
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Small chromium-doped silicon clusters CrSin: structures, IR spectra, charge effect, magnetism and chirality†
Bao-Ngan
Nguyen-Ha
 ab,
Ngoc Thach
Pham
c,
Pieterjan
Claes
d,
Peter
Lievens
d,
André
Fielicke
e,
Vu Thi
Ngan
c,
Minh Tho
Nguyen
*ab and
Ewald
Janssens
*d
ab,
Ngoc Thach
Pham
c,
Pieterjan
Claes
d,
Peter
Lievens
d,
André
Fielicke
e,
Vu Thi
Ngan
c,
Minh Tho
Nguyen
*ab and
Ewald
Janssens
*d
aLaboratory for Chemical Computation and Modeling, Institute for Computational Science and Artificial Intelligence, Van Lang University, Ho Chi Minh City, Vietnam. E-mail: ngan.nguyenhabao@vlu.edu.vn; minhtho.nguyen@vlu.edu.vn
bFaculty of Applied Technology, School of Technology, Van Lang University, Ho Chi Minh City, Vietnam
cLaboratory of Computational Chemistry and Modeling (LCCM), Department of Chemistry, Faculty of Natural Sciences, Quy Nhon University, Quy Nhon, Vietnam
dQuantum Solid State Physics, KU Leuven, Celestijnenlaan 200D, B-3001 Leuven, Belgium. E-mail: ewald.janssens@kuleuven.be
eFritz-Haber-Institut der Max-Planck-Gesellschaft, Faradayweg 4-6, 14195 Berlin, Germany
First published on 17th May 2023
Abstract
A series of small chromium-doped silicon clusters CrSin with n = 3–10 in the cationic, neutral and anionic charge states were investigated using quantum chemical methods. The CrSin+ cations with n = 6–10 were produced in the gas phase and characterized by far-IR multiple photon dissociation (IR-MPD) spectroscopy. Good agreement between experimental spectra in the 200–600 cm−1 frequency range and those determined for the lowest-energy isomers by density functional theory calculations (B3P86/6-311+G(d)) provide a strong support for the geometrical assignments. An extensive structural comparison for the three different charge states shows that the structural growth mechanism inherently depends on the charge. While the structures of the cationic clusters are preferentially formed by addition of the Cr dopant to the corresponding pure silicon cluster, it favors substitution in both the neutral and anionic counterparts. The Si–Cr bonds of the studied CrSin+/0/− clusters are polar covalent. Apart from a basket-like Cr@Si9− and an endohedral Cr@Si10− cage, the Cr dopant takes an exohedral position and bears a large positive charge in the clusters. The exohedrally doped clusters also have a high spin density on Cr, manifesting the fact that the intrinsic magnetic moment of the transition metal dopant is well conserved. Three CrSin clusters have a pair of enantiomeric isomers in their ground state, namely the cationic n = 9 and the neutral and anionic n = 7. Those can be distinguished from each other by their electronic circular dichroism spectra, calculated using time-dependent density functional theory. Those enantiomers, being intrinsically chiral inorganic compounds, might be used as building blocks of optical-magnetic nanomaterials because of their high magnetic moments and ability to rotate the plane of polarization.
1. Introduction
Miniaturization plays an important role in current technological developments, and attracts much attention in both fundamental research and industrial applications.1 The demand of consumers for novel and cost-effective materials with salient, tailored properties has been, and still is, triggering an explosive advancement in the use of nanomaterials including assemblies of nanoparticles and small clusters. In this context silicon clusters have been studied extensively. However, due to the intrinsic dangling bonds,2 the thermodynamic stability of pure silicon clusters turns out to be relatively low. Since the silicon atom favors sp3 hybridization, pure silicon clusters exhibit geometries much different from their carbon analogues, and their inherent low stability means they are not usable as building blocks for nanomaterials.3 Due to the peculiar properties of electrons in nd subshells, transition metal dopants can stabilize silicon clusters by fundamentally transforming their geometric and electronic structures.4 Of the 3d transition metals, chromium emerges as an interesting dopant thanks to its high magnetic moment caused by its half-filled 3d electron shell. This, for instance, has led to the use of Cr–Si alloys in resistors and microelectronic devices.5–8The structures of several small and medium anionic CrSin− clusters were assigned by a combination of photoelectron spectroscopy and density functional theory (DFT) calculations. The magnetic moment of the Cr atom in Cr@Si10− is proven to be quenched due to its encapsulation by a Si10 cage, whereas the stable exohedral geometries for CrSin− with n = 3–9 tend to exhibit significant magnetic moments ranging from 3 to 5μB.9 The endohedral Cr@Si14− anion in a doublet state with a C2v geometry is the most stable cluster of the multiply Cr-doped CrnSi15−n− (n = 1–3) series, whereas the Cr2Si13− and the Cr3Si12− possess Cs and D6d symmetric structures with magnetic moments of 3 and 7μB, respectively.10 While the CrSin− anions with n = 14 and 15 exhibit fully endohedral structures, larger CrSin−n = 16–18 anions prefer a fullerene-type Si14 cage encapsulating a Cr atom, with the extra Si atoms added on the fullerene surface.11 Due to the lack of experimental studies and dedicated computations, the structures and properties of neutral CrSin and cationic CrSin+ clusters are not yet well established, especially their magnetic moments and the size at which an exohedral–endohedral transition occurs.
Earlier studies of neutral CrSin clusters, under a severe constraint of the lowest spin singlet state, claimed that a fully endohedral structure of CrSin does not exist with n < 12.9,12,13 The magic neutral Cr@Si12 cluster was determined to have an endohedral hexagonal prism structure with a completely quenched magnetic moment.14,15 This structure was also confirmed for the anionic CrSi12− by photoelectron spectroscopic study (PES),16 but the interpretation of its electronic properties has not reached a consensus yet.14,17–19
For the cations, Beck20 conducted in 1987 the first experiments generating transition metal doped silicon cluster cages M@Sin, with M = Cr, Mo, W, in a supersonic molecular beam20 and drew a conclusion about a particular stability of the Cr@Si15+ and Cr@Si16+ cations,21 which was confirmed in 2006 for related clusters by photofragmentation mass spectrometry experiments.22 In another experiment using mass-selection in a time-of-flight mass spectrometer, the Cr@Si15+ and Cr@Si16+ clusters were reported that they dissociate via the loss of silicon, producing smaller Cr–Si species, enhancing for encapsulated Cr structures, whereas CrSi7+ prefers a primary loss of the Cr+ ion and a neutral Si7 unit, thus enhancing for an exohedral structure.23 A Frank–Kasper polyhedral structure was suggested for the Cr@Si16+ cation based on X-ray absorption spectroscopy.24 This Cr@Si16+ structure re-confirms and further points out that an occupied 1H state in the case of Cr@Si16+ compensates for the “non-magic” part within the spherical potential model; hence an electron shell closure is not a necessary condition to stabilize a cage-like structure as ground state.25 Cr@Si11+ was identified as the smallest endohedral CrSin+ cluster,26 but the structures or smaller cationic CrSin+ clusters are still elusive.
Previous joint experimental and computational studies using infrared multiple photon dissociation (IR-MPD) spectroscopy, far-infrared-vacuum-ultraviolet two color ionization spectroscopy, and DFT calculations have successfully assigned the geometrical structures of different metal clusters such as the Au clusters,27–29 the recently discovered NbAl8H8+ cluster,30 cationic and neutral metal-doped silicon clusters M@Sin with Cu, V, Mn, Co, Ag, Au, Nb…31–41 and pure cationic (n = 6–11, 13–15, 18)42 and neutral Sin (n = 6–10, 15)43,44 clusters.
