
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCoordination and thermodynamic properties of aqueous protactinium(V) by first-principle calculations†
Hanna
Oher
 ab,
Jérémy
Delafoulhouze
c,
Eric
Renault
c,
Valérie
Vallet
d and
Rémi
Maurice
*ab
ab,
Jérémy
Delafoulhouze
c,
Eric
Renault
c,
Valérie
Vallet
d and
Rémi
Maurice
*ab
aSUBATECH, UMR CNRS 6457, IN2P3/IMT Atlantique/Université de Nantes, 4 rue Alfred Kastler, BP 20722, 44307 Nantes Cedex 3, France
bUniv Rennes, CNRS, ISCR (Institut des Sciences Chimiques de Rennes) – UMR 6226, 35000 Rennes, France. E-mail: remi.maurice@univ-rennes.fr
cNantes Université, CNRS, CEISAM UMR 6230, F-44000 Nantes, France
dUniv. Lille, CNRS, UMR 8523 – PhLAM – Physique des Lasers, Atomes et Molécules, F-59000 Lille, France
First published on 20th March 2023
Abstract
Protactinium (Z = 91) is a very rare actinide with peculiar physico-chemical properties. Indeed, although one may naively think that it behaves similarly to either thorium or uranium by its position in the periodic table, it may in fact follow its own rules. Because of the quite small energy gap between its valence shells (in particular the 5f and 6d ones) and also the strong influence of relativistic effects on its properties, it is actually a challenging element for theoretical chemists. In this article, we combine experimental information, chemical arguments and standard first-principle calculations, complemented by implicit and explicit solvation, to revisit the stepwise complexation of aqueous protactinium(V) with sulfate and oxalate dianionic ligands (SO42− and C2O42−, respectively). From a methodological viewpoint, we notably conclude that it is necessary to at least saturate the coordination sphere of protactinium(V) to reach converged equilibrium constant values. Furthermore, in the case of single complexations (i.e. with one sulfate or oxalate ligand bound in the bidentate fashion), we show that it is necessary to maintain the coordination of one hydroxyl group, present in the supposed [PaO(OH)]2+ precursor, to obtain coherent complexation constants. Therefore, we predict that this hydroxyl group is maintained in the formation of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 complexes while we confirm that it is withdrawn when coordinating three sulfate or oxalate ligands. Finally, we stress that this work is a first step toward the future use of theoretical predictions to elucidate the enigmatic chemistry of protactinium in solution.
:1 complexes while we confirm that it is withdrawn when coordinating three sulfate or oxalate ligands. Finally, we stress that this work is a first step toward the future use of theoretical predictions to elucidate the enigmatic chemistry of protactinium in solution.
1 Introduction
Protactinium (Z = 91) is an enigmatic element for the chemists.1 While it is located in between thorium and uranium in the periodic table, it may display unique properties that severely hinder experimental studies and interpretations. For instance, the five valence electrons of its free atom may spread in numerous ways on the valence 5f, 6d, 7s and 7p shells, which generates a quantum chaos of states even well before ionization.2–4 In solution, protactinium chemistry is expected to be dominated by its +V oxidation state (formally free of valence electron), with potential occurrence under specific reducing conditions of the unstable +IV one (displaying one unpaired 5f electron in the ground state).5 Therefore, the aqueous chemistry of protactinium could well have been dominated by easily identifiable and computable species.Unfortunately, Pa(V) and Pa(IV) are prone to hydrolysis, polymerization and precipitation and other issues such as a strong tendency to adsorb into glass, etc.6 Furthermore, the intrinsic radiations of its isotopes imply specific safety regulations to be followed. Consequently, the speciation of Pa(V) and Pa(IV) in aqueous solution is largely unknown, which further complicates the performance of a theoretical study dedicated to the coordination and thermodynamic properties of these; thus, one has to rely on ad hoc research hypotheses.
In this work, we will focus on protactinium(V) since it is expected to have a closed-shell electronic ground state in most of its associated chemical species. Therefore, single-reference approaches such as Kohn–Sham density functional theory (DFT) or second-order Møller–Plesset perturbation theory (MP2) may be a priori safely used, contrary to Pa(IV) systems. Despite this apparent simplicity in the electronic structure theory part, challenges remain: solvation and/or hydrolysis issues must be tackled and relativistic effects, especially the scalar ones, should be accounted for. In fact, since little experimental data regarding speciation and complexation constants available, the choice of the Pa(V) chemical complexes will be severely restrained. Anyway, we will show that it is necessary to combine experimental information, chemical arguments and outcomes of calculations to reach conclusions.
The paper is organized as follows. First, the choice of the systems and our working hypotheses will be explained. Then, computational details will be given, prior to results and discussion, organized by increasing solvation and/or chemical complexity.
2 Choice of the systems and working hypotheses
The chemistry of Pa(V) is characterized by a general lack of experimental and/or computational data.7 In this work, we aim at showing that standard quantum chemical calculations may be of help to predict the relative stabilities of Pa(V) complexes, based on comparisons between theoretical and experimental data. Therefore, it is necessary to make a quick survey of known experimental data to justify our choice of the systems under study. In particular, the available experimental data should ideally help us to formulate relevant hypotheses concerning the nature of the complexes that are involved in the equilibrium processes at play. After this, we will introduce additional working hypotheses that are required to define a practical computational strategy, systematically improvable in terms of solvation and/or chemical models.The identification of Pa(V) species is a difficult task. Several types of complementary experiments may be performed, none of them being independently conclusive. We may classify them in two main types, (i) experiments at tracer scale and (ii) spectroscopy experiments (ponderable scale). In (i), we may encounter methods to determine the molecular charge (ion chromatography and/or migration experiments under an electric field such as capillary electrophoresis) and also methods to determine equilibrium constants, based on competition between two distinct phases (e.g. liquid–liquid extraction). In (ii), we may find X-ray absorption data for instance, based on near-edge or extended structures (XANES and EXAFS, respectively). By taking profit of the X-ray absorption spectrum dependence on the molecular structure, especially in the EXAFS regime, one may refine guessed structural models by a fitting procedure, leading in the end to an experimental structure.
