
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceExciton diffusion in poly(3-hexylthiophene) by first-principles molecular dynamics†
Cheick Oumar
Diarra
 a,
Mauro
Boero
b,
Emilie
Steveler
a,
Thomas
Heiser
a and
Evelyne
Martin
*a
a,
Mauro
Boero
b,
Emilie
Steveler
a,
Thomas
Heiser
a and
Evelyne
Martin
*a
aUniversité de Strasbourg, CNRS, Laboratoire ICube, UMR 7357, F-67037 Strasbourg, France. E-mail: evelyne.martin@unistra.fr
bUniversité de Strasbourg, CNRS, Institut de Physique et Chimie des Matériaux de Strasbourg, UMR 7504, Strasbourg F-67034, France
First published on 15th May 2023
Abstract
Poly(3-hexylthiophene) (P3HT) is a polymer used in organic solar cells as a light absorber and an electron donor. Photogenerated excitons diffuse and dissociate into free charge carriers provided they reach the absorber boundaries. The device efficiency is therefore dependent on the exciton diffusion. Although measurements can be performed for example by time-resolved photoluminescence, a quantitative modeling is highly desirable to get an insight into the relationship between the atomic structure at finite temperature and the diffusion coefficient of the exciton. This is the objective of the present work, achieved by resorting to first-principles molecular dynamics in combination with the restricted open-shell approach to model the singlet excited state. The maximally localized Wannier functions and their centers are used to monitor and localize the electron and the hole along the dynamics. The resulting diffusion coefficient is in close agreement with available measurements.
1. Introduction
The use of organic materials such as polymers or small molecules in solar cells enables the reduction of the fabrication cost and realization of flexible modules. By changing the constituent molecules, a higher tunability than the one provided by inorganic materials is possible, and recently efficiencies close to 20% have been obtained with polymer donors and non-fullerene acceptors, with record values for ternary bulk-heterojunction (BHJ)1 and solid additive-treated BHJ2 devices. The active layers of BHJ solar cells are blends of two families of organic materials, electron donors and electron acceptors. When light is absorbed, an exciton is generated and diffuses. At the donor/acceptor interface, the exciton dissociates into a free electron and a hole, which are subsequently collected to generate an electrical current. However if the diffusion length of the exciton is too short to reach the interface, the exciton undergoes recombination before being able to generate any current, thus not contributing to any energy harvesting. Our goal is to provide a guideline from atomic scale modeling for the rational design of organic materials with the highest possible exciton diffusion length.The exciton diffusion length is expressed as  , where D is the diffusion coefficient and τ is the exciton lifetime. In the present work, an important step toward our ultimate goal (quantify L) is made by focusing on the diffusion coefficient D. The exciton diffusion coefficient can be measured experimentally, for instance by time resolved photoluminescence.3 From a theoretical standpoint, the Frenkel–Holstein Hamiltonian scheme has been applied to various organic polymers4 and more recently to 2D systems.5 First-principles molecular dynamics (FPMD) can provide alternatively a valuable atomic-scale picture of the dynamical process at finite temperature and link the atomic and electronic structures to the efficiency of a diffusion process. Vibronic effects are included a priori and do not need to be added, while inclusion of ad hoc contributions from intermolecular charge transfer6 is not necessary. Moreover, our dynamical approach being based on density-functional theory (DFT) as in any FPMD scheme, possible couplings between atomic and electronic motions are intrinsically accounted for, thus including the cases of trapping and polarons implying coupling with atomic vibrations. The assessment of the degree of the predictive power of a computational scheme is a stringent comparison with the available experimental data. To this aim, we focus on poly(3-hexylthiophene) (P3HT), a polymer widely used in BHJ solar cells. The structure of P3HT has been extensively characterized,7,8 and the exciton diffusion coefficient has been measured in the crystalline domains (see Table 2).9
, where D is the diffusion coefficient and τ is the exciton lifetime. In the present work, an important step toward our ultimate goal (quantify L) is made by focusing on the diffusion coefficient D. The exciton diffusion coefficient can be measured experimentally, for instance by time resolved photoluminescence.3 From a theoretical standpoint, the Frenkel–Holstein Hamiltonian scheme has been applied to various organic polymers4 and more recently to 2D systems.5 First-principles molecular dynamics (FPMD) can provide alternatively a valuable atomic-scale picture of the dynamical process at finite temperature and link the atomic and electronic structures to the efficiency of a diffusion process. Vibronic effects are included a priori and do not need to be added, while inclusion of ad hoc contributions from intermolecular charge transfer6 is not necessary. Moreover, our dynamical approach being based on density-functional theory (DFT) as in any FPMD scheme, possible couplings between atomic and electronic motions are intrinsically accounted for, thus including the cases of trapping and polarons implying coupling with atomic vibrations. The assessment of the degree of the predictive power of a computational scheme is a stringent comparison with the available experimental data. To this aim, we focus on poly(3-hexylthiophene) (P3HT), a polymer widely used in BHJ solar cells. The structure of P3HT has been extensively characterized,7,8 and the exciton diffusion coefficient has been measured in the crystalline domains (see Table 2).9
The structure of P3HT (at T = 0 K) is presented in Fig. 1 (at the top). In this figure, the structures are relaxed within the framework of DFT for the calculation of the electronic structure as described below. The P3HT polymer is usually modeled by a segment containing a finite number of thiophenes and methyl groups instead of the hexyl chains, to reduce the computational workload. Penta(3-methylthiophene) (penta3MT) (Fig. 1, bottom) is an example of such molecules and was chosen by Schwermann et al.10 to study the exciton diffusion. At room temperature, however, penta3MT molecules fold and the heterocycles at the extremities are free to rotate (see Fig. 2). This conformational change complicates the observation of an exciton transfer between molecules. To overcome this difficulty, Schwermann et al.10 used a restraining potential to obtain the free energy of the exciton transfer as a function of the distance between two molecules.