In the current work, a comprehensive analysis of cationic Cr-doped silicon clusters through experimental and computational methods is carried out. IR-MPD experiments are conducted on the CrSin+n = 6–10 cations, providing experimental benchmark data for the structural assignment of cationic Cr-doped silicon clusters. DFT computations are employed to investigate the electronic and geometric structures of cationic as well as neutral chromium doped silicon clusters for sizes n = 3–10. The anions, whose structures have already been experimentally and theoretically established in literature,9 are re-optimized in this study at the same theoretical level as used for the cationic and neutral clusters, allowing a consistent comparison in energetic parameters, growth mechanisms, and other properties for all three charge states. In addition, this study reports the observation of chiral structures in some low-lying Cr-doped Si clusters. Although asymmetric geometries of clusters have been found in several studies by both experimental and theoretical methods alike,45–52 they remain elusive until now,53 especially chiral species with semiconductor relevance.54 This finding motivates the investigation of the properties of chiral chromium-doped silicon clusters.
2. Methods
2.1 Experimental methods
All spectroscopic measurements are performed in a molecular beam setup55 coupled to a beamline of the Free Electron Laser for Infrared experiments (FELIX) user facility.56 The clusters are produced in a dual-target dual-laser vaporization cluster source by pulsed ablation of chromium and silicon plate targets. Cluster–argon complexes are formed by condensation of the vaporized material in a short pulse of He gas containing a fraction (5%) of Ar and cooled in a thermalization channel attached to the source (80 K). Cluster growth in such type of a gas aggregation source does not take place under thermal equilibrium conditions and kinetic trapping of structural isomers in local minima on the potential energy surface is possible.57,58The experimental conditions (laser powers, gas pressure, timing) were optimized to produce silicon clusters containing 5–30 atoms with a low concentration of chromium (no or one chromium atom per cluster). Of these produced clusters, those with an exohedral Cr atom are found to absorb Ar most easily, i.e. CrSin+ with n = 6–10.26 It is on those Ar-tagged Cr-doped silicon clusters that the IR-MPD experiments are performed. A typical mass spectrum of CrSin+ is presented in the ESI.†
Resonant absorption of infrared light heats the cluster–argon complexes through internal vibrational redistribution, which may result in desorption of weakly bound Ar atoms from the complexes. Mass, and hence composition specific IR-MPD spectra are constructed by recording the mass spectrometric intensities of the ionic complexes as a function of the FELIX frequency in the 230–560 cm−1 range using a time-of-flight mass spectrometer. From the recorded depletion spectra, IR absorption spectra are established as described previously.55
2.2 Theoretical methods
While density functional theory (DFT) methods are widely used in the determination of cluster structures and properties, no single functional consistently describes all their properties accurately. A specific type of clusters should be studied by using appropriate functionals. In previous studies on singly transition metal doped silicon clusters,18,32,34,59 we found that the hybrid B3P86 functional in conjunction with the 6-311+G(d) basis set is able to well describe their geometric and electronic structures as well as to provide their vibrational spectra in good agreement with experiments. For the sake of facilitating a comparison with these previous studies, we again choose the B3P86 functional and the 6-311+G(d) basis set to investigate the geometries and vibrational spectra for the CrSin series in the singly charged cationic and anionic as well as neutral states.Because the energy differences between isomers are often small, a further comparison of relative energies is performed using the correlation consistent aug-cc-pVTZ basis set with the same B3P86 functional in some cases. Results presented in Table S1 of the ESI† show that most of the series of clusters considered hold the same energetic ordering of isomers at both levels of theory, except for the cationic CrSi6+, neutral CrSi7, CrSi10 and anionic CrSi7− in which some small changes in the energetic ordering occur. Unless otherwise noted, the relative energy values quoted in the following sections are obtained at the B3P86/6-311+G(d)+ZPE level. Due to abundance of local minima on the potential energy surface of each cluster size, we include only the lowest-lying isomers whose relative energies are in proximity of the most stable isomer (with a relative energy of <1 eV).60,61
For the location of the cationic CrSin+ and neutral CrSin isomers, initial guess structures are generated by considering stable geometries of cationic, anionic, neutral metal and non-metal doped Si clusters reported in the literature.3,9,31,32,34,39,40,59,62–69 Additionally, geometries of the pure Si clusters in different charge states taken from previous studies31,34,37,42,44,70–73 are used as starting structures on which the Cr atom is added at various positions or an atom of the Si frame is replaced by a Cr atom. These structures are subsequently optimized in different multiplicities, from the singlet to the septet state for systems with an even number of electrons, and from the doublet to the octet state for systems with an odd number of electrons. In case that a geometry optimization converges in a new structure in a certain spin state, it is then taken for a new optimization cycle in other spin states. In case that similar structures have very small energy differences, they are re-optimized under symmetry constraints to verify the stable geometries. Although a large number of initial structures are created, only a small number, being <15, of true energy minimum structures are attained for each size, confirming that both cationic CrSin+ and neutral CrSin clusters are only stable at some specific geometries (cf. the following section). Geometry optimizations are followed by calculations of harmonic vibrational frequencies to confirm the identity of the optimized stationary points. The latter are then used, without applying any scaling factor, to simulate their vibrational spectra by convoluting each harmonic mode with a Gaussian line shape with a full width at half maximum of 3 cm−1. Harmonic vibrational frequencies are also used for evaluating the zero-point energies (ZPE) that are needed as corrections to relative energies between isomers based on the total energies. Although the ZPE corrections seem to be canceled out since ZPEs of isomers at the same size are roughly similar,74 for the sake of completeness, they are included in the present evaluation.
For the identification of the two enantiomers of a chiral cluster's pair, the time-dependent density functional theory method (TD-DFT) at the B3P86/6-311+G(d) level is used to construct the corresponding electronic circular dichroism (CD) spectra. The L- and the R-notations are used for optical isomers with an up-direction and a down-direction spectrum, respectively. All the quantum chemical calculations are conducted by using the Gaussian 09 package.75
3. Results and discussion
Conventionally, the isomers discussed hereafter are denoted by Xn-isoY where X = C (cation), N (neutral) and A (anion), n = 3–10 identifies the number of Si atoms, and Y = 1, 2, 3… enumerates the isomers according to their increasing relative energy. Accordingly, Xn-iso1 invariably refers to the lowest-lying isomer of the Xn series, and relative energies of other isomers are given with respect to its reference.3.1 CrSi3+/0/−, CrSi4+/0/− and CrSi5+/0/−
Fig. 1 and Fig. S2, S3, S14 (ESI† file) show structures, relative energies and electronic states of the low-lying cationic, neutral and anionic isomers for n = 3, 4 and 5. The most stable isomers of CrSi3+, CrSi3 and CrSi3− share a common planar rhombic shape with the Cr atom placed at an acute angle. Similarly, the lowest-lying isomer of CrSi4+ also has a planar structure with the Cr atom capping an edge of a rhombic Si4 cluster. However, the lowest-lying neutral and anionic CrSi40/− isomers adopted a 3D distorted trigonal bipyramidal structure with the Cr atom situated at an equatorial position. At n = 5, all three charge states of the clusters adopt 3D shapes, and the lowest-lying structure of CrSi5+ has Cr placed at an axial position, while in both neutral and anionic states Cr is located at an equatorial position of the distorted octahedron. | ||
| Fig. 1 Diagram illustrating the structural evolution of cationic, neutral and anionic chromium doped silicon clusters in comparison with the pure silicon clusters. Structures of cationic, neutral, anionic pure silicon clusters are taken from ref. 42–44, 67, 70–73 and re-calculated at the B3P86/6-311+G(d) level. | ||
3.2 CrSi6+/0/−
 | ||
| Fig. 2 IR-MPD spectrum of CrSi6+·Ar (upper panel) and simulated harmonic infrared spectra of low-energy isomers of CrSi6+ (B3P86/6-311+G(d)). Geometric structures of the low-energy isomers are shown on the right. Experimental data points are shown as red crosses, while the full black line corresponds to a three-point running average. | ||
On the other hand, the CsC6-iso2 (6A′, +0.03 eV) shows a better agreement with the experimental IR-MPD spectrum (Fig. 2) because its calculated 383 cm−1 band corresponds to the experimental ∼375–400 cm−1 feature, and the combination of calculated signals at 433, 447 and 451 cm−1 also fits the broad experimental band from ∼425 to ∼460 cm−1. At the B3P86/aug-cc-pVTZ level, the C6-iso2-sextet isomer turns out to be 0.008 eV lower in energy than C6-iso1-sextet. In view of their tiny energy difference, these two isomers are basically degenerate and competitive for the global minimum isomer of the CrSi6+ cluster, in which C6-iso2 appears to match better with the experimental IRMPD spectrum. The C6-iso3 (+0.10 eV) and C6-iso4 (+0.11 eV) isomers in the sextet state also are taken into account since they possess specific IR signals within the observed regions (the broad bands of ∼375–400 cm−1 and ∼425–460 cm−1). However, their spectra insufficiently describe the experiment in the ∼200–350 cm−1 range and therefore are unlikely to contribute to the experimental spectrum.