It is important to understand that equilibrium constants are determined by fitting procedures, implying that an underlying thermodynamic model is selected. Because the activity of water in water is set to 1, all the reactions are defined up to a given number of water molecules. This is a strong limitation of the liquid–liquid extraction approach. For instance, Mendes et al.8 have shown that Pa(V) can form a 1:1 complex with DTPA (diethylenetriaminepentaacetic acid) and have determined the associated formation constant for the following reaction:
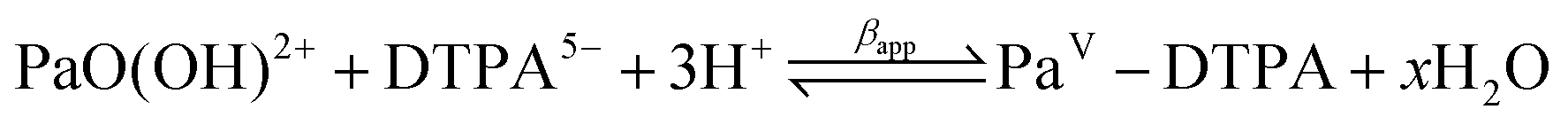 | (1) |
EXAFS data of Pa(V) have been acquired in fluoric acid (HF), sulfuric acid (H2SO4) and oxalic acid (H2C2O4) concentrated media.9–11 Concentrated media are interesting because we may expect to saturate the first coordination sphere of the Pa(V) ion and end up with a simpler speciation than at intermediate concentrations, i.e. we may observe single well-defined chemical species. While in fluoric acid the mono–oxo bond of the [PaO(OH)]2+ starting species is cleaved (up to eight fluorides being coordinated11,12), it is observed in both the sulfuric and oxalic media. Therefore, we may expect similar complexes to be formed. Since in the oxalic acid case, the analysis of the data led to the firm identification of the [PaO(C2O4)3]3− complex, we thus hypothesize the formation of the [PaO(SO4)3]3− complex in sulfuric acid medium. We recall here that our objective is to determine relative complexation constants, meaning that it is important to compare systems of similar natures to obtain accurate values by error cancellations, as was done in the past for exploring the astatine (Z = 85) basic chemistry.13–17
Having suspected that complexes of similar natures may be formed at high H2SO4 and H2C2O4 concentrations, in particular of the [PaOX3]3− type, X being either (SO4) or (C2O4), we continue our survey with the corresponding liquid–liquid extraction data.7,9,10,18 In these studies, the formations of 1:1, 1:2 and 1:3 complexes of Pa(V) with sulfate or oxalate ligands were hypothesized and described by the following chemical equilibria:
 | (2) |
:1 to 1:3 complexes were confirmed in both the cases and the apparent formation constants have been determined at 25 °C by a fitting procedure10,18 (see Table 1).
From the logβapp,i values reported in Table 1, we observe a similar trend in both the series, with the logβapp,2 values being nearly twice the logβapp,1 ones, and the logβapp,3 ones being nearly 2.5 times larger than the logβapp,1 ones. Because of this, we hypothesize that for each i value, the two products, i.e. [PaO(SO4)i]3−2i and [PaO(C2O4)i]3−2i, have similar natures. Therefore, even if the nature of the ending complexes is not (yet) fully known, we propose to focus on three ligand-exchange reactions:
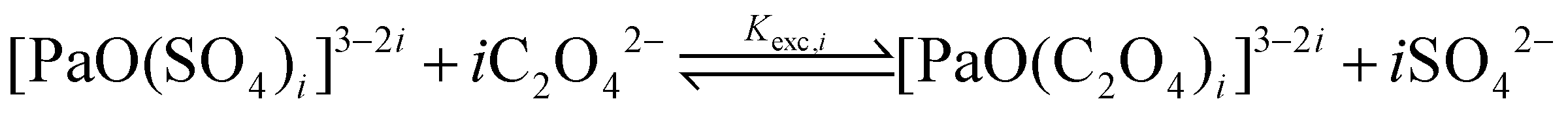 | (3) |
| logKexpexc,i = logβapp,i[C2O42−] − logβapp,i[SO42−] | (4) |
Our computational study aims at fairly reproducing the 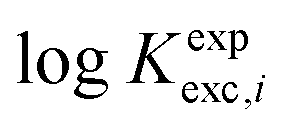 values by using solvation and chemical models of increasing complexities. To achieve this goal, we formulate the following working hypotheses:
values by using solvation and chemical models of increasing complexities. To achieve this goal, we formulate the following working hypotheses:
(1) In all the complexes, the ligands bind in a bidentate fashion (see Fig. 1);
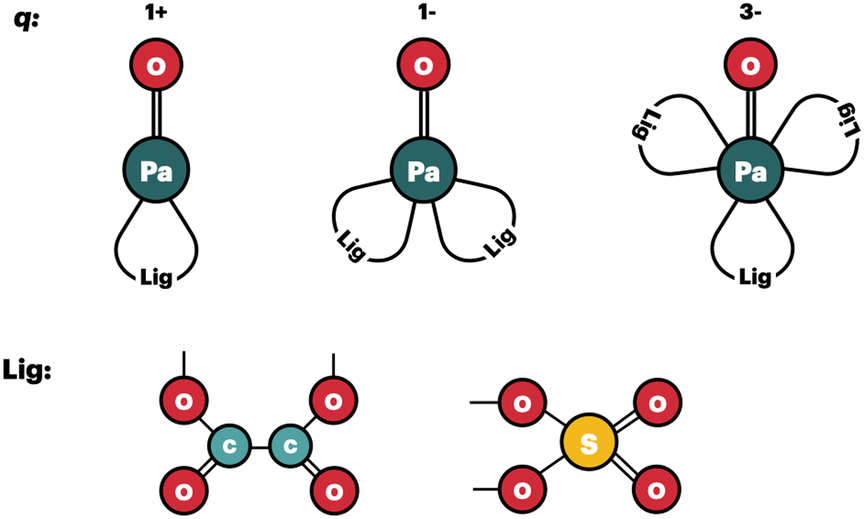 | ||
| Fig. 1 Schematic representation of the geometrical models for the Pa(V) complexes that are used to compute the logKexc,i values. | ||
(2) We can compute the log of the exchange constants by means of a thermodynamic cycle,19 with computation of the log of the gas phase constants at our reference level of theory and computation of the solvation energies of the species with a polarizable continuum model (PCM);
(3) We assume that the errors done by our PCM approach concerning the solvation of the free SO42− and C2O42− ligands are similar, meaning that explicit solvation will not be applied on the free ligands (note that the opposite would potentially turn out to be a dead end owing the complexity of the generated microsolvated structures20,21).
