
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThe SARS-CoV-2 spike protein structure: a symmetry tale on distortion trail†
Inbal
Tuvi-Arad
 * and
Yaffa
Shalit
* and
Yaffa
Shalit
Department of Natural Sciences, The Open University of Israel, Raanana, Israel. E-mail: inbaltu@openu.ac.il
First published on 15th May 2023
Abstract
A preliminary step in the SARS-CoV-2 human infection process is a conformational change of the receptor binding domain (RBD) of its spike protein, characterized by a significant loss of symmetry. During this process, the residues which later on bind to the human angiotensin converting enzyme 2 (ACE2) receptor, become exposed at the surface of the protein. Symmetry analysis of a data set of 33 protein structures from experimental measurements and 32 structures from molecular dynamics simulation, show that the initial state carries clear indications on the structure of the final state, with respect to the local distortion along the sequence. This surprising finding implies that this type of analysis predicts the mechanism of change. We further show that the level of local distortion at the initial state increases with variant's transmissibility, for the wild type (WT) along with past and present variants of concern (WT ∼ alpha < beta < delta < Omicron BA.1), in accordance with the trend of their evolutionary path. In other words, the initial structure of the variant which is most infectious is also the most distorted, making its path to the final state shorter. It has been claimed that the RBD migration of the spike protein is allosterically controlled. Our analysis provides a quantitative support to a major theorem in this respect – that information about an allosteric process is encoded in the structure itself, suggesting that the path of local distortion is related to an allosteric information network.
Introduction
Understanding the structure and course of action of the severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) spike protein is an important scientific challenge. Research in the last three years focused on several variants characterized by different mutations which affect their transmissibility.1–4 All variants of the spike protein demonstrate 3-fold symmetry, and a common mechanism for breaking it. Starting from a closed symmetric structure, one or two receptor binding domains (RBD), at the tip of the spike protein, migrate upwards, and create an open structure that can bind to the human angiotensin converting enzyme 2 (ACE2) receptor.5–8 Consequently, the RBD region becomes asymmetric, while the rest of the protein maintains its original symmetry. Quantifying the changes to the symmetry of the protein during this process is the focus of the current study.In its closed state, the spike protein presented in Fig. 1, is a symmetric homotrimer constructed of two subunits, S1 and S2, covered by glycans. The S1 subunit is responsible for recognizing and binding to the human ACE2 receptor and stabilizing the S2 core. The S2 core contains the fusion machinery of the spike. Binding of S1 to ACE2 exposes the core of S2 and leads to substantial conformational change and membrane fusion. The S1 subunit is constructed of several domains: N-terminal domain (NTD), RBD, and two subdomains (SD1 and SD2), also referred to as C-terminal domains (CTD1 and CTD2).6,7 Short N2R (NTD to RBD) linkers connect the NTD and RBD domains.9 Table S1 in the (ESI†) specifies the residues in each domains. Here we focus on the first stage of the process in which the conformation changes from closed and symmetric (3-Down state) to open and asymmetric (1-Up state). Understanding this process is a key factor in preventing the binding of the spike protein to ACE2. Raghuvamsi et al.10 studied the interaction of the spike protein with ACE2 and suggested that locking the RBD in its closed state, by binding small molecules, would have a therapeutic value. Dokainish et al.,11 found cryptic pockets at the RBD interface. Applying virtual screening methodology, they identified several molecules that may serve as drugs. These can stabilize the protein in an intermediate conformation and prevent the conversion to the 1-Up state.