Overall, of all the studied isomers of CrSi6+, the two degenerate and competitive isomers C6-iso1 and C6-iso2 in their sextet state give the best agreement to the experimental spectrum and may coexist in the experiment.
The lowest-lying neutral CrSi6 isomer is N6-iso1 (C2v, 5B2), which has a shape similar to the anionic A6-iso2 (cf.Fig. 1). Other neutral n = 6 isomers are at least 0.36 eV higher in energy with respect to N6-iso1 (cf. Fig. S9, ESI†). Hence, no other structure effectively competes with N6-iso1 for the ground-state of the neutral CrSi6 cluster.
3.3 CrSi7+/0/−
 | ||
| Fig. 3 IR-MPD spectrum of CrSi7+·Ar (upper panel) and calculated harmonic infrared spectra (B3P86/6-311+G(d)) of low-energy isomers of CrSi7+. | ||
The simulated IR spectra of these three lowest-lying isomers share some similarities. The most striking common feature is the presence of two prominent peaks in the 390–430 cm−1 range, which correspond to normal vibrations of the Si7 frame and fit very well with the intense bands centered at ∼410 and ∼425 cm−1 in the experimental spectrum. However, two other experimental features at ∼340 and ∼375 cm−1 only appear in the calculated spectrum of C7-iso1, while only the spectrum of C7-iso3 displays a sign of the low-intensity experimental signal at ∼485 cm−1.
The infrared spectrum of the global minimum of CrSi7+, the Cr-edge-capped C7-iso1 in a sextet state, matches very well with the experimental IR spectrum (Fig. 3). In view of their similar IR spectra, both C7-iso2 and C7-iso3 isomers, we cannot exclude that they contribute to the molecular beam with a smaller extent. We note that there may be unimolecular rearrangements between these isomers by the hopping of the Cr atom around the core Si cluster.
Concerning the anions that have been analyzed before,9 a pair of stable enantiomers at n = 7 is now discovered, including A7-iso2-L and A7-iso2-R whose structures are similar to the neutral pair N7-iso2-R and N7-iso2-L, respectively (cf. Fig. S10 and S14, ESI†). When recalculating their energies at the B3P86/aug-cc-pVTZ level, there is again a reversed energetic order in such a way that the pair of enantiomers A7-iso2-L/R has the lowest energy, marginally below A7-iso1 in both quartet and sextet states (by only 0.04 eV). As in the neutral state, this anion basically is characterized by a degenerate ground state.
3.4 CrSi8+/0/−
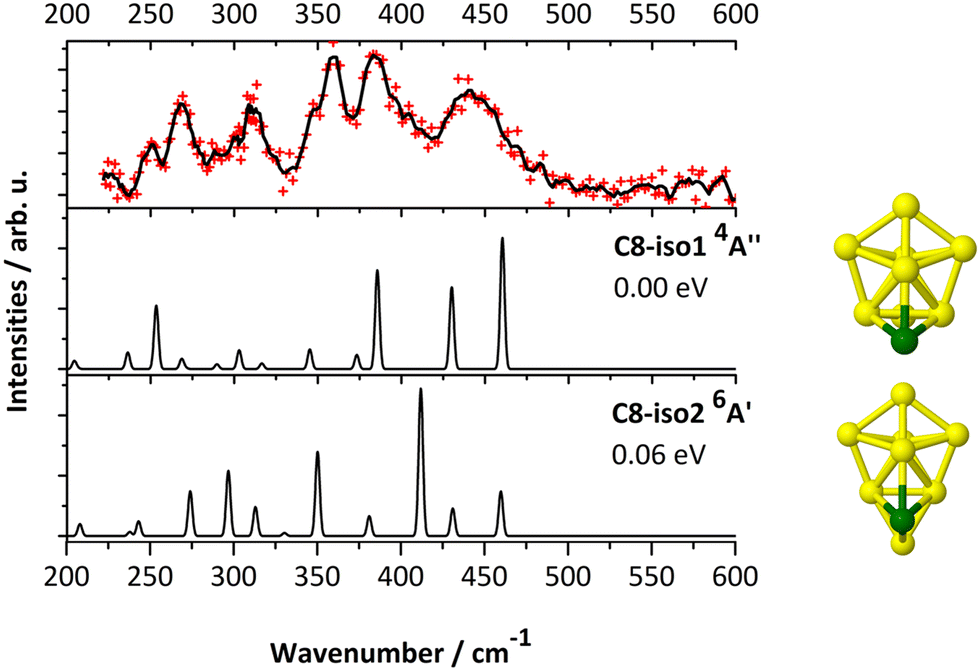 | ||
| Fig. 4 IR-MPD spectrum of CrSi8+Ar (upper panel) and calculated harmonic infrared spectra (B3P86/6-311+G(d)) of low-energy isomers of CrSi8+. | ||
In summary, while the stable structure C8-iso1 (Cs, 4A′′) contributes to the experimental IR-MPD spectrum of the CrSi8+ cation, the presence of the higher spin isomer C8-iso2 (Cs, 6A′) cannot be ruled out.