 | ||
| Fig. 1 Atomic models of the polymers P3HT and P3MT and of the molecule of penta3MT. Carbon atoms are represented in brown, sulfur in yellow, and hydrogen in white. The simulation cell boundaries are indicated by dashed red lines. | ||
 | ||
| Fig. 2 Representative snapshot of the structure at 300 K of the penta3MT molecule shown in Fig. 1. The molecule underwent a rotation along the x axis, a bending, and one thiophene group at the extremity of the molecule rotated (black arrow). These conformational changes break the alternation in the thiophene orientation. | ||
In the present work, we propose an alternative modeling strategy relying on the use of periodic boundary conditions to model a virtually infinite polymer instead of a small molecule. At finite temperature, we show that the polymer keeps a crystalline arrangement, with a structure that depends on the length of the lateral chains, hexyl in P3HT or methyl in poly(3-methylthiophene) (see Fig. 1, middle). The type of structure (conformers) and the lattice parameters obtained by our simulation results are in good agreement with known experimental structures, specifically Form I of P3HT; this specific structure is the thermodynamically stable one generally obtained during the fabrication of organic solar cells, and the planar configuration is the one resulting upon doping (see Fig. 3). Concerning the diffusion coefficient D, we track the exciton by calculating the maximally localized Wannier functions and related Wannier centers11 of the bound electrons and holes. The exciton diffusion coefficient is estimated by computing the mean square displacement using the generated trajectory. In this way, our procedure is free from any hypothesis regarding the mechanism of the exciton transfer, for example, Förster12 or Dexter.13 The resulting diffusion coefficient is in quantitative agreement with the measurements.
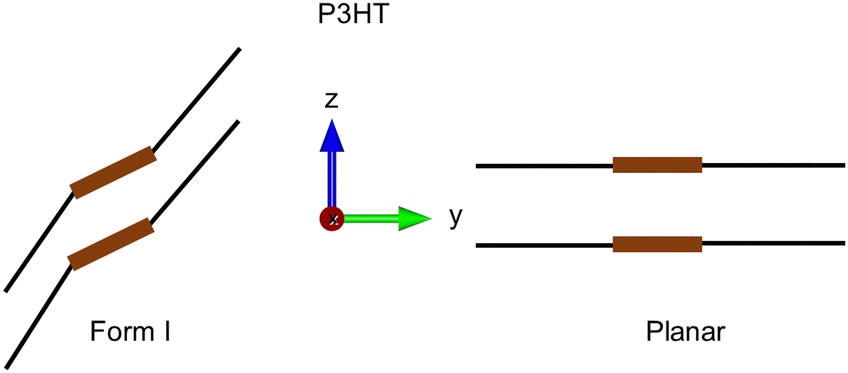 | ||
| Fig. 3 Schematic representation of the crystalline structures of P3HT: Form I in pristine P3HT7 (left side) and planar geometry of doped P3HT14 (right side). | ||
The paper is organized as follows. In Section 2, we provide a description of the first-principles molecular dynamics method applied to describe the first excited singlet state. Section 3 presents the approach used to localise the exciton via the Wannier centers, with an illustration in the case of the penta3MT molecule. Section 4 and Section 5 are focused on the exciton diffusion in P3MT and P3HT, respectively. Conclusions are drawn in the final Section 6.
2. Computational method
Molecular dynamics (MD) simulations are performed using the Car–Parrinello (CP) method15 as implemented in the developers version 4.3 of the CPMD code.16 We select for the exchange–correlation part of the Kohn–Sham total energy, the formulation proposed by Perdew, Burke and Ernzerhof (PBE).17 The valence-core interactions are described by norm-conserving Troullier–Martins18 pseudopotentials. Valence electrons are represented on a plane-wave basis set with a cutoff of 70 Ry, and the Brillouin zone sampling is limited to the Γ point. A fictitious electron mass of 400 a.u. and a time step Δt = 0.1 fs (4 a.u.) are chosen to ensure optimal conservation of the constants of motion. The ionic temperature is controlled with a Nosé–Hoover19–21 thermostat chain,22 whereas for the fictitious electronic kinetic energy we use a Blöchl–Parrinello thermostat.23 Masses of the thermostats (in units of corresponding frequencies) are taken equal to 2500 cm−1 for the ions and 5000 cm−1 for the fictitious electronic degrees of freedom.Excited-state processes in organic molecules most often proceed in the lowest excited singlet (S1) or triplet (T1) states. Higher excited states have short lifetimes24–26 and undergo radiationless relaxation to S1, as per Kasha's rule.27 Therefore, in our study we only considered the S1 state, which can be described by the restricted open-shell Kohn–Sham (ROKS) method. The ROKS28–30 formalism has been successfully applied for the modeling of photoreactions31 and carefully benchmarked on experimental data.32 Within the ROKS, the exciton remains in the S1 state without the possibility to relax into S0, unless resorting to a specific approach of non-adiabatic dynamics (surface hopping).33 For regioregular P3HT films, the exciton lifetimes reported in the literature range from 400 to 850 ps,34–36 and hence are considerably longer than the time span of our trajectories. The ROKS approach is therefore perfectly suited to monitor the diffusion of the exciton in the S1 state representative of the exciton prior its recombination to S0. We recall that the ROKS approach used here has been consistently formulated within the DFT framework28–30 and, as such, its transferability is the one granted by this underlying theory provided that electronic states do not involve transitions above the first excited state.