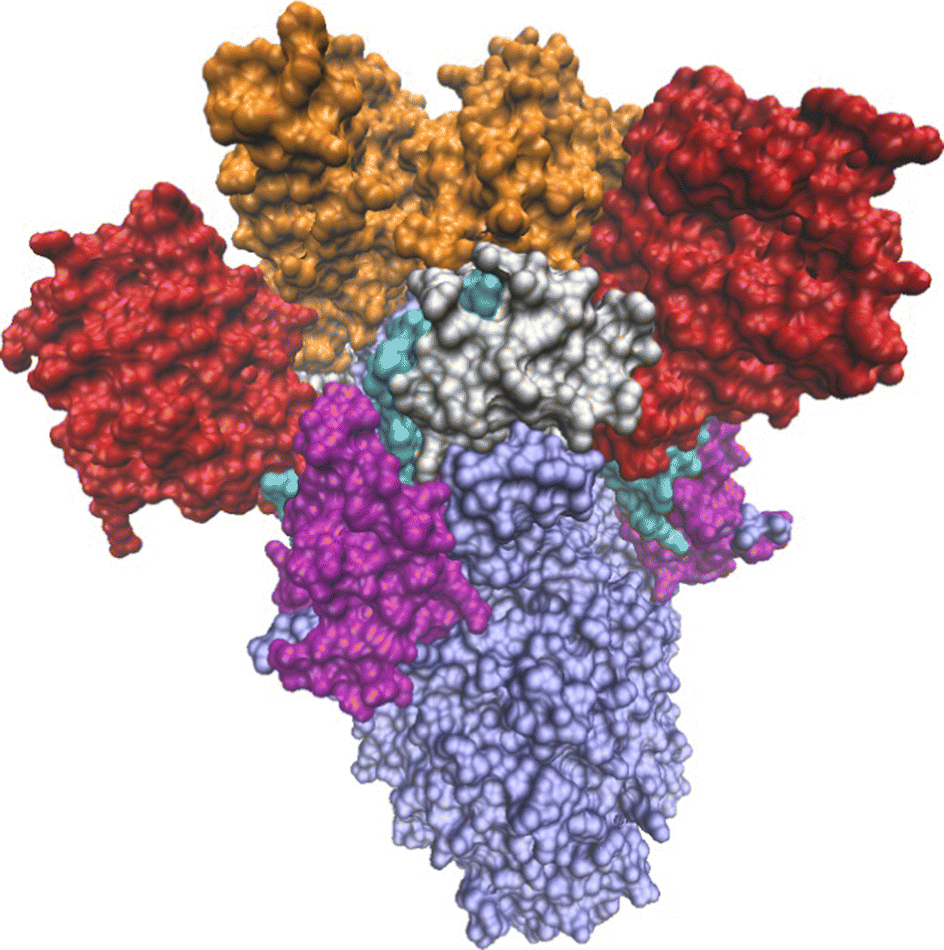 | ||
| Fig. 1 The 3-Down state of the SARS-CoV-2 spike protein of the Omicron BA.1 variant (PDB-ID: 7TF8). The S1 subunit is colored by domains: red-NTD, cyan-N2R, orange-RBD, silver-SD1, purple-SD2. The S2 subunit is colored light blue. | ||
Several studies suggested that the 3-Down to 1-Up conformational change of the Sars-CoV-2 spike protein is allosterically controlled, but the complete mechanism hasn't been fully explored.12–15 Gobeil et al. showed that the D614G mutation in the SD2 domain has an allosteric effect leading to the alteration of the Up/Down RBD conformation.15 They further claimed that variations in remote regions, i.e., the SD1 and SD2 domains, play an essential role in modulating the spike allostery and affecting the conformational change.6 Molecular dynamics (MD) simulations conducted by Verkhivker12 confirmed the role of the D614G mutation in modulating the allosteric communication, and suggested that allosteric coupling between stable regulatory centers and conformationally adaptable hotspots determine the binding affinity and long-range communications of the SARS-CoV-2 complexes with nanobodies.
In a recent review, Thirumalai et al.16 focused on the relation between symmetry and allostery and stressed that while there is high variability of pathways that connect allosteric states, the sensitivity of allosteric signaling to the structure of the specific system, implies that the ability for allostery is encoded in the structure itself. The network of the most dominant residues that transmit the allosteric signal defines an allosteric wiring diagram, that controls the information transfer between the two sites.16 While the variability of pathways that connect the binding site and the allosteric site can be high, the sensitivity of allosteric signaling to the structure of the specific system, implies that the ability for allostery is generally encoded in the apo state. In other words, the structural elements that construct the network of information could potentially be deduced by the structure of the protein in the absence of a ligand.16 Thirumalai et al. describe allosteric signaling as a strain propagation process, and postulate that specific regions in the protein must be stiff enough to absorb this strain. In addition, allosteric states must have lower symmetry than the disordered regions in order to transmit the signal across the structure.16 This view is in line with Papaleo et al.17 that summarized multiple evidences that flexible regions in the protein, such as loops and linkers play an important part in modulating allosteric processes. On the one hand, if the linkers are flexible and pre-encoded conformational sub-states of the linkers exist, they can lower the barriers for conformational transitions between connected domains and accelerate the allosteric process. On the other hand, when these linkers are stiff, they can prevent unfavorable inter-domain contacts, help separate between the domains, and affect the biological function.