3.5 CrSi9+/0/−
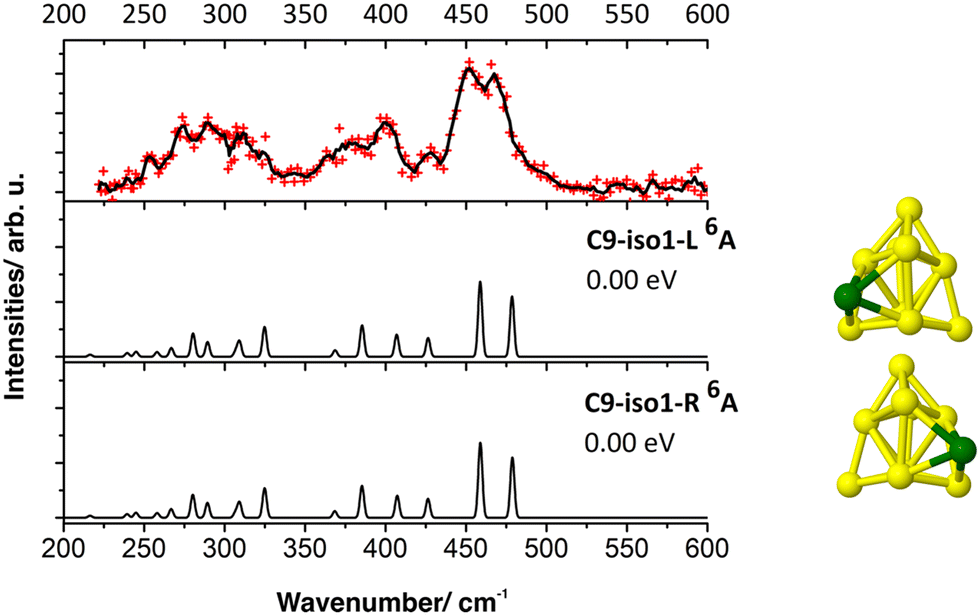 | ||
| Fig. 5 IR-MPD spectrum of CrSi9+·Ar (upper panel) and calculated harmonic infrared spectra (B3P86/6-311+G(d)) of low-energy isomers of CrSi9+. | ||
3.6 CrSi10+/0/−
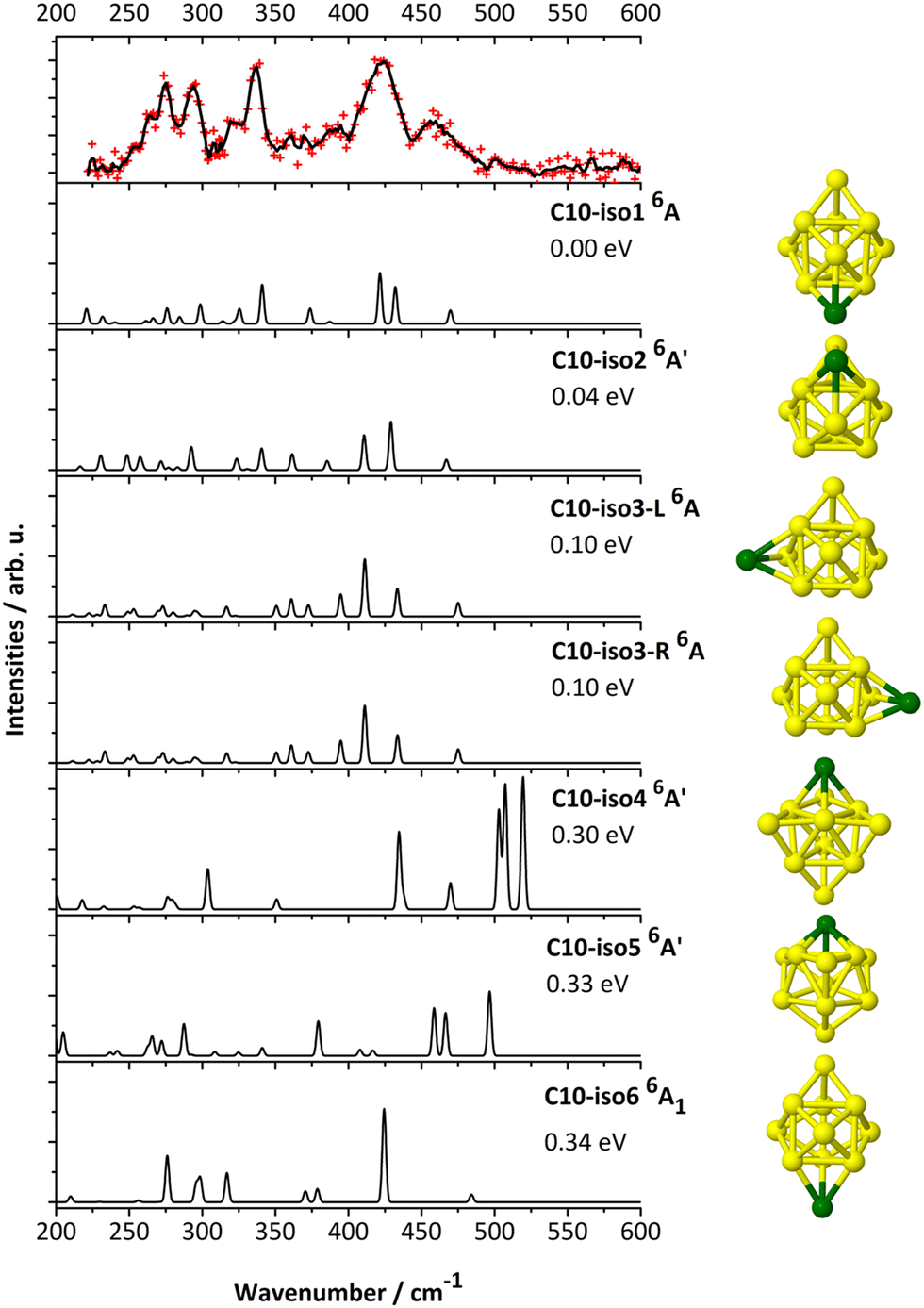 | ||
| Fig. 6 IR-MPD spectrum of CrSi10+·Ar (upper panel) and calculated harmonic infrared spectra (B3P86/6-311+G(d)) of low-energy isomers of CrSi10+. | ||
The computed spectra of both C10-iso1 and C10-iso2 (+0.04 eV) agree best with the experimental data. The highest intensity experimental band, peaking at ∼420 cm−1 corresponds to the two modes centered at ∼422 and ∼432 cm−1 of C10-iso1, ∼410 and ∼429 cm−1 of C10-iso2, ∼411 and ∼433 cm−1 of C10-iso3-L/R, or with the degenerate peak at ∼425 cm−1 in C10-iso6. All isomers have vibrational modes that agree with the broad experimental band between ∼445 and ∼470 cm−1. Only the C10-iso1 and C10-iso2 sufficiently reproduce the 225 to 400 cm−1 low-frequency range. In view of the small relative energy difference between C10-iso1 and C10-iso2 and their comparable spectra, it is reasonable to again assume a co-existence of both isomers in the molecular beam.
While the assigned cationic and neutral isomers follow an exohedral motif retaining the Si10 TTP pattern plus a capping Cr atom onto different faces, the anionic A10-iso1 possesses now an endohedral structure in which the Cr dopant is fully encapsulated by a Si cage.9
To get more insight into the similarity of the structure of C10-iso1 and N10-iso1 isomers, their molecular orbitals (MOs) are analyzed. The electron configuration of C10-iso1 can be described as: [(1S)2(1P)6(1Da)2(1Db)4(1Dc)4(2S)2(1Fa)4(2Da)4(1Fb)6(2P)6//(2Db)1(2Dc)1(2Dc)1(1Fc)1(1Fc)1] or [(1S)2(1P)6(1D)10(2S)2(2P)6(1F)12(2D)7] while that of N10-iso1 as:
[(1S)2(1Pa)2(1Pb)4(1Da)2(1Db)8(2S)2(1Fa)2(2Da)2(2Db)2(1Fb)4(2Pa)4(1Fc)4(2Pb)2(2Dc)2//(2Dc)1(2Dd)1(1Fd)1(1Fd)1] or [(1S)2(1P)6(1D)10(2S)2(2P)6(1F)12(2D)8] (cf.Fig. 7), indicating that an electron on a 2D shell of the neutral isomer is removed to form the cationic counterpart. (2Db)1(2Dc)1(2Dc)1(1Fc)1(1Fc)1 and (2Dc)1(2Dd)1(1Fd)1(1Fd)1 are singly occupied MOs of the cationic and neutral CrSi10 cluster, respectively. The subscripts a, b, c and d denote for the lifting of the degeneracy of the high angular momentum states (1P, 1D, 2P, 2D and 1F) due to the cluster's non-spherical symmetry.
 | ||
| Fig. 7 Calculated density of states (DOS) of cationic CrSi10+ (A) and neutral CrSi10 (B) and the projection of the DOS onto atomic orbitals (AO) of Si and Cr. Positive and negative DOS represent spin-up and spin-down electrons, respectively. | ||
3.7 Wavenumber region for motions of Cr dopant
Given the higher mass of the Cr dopant, vibrational modes involving the motion of Cr are mainly located in the wavenumber region below 200 cm−1. Vibrations in the wavenumber range from 200 to 600 cm−1 are mainly complex combinations of Si–Si vibrations. But the exact location of the Cr dopant atoms influences the nuclear movement of the Sin frame as a whole. Therefore the measured IR spectra are sensitive to the position of the dopant atom and the spectra of the chromium doped clusters as different from both the pure Sin+ (n = 6–10) cations42 and Sin (n = 6, 7,10) neutral43 clusters.3.8 Growth mechanisms
Fig. 1 compares the structures of CrSin+, CrSin and CrSin− with each other and with the corresponding pure silicon clusters. Overall, the most stable structures follow a consistent trend in which the Cr dopant is either adding onto, or replacing a Si atom in the stable pure silicon cluster with the same number of Si atoms. A few sizes do not follow this pattern, and they are presented at the bottom row of the figure.All assigned cationic clusters whose IR spectra match well with experiment, prefer a Cr addition onto either an edge, a face or a vertex of the pure Sin0/+. In view of the expected small differences in the interaction energies arising from these additions, several quasi-energetic isomers emerge.