Also, we recall the previously formulated hypotheses still apply, meaning that we assume that the mono–oxo bond is present in all the complexes and also that for each i reaction, the corresponding sulfate and oxalate complexes have a similar nature.
Finally, we define the log of the exchange constants as follows (T is fixed at 298.15 K and P at 1 atm):
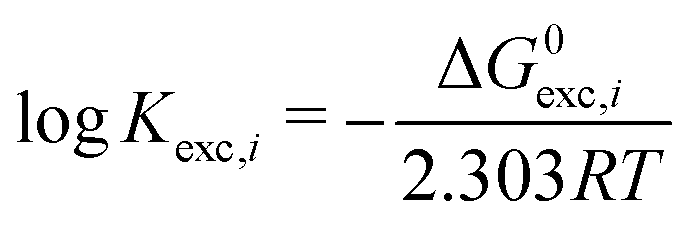 | (5) |
3 Computation details
Geometries and electronic structures of Pa(V) are expected to be well described by single-reference approaches, due to the closed-shell nature of the valence-free reference Pa5+ ion. We have thus directly restricted our preparatory study to the MP2 and Kohn–Sham DFT methods, with inclusion of scalar relativistic effects (via the Douglas–Kroll–Hess Hamiltonian22–24 or a suited relativistic pseudopotential), and in the latter case the use of a hybrid exchange–correlation functional. We have actually compared results obtained with the MP2,25 DFT/B3LYP26 and DFT/PBE027,28 methods, with different basis sets (with an all-electron basis29vs. bases30–32 combined with a relativistic pseudopotential for Pa33), and different codes (Gaussian,34 ORCA 4.2.135 and OpenMolcas36). The results and a more complete discussion can be found in ESI.† Our main findings are:(1) Basis sets combined with pseudopotentials generally give geometries in good agreement with the employed all-electron basis, unless a specific and unrecommended implementation is used.
(2) The use of the def2-TZVP and aug-cc-pVTZ basis sets on the light atoms (i.e. all atoms besides Pa) leads to a significant improvement over the previous results that were based on the former 6-31+G* ones.10
(3) All the tested approaches are in good agreement with the EXAFS structure, with perhaps a slight advantage of the DFT/PBE0 approach, essentially concerning the mono–oxo bond distance.
In the remainder of the article, the gas-phase geometries, harmonic frequencies, and gas-phase free energies of the oxalate and sulfate complexes with Pa(V) were obtained using the Gaussian 16.A.03 software.34 All the reported structures correspond to minima of the energy (no imaginary frequency). The Kohn–Sham equation was solved by using the hybrid PBE0 exchange–correlation functional.27,28 In these calculations, the def2-TZVP (second generation of triple-ζ plus polarization quality) basis set32 has been used for all the light elements (H, C, O, S). For the protactinium atom, the small-core scalar relativistic effective core potential ECP60MWB was used33 along with its associated basis set, formed by contracting a 14s13p10d8f set into a 10s9p5d4f one.31
The aqueous solvation free energies were computed at the Hartree–Fock (HF) level with the same basis sets and pseudopotentials as used in the DFT/PBE0 calculations. To mimic the long-range solvent effects, the conductor-like polarizable continuum model (CPCM)37,38 was used as it is implemented in the Gaussian 16 program package. In the absence of any alternative, the UFF radius was set for the Pa atom (1.712 Å), while the rest of the cavity was based on UAHF radii39 for the other atoms/groups (Ooxo = 1.59 Å, Oligand = 1.35 Å, C = 1.68 Å, S = 1.98 Å, OH− = 1.59 Å, H2O = 1.68 Å). We recall here that the UAHF radii depend on the charge and environment of the atom of interest. For the bidentate SO42− and C2O42− ligands, we have considered ad hoc equal −1/2 charges on the oxygen atoms, hence the Oligand radius value. Also, note that no sphere is created around the H atoms (united-atom approach), which explains the OH− and H2O notations. Since those radii were initially optimized with the alpha scaling factor of 1.2, it was manually set to this value in our calculations.
Choice was made to apply the UAHF model instead of the UAKS after initial calculations concerning the geometry of the [PaO(C2O4)3]3− complex and the determination of the logKexc,3 value. In fact, a positive solvation contribution to logKexc,3 was obtained with the UAKS model, resulting in an unphysical negative logKexc,3 value. In a previous study related to astatine chemistry,15 it was also found that the UAHF model was more accurate than the UAKS one to compute ligand-exchange reaction constants, the UAKS one being more accurate only if applied in a single-point fashion at the UAHF geometries. In view of finding a compromise between accuracy and simplicity of application (no need for extra single-point calculations), we have thus here retained the UAHF model for the production calculations.
4 Results and discussion
In this section, we will introduce models of increasing complexity. First, we will look at the gas-phase ligand-exchange reaction constants, and complement these by an implicit solvation model. Then, we will also include explicitly water molecules in the quantum chemical framework, meaning that we will use a combined implicit and explicit solvation model. Finally, we will discuss the possibility for maintaining the hydroxyl group on both the left-hand and right-hand sides of the ligand-exchange reactions. At each stage, we will compare the computed logKexc,i values to the logKexpexc,i ones.