Since the exciton transfer between polymer chains is specifically targeted, the inclusion of appropriate dispersion forces becomes crucial. We have chosen to disentangle these van der Waals (vdW) dispersion interactions from the main body of the Kohn–Sham Hamiltonian following the semiempirical vdW “D3” approach by Grimme et al.,37,38 as opposed to a first-principles treatment of the van der Waals (vdW) interactions with a Wannier scheme.39,40 The intra- and inter-molecular forces resulting from the combination of DFT and the Grimme-D3 model implicitly contain all the ingredients that led to the development of the HJ-aggregate models for the P3HT.41,42
The simulation protocol used is the following. The electronic structure of the initial configuration formed by one or several molecules is first converged to the ground state S0 in the standard Kohn–Sham formalism with a convergence criterion of the accuracy of 10−6 a.u. on the total energy. The ionic structure is subsequently relaxed to a local minimum, which serves as the initial configuration for the second part of the calculation within the ROKS approach. Hence, the electronic structure is converged in the S1 excited state at the same accuracy (10−6 a.u) and the ionic structure is relaxed. A short dynamical run in the microcanonical NVE ensemble is done to check the stability of the configuration before controlling the temperature. A rescaling of the velocities is applied during the initial 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 steps (∼1 ps) to bring the system to 10 K before moving to canonical NVT simulations. The temperature is then gradually increased with a Nose–Hoover thermostat during 6 ps, first to T = 100 K and finally to T = 300 K during 40 ps. During the step at T = 300 K, the maximally localized Wannier functions (MLWFs) minimizing the spread11 are computed upon Kohn–Sham unitary transformations of the non-localized Kohn–Sham orbitals. To minimize the Vanderbilt spread functional, the iterative steepest descent algorithm is used and the iterations are done until the convergence criterion (residual error lower than 8 × 10−5) is reached. The centers of mass of the obtained localized orbitals, the Wannier function centers (WFCs), are then used to provide a clear picture of the electronic degrees of freedom. WFC trajectories are obtained by sampling the dynamical trajectory every ten simulation steps (Fig. 4). The CPMD input file for the calculation of the WFC trajectory is provided in the ESI.†
000 steps (∼1 ps) to bring the system to 10 K before moving to canonical NVT simulations. The temperature is then gradually increased with a Nose–Hoover thermostat during 6 ps, first to T = 100 K and finally to T = 300 K during 40 ps. During the step at T = 300 K, the maximally localized Wannier functions (MLWFs) minimizing the spread11 are computed upon Kohn–Sham unitary transformations of the non-localized Kohn–Sham orbitals. To minimize the Vanderbilt spread functional, the iterative steepest descent algorithm is used and the iterations are done until the convergence criterion (residual error lower than 8 × 10−5) is reached. The centers of mass of the obtained localized orbitals, the Wannier function centers (WFCs), are then used to provide a clear picture of the electronic degrees of freedom. WFC trajectories are obtained by sampling the dynamical trajectory every ten simulation steps (Fig. 4). The CPMD input file for the calculation of the WFC trajectory is provided in the ESI.†
 | ||
| Fig. 4 Atomic coordinates (r) and electronic orbitals (ψ) are propagated according to the Car–Parrinello equations of motion. The spread minimization for the calculation of the Wannier functions and centers is done on selected configurations along the dynamical trajectory. | ||
3. Exciton localisation in penta3MT
We first focus on penta3MT, a small molecule containing five thiophene units (52 atoms, i.e. 5 S, 25 C, and 22 H) of the P3HT polymer as shown in the bottom panel of Fig. 1. The WFCs are sketched in Fig. 5 for the ground state S0 and for the first excited singlet state S1 and superimposed on the atomic structure. In the case of a double bond, i.e. two Wannier centers in the middle of a C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bond, the WFCs are highlighted in green, while they are plotted in grey otherwise. A perfect alternation of double and single bonds characterizes the S0 state. An example of MLWFs is shown to illustrate the character of these localized orbitals. In the S1 state, the Wannier center of the hole is shown in red, while the blue color is chosen for the electron. The two WFCs are separated by a distance of 6 Å. The MLWFs of both the electron and the hole, although localized according to the procedure indicated, have still a rather large spread, as shown by the isosurface representation in the bottom panel of Fig. 5, contrary to other MLWFs corresponding to bonding states. Because of the topology of the molecule, the MLWFs spread along the backbone of the system. The MLWF dispersions of both the hole and the electron amount to 6.5 Å. This value being larger than the separation between the two WFCs (6 Å), we can infer that the electron and the hole form a bound pair. Another feature worthy of note is the fact that in this S1 state, the alternation of single and double bonds is disrupted in the vicinity of the electron and the hole.
C bond, the WFCs are highlighted in green, while they are plotted in grey otherwise. A perfect alternation of double and single bonds characterizes the S0 state. An example of MLWFs is shown to illustrate the character of these localized orbitals. In the S1 state, the Wannier center of the hole is shown in red, while the blue color is chosen for the electron. The two WFCs are separated by a distance of 6 Å. The MLWFs of both the electron and the hole, although localized according to the procedure indicated, have still a rather large spread, as shown by the isosurface representation in the bottom panel of Fig. 5, contrary to other MLWFs corresponding to bonding states. Because of the topology of the molecule, the MLWFs spread along the backbone of the system. The MLWF dispersions of both the hole and the electron amount to 6.5 Å. This value being larger than the separation between the two WFCs (6 Å), we can infer that the electron and the hole form a bound pair. Another feature worthy of note is the fact that in this S1 state, the alternation of single and double bonds is disrupted in the vicinity of the electron and the hole.