Different from the cations, the neutral and anionic counterparts tend to a characteristic substitution. At the sizes of n = 3–9, the Cr atom prefers to replace various Si positions of the Sin+1 framework in both neutral and anionic states. However, at n = 10, while the neutral acts to retain the TTP frame, an exohedral-to-endohedral structural change occurs for the anion. The different growth mechanisms of the three charge states strengthens the viewpoint that the structural motif of a cluster is strongly impacted by, or even a manifest of, the total charge.
3.9 Relationship between charge effects, magnetisms and bonding
Natural electron configurations (NEC) and spin densities are evaluated by using the NBO 5.G program76 at the B3P86/6-311+G(d) level. The average binding energies and fragmentation energies are calculated at the same level to probe the stability of clusters. The formulas employed are set forth as follows:| Eb-C = [nE(Si) + E(Cr+) − E(SinCr+)]/(n + 1) | (1) |
| Eb-N = [nE(Si) + E(Cr) − E(SinCr)]/(n + 1) | (2) |
| Eb-A= [(n − 1)E(Si) + E(Si−) + E(Cr) − E(SinCr−)]/(n + 1) | (3) |
| DC(Cr+) = E(Sin,non-relaxed) + E(Cr+) − E(SinCr+) | (4) |
| DN(Cr+) = E(Sin−,non-relaxed) + E(Cr+) − E(SinCr) | (5) |
| DN(Cr) = E(Sin,non-relaxed) + E(Cr) − E(SinCr) | (6) |
| DA(Cr) = E(Sin−,non-relaxed) + E(Cr) − E(SinCr−) | (7) |
| DC/N/A(Si) = E(Si(n−1)Cr+/0/−) + E(Si) − E(SinCr+/0/−) | (8) |
The Wigner–Witmer spin conservation rule is applied to calculate both the average binding energy and fragmentation energy as it was suggested to give reasonable results in the case of CrSi14.77 The removed Si atoms have similar positions in the corresponding structures of the cationic, neutral and anionic clusters considered. The Cr+ cation and Cr atom are removed from the cationic and anionic isomers, respectively. In case of the neutral clusters, both removal of the Cr+ cation and the Cr atom are considered.
| Isomer | State | D C/N(Cr+) | D N/A(Cr) | D C/N/A(Si) | E b-C/N/A | Atomic charge on Cr | Electron population on Cr's 3d shell | Electron population on Cr's 4s shell | Spin density of Cr | Total Wiberg bond of Cr | Total Wiberg bond of cleaved Si | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Cation | C3-iso1 | 4B2 | 4.77 | 5.02 | 2.40 | 0.83 | 4.86 | 0.30 | 4.54 | 1.36 | 2.32 | |
| C4-iso1 | 6A′ | 2.10 | 5.70 | 2.74 | 0.84 | 4.92 | 0.23 | 4.82 | 0.66 | 2.23 | ||
| C5-iso1 | 6A′ | 2.58 | 5.98 | 2.95 | 0.77 | 4.90 | 0.29 | 4.62 | 1.04 | 2.79 | ||
| C6-iso2 | 6A′ | 2.19 | 6.55 | 3.11 | 0.82 | 4.94 | 0.21 | 4.73 | 0.74 | 2.41 | ||
| C7-iso1 | 6A1 | 2.32 | 6.20 | 3.27 | 0.83 | 4.96 | 0.20 | 4.87 | 0.55 | 2.83 | ||
| C8-iso1 | 4A′’ | 5.20 | 5.72 | 3.23 | 0.78 | 4.97 | 0.22 | 4.34 | 1.45 | 2.48 | ||
| C9-iso1-L/R | 6A | 2.42 | 6.33 | 3.33 | 0.75 | 5.03 | 0.18 | 4.54 | 0.91 | 2.76 | ||
| C10-iso1 | 6A | 2.52 | 7.07 | 3.42 | 0.82 | 4.96 | 0.20 | 4.71 | 0.74 | 2.98 | ||
| Neutral | N3-iso1 | 5A1 | 6.78 | 3.69 | 4.96 | 2.59 | 0.66 | 4.94 | 0.38 | 4.67 | 1.22 | 2.74 |
| N4-iso1 | 5B2 | 6.76 | 2.87 | 5.62 | 2.89 | 0.61 | 4.98 | 0.33 | 4.56 | 1.32 | 2.65 | |
| N5-iso1 | 5B1 | 7.27 | 4.22 | 6.07 | 3.12 | 0.58 | 4.99 | 0.36 | 4.47 | 1.50 | 2.85 | |
| N6-iso1 | 5B2 | 6.77 | 3.25 | 5.92 | 3.25 | 0.64 | 5.05 | 0.25 | 4.44 | 1.34 | 2.79 | |
| N7-iso1 | 5A′ | 7.44 | 4.14 | 5.67 | 3.26 | 0.44 | 5.04 | 0.40 | 4.32 | 1.76 | 2.63 | |
| N8-iso1 | 5A′′ | 6.40 | 3.85 | 5.67 | 3.30 | 0.58 | 5.14 | 0.22 | 4.31 | 1.38 | 2.73 | |
| N9-iso1 | 5A | 6.82 | 4.15 | 6.40 | 3.36 | 0.56 | 5.10 | 0.23 | 4.18 | 1.60 | 3.02 | |
| N10-iso1 | 5A′ | 6.14 | 2.80 | 6.09 | 3.43 | 0.67 | 5.04 | 0.27 | 4.59 | 1.02 | 2.68 | |
| Anion | A3-iso1 | 6A1 | 3.06 | 5.69 | 2.80 | 0.30 | 4.99 | 0.66 | 5.08 | 1.04 | 2.96 | |
| A4-iso1 | 4A2 | 5.34 | 6.15 | 3.00 | 0.42 | 5.09 | 0.40 | 4.38 | 1.81 | 2.78 | ||
| A5-iso1 | 6A′ | 3.37 | 7.07 | 3.29 | 0.46 | 5.05 | 0.44 | 4.87 | 1.03 | 2.84 | ||
| A6-iso1 | 4A1 | 7.42 | 7.29 | 3.37 | 0.24 | 5.04 | 0.57 | 4.15 | 2.41 | 3.07 | ||
| A7-iso1 | 6A′ | 4.40 | 6.39 | 3.38 | 0.28 | 5.14 | 0.41 | 4.51 | 1.48 | 3.06 | ||
| A8-iso1 | 6A1 | 3.24 | 6.33 | 3.45 | 0.52 | 5.21 | 0.23 | 4.54 | 1.03 | 2.98 | ||
| A9-iso2 | 6A | 3.75 | 6.19 | 3.50 | 0.43 | 5.23 | 0.23 | 4.43 | 1.21 | 3.06 | ||
| A10-iso1 | 2A | 10.73 | 6.33 | 3.55 | -2.64 | 7.77 | 0.45 | 0.81 | 4.22 | 2.91 | ||
 | ||
| Fig. 8 Fragmentation energy to remove either Cr or Cr+ (A) and total Wiberg bond index of Cr (B), in the lowest-energy cationic, neutral and anionic lowest-lying CrSin+/0/− isomers with n = 3–10. Calculated values are obtained from B3P86/6-311+G(d) + ZPE computations. | ||
The Si–Si bond dissociation energy depends mainly on the charge and structure of the cluster. Overall, for the small sizes n = 3–5 the trends for the dissociation of a Si atom are similar in the cationic, anionic and neutral species (cf.Fig. 9A). These small-sized clusters have low binding energies per atom. In the n = 6–8 size range, either neutral or anionic, the Si–Si dissociation energy is related to the strong binding of the pentagonal bipyramidal frame. Removal of a Si atom located at a vertex of the pentagonal bipyramid turns out to be more difficult than a Si atom capped onto a face or an edge-doped position. Properties of a pair of neutral and anionic clusters become totally different from each other at the size of n = 10, due to the large structural difference between the exohedral (neutral) and the endohedral (anion) cluster.
 | ||
| Fig. 9 Fragmentation energy to remove a Si atom (A) and total Wiberg bond index of dissociated Si (B), in the lowest-energy cationic, neutral and anionic lowest-lying CrSin+/0/− isomers with n = 3–10. Calculated values are obtained from B3P86/6-311+G(d) + ZPE computations. | ||
In the case of cationic clusters, there are two opposite tendencies in the DC(Cr+) and the DC(Si) curves (cf.Fig. 8A and 9A), demonstrating that if the Si–Si bond is getting stronger, it is easier to remove a Cr atom from the Si cluster.