4.1 Gas phase (GP) and implicit solvation (PCM)
The zeroth-order description of solvation consists in fully neglecting any solvent effect, implying that gas-phase reaction constants are actually first sought. In the process, the geometries of the sulfate and oxalate complexes of interest are determined, together with the structures of the free ligands. For all the complexes, we find that binding in bidentate fashion is by far preferable (which justifies our previous hypothesis number #1).As already mentioned, the EXAFS structure of the [PaO(C2O4)3]3− complex has been reported by Mendes et al.10 We note that attempts to determine the structure of the analogous [PaO(SO4)3]3− complex have been reported,9,40 but that information concerning the binding modes is still lacking, meaning that even if the presence of the short mono–oxo bond was confirmed, it is hard to know how accurate the other Pa–Oligand bond distances are. Therefore, we only retain the EXAFS structure of the [PaO(C2O4)3]3− complex for comparison purposes.
As shown in ESI,† the DFT/PBE0 approach leads to a good agreement with the experimental structure, despite a moderate discrepancy on the Pa–Ooxo bond distance (1.86 Å vs.1.75 Å in the experiment). At this stage, the computational/experimental origin of this discrepancy is not known, and it may relate to experimental and/or computational uncertainty. Addressing this issue would require generating new EXAFS experiments and/or new fits of existing data and also performing an extensive electronic structure theory benchmark, which is out of the scope of the present paper. In fact, we remark that for all the Pa–Oligand bond distances of interest (i.e. the ones of the bound atoms) as well as for the Pa–C bond distances, the mean absolute deviation between the computed and experimental values is below 0.02 Å. We thus assume that the retained DFT/PBE0 structure are overall accurate enough for further proceeding with the computation of ligand-exchange reaction constants. Moreover, we remark that a Pa–Ooxo bond distance of 1.86 Å seems significantly longer than the one in bare PaO3+ (1.68 Å) and in the PaO(OH)2+ (1.71 Å) precursor. The three studied reactions have been renamed as R1, R2 and R3, depending on the number of ligands involved in the reaction. The gas-phase structures of the complexes involved in these reactions are displayed in Fig. S1 (ESI†). Results are reported in Table 2 (see more specifically the “GP” column).
| logKexc,i |
GP | PCM | PCM + nH2O | Expt. | |
|---|---|---|---|---|---|
| FCS | FCS + 1 | ||||
| R1 | 6.0 | 9.4 | — | — | 3.5 |
| R2 | 2.6 | 19.9 | 12.0 | 8.8 | 7.5 |
| R3 | 2.8 | 16.1 | 16.1 | 14.6 | 10.5 |
For all the computed species, the obtained lowest-energy structure was separated by at least 10 kJ mol−1 with any other conformer or isomer (in terms of Gibbs free energy at 298.15 K). Therefore, we have used only one structure per species to compute the reaction constants. As can be seen in Table 2, the gas-phase ligand-exchange reaction constants do not follow at all the experimental trend, going from logKexc,1 = 6.0 (R1) to logKexc,3 = 2.8 (R3). This may be a net indication that solvent effects may be at play, suggesting that important solute–solvent interactions must somehow be accounted for.38
As a first solvation model, we have included a correction in the free energy of each species arising from a PCM model (see the Computational details section). In computing those solvation-induced corrections to the free energies, it is important to check that the geometry that is optimized with application of the PCM is of the same nature as the gas-phase one. Furthermore, in our calculations, we have employed the UAHF model, meaning that both the gas phase and PCM geometries are obtained at the HF level of theory. Therefore, we not only need to check that those two match well, we also need to check that the gas phase DFT/PBE0 and HF geometries are also in qualitative accord. In all the cases, the GP(DFT/PBE0), GP(HF) and PCM(HF) structures qualitatively matched, meaning that consistent corrections to the free energies of the species of interest were obtained. The deduced values of the logarithms of the reaction constants are listed in Table 2 (see in particular the “PCM” column).
Again, even if now logKexc,1 is the weakest of the three, the computational trend differs drastically from the experimental one. Since we observe important changes by introducing the PCM, we may already conclude that solvation is crucial in this case, even if its treatment remains to be more precise. Within the framework of a static quantum chemical approach, we have two main degrees of freedom, we may (i) explicitly treat some of the solvent (here water) molecules or (ii) change the nature of the chemical species that are involved in the studied equilibria, which will be done in the next subsections. Note that the addition of explicitly treated water molecules in the first coordination sphere of the Pa(V) ions may also lead to better geometrical models for the complexes of interest.
4.2 Implicit plus explicit solvation (PCM + nH2O)
The combination of implicit and explicit solvation usually improves the quality of thermodynamic results at the price of an increased complexity in the conformational search of the solute geometries.38 A key point lies in the choice of the number of water molecules. At each addition of one water molecule, one may introduce key interactions such as a coordination or a hydrogen bond. Since we compute ligand-exchange reaction constants, it is also important to ensure that the added interactions on the left-hand and right-hand sides of the reaction are of similar nature.16 As a consequence, the computed logKexc,i values may not monotonously evolve with the number of explicitly treated water molecules, and it may not be worth reporting each and every step.