Conformational modifications during an allosteric process naturally change the proteins' level of symmetry. This effect can be examined globally (for the whole protein) or locally (for domains or fragments of them). Experimental measurements by Zhang et al.2 showed that the D614G mutation, which is common to all the variants, increases the distortion of the 3-Down conformation of the spike protein, that is, creates a slightly more open conformation as compared with the WT trimer. Quantifying the distortion and the change of symmetry, may improve our ability to understand and control the allosteric mechanism. Continuous symmetry measures (CSMs)18–21 and the related continuous chirality measure (CCM)22 are particularly useful for this purpose. These molecular descriptors estimate the level of distortion of molecules and proteins with approximate symmetry by calculating the distance between a given molecule and its nearest symmetric (or achiral) structure. Recently we modified the method by utilizing the connectivity map of the molecules at hand in order to reduce the number of permutations that the code needs to scan. This modification created a fast, extensive and adequate method to calculate this set of three-dimensional descriptors, that are applicable for both molecules and proteins.21,23 These measures have the capability to capture the most subtle conformational changes during chemical processes.24–28 In recent years the method was used in various studies on protein structure. Bonjack and Avnir29 studied domain swapping with CSMs. They used a running ruler approach to calculate the deviation from C2-symmetry of homodimer fragments in order to identify the hinge region in each protein. Wang et al.30 introduced a chirality spectrum that presents the distance of each residue along the sequence from its nearest achiral structure. They showed that peaks in this spectrum indicate local distortive regions along the protein sequence due to helix kinks, β-sheets twists and secondary structure junctions. Recently, we used the CCM of residues as a conformational similarity descriptor to classify the tendency of amino acids to distort protein homodimers.31 Here we build on the above studies and apply the CSM and CCM methodology to analyze the changes of symmetry during the RBD migration of the SARS-CoV-2 spike protein, in an attempt to reveal the relation between symmetry, structure and function of this protein.
Results and discussion
A set of 63 PDB files were downloaded from the RCSB-PDB,32 representing different electronic microscopy measurements of WT as well as four variants of the SARS-CoV-2-spike protein: Alpha, Beta, Delta and Omicron BA.1 for which reliable data on both 3-Down and 1-Up conformers exist. These were filtered to create a smaller set of 33 spike proteins. Proteins with significant percentages of missing residues were excluded. The Methods section specifies the details of our selection and filtering strategy. Several descriptors were used to analyze the structures: S(C3) of the whole protein and its domains, representing the overall distortion level with respect to the C3 point group, S(C3) of consecutive trimeric fragments of 10 residues along the RBD taken together from the three RBD chains representing local distortion along the RBD sequence, CCM of single residues of single chains along the RBD representing the local conformation of each residue. Glycans and solvent molecules were excluded from the CSM calculation. It should be noted that while the protein is generally covered by glycans, experimental data show that only few glycan molecules exist in the vicinity of the RBD. Table S2 of the ESI† specifies the variants and conformers of our final data set. In what follows we present the CSM analysis of these proteins.Asymmetry is mainly in the RBD
The experimental measurements of the different variants in our set of 33 proteins were not performed in the exact same conditions, and the proteins include different mutations. However, they all display common levels of distortion related mainly to their conformational state. Table 1 presents the mean and standard deviation (SD) of S(C3) for our set of proteins, grouped by conformational state and regardless of the variant. The fact that there are distorted regions was reported in several publications.6–8 Our purpose was to quantify the level of distortion on a normalized continuous scale, and provide means to compare it with respect to the states, subunits and domains.| State | All spike | S1 | S2 | RBD | NTD | SD1 | SD2 | |
|---|---|---|---|---|---|---|---|---|
| 3-Down (15) | Mean | 0.047 | 0.086 | 0.014 | 0.126 | 0.080 | 0.044 | 0.084 |
| SD | 0.044 | 0.082 | 0.009 | 0.112 | 0.079 | 0.052 | 0.080 | |
| 1-Up (18) | Mean | 1.295 | 2.622 | 0.022 | 11.727 | 0.083 | 0.076 | 0.071 |
| SD | 0.178 | 0.420 | 0.009 | 1.000 | 0.037 | 0.033 | 0.064 | |