In summary, while the Cr and Cr+ dissociation energies are impacted by the total Wiberg bond index and the multiplicity, the strength of Si–Si bonds is more affected by the cluster's charge and structure. A cluster becomes thermodynamically more stable when its size increases. Generally, the average binding energies of cationic, neutral and anionic clusters become larger from n = 3 to n = 10 except for C8-iso1, C8-iso2 (Fig. 10A and Table S3, ESI†), showing that the cationic CrSi8+ is slightly less stable.
 | ||
| Fig. 10 Average binding energy per atom (A) and charges on Cr atom (B), in the lowest-energy cationic, neutral and anionic lowest-lying CrSin+/0/− isomers with n = 3–10. Calculated values are obtained from B3P86/6-311+G(d) + ZPE computations. | ||
In Fig. 10A, the ordering Eb-A > Eb-N > Eb-C demonstrates the important role of the charge state on the stability. Moreover, the adiabatic ionization energies (IEa) and the adiabatic electron affinities (EAa) of the lowest-lying neutral isomers in Table 2 (cf. Table S4 for other structures, ESI†) are also significantly size-dependent. While the IEa values amount to 7–8 eV, the EAa are relatively large, being in the range of 2.5 to 3.3 eV, indicating the high stability of the anions.
| Neutral structure | IEa (eV) | AEa (eV) |
|---|---|---|
| N3-iso1 | 8.15 | 2.80 |
| N4-iso1 | 7.23 | 2.54 |
| N5-iso1 | 7.41 | 3.10 |
| N6-iso1 | 7.50 | 2.52 |
| N7-iso1 | 7.52 | 2.79 |
| N8-iso1 | 7.41 | 3.28 |
| N9-iso1 | 7.87 | 3.31 |
| N10-iso1 | 6.62 | 3.05 |
The spin densities on the Si atoms turn out to be very small or even insignificant (∼ 0.0–0.3 e) in comparison to that of Cr, indicating that electrons on Si centers are actually paired. The number of electrons on the atomic orbital (AO) of AO-3s(Si) decrease by ∼0.3–0.5 e whereas they increase in AO-3p(Si) by ∼0.3–0.7 e, indicating the typical hybridization between the 3s and 3p orbitals of Si atoms.10
In terms of magnetic moments, charge distribution calculations (cf.Table 1) show that the index of electron occupation on Cr(3d) shells of the cationic cluster amounts to ∼5 e, illustrating an occupancy of five single electrons on the 3d-AOs, while the electron density on the Cr (4s) shells are equal to only ∼0.2 e, showing the near absence of electrons in the 4s orbitals. Such an electron configuration, combined with the local spin densities on Cr (∼ 4.5–5.0), elucidates why these CrSin+ cations are more stable in the sextet state and it confirms that the magnetic moments are mainly located on Cr. The magnetic moments are equally concentrated mainly on the Cr atom for exohedral structures of neutral and anionic isomers as well (the local spin densities on the Cr atom being ∼4.2–4.6 for neutral isomers with sizes n = 3–10 and ∼4.2–5.1 for anionic isomers with sizes n = 3–9), highlighting the high magnetic moments majorly located on Cr counterparts, except for the endohedral A9-iso2 and A10-iso2 whose local spin densities on Cr are approximately equal to 1.
The net atomic charges of Cr (0.7–0.8 e for cations, 0.4–0.6 e for neutral, 0.2–0.5 e for anions excepted A10-iso1) (cf.Table 1) show that the total charges are not completely localized on Cr atoms but rather dispersed over the cluster.
3.10 Chirality
It has been established that a molecule may be chiral, and thereby optically active, only if it does not possess an improper rotation axis.78 There are four different types of chirality in inorganic nanostructures,79 namely, (1) asymmetry of the inorganic core of the nanoparticles, (2) chirality of the inorganic surface, (3) chirality of the stabilizer shell, and (4) chirality by a chiral field effect. It appears that in the series considered here, the Cr-doping leads to chiral enantiomers that exhibit an intrinsic chirality of the cluster core, and can thus be assigned as a first type inorganic nanoscale chirality. Exohedral geometries combined with bond length difference between Si–Cr and Si–Si often lead to a symmetry breaking.A cluster at different multiplicity may have different geometric symmetries. In some cases, its structure is lowered to a C1 point group inducing chiral structures. For example, in the case of the pair of C8-iso5-L and C8-iso5-R isomers, they are stable enantiomers of each other at the sextet state, but they converge to the same structure at the quartet state bearing a Cs point group (cf. Fig. S6, ESI†).
As presented above, we discover in the present study three ground state enantiomeric pairs corresponding to the cationic CrSi9+ (C9-iso1-L/R), the neutral CrSi7 (N7-iso2-L/R) and the anionic CrSi7− (A7-iso2-L/R) (cf.Fig. 11). These exohedral enantiomers can be built up by interlocking the trigonal bipyramidal blocks together and the Cr atom is located at an equatorial vertex of one of these interlocking trigonal bipyramidal blocks.
 | ||
| Fig. 11 Electronic circular dichroism (ECD) spectra of the lowest-lying energy enantiomers (A): CrSi9+, (B): CrSi7 and (C): CrSi7− calculated by the TD-B3P86/6-311+G(d) method. | ||
Evidently, the two clusters of an enantiomeric pair share with each other an identical IR spectrum as well as undistinguishable UV-Vis spectra (cf. Fig. S17, ESI†). In order to distinguish them, the electronic circular dichroism (ECD) spectra are calculated by using the TD-DFT method at the B3P86/6-311+G(d) level. The ECD spectra of three ground state enantiomers are presented in Fig. 11, while the ECDs of the other pairs are presented in Fig. S18 (ESI†). As these exohedral enantiomeric structures exhibit both typical optical and magnetic properties, they can be regarded as possible building blocks for opto-magnetic nanomaterials.
4. Concluding remarks
The present combined experimental and theoretical study has led to a number of conclusions:i. First, the exohedral structures with large magnetic moments of the cationic chromium-doped silicon clusters CrSin+ with n = 6–10 have successfully been assigned by combining the IR-MPD spectra of the CrSin+·Ar complexes in the 200 to 600 cm−1 range with DFT calculations using the hybrid B3P86 functional and the 6-311+G(d) basis set.
ii. Second, an extensive structural comparison of the cationic, neutral and anionic clusters shows that the total charge significantly affects their structural evolution. While the Cr atom prefers an addition onto a pure silicon cluster in case of cations CrSin+ (n = 3–10), the metal dopant favors a substitution in the neutral SinCr and anionic SinCr− counterparts, except for the CrSi10− anion which adopts an endohedral structure.
iii. Third, the large magnetic moments of the exohedral clusters (the local spin density of cationic, neutral and anionic clusters is 4.2–5.0) are majorly located on Cr, indicating the highly intrinsic magnetic moment of the metallic atom is well conserved following doping, i.e. a sextet electronic state for cationic and anionic isomers and a quintet electronic state for neutral isomers. The Si–Cr bonds of CrSin+/0/− clusters in three charge states with n = 3–10 are polar covalent bonds. Apart from the endohedral anionic A9-iso2 and A10-iso1 clusters that have a negative charge on the Cr atom, the other considered clusters have exohedral structures with a positive charge centered on the Cr position.
iv. Last but not least, three pairs of lowest-lying energy enantiomers C9-iso1-L/R, N7-iso2-L/R and A7-iso2-L/R whose structures are interlocking the trigonal bipyramidal blocks, are discovered. These stable enantiomers inherently possessing both typical optical and magnetic properties are assigned as intrinsically chiral nanomaterials.
Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
The work of BNNH and MTN is funded by VinGroup (Vietnam) and supported by VinGroup Innovation Foundation (VinIF) under project code VinIF.2020.DA21; they are also grateful to Van Lang University. Quantum chemical computations were carried out using the facilities of Quy Nhon University (VTN). The experimental work was supported by the KU Leuven Research Council (C1 Grant C14/22/103) and the Research Foundation-Flanders (FWO-Vlaanderen, G0A05.19N). The authors gratefully acknowledge the Nederlandse Organisatie voor Wetenschappelijk Onderzoek (NWO) for the experiments carried out at the FELIX Laboratory. AF thanks Gerard Meijer for his continuing support.References
- S. M. Bhagyaraj, O. S. Oluwafemi, N. Kalarikkal and S. Thomas, Synthesis of inorganic nanomaterials: advances and key technologies, Woodhead Publishing, Duxford England, Cambridge, MA, 2018 Search PubMed.
- M. T. Nguyen and B. Kiran, Clusters: Structure, Bonding and Reactivity, Springer International Publishing, Cham, 1st edn, 2017 Search PubMed.
- P. Claes, E. Janssens, V. T. Ngan, P. Gruene, J. T. Lyon, D. J. Harding, A. Fielicke, M. T. Nguyen and P. Lievens, Phys. Rev. Lett., 2011, 107, 173401 CrossRef CAS PubMed.
- S. Neukermans, X. Wang, N. Veldeman, E. Janssens, R. E. Silverans and P. Lievens, Int. J. Mass Spectrom., 2006, 252, 145–150 CrossRef CAS.
- R. K. Waits, Thin Solid Films, 1973, 16, 237–247 CrossRef CAS.
- R. K. Waits, Proc. IEEE, 1971, 59, 1425–1429 CAS.
- D. Caputo, G. de Cesare, A. Nascetti and M. Tucci, Thin Solid Films, 2007, 515, 7517–7521 CrossRef CAS.
- M. Jhabvala, S. Babu, C. Monroy, M. M. Freund and C. D. Dowell, Cryogenics, 2002, 42, 517–526 CrossRef CAS.
- X.-Y. Kong, H.-G. Xu and W. Zheng, J. Chem. Phys., 2012, 137, 064307 CrossRef PubMed.
- B. Yang, H. Xu, X. Xu and W. Zheng, J. Phys. Chem. A, 2018, 122, 9886–9893 CrossRef CAS PubMed.
- K. Wang, H.-Y. Zhao, L. Miao, Z.-Z. Jia, G.-J. Yin, X.-D. Zhu, R. Moro, B. von Issendorff and L. Ma, J. Phys. Chem. A, 2022, 126, 1329–1335 CrossRef CAS PubMed.
- H. Kawamura, V. Kumar and Y. Kawazoe, Phys. Rev. B: Condens. Matter Mater. Phys., 2004, 70, 245433 CrossRef.
- L.-j Guo, G.-f Zhao, Y.-z Gu, X. Liu and Z. Zeng, Phys. Rev. B: Condens. Matter Mater. Phys., 2008, 77, 195417 CrossRef.
- S. N. Khanna, B. K. Rao and P. Jena, Phys. Rev. Lett., 2002, 89, 016803 CrossRef CAS PubMed.
- U. Farooq, S. Naz, H.-G. Xu, B. Yang, X. Xu and W.-J. Zheng, Coord. Chem. Rev., 2020, 403, 213095 CrossRef CAS.
- W. Zheng, J. M. Nilles, D. Radisic and K. H. Bowen Jr., J. Chem. Phys., 2005, 122, 071101 CrossRef PubMed.
- M. B. Abreu, A. C. Reber and S. N. Khanna, J. Phys. Chem. Lett., 2014, 5, 3492–3496 CrossRef CAS PubMed.
- N. D. Phi, N. T. Trung, E. Janssens and V. T. Ngan, Chem. Phys. Lett., 2016, 643, 103–108 CrossRef CAS.
- J. Ulises Reveles and S. N. Khanna, Phys. Rev. B: Condens. Matter Mater. Phys., 2005, 72, 165413 CrossRef.
- S. M. Beck, J. Chem. Phys., 1987, 87, 4233–4234 CrossRef CAS.
- S. M. Beck, J. Chem. Phys., 1989, 90, 6306–6312 CrossRef CAS.
- S. Neukermans, X. Wang, N. Veldeman, E. Janssens, R. E. Silverans and P. Lievens, Int. J. Mass Spectrom., 2006, 252, 145–150 CrossRef CAS.
- J. B. Jaeger, T. D. Jaeger and M. A. Duncan, J. Phys. Chem. A, 2006, 110, 9310–9314 CrossRef CAS PubMed.
- J. T. Lau, K. Hirsch, P. Klar, A. Langenberg, F. Lofink, R. Richter, J. Rittmann, M. Vogel, V. Zamudio-Bayer, T. Möller and B. V. Issendorff, Phys. Rev. A, 2009, 79, 053201 CrossRef.
- D. Palagin, M. Gramzow and K. Reuter, J. Chem. Phys., 2011, 134, 244705 CrossRef PubMed.
- E. Janssens, P. Gruene, G. Meijer, L. Wöste, P. Lievens and A. Fielicke, Phys. Rev. Lett., 2007, 99, 063401 CrossRef PubMed.
- P. V. Nhat, N. T. Si, V. G. Kiselev, A. Fielicke, H. T. Pham and M. T. Nguyen, Chem. Commun., 2022, 58, 5785–5788 RSC.
- P. Ferrari, G.-L. Hou, O. V. Lushchikova, F. Calvo, J. M. Bakker and E. Janssens, Phys. Chem. Chem. Phys., 2020, 22, 11572–11577 RSC.
- P. V. Nhat, N. T. Si, N. T. N. Hang and M. T. Nguyen, Phys. Chem. Chem. Phys., 2022, 24, 42–47 RSC.
- P. Ferrari, H. T. Pham, J. Vanbuel, M. T. Nguyen, A. Fielicke and E. Janssens, Chem. Commun., 2021, 57, 9518–9521 RSC.
- V. T. Ngan, P. Gruene, P. Claes, E. Janssens, A. Fielicke, M. T. Nguyen and P. Lievens, J. Am. Chem. Soc., 2010, 132, 15589–15602 CrossRef CAS PubMed.
- Y. Li, N. M. Tam, A. P. Woodham, J. T. Lyon, Z. Li, P. Lievens, A. Fielicke, M. T. Nguyen and E. Janssens, J. Phys. Chem. C, 2016, 120, 19454–19460 CrossRef CAS.
- X. Li, P. Claes, M. Haertelt, P. Lievens, E. Janssens and A. Fielicke, Phys. Chem. Chem. Phys., 2016, 18, 6291–6300 RSC.
- V. T. Ngan, E. Janssens, P. Claes, J. T. Lyon, A. Fielicke, M. T. Nguyen and P. Lievens, Chem. – Eur. J., 2012, 18, 15788–15793 CrossRef CAS PubMed.
- V. T. Ngan, M. Haertelt, J. Lyon, A. Fielicke, M. T. Nguyen, P. Lievens and E. Janssens, J. Chem. Phys., 2013, 138, 194301 CrossRef PubMed.
- Y. Li, J. T. Lyon, A. P. Woodham, P. Lievens, A. Fielicke and E. Janssens, J. Phys. Chem. C, 2015, 119, 10896–10903 CrossRef CAS.
- P. Gruene, A. Fielicke, G. Meijer, E. Janssens, V. T. Ngan, M. T. Nguyen and P. Lievens, Chem. Phys. Chem., 2008, 9, 703–706 CrossRef CAS PubMed.