At this stage, it is important to come back to our computational hypothesis #3, according to which error cancellations allow us to avoid performing a tricky microsolvation study of the free oxalate and sulfate ligands (many water molecules being required to fill their first solvation spheres20,21). In fact, two independent PCM studies were successful in reproducing an experimental trend that involves the free oxalate or the sulfate ligand: Aquino et al.41 used a −986.6 kJ mol−1 solvation energy for the free oxalate ligand to study its successive complexation with the Al3+ ion and Lee and McKee42 recommended a −1028.8 kJ mol−1 solvation energy for the free sulfate ligand to study the dissolution of salts. We note that the resulting difference in solvation energies, −42.2 kJ mol−1, is pretty close to the −40.0 kJ mol−1 that we have obtained with the UAHF model, which supports our hypothesis. As a result, water molecules are only added to the protactinium complexes. We thus rewrite our generic ligand-exchange reaction as:
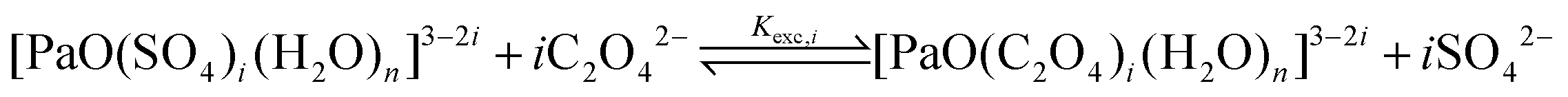 | (6) |
In practice, a stepwise water addition was performed, with a conformational search based on multiple starting structures, having for instance ligands bound in monodentate or bidentate fashions, and also water molecules interacting with either the Pa(V) ion (coordination bonds) or the ligands (hydrogen bonds). Out of this stepwise study, we concluded that we needed to at least saturate the first coordination sphere of the Pa(V) ion to obtain similar structures for each pair of sulfate and oxalate complexes. Therefore, the intermediate steps with an incomplete first coordination sphere are not reported here.
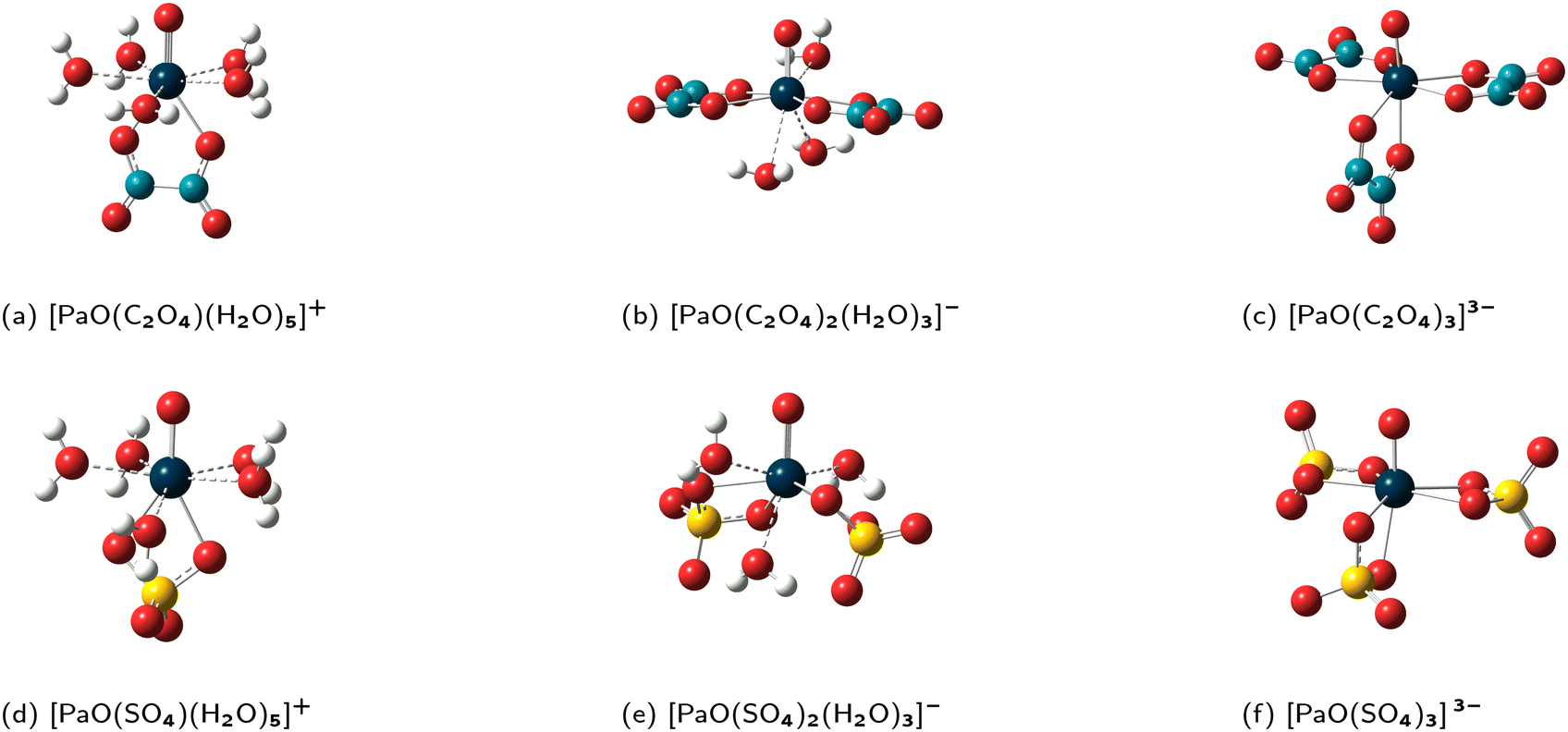 | ||
| Fig. 2 Representations of the GP(DFT/PBE0) structures of the Pa(V) complexes, obtained with just-saturated first-coordination spheres (FCS) (a)−(f). Color code: dark blue (Pa), yellow (S), red (O), light blue (C) and white (H). | ||
It is clear from Fig. 2 that for each reaction, both the complexes display similar interactions. Thus, we can readily proceed with the determination of the reaction constants (see “PCM + nH2O”/“FCS” column in Table 2). For the sake of pedagogy, we will now discuss the results in reversed order, i.e. from R3 till R1. Since there is no added water molecule in the [PaOX3]3− complexes, note that the value logKexc,3 of 16.1 is simply reported from the (previous) “PCM” column.