Table 1 highlights several observations. (1) Looking at the overall protein (“All spike” column), the symmetry of the 3-Down state is higher (smaller CSM values) than the 1-Up state. While this is expected, we note that the first is not zero, meaning that even at the closed state the protein is not perfectly symmetric. This result is in accordance with pervious findings regarding the CSM levels of symmetric proteins.23,31,33 (2) The S1 subunit is more distorted then the S2 subunit, and within the S1 subunit, it is the trimer of the RBD domains that exhibits the highest distortion. This is clearly evident for the open state, and to a smaller extent for the closed, 3-Down, state as well. As will be shown below, the distortion of the RBD trimer carries information about the migration process even before the process started. (3) The distortion levels of both states are similar in regions that maintain high symmetry (S2 subunit and the NTD, SD1 and SD2 domains) teaching that the symmetry of regions outside the RBD are mostly unaffected by the migration process. It should be noted that the CSM is not an additive function. Therefore, the CSM of the whole spike protein need not be equal to the sum of the CSMs of its subunits or domains. Indeed, it displays much more moderate levels of distortion, since the RBD is only a small portion of the overall structure. (4) When the CSM is small, the SD is relatively high. This is a known property of the CSM. As was demonstrated previously, the CSM distribution is commonly asymmetric around the mean, and tends to have a Gamma or Log-Normal distribution with a long tail, portraying structures with relatively high distortion.34 This by definition increases the SD of the CSM, particularly when the range of the CSM is small. Since this happens for relatively symmetric structures, it has a minor affect on the rest of our analysis.
Conformational similarity analysis of the RBD
In order to gain insight into the residues that contribute most to the distortion in each state, we start by analyzing the structure of the RBD domains (between residues N330 and P527)9 of two conformers of the Omicron BA.1 variant measured by electron microscopy, with PDB-ID: 7TF89 (3-Down) and 7TO435 (1-Up), refined by two different laboratories. A snapshot of the 1-Up conformer in and around the RBD trimer is presented in Fig. 2, showing that the boundary residues, P330 and P527 are in close proximity, and remain close to their linked domains even at the open state. Looking at these snapshots, it is clear that the RBD migration involves rotation of the whole domain. As we explain next, this is not the only geometrical change that occurs. | ||
| Fig. 2 A partial snapshot of the Omicron BA.1 spike protein near the RBD region. (a) 3-Down conformation (PDB-ID: 7TF8). (b) 1-Up conformation (PDB-ID: 7TO4). Orange: the migrating RBD (chain A). Residues P330 and P527 (in blue) mark the boundaries of the RBD domain on chain A (P527 is hidden in the 1-Up conformer). Pink and dark red: The other two RBD domains. Silver: SD1 domains. Cyan: N2R linkers between the NTD and RBD domains. | ||
We applied the CCM of each residue (including its side chain), as a three-dimensional conformational similarity descriptor, and analyzed the conformations of the residues along the RBD in order to characterize the changes due to the migrating domain. In previous studies we showed that the variability of the CCM along the sequence of a protein stems from differences in the secondary structures and the side chains.30,31Fig. 3 shows the correlation plot between the CCM per residue on chain A and chain B, for the 1-Up state of the Omicron BA.1 variant shown in Fig. 2. Chain A is the migrating chain. The Pearson correlation factor between the two chains is 0.76. Pearson correlation factors of 0.81 and 0.80 were obtained between chains A and C, and chains B and C respectively. The correlation factors for the 3-Down state were similar: 0.75, 0.76 and 0.79 for A–B, A–C and B–C respectively (see Fig. S1 in the ESI† for the correlation plots). The fact that the correlation is not perfect teaches us that the chain opening process may involve conformational change of the residues apart for the rotation of the whole domain. To draw more conclusive conclusions on this issue, we applied a statistical approach and calculated the Pearson correlation factors to our set of 33 spike proteins. The results are presented in Fig. 4 in the form of box and whisker plots. At the 3-Down state, no significant difference is shown between the different chains and the median correlation factor is 0.84. While the conformation of pairs of equivalent residues on each two chains may differ, these differences spread equally between the three chains. For the 1-Up state, this is not the case. Prior to this analysis the chain names were set such that A is always the migrating chain. Fig. 4 shows that the conformational similarity of residues on chains B and C, which do not migrate, is comparable to the 3-Down state. The correlation factor decreases when the migrating chain A is considered, teaching that the RBD migration involves local conformational changes apart for the domain rotation. CCM analysis at the backbone level, presented in Fig. S2 (ESI†), shows similar trends.