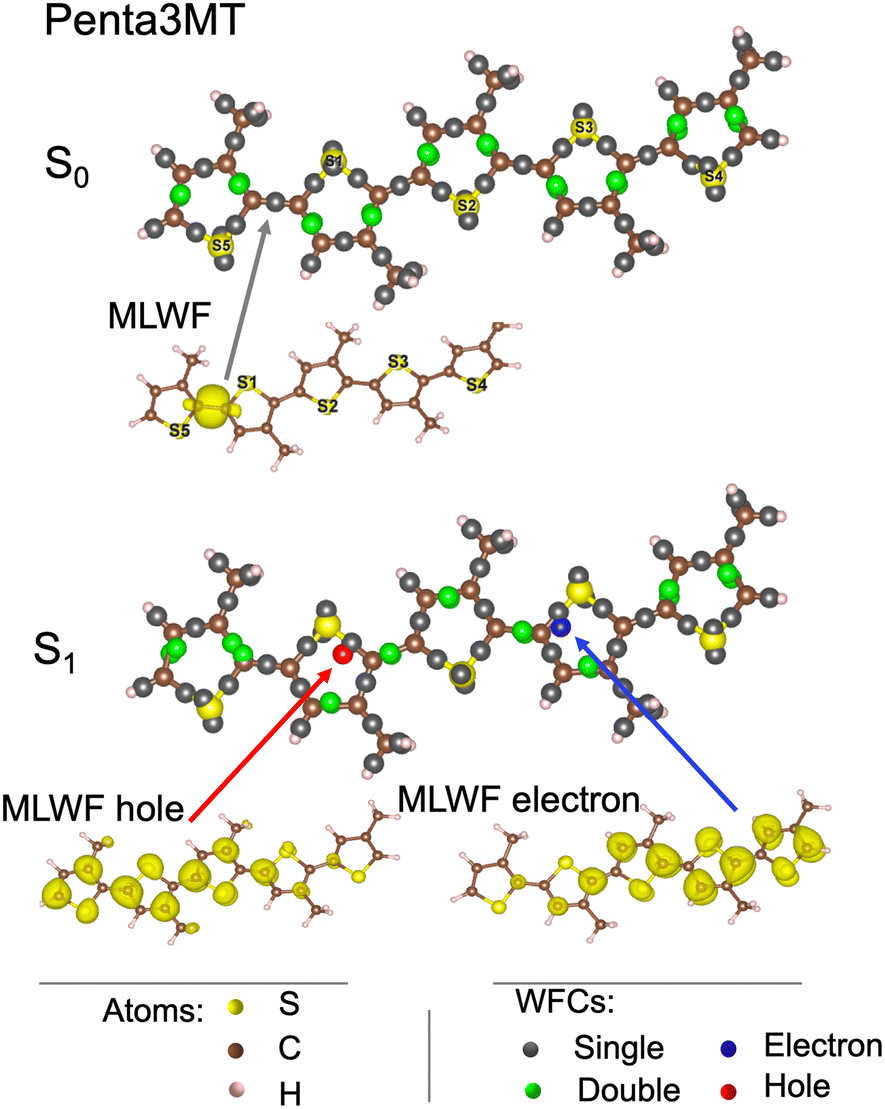 | ||
| Fig. 5 Wannier function centers (WFCs) of the S0 and S1 states of penta3MT at 0 K, and examples of maximally localized Wannier functions (MLWFs). | ||
As mentioned above, at room temperature, the structure of a penta3MT molecule departs from its initial planarity and the thiophene groups at the boundary undergo barrierless rotations. The penta3MT molecule is therefore not suited to the modeling of the exciton transfer in a system expected to maintain a better degree of structural organization. For this reason, we consider virtually infinite polymers subject to periodic boundary conditions (PBCs) in the following sections.
4. Exciton diffusion in P3MT
4.1 Exciton in a single P3MT polymer
Our first target is P3MT, a polymer characterized by shorter lateral chains than P3HT, as shown in Fig. 1. Since the number of atoms is smaller, the computational workload is reduced while preserving the main structural features related to our study. The simulation cell is adapted to host six thiophenes in the x direction, which corresponds to Lx = 23.26 Å, whereas the dimensions Ly and Lz in the other directions are large enough to prevent any interactions between the molecules. The number of atoms in this polymer is equal to 60 (6 sulfur, 30 C and 24 H). A major difference with respect to the penta3MT is that the electron and hole MLWFs extend over all the thiophenes (Fig. 6), instead of half the molecule as in the case of penta3MT. This result is in line with the existence of delocalised electronic modes (polarons) in P3HT.43 In Fig. 6, the WFCs of the electron and the hole are separated by 9.5 Å and their spreads are equal to 7.6 Å.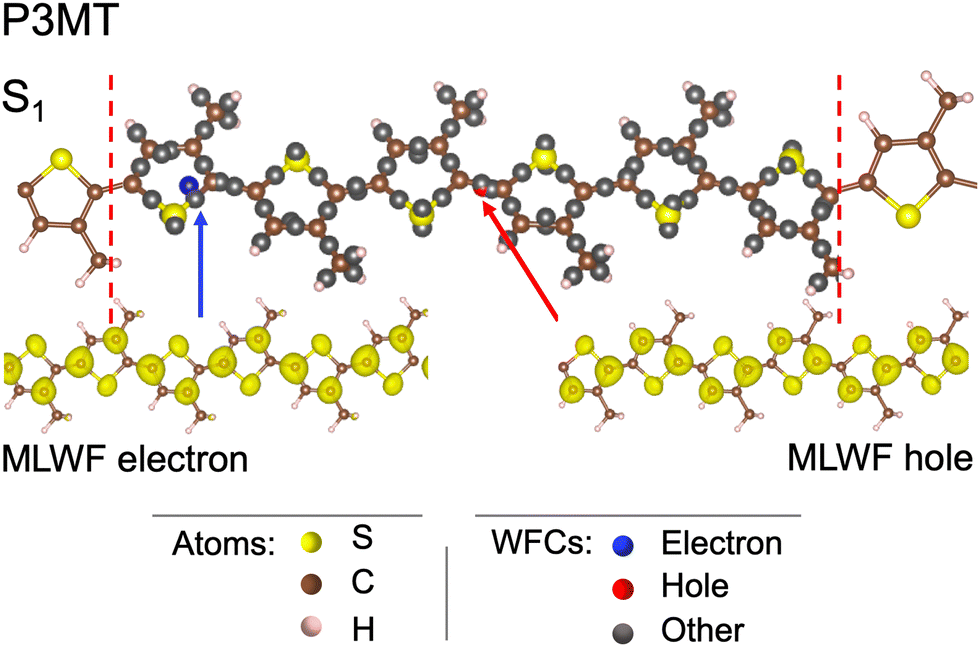 | ||