Things start to be less trivial with the [PaOX2]− complexes. In both the sulfate and oxalate complexes, three water molecules are introduced. As before, we have checked that each series of three structures, namely GP(DFT/PBE0), GP(HF) and PCM(HF) qualitatively match, meaning that these correspond to a same isomer and conformer. Also, we have excluded the possibility for the need of considering an ensemble of conformers, since no other conformer was found at less than 10 kJ mol−1 above the retained one in terms of Gibbs free energy (both in the gas phase and after introducing the PCM correction). We now see a significant improvement of the computed logKexc,2 value, since it is now for the first time below the logKexc,3 one. In fact, the logKexc,3 − logKexc,2 value is already in good agreement with the experiment (4.1 vs. 3.0). Therefore, we conclude that the improvement of our computational description goes together with a better accord with the experiment, which is quite encouraging for what is to follow.
In the case of R1, we have not reported any value in Table 2. In fact, depending on the starting conformer, we have obtained logKexc,1 values ranging from 10.0 to −11.6, and we have been unable to find couples of GP(HF) and PCM(HF) structures that match. In this particular case, the static approach combined with a PCM correction seems to act as a random number generator and it is not recommended to report any data. We thus need to figure out how to obtain more stable value in the R1 case, which may come with a change in the chemical process (more water molecules, change in chemical composition) or even of paradigm (by going beyond the static approach for instance). The latter possibility being out of the scope of the present paper, we will continue by proceeding with the other two possibilities.
Despite the relative favorable correspondence with the experimental data for R2 and R3, it is still important to check that a potential change in the process does not destroy this mighty agreement. In particular, it is important to show that addition of one water molecule outside the first coordination shell (denoted FCS + 1 in the remainder of the article), which may physically lead to a better stabilization of one of the dianionic ligands, maintains this successful trend.
Kexc,2 and logKexc,3 values become practically closer to the experimental ones while the logKexc,3 − logKexc,2 difference remains in fair agreement with experiment. We thus conclude that the previous encouraging results obtained for R2 and R3 were not fortuitous, since improvement of the description maintains them. One should point out that using FCS + 2 configurations did not bring any substantial changes to the computed logKexc,n values, further confirming the protactinium(V) coordination number and the thermodynamic stability of considered species. Moreover, since addition of one or two water molecules does not solve the pathological situation encountered in the R1 case, we pursue by increasing the chemical complexity. Since we have seen that the FCS + 1 results are overall better than the FCS ones, we stick to the FCS + 1 level in this complementary study.
4.3 Probing the relevance of the hydroxyl-group scenario
All the previous results were based on the hypothesis that the hydroxyl group, present in the [PaO(OH)]2+ starting species, is withdrawn by any complexation. While this is quite clear to us in the R3 case (see the Choice of the systems and working hypotheses section), for which we have obtained a good agreement with the experiment, this hypothesis may be questioned in the R1 and R2 cases, in particular the R1 one for which we were so far unable to report any reliable value. If we make the opposite hypothesis, i.e. that the hydroxyl group is maintained, the global formation reactions of the complexes of interest now write: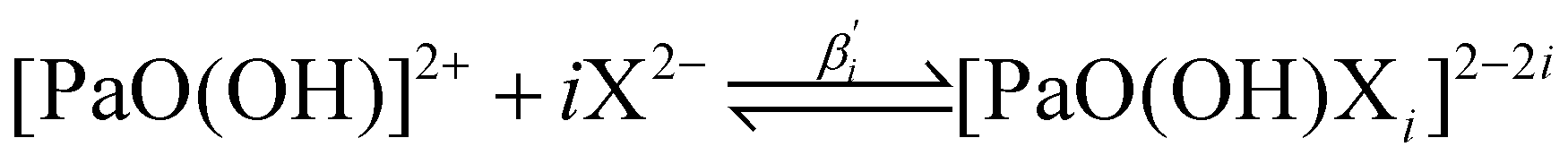 | (7) |
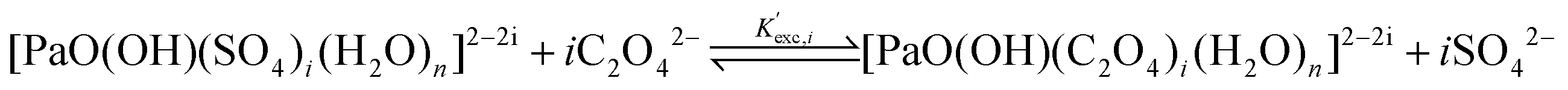 | (8) |
The four new FCS + 1 GP(DFT/PBE0) structures are displayed in Fig. 3. The hydroxyl group is always found to be in trans position with respect to the mono–oxo one, and the sulfate and oxalate ligands are still bound in the bidentate fashion, as in all the previous structures. In those four new structures, the CN of the Pa(V) ion is 7. Luckily, this reduces the possibilities for arranging the water molecules within the FCS and also further away from it. In practice, we now have the expected correspondence between the GP(DFT/PBE0), GP(HF) and PCM(HF) structures even in the complexes that are involved in R1-OH, meaning that we may now propose properly determined logKexc,i values for all reactions. Also, we now have another logKexc,2 value, associated with the R2-OH equilibrium. Those two new values, together with our previous best estimates (FCS + 1) are reported in Table 3.