 | ||
| Fig. 3 Correlation between the residues' CCM on chain A and chain B for the 1-Up state of the Omicron BA.1 variant. Red line represents the correlation line. Chain A is the migrating RBD domain. PDB-ID: 7TO4. | ||
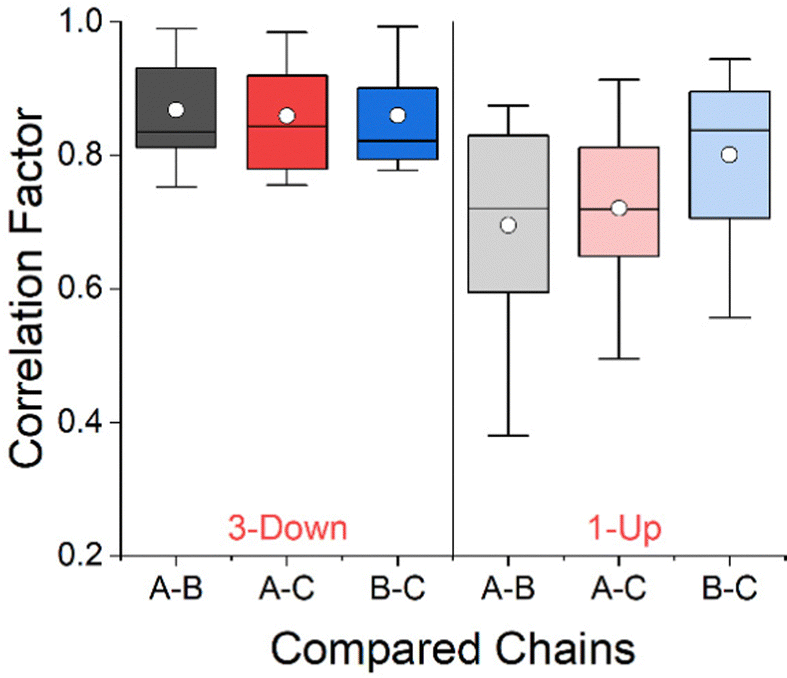 | ||
| Fig. 4 Box and whisker plots of Pearson correlation factors for residues CCM between chains of the RBD. The 3-Down and 1-Up states are represented by 15 and 18 proteins respectively. Box boundaries represent 25–75% of the data, horizontal line within the box is the median and the white circle is the mean value in each box. | ||
The conformational changes are manifested in the secondary structure as well, as can be seen in Fig. S3 in the ESI† that displays similar trends to those in Fig. 4. Another perspective of the structural change comes from looking at the distribution of residues within the different secondary structures. Taking the full set of proteins in our study, regardless of the variant, we found that for the 3-Down conformer, on average, 60% of the residues in the RBD are characterized with flexible secondary structures (loops and irregular elements, bends and turns) that are more likely to deliver structural changes.17 The rest of the residues are more confined: 25% are in β-strands structures and 14% are in different types of helices. For the 1-Up the numbers changes to 65%, 24% and 11% respectively, showing that the conformational change is accompanied by increased flexibility of the RBD backbone on the expanses of decrease in helix type structures. The increase in flexibility supports our finding that the RBD opening mechanisms is more than a pure rotation over a hinge, and involves local conformational changes as well.