| Fig. 6 Wannier function centers (WFCs) and maximally localized Wannier functions (MLWFs) of the electron and hole in the S1 state in a single polymer of P3MT at 0 K. The boundaries of the simulation cell are indicated by dashed red lines. | ||
4.2 Exciton diffusion in a system of two P3MT polymers
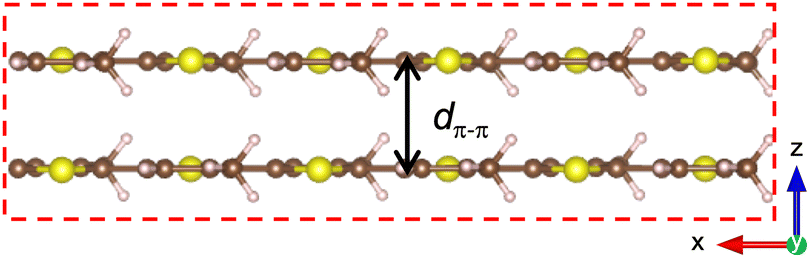 | ||
| Fig. 7 Simulation cell hosting two P3MT polymers of infinite length, ensured by the periodic boundary conditions. The distance between the two polymers is dπ–π = 3.57 Å in the z direction. The cell boundaries are indicated by dashed red lines. | ||
 | ||
| Fig. 8 Snapshots of the two P3MT polymers equilibrated at 300 K in the S1 state along the y–x, x–y and x–z planes. | ||
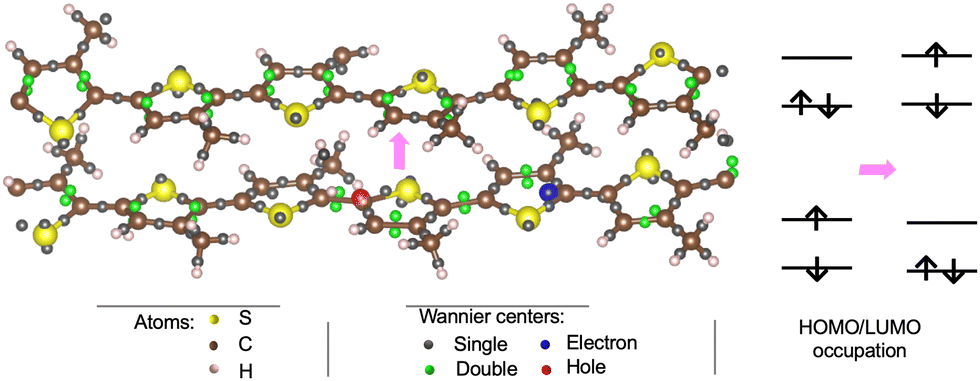 | ||
| Fig. 9 Left: Wannier centers (WFCs) in the P3MT polymer hosting the exciton (bottom molecule) and in the second polymer (top molecule). Right: Schematic of the HOMO (highest occupied molecular orbital) and LUMO (lowest unoccupied molecular orbital) occupation associated with the exciton transfer (schematized by a pink arrow). | ||
 | (1) |
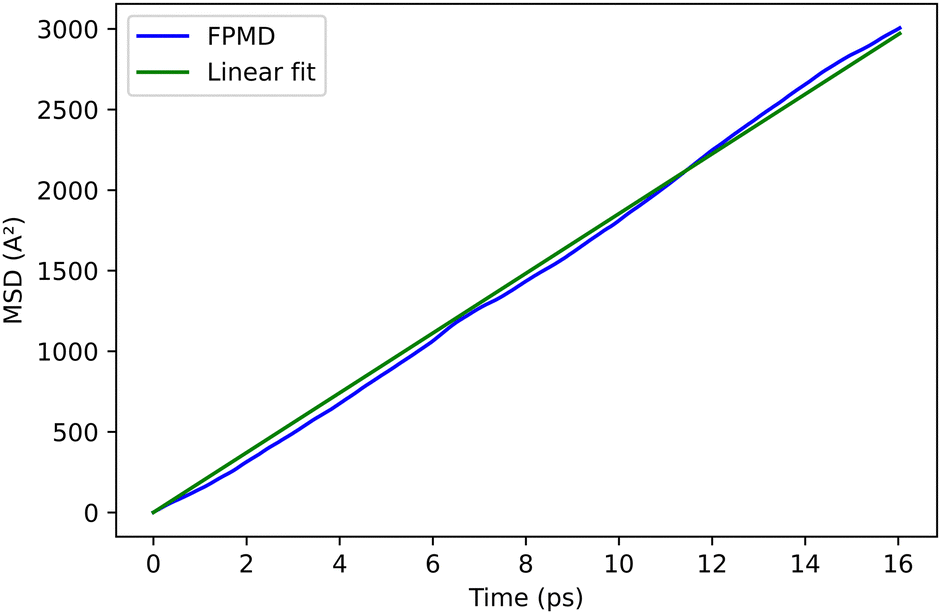 | ||
| Fig. 10 Mean square displacement of the exciton WFC in the dimer of P3MT. | ||
5. Exciton diffusion in P3HT
In the former paragraph, we have shown that following the trajectory of the WFCs of both the electron and the hole is a practical tool to determine the exciton diffusion coefficient in appreciable agreement with the data reported in the literature. We remark, however, that the experimental measurements were done on P3HT, a polymer carrying hexyl groups instead of methyl ones as lateral chains. One could argue that the impact of shorter chains should be negligible since the transfer does not proceed in the (y) direction