- V. Khanna, R. Singh, P. Claes, M. T. Nguyen, A. Fielicke, E. Janssens, P. Lievens and J. E. McGrady, J. Phys. Chem. A, 2022, 126, 1617–1626 CrossRef CAS PubMed.
- J. Zhao, Q. Du, S. Zhou and V. Kumar, Chem. Rev., 2020, 120, 9021–9163 CrossRef CAS PubMed.
- Y. Li, J. T. Lyon, A. P. Woodham, A. Fielicke and E. Janssens, Chem. Phys. Chem., 2014, 15, 328–336 CrossRef CAS PubMed.
- Y. Li, Geometric, Electronic and Magnetic Properties of Transition Metal Doped Silicon Clusters, and Development of a Magnetic Deflection Setup, PhD thesis, KU Leuven, 2016.
- J. T. Lyon, P. Gruene, A. Fielicke, G. Meijer, E. Janssens, P. Claes and P. Lievens, J. Am. Chem. Soc., 2009, 131, 1115–1121 CrossRef CAS PubMed.
- A. Fielicke, J. T. Lyon, M. Haertelt, G. Meijer, P. Claes, J. de Haeck and P. Lievens, J. Chem. Phys., 2009, 131, 171105 CrossRef PubMed.
- M. Haertelt, J. T. Lyon, P. Claes, J. de Haeck, P. Lievens and A. Fielicke, J. Chem. Phys., 2012, 136, 064301 CrossRef PubMed.
- K. R. Krishnadas, L. Sementa, M. Medves, A. Fortunelli, M. Stener, A. Fürstenberg, G. Longhi and T. Bürgi, ACS Nano, 2020, 14, 9687–9700 CrossRef CAS PubMed.
- Q.-Y. Zhang and L. Zhao, Tetrahedron Lett., 2018, 59, 310–316 CrossRef CAS.
- Y. Wang, B. Nieto-Ortega and T. Bürgi, Nat. Commun., 2020, 11, 4562 CrossRef CAS PubMed.
- D. Zerrouki, J. Baudry, D. Pine, P. Chaikin and J. Bibette, Nature, 2008, 455, 380–382 CrossRef CAS PubMed.
- Y. Sato, M. Mitani and H. Yao, Phys. Chem. Chem. Phys., 2019, 21, 14984–14991 RSC.
- A. Lechtken, D. Schooss, J. R. Stairs, M. N. Blom, F. Furche, N. Morgner, O. Kostko, B. von Issendorff and M. M. Kappes, Angew. Chem., Int. Ed., 2007, 46, 2944–2948 CrossRef CAS PubMed.
- X. Gao, B. Han, X. Yang and Z. Tang, J. Am. Chem. Soc., 2019, 141, 13700–13707 CrossRef CAS PubMed.
- G. Deng, B. K. Teo and N. Zheng, J. Am. Chem. Soc., 2021, 143, 10214–10220 CrossRef CAS PubMed.
- J. Kumar, K. G. Thomas and L. M. Liz-Marzán, Chem. Commun., 2016, 52, 12555–12569 RSC.
- Y. Li, T. Higaki, X. Du and R. Jin, Adv. Mater., 2020, 32, 1905488 CrossRef CAS PubMed.
- A. Fielicke, G. von Helden and G. Meijer, Eur. Phys. J. D, 2005, 34, 83–88 CrossRef CAS.
- D. Oepts, A. F. G. van der Meer and P. W. van Amersfoort, Infrared Phys. Technol., 1995, 36, 297–308 CrossRef CAS.
- R. Ferrando, J. Jellinek and R. L. Johnston, Chem. Rev., 2008, 108, 845–910 CrossRef CAS PubMed.
- G. Krishnan, M. A. Verheijen, G. H. ten Brink, G. Palasantzas and B. J. Kooi, Nanoscale, 2013, 5, 5375–5383 RSC.
- Y. Li, N. M. Tam, P. Claes, A. P. Woodham, J. T. Lyon, V. T. Ngan, M. T. Nguyen, P. Lievens, A. Fielicke and E. Janssens, J. Phys. Chem. A, 2014, 118, 8198–8203 CrossRef CAS PubMed.
- E. J. Baerends, V. Branchadell and M. Sodupe, Chem. Phys. Lett., 1997, 265, 481–489 CrossRef CAS.
- S. Pittalis, S. Kurth and E. K. U. Gross, J. Chem. Phys., 2006, 125, 084105 CrossRef CAS PubMed.
- Y. Chen, J. Yang and C. Dong, Comput. Theor. Chem., 2019, 1170, 112635 CrossRef CAS.
- Y. Chen, Y. Liu, S. Li and J. Yang, J. Cluster Sci., 2019, 30, 789–796 CrossRef CAS.
- N. A. Borshch, N. S. Pereslavtseva and S. I. Kurganskii, J. Phys. Conf. Ser., 2019, 1203, 012056 CrossRef CAS.
- C. Dong, L. Han, J. Yang and L. Cheng, Int. J. Mol. Sci., 2019, 20, 2933 CrossRef CAS PubMed.
- Y. Gu, J. Yang and L. Cheng, Int. J. Quant. Chem., 2020, 120, e26087 CrossRef CAS.
- Y. R. Zhao, T. T. Bai, L. N. Jia, W. Xin, Y. F. Hu, X. S. Zheng and S. T. Hou, J. Phys. Chem. C, 2019, 123, 28561–28568 CrossRef CAS.
- L. V. Duong, N. N. Tri, N. P. Hung and M. T. Nguyen, J. Phys. Chem. A, 2022, 126, 3101–3109 CrossRef CAS PubMed.
- S.-J. Lu, X.-L. Xu, G. Feng, H.-G. Xu and W.-J. Zheng, J. Phys. Chem. C, 2016, 120, 25628–25637 CrossRef CAS.
- A. Shvartsburg, B. Liu, M. Jarrold and K.-M. Ho, J. Chem. Phys., 2000, 112, 4517–4526 CrossRef CAS.
- A. Zdetsis, J. Chem. Phys., 2008, 127, 244308 CrossRef PubMed.
- S. Nigam, C. Majumder and S. K. Kulshreshtha, J. Chem. Phys., 2004, 121, 7756–7763 CrossRef CAS PubMed.
- N. M. Tam and M. T. Nguyen, Chem. Phys. Lett., 2013, 584, 147–154 CrossRef CAS.
- E. G. Lewars, in Computational Chemistry: Introduction to the Theory and Applications of Molecular and Quantum Mechanics, ed. E. G. Lewars, Springer International Publishing, Cham, 2016, pp. 9–49 Search PubMed.
- G. W. T. M. J. Frisch, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. Marenich, J. Bloino, et al., Gaussian 09, Rev. A.01, Gaussian, Inc., Wallingford CT, 2009 Search PubMed.
- E. D. Glendening, J. K. Badenhoop, A. E. Reed, J. E. Carpenter, J. A. Bohmann, C. M. Morales and F. Weinhold, Theoretical Chemistry Institute, University of Wisconsin, Madison, WI, 2004 Search PubMed.
- J. He, K. Wu, C. Liu and R. Sa, Chem. Phys. Lett., 2009, 483, 30–34 CrossRef CAS.
- P. W. Atkins and J. De Paula, Atkins' Physical Chemistry, Oxford University Press, Oxford, 2014 Search PubMed.
- W. Ma, L. Xu, A. F. de Moura, X. Wu, H. Kuang, C. Xu and N. A. Kotov, Chem. Rev., 2017, 117, 8041–8093 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: (i) Structures and relative energies of the cationic, neutral and anionic CrSin+/0/− clusters (n = 3–10), (ii) average binding energies, fragmentation energies, ionization energies (IEa), electron affinities (EAa) and natural population analysis for ground states of CrSin+/0− clusters, and (iii) calculated IR, UV-Vis and ECD spectra of low-lying enantiomers of CrSin+/0/− clusters. See DOI: https://doi.org/10.1039/d3cp00317e |
| This journal is © the Owner Societies 2023 |