Distortion along the RBD sequence
We now turn to analyzing the deviation from symmetry of the three RBD domains. For this purpose, we applied a running ruler approach and calculated S(C3) for consecutive trimeric fragments of 10 residues from all RBD chains (see Methods). Plotting the CSM of these fragments along the sequence creates a CSM spectrum that highlights regions with local distortion.29,30Fig. 5 presents these spectra for the 3-Down (left Y scale) and 1-Up (right Y scale) states of the Omicron BA.1 variant presented in Fig. 2. The CSM calculations were performed with all atoms. Fig. S4 (ESI†) with CSM analysis at the backbone level, shows a similar trend with slightly smaller scale for the 3-Down conformer. Peaks in these spectra represent highly distorted regions while valleys represent regions of relatively conserved symmetry. It is surprising to see that the highest peaks are approximately centered around the same residues in both states. The two peaks in the left part of the spectra are particularly conserved at the open state, while the others are either split to two peaks, or move to the right of the spectrum, resembling a distortion wave that travels from a specific residue to its nearby residues.27 Generally, the RBD migration leads to increased distortion along the whole RBD sequence, but the strength of the distortion varies. Regions that were distorted at the 3-Down state become highly distorted at the 1-Up state, while symmetric regions at the 3-Down state are much less affected by the domain migration and display higher symmetry conservation. This trend is supported both by the general theory of Thirumalai et al.16 on allosteric processes, that stated that the capacity to change is encoded in the original structure, and by the view of Papaleo et al.17 regarding the role of flexible and stiff fragments in transferring information. Applying these concepts here provide an interesting interpretation: Specific residues of the RBD domain, while in the 3-Down state, already carry the information about the conformation that could interact with ACE2, and can thus direct and lead the change.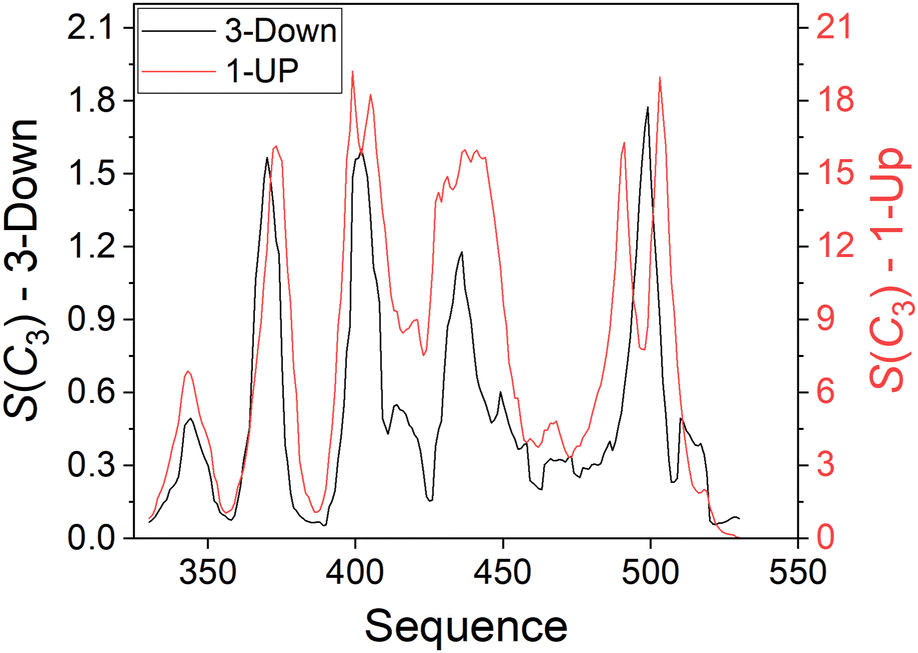 | ||
| Fig. 5 Distortion with respect to C3 symmetry of 10-residues fragments (with all atoms included) along the sequence of the RBD domain of the Omicron BA.1 variant of the SARS-CoV-2 spike protein. Black: 3-Down state, left Y scale (PDB-ID: 7TF8). Red: 1-Up state, right Y scale (PDB-ID: 7TO4). | ||
The nature of the CSM parameter is such that when branches of a symmetric structure drift apart as a whole, the CSM decreases, due to the normalization factor in eqn (1). In Fig. 5 we see the opposite trend – one RBD chain drifts apart from the other two, but the CSM increases, approximately by an order of magnitude. This can be explained by noting that the geometrical change is not a simple translation with respect to the original C3 rotation axis, but involves rotation (as suggested by the snapshots of Fig. 2) along with local conformational changes of the side chains as discussed above.