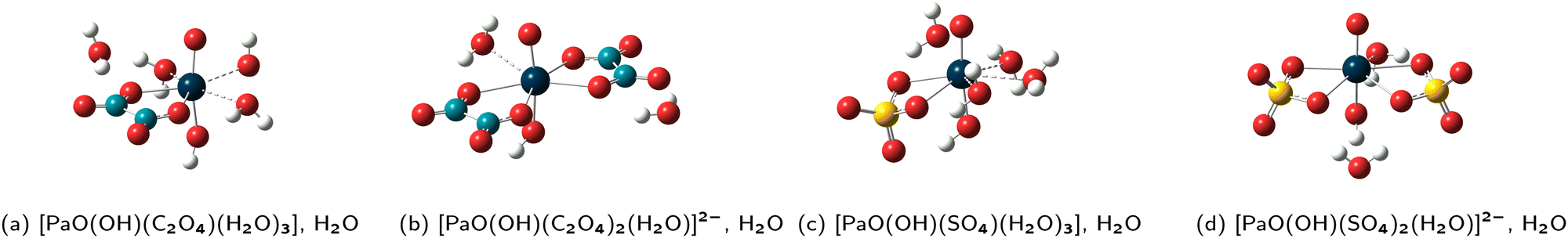 | ||
| Fig. 3 Representations of the GP(DFT/PBE0) structures of the complexes involved in the R1-OH and R2-OH reactions, obtained with one more water than just saturated first-coordination spheres (FCS + 1) (a)−(d). Color code: dark blue (Pa), yellow (S), red (O), light blue (C) and white (H). | ||
|
|
Comput. | Expt. |
|---|---|---|
| R1/R1-OH | —/7.0 | 3.5 |
| R2/R2-OH | 8.8/12.1 | 7.5 |
| R3/R3-OH | 14.6/— | 10.5 |
As can be seen in this table, the values obtained for R1-OH and R2-OH are both quite consistent with the experiment. Moreover, the logKexc,2 − logKexc,1 difference is also in good agreement with the experiment. Even if this agreement, based on the analysis of apparent constants for the experimental values, may not be a definitive proof for the occurrence of the hydroxyl-group scenario in the R1 case (more experimental information may be required), we conclude by predicting that this scenario must be at play since our static approach is found to fail at reproducing the trends otherwise.
Concerning the R2 reaction, both scenarios lead to computed logKexc,2 values in decent accord with the experiment. Therefore, it is hard to know a priori if the hydroxyl group is maintained or withdrawn in the 1:2 complexation. Moreover, being in fact an intermediate situation sandwiched between the two clear R1 and R3 cases, it could happen that the hydroxyl group is maintained in the sulfate complex and not in the oxalate one. Unfortunately, this cannot be well probed with our calculations since we need to have similar pictures in both the left-hand and right-hand sides of the reactions to favor error cancellations, especially on the solvation contributions.
5 Concluding remarks
In this work, we performed a step-by-step study to deduce ligand-exchange reaction constants. The studied equilibria involved Pa(V) complexes with sulfate and oxalate ligands, displaying the characteristic Pa mono–oxo bond, and in some cases hydroxyl groups. Our computational approach is based on the DFT/PBE0 level to determine the gas-phase complexation constants, and on the UAHF model for the solvation corrections. In the absence of an alternative, note that the UFF radius has been used for protactinium.We have seen that solvation of Pa(V) complexes may be tricky, in the sense that the implicit model alone is not sufficient to reach good values, even if relative constants are looked for. Instead, we show that it is needed to at least saturate the coordination sphere of the Pa(V) ion by water addition. Also, in the case of low-coordinated complexes, we reveal that the speciation may be more complex than expected and that the relevant complexes may still display one hydroxyl group.
The determination of the speciation of Pa(V) in non-complexing and in complexing media is still in its infancy. It is extremely hard to firmly identify new chemical species, and we believe that our work, which has led to the validation of a computational protocol, can open the way for future studies assisted by computations. In particular, we have shown that calculations of ligand-exchange reaction constants can be quite informative concerning trends (which ligands is a stronger complexant) and also regarding the occurrence of given chemical groups and/or bonds in the formed complexes.
Author contributions
H. O. and R. M. defined the working hypotheses and the computational strategy. H. O. was responsible for performing and/or checking all the calculations. J. D. performed preliminary calculations concerning the hydroxyl-group scenario. The first draft of the manuscript was written by H. O. and R. M. All the authors contributed to the scientific development of the work and were given the opportunity to amend the manuscript.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
This work was supported by the Région des Pays de la Loire (RCT-PaPAn project) and by the French National Research Agency (ANR-21-CE29-0027, LABEX CaPPA/ANR-11-LABX-0005-01 and I-SITE ULNE/ANR-16-IDEX-0004 ULNE). HPC resources from CCIPL (Centre de Calcul Intensif des Pays de la Loire) were used.References
- R. Wilson, Nat. Chem., 2012, 4, 586 CrossRef CAS PubMed.
- A. V. Viatkina, M. G. Kozlov and V. V. Flambaum, Phys. Rev. A, 2017, 95, 022503 CrossRef.
- P. Naubereit, T. Gottwald, D. Studer and K. Wendt, Phys. Rev. A, 2018, 98, 022505 CrossRef CAS.
- P. Naubereit, D. Studer, A. V. Viatkina, A. Buchleitner, B. Dietz, V. V. Flambaum and K. Wendt, Phys. Rev. A, 2018, 98, 022506 CrossRef CAS.
- C. M. Marquardt, P. J. Panak, C. Apostolidis, A. Morgenstern, C. Walther, R. Klenze and T. Fanghänel, Radiochim. Acta, 2004, 92, 445–447 CrossRef CAS.
- C. Le Naour, M. Maloubier and J. Aupiais, Radiochim. Acta, 2022, 110, 481–493 CrossRef CAS.
- C. Le Naour, J. Roques, C. Den Auwer, P. Moisy and J. Aupiais, Radiochim. Acta, 2019, 107, 979–991 CrossRef CAS.
- M. Mendes, S. Leguay, C. Le Naour, S. Hamadi, J. Roques, P. Moisy, D. Guillaumont, S. Topin, J. Aupiais, C. Den Auwer and C. Hennig, Inorg. Chem., 2013, 52, 7497–7507 CrossRef CAS PubMed.
- C. Le Naour, D. Trubert, M. V. Di Giandomenico, C. Fillaux, C. Den Auwer, P. Moisy and C. Hennig, Inorg. Chem., 2005, 44, 9542–9546 CrossRef CAS PubMed.
- M. Mendes, S. Hamadi, C. Le Naour, J. Roques, A. Jeanson, C. Den Auwer, P. Moisy, S. Topin, J. Aupiais, C. Hennig and M. V. Di Giandomenico, Inorg. Chem., 2010, 49, 9962–9971 CrossRef CAS PubMed.
