7,10,13,16,19-heptaazacyclohenicosane ([21]aneN7) [dagger], J. Chem. Soc., Perkin Trans. 2, 1999, 23–32 RSC.
- Z. Wu, W. Jing and E. Wang, [JW1]Oxidation of NADH by dopamine incorporated in lipid film cast onto a glassy carbon electrode, Electrochem. Commun., 1999, 1(11), 545–549 CrossRef CAS.
- C. E. Banks and R. G. Compton, Exploring the electrocatalytic sites of carbon nanotubes for NADH detection: an edge plane pyrolytic graphite electrode study, Analyst, 2005, 130(9), 1232–1239 RSC.
- S. Prasannakumar, R. Manjunatha, C. Nethravathi, G. S. Suresh, M. Rajamathi and T. V. Venkatesha, Graphene-carbon nanotubes modified graphite electrode for the determination of nicotinamide adenine dinucleotide and fabrication of alcohol biosensor, J. Solid State Electrochem., 2012, 16(10), 3189–3199 CrossRef CAS.
- W. M. Haynes, D. R. Lide and T. J. Bruno, CRC handbook of chemistry and physics, CRC press, 2016 Search PubMed.
- B. Tjaden, S. J. Cooper, D. J. L. Brett, D. Kramer and P. R. Shearing, On the origin and application of the Bruggeman correlation for analysing transport phenomena in electrochemical systems, Curr. Opin. Chem. Eng., 2016, 12, 44–51 CrossRef.
- B. Tjaden, D. J. L. Brett and P. R. Shearing, Tortuosity in electrochemical devices: a review of calculation approaches, Int. Mater. Rev., 2018, 63(2), 47–67 CrossRef CAS.
- L. Shen and Z. Chen, Critical review of the impact of tortuosity on diffusion, Chem. Eng. Sci., 2007, 62(14), 3748–3755 CrossRef CAS.
- H. H. Heenen, H. Shin, G. Kastlunger, S. Overa, J. A. Gauthier, F. Jiao and K. Chan, The mechanism for acetate formation in electrochemical CO(2) reduction on Cu: selectivity with potential, pH, and nanostructuring, Energy Environ. Sci., 2022, 15(9), 3978–3990 RSC.
- R. A. Herold, R. Reinbold, C. J. Schofield and F. A. Armstrong, NADP(H)-dependent biocatalysis without adding NADP(H), Proc. Natl. Acad. Sci. U. S. A., 2023, 120(1), e2214123120 CrossRef CAS PubMed.
- G. Wen, B. Ren, X. Wang, D. Luo, H. Dou, Y. Zheng, R. Gao, J. Gostick, A. Yu and Z. Chen, Continuous CO2 electrolysis using a CO2 exsolution-induced flow cell, Nat. Energy, 2022, 7, 978–988 CrossRef CAS.
| This journal is © the Owner Societies 2023 |