Several experimental studies and MD calculations suggested that the RBD domain rotates through a hinge, although its specific location is not always specified and varies between measurements and variants6,7,36,37 For the Omicron BA.1 variant, Verkhivker12 showed that several residues act as rigid hinges, i.e., F318, L387, and F429. Our calculations show that these residues are in regions of relatively conserved symmetry even at the 1-Up state. F318 is outside the RBD domain, in region of high symmetry like the rest of the protein. The other two residues are at the vicinity of the second and third minimum points on the left part of the curves in Fig. 5. The numbers on the x axis of the CSM spectrum mark the first index of a 10-residues fragment. Therefore, residues at the minimum points are not the only source for a fragment's CSM value and one shouldn't expect a perfect match. The MD study of Fallon et al.7 on the WT variant showed that a salt bridge between D389 and K528 on the RBD is broken upon RBD rotation. They defined the RBD between residues 335 and 530 which is slightly different then the range used here, but this small difference does not change the shape of the CSM spectra in Fig. 5. D389 is close to L387 found by Verkhivker,12 supporting the concept of a hinge in this region. Analysis of the 3-Down structure in Fig. 5 (PDB-ID: 7TF8) with VMD38 did not reveal a salt bridge between these or adjacent residues, suggesting that the specific locations of such bridges vary between variants. In this respect our approach of analyzing fragments of residues rather than specific residues seems more inclusive. All mentioned residues generally reside in CSM valleys, i.e., with relatively conserved symmetry. Similar trends are seen for the CSM spectra of other variants as well, as discussed below. Finally, we note that the distortion near D389 is higher than near K528, suggesting that movement around D389 on the migrating domain is more significant than around D528.
Having discussed the valleys in the CSM spectrum, we turn to explore its peaks in which the distortion is relatively high. Fig. 6 shows snapshots of the RBD domains for the two states in which residues at the tip of each peak are presented with van der Waals spheres colored by their secondary structure, as determined by VMD.38 Many of the residues in peak regions are located on the surface of the RBD domains in regions responsible for the interaction with the ACE2 receptor. Broadly considered, these regions are bound between G446 (S446 for Omicron BA.1) and V510.3 Generally, the secondary structure in peak regions is similar in both conformers, and as discussed above, they tend to be flexible. It is interesting to note that the peaks spread every ca. 30 residues that lie in close proximity to each other as shown in Fig. 6. This finding suggests that non-bonding interactions between these residues may form an information network, or an allosteric wiring diagram. Further research is needed to explore this direction.