- S. M. De Sio and R. E. Wilson, Inorg. Chem., 2014, 53, 12643–12649 CrossRef CAS PubMed.
- R. E. Wilson, S. De Sio and V. Vallet, Eur. J. Inorg. Chem., 2016, 5467–5476 CrossRef CAS.
- J. Champion, M. Seydou, A. Sabatié-Gogova, E. Renault, G. Montavon and N. Galland, Phys. Chem. Chem. Phys., 2011, 13, 14984–14992 RSC.
- J. Champion, A. Sabatié-Gogova, F. Bassal, T. Ayed, C. Alliot, N. Galland and G. Montavon, J. Phys. Chem. A, 2013, 117, 1983–1990 CrossRef CAS PubMed.
- D.-C. Sergentu, G. David, G. Montavon, R. Maurice and N. Galland, J. Comput. Chem., 2016, 37, 1345–1354 CrossRef CAS PubMed.
- D.-C. Sergentu, D. Teze, A. Sabatié-Gogova, C. Alliot, N. Guo, F. Bassal, I. Da Silva, D. Deniaud, R. Maurice, J. Champion, N. Galland and G. Montavon, Chem. – Eur. J., 2016, 22, 2964–2971 CrossRef CAS PubMed.
- N. Guo, D.-C. Sergentu, D. Teze, J. Champion, G. Montavon, N. Galland and R. Maurice, Angew. Chem., Int. Ed., 2016, 128, 15595–15598 CrossRef.
- M. Di Giandomenico and C. Le Naour, Inorg. Chim. Acta, 2009, 362, 3253–3258 CrossRef CAS.
- J. Ho, Phys. Chem. Chem. Phys., 2015, 17, 2859–2868 RSC.
- B. Gao and Z.-f Liu, J. Chem. Phys., 2004, 121, 8299–8306 CrossRef CAS PubMed.
- B. Gao and Z.-f Liu, J. Phys. Chem. A, 2005, 109, 9104–9111 CrossRef CAS PubMed.
- M. Douglas and N. M. Kroll, Ann. Phys., 1974, 82, 89–155 CAS.
- B. A. Hess, Phys. Rev. A: At., Mol., Opt. Phys., 1986, 33, 3742–3748 CrossRef CAS PubMed.
- G. Jansen and B. A. Hess, Phys. Rev. A: At., Mol., Opt. Phys., 1989, 39, 6016–6017 CrossRef PubMed.
- C. Møller and M. S. Plesset, Phys. Rev., 1934, 46, 618–622 CrossRef.
- A. D. Becke, J. Chem. Phys., 1998, 109, 2092–2098 CrossRef CAS.
- J. P. Perdew, M. Ernzerhof and K. Burke, J. Chem. Phys., 1996, 105, 9982–9985 CrossRef CAS.
- C. Adamo and V. Barone, J. Chem. Phys., 1999, 110, 6158–6170 CrossRef CAS.
- D. A. Pantazis and F. Neese, J. Chem. Theory Comput., 2011, 7, 677–684 CrossRef CAS.
- R. A. Kendall, T. H. Dunning and R. J. Harrison, J. Chem. Phys., 1992, 96, 6796–6806 CrossRef CAS.
- X. Cao and M. Dolg, THEOCHEM, 2004, 673, 203–209 CrossRef CAS.
- F. Weigend and R. Ahlrichs, Phys. Chem. Chem. Phys., 2005, 7, 3297–3305 RSC.
- X. Cao, M. Dolg and H. Stoll, J. Chem. Phys., 2003, 118, 487–496 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery, Jr., J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 16 Revision A.03, Gaussian Inc., Wallingford CT, 2016 Search PubMed.
- F. Neese, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2017, 8, e1327 Search PubMed.
- F. Aquilante, J. Autschbach, A. Baiardi, S. Battaglia, V. A. Borin, L. F. Chibotaru, I. Conti, L. De Vico, M. Delcey, I. Fdez. Galván, N. Ferré, L. Freitag, M. Garavelli, X. Gong, S. Knecht, E. D. Larsson, R. Lindh, M. Lundberg, P. Å. Malmqvist, A. Nenov, J. Norell, M. Odelius, M. Olivucci, T. B. Pedersen, L. Pedraza-González, Q. M. Phung, K. Pierloot, M. Reiher, I. Schapiro, J. Segarra-Mart, F. Segatta, L. Seijo, S. Sen, D.-C. Sergentu, C. J. Stein, L. Ungur, M. Vacher, A. Valentini and V. Veryazov, J. Chem. Phys., 2020, 152, 214117 CrossRef CAS PubMed.
- V. Barone and M. Cossi, J. Phys. Chem. A, 1998, 102, 1995–2001 CrossRef CAS.
- M. Cossi, N. Rega, G. Scalmani and V. Barone, J. Comput. Chem., 2003, 24, 669–681 CrossRef CAS PubMed.
- V. Barone, M. Cossi and J. Tomasi, J. Chem. Phys., 1997, 107, 3210–3221 CrossRef CAS.
- M. V. Di Giandomenico, M. Mendes, C. Le Naour, C. DenAuwer, P. Moisy and C. Hennig, Behaviour of Protactinium(v) in complexing media, Conference: Atalante 2008: Nuclear fuel cycle for a sustainable future, Montpellier (France), 19–23 May 2008, 2008.
- A. J. A. Aquino, D. Tunega, G. Haberhauer, M. Gerzabek and H. Lischka, Phys. Chem. Chem. Phys., 2000, 2, 2845–2850 RSC.
- T. B. Lee and M. L. McKee, Inorg. Chem., 2011, 50, 11412–11422 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Preparatory study on molecular geometries, which includes comparisons of methods, basis sets and codes, and supplemental figure representing the structures of the bare complexes. See DOI: https://doi.org/10.1039/d3cp00323j |
| This journal is © the Owner Societies 2023 |