of the chains. However, we show in the present section that considering hexyl chains results in a significant modification of the lattice structure, in close agreement with Form I observed experimentally8 in the absence of doping. This, in turn, might have an influence on the electronic properties and, ultimately, on the dynamical behavior of an exciton.The starting system in our simulations is a simulation cell containing two P3HT polymers in a planar configuration. For this specific system, the total number of atoms is 300, namely 12 S, 120 C, and 168 H atoms. The planar configuration is kept during a simple geometry relaxation, but when dynamics are allowed and the temperature rises to T = 10 K (by an NVT thermostat control), a conformational change toward Form I occurs spontaneously. This conformation persists at T = 300 K (Fig. 11 and 12).
 | ||
| Fig. 11 Evolution of the structure of the periodic system of two P3HT polymers from a planar configuration at T = 0 K to Form I at room temperature (representative snapshot, the alkyl chains fluctuating dynamically). | ||
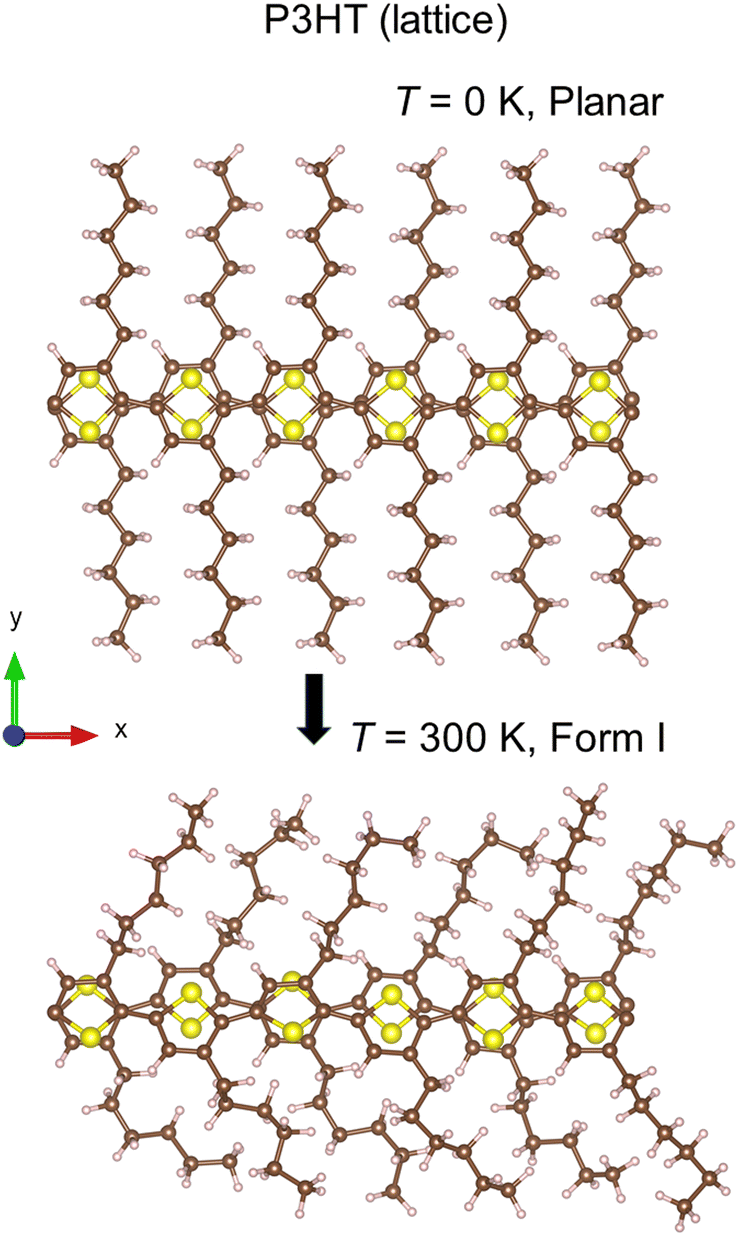 | ||
| Fig. 12 Evolution of the structure of the periodic system of two P3HT polymers from planar at T = 0 K to Form I at room temperature (representative snapshot, the alkyl chains fluctuating dynamically). View of the system of Fig. 11 on the x–y plane. | ||
The π–π distance, defined as the shortest distance between the two polymer chains, is dπ–π = 3.5 ± 0.1 Å. The stacking distance is different in this lattice structure and equal to dstack = 3.7 ± 0.1 Å. The two polymers are tilted of θ = 23 ± 3°. These values are in quantitative agreement with the measurements by electron and X-ray diffraction7,8 as reported in Table 1.
| Present work | Experiments7,8 | |
|---|---|---|
| d π–π (Å) | 3.5 ± 0.1 | 3.4 |
| d stack (Å) | 3.7 ± 0.1 | 3.8 |
| θ (°) | 23 ± 3 | 26 ± 5 |
The trajectories of the electron and hole WFCs are monitored at T = 300 K. We remark that the average distance between the electron and hole (7.6 Å) during this trajectory is typical of a Frenkel exciton.26 The values of the diffusion coefficient, obtained from the mean square displacement of the exciton WFCs, as explained above, are reported in Table 2 along with the experimental value, in fairly good agreement with the present calculations. The reduction of the diffusion coefficient in the P3HT compared to the P3MT can be rationalized by the reduction of the effective cross-section of the π–π interaction in Form I compared to the planar configuration, due to a geometric effect. The simulation cell size, containing two molecules of six thiophenes periodically repeated, is sufficiently large to capture all the features of the excitonic diffusion process.
| D (10−3 cm2 s−1) | |
|---|---|
| P3MT | 9.3 |
| P3HT | 8.0 |
| P3HT, exp.9 | 7.9 |
The results obtained provide convincing support to our computational first principles approach for the polymer carrying hexyl chains, spontaneously reverting to its natural Form I at room temperature. Structural parameters are in excellent quantitative agreement with available measurements. At finite temperature, our model is also able to reproduce with appreciable accuracy the diffusion coefficient characterizing the motion of the exciton in the S1 state. The result is in quantitative agreement with data from time-resolved spectroscopy, thus serving as a benchmark not only for structural but also for the excitonic properties of the system.
6. Conclusions
The present work proposes a methodology for the calculation of the diffusion coefficient of an exciton in the S1 state. The reliability and predictivity of the method are demonstrated in the case of P3HT, a conjugated polymer used worldwide for applications in organic photovoltaic materials. We rely on dynamical simulations able to produce relatively long-lasting atomic trajectories at room temperature, within the FPMD framework. The ROKS formalism is then applied to describe the S1 excited state. The motion of the exciton is obtained in terms of the WFCs of the electron and hole thus providing a practical tool to visualize such an evolving electronic structure bypassing the calculation and storage workload that accumulation of extended wavefunctions would imply.In the case of the P3HT system targeted in this work, we show that two important characteristics of the material have to be considered. The first one is its virtually infinite length, accounted for by appropriate use of the periodic boundary conditions. This avoids uncontrolled conformational changes of the polymer structure and preserves an ordered structure, along with the π–π interactions, also at finite temperature, compatible with a realistic system in which an exciton transfer is expected to occur. The second important feature is represented by the chains, in the sense that their full length allows observing the Form I lattice. If methyl groups replace the original hexyl ones, the resulting structure is nonetheless still realistic, but models a planar arrangement observed experimentally in doped P3HT.
The Wannier functions and centers (along with their associated spread) show that the exciton is delocalised along the polymer chain and transfers from one polymer monomer to the next one with a diffusion coefficient in good agreement with reported experiments. Our approach paves the route to thorough and reliable computational characterization of the exciton transfer that will serve as a guideline for the comprehension of advantages and limitations of other molecular systems, thus offering a guideline for a rational design of organic materials with exciton diffusion lengths sufficient to grant a good efficiency of next-generation photovoltaic devices.
Author contributions
C. O. Diarra: investigation, software, validation, and visualization. M. Boero: methodology, writing – review and editing. E. Steveler: supervision, writing – review and editing. T. Heiser: conceptualization, writing – review and editing. E. Martin: supervision, methodology, and writing – original draft.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge the High Performance Computing Center of the University of Strasbourg for supporting this work by providing scientific support and access to computing resources. Part of the computing resources were funded by the Equipex Equip@Meso project (Programme Investissements d'Avenir) and the CPER Alsacalcul/Big Data. Calculations on the larger systems (oligomers) were performed by using resources from GENCI (Grand Equipement National de Calcul Intensif) (grant no. 0910296 and 0905071). This work of the Interdisciplinary Institute HiFunMat, as part of the ITI 2021-2028 program of the University of Strasbourg, CNRS and Inserm, was supported by IdExUnistra (ANR-10-IDEX-0002) and SFRI (STRAT’US project, ANR-20-SFRI-0012) under the framework of the French Investments for the Future Program.Notes and references
- H. Chen, S. Y. Jeong, J. Tian, Y. Zhang, D. R. Naphade, M. Alsufyani, W. Zhang, S. Griggs, H. Hu, S. Barlow, H. Y. Woo, S. R. Marder, T. D. Anthopoulos, I. McCulloch and Y. Lin, Energy Environ. Sci., 2023, 16, 1062–1070 RSC.
- C. Li, X. Gu, Z. Chen, X. Han, N. Yu, Y. Wei, J. Gao, H. Chen, M. Zhang, A. Wang, J. Zhang, Z. Wei, Q. Peng, Z. Tang, X. Hao, X. Zhang and H. Huang, J. Am. Chem. Soc., 2022, 144, 14731–14739 CrossRef CAS PubMed.
- J. D. A. Lin, O. V. Mikhnenko, J. Chen, Z. Masri, A. Ruseckas, A. Mikhailovsky, R. P. Raab, J. Liu, P. W. M. Blom, M. A. Loi, C. J. García-Cervera, I. D. W. Samuel and T.-Q. Nguyen, Mater. Horiz., 2014, 1, 280–285 RSC.
- W. Barford, E. R. Bittner and A. Ward, J. Phys. Chem. A, 2012, 116, 10319–10327 CrossRef CAS PubMed.
- S. M. Janke, M. B. Qarai, V. Blum and F. C. Spano, J. Chem. Phys., 2020, 152, 144702 CrossRef CAS PubMed.
- N. J. Hestand and F. C. Spano, Chem. Rev., 2018, 118, 7069–7163 CrossRef CAS PubMed.
- N. Kayunkid, S. Uttiya and M. Brinkmann, Macromolecules, 2010, 43, 4961–4967 CrossRef CAS.
- M. Brinkmann, J. Polym. Sci., Part B: Polym. Phys., 2011, 49, 1218–1233 CrossRef CAS.
- H. Ohkita, Y. Tamai, H. Benten and S. Ito, IEEE J. Sel. Top. Quantum Electron., 2016, 22, 100–111 Search PubMed.
- C. Schwermann and N. L. Doltsinis, Phys. Chem. Chem. Phys., 2020, 22, 10526–10535 RSC.
- N. Marzari, A. A. Mostofi, J. R. Yates, I. Souza and D. Vanderbilt, Rev. Mod. Phys., 2012, 84, 1419–1475 CrossRef CAS.
- T. Förster, Ann. Phys., 1948, 437, 55–75 CrossRef.
- D. L. Dexter, J. Chem. Phys., 1953, 21, 836–850 CrossRef CAS.
- V. Untilova, H. Zeng, P. Durand, L. Herrmann, N. Leclerc and M. Brinkmann, Macromolecules, 2021, 54, 6073–6084 CrossRef CAS.
- R. Car and M. Parrinello, Phys. Rev. Lett., 1985, 55, 2471–2474 CrossRef CAS PubMed.
- Jointly by IBM Corporation and by Max Planck Institute, Stuttgart, CPMD code, 2022, https://github.com/CPMD-code.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed.
- N. Troullier and J. L. Martins, Phys. Rev. B: Condens. Matter Mater. Phys., 1991, 43, 1993–2006 CrossRef CAS PubMed.
- S. Nosé, Mol. Phys., 1984, 52, 255–268 CrossRef.
- S. Nosé, J. Chem. Phys., 1984, 81, 511–519 CrossRef.
- W. G. Hoover, Phys. Rev. A, 1985, 31, 1695–1697 CrossRef PubMed.
- G. J. Martyna, M. L. Klein and M. Tuckerman, J. Chem. Phys., 1992, 97, 2635–2643 CrossRef.
- P. E. Blöchl and M. Parrinello, Phys. Rev. B: Condens. Matter Mater. Phys., 1992, 45, 9413–9416 CrossRef PubMed.
- N. Turro, Modern Molecular Photochemistry, University Science Books, 1991 Search PubMed.
- A. H. Zewail, Femtochemistry: Ultrafast Dynamics of the Chemical Bond (World Scientific Series in 20th Century Chemistry), World Scientific, 1994 Search PubMed.
- A. Köhler and H. Bässler, Electronic Processes in Organic Semiconductors: An Introduction, Wiley, 1st edn, 2015 Search PubMed.
- M. Kasha, Discuss. Faraday Soc., 1950, 9, 14–19 RSC.
- I. Frank, J. Hutter, D. Marx and M. Parrinello, J. Chem. Phys., 1998, 108, 4060–4069 CrossRef CAS.
- S. Grimm, C. Nonnenberg and I. Frank, J. Chem. Phys., 2003, 119, 11574–11584 CrossRef CAS.
- C. Nonnenberg, S. Grimm and I. Frank, J. Chem. Phys., 2003, 119, 11585–11590 CrossRef CAS.
- I. Frank, Chem. Phys. Lett., 2018, 702, 76–81 CrossRef CAS.
- I. Frank, J. Hutter, D. Marx and M. Parrinello, J. Chem. Phys., 1998, 108, 4060–4069 CrossRef CAS.
- N. L. Doltsinis and D. Marx, Phys. Rev. Lett., 2002, 88, 166402 CrossRef PubMed.
- S. Cook, H. Liyuan, A. Furube and R. Katoh, J. Phys. Chem. C, 2010, 114, 10962–10968 CrossRef CAS.
- W. Beek, M. Wienk and R. Janssen, Adv. Funct. Mater., 2006, 16, 1112–1116 CrossRef CAS.
- S. Cook, A. Furube and R. Katoh, Energy Environ. Sci., 2008, 1, 294–299 RSC.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed.
- S. Grimme, J. Comput. Chem., 2006, 27, 1787–1799 CrossRef CAS PubMed.
- P. L. Silvestrelli, Phys. Rev. Lett., 2008, 100, 053002 CrossRef PubMed.
- T. Ikeda and M. Boero, J. Chem. Phys., 2015, 143, 194510 CrossRef PubMed.
- F. Paquin, H. Yamagata, N. J. Hestand, M. Sakowicz, N. Bérubé, M. Côté, L. X. Reynolds, S. A. Haque, N. Stingelin, F. C. Spano and C. Silva, Phys. Rev. B: Condens. Matter Mater. Phys., 2013, 88, 155202 CrossRef.
- F. C. Spano and C. Silva, Annu. Rev. Phys. Chem., 2014, 65, 477–500 CrossRef CAS PubMed.
- O. P. Dimitriev, D. A. Blank, C. Ganser and C. Teichert, J. Phys. Chem. C, 2018, 122, 17096–17109 CrossRef CAS.
- C. A. Hunter and J. K. M. Sanders, J. Am. Chem. Soc., 1990, 112, 5525–5534 CrossRef CAS.
- C. R. Martinez and B. L. Iverson, Chem. Sci., 2012, 3, 2191 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3cp00533j |
| This journal is © the Owner Societies 2023 |