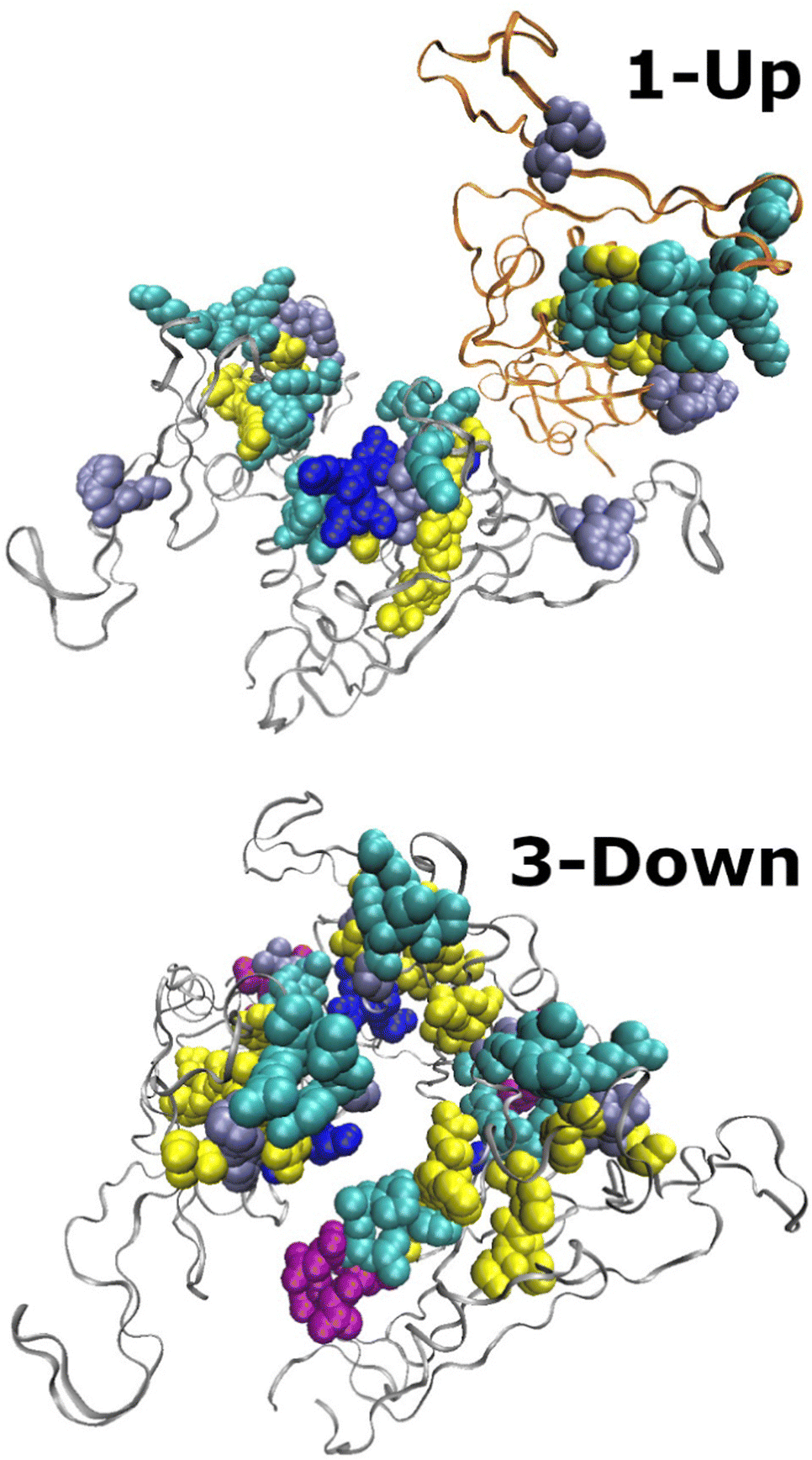 | ||
| Fig. 6 Snapshots of the RBD domains of the Omicron BA.1 spike protein. Bottom: 3-Down (PDB-ID: 7TF8), residues with CSM ≥ 1 are colored. Top: 1-Up state (PDB-ID: 7TO4), residues with CSM ≥ 15 are colored. Colors follow the secondary structure: blue-310-helix, cyan-turns, purple-α-helix, ice-blue-Loops and disordered elements, yellow-β-strands. Ribbons mark the RBD chains. The migrating chain in the 1-Up model is marked with orange ribbons. | ||
Variants analysis
The above analysis was repeated for 33 proteins specified in Tables 1 and S2 (ESI†). Fig. 7 presents the average spectra per variant for the 3-Down state (Fig. 7A, 15 proteins) and 1-Up state (Fig. 7B, 18 proteins). These were calculated with all the atoms of the side chains. Analysis at the backbone level shows similar trends (Fig. S5, ESI†). Naturally the level of distortion at the backbone level is smaller as compared with the complete set of atoms. This is particularly evident for the 3-Down state (the differences for the 1-Up state are insignificant at this scale). | ||
| Fig. 7 CSM spectra of 10-residues fragments along the sequence of the RBD for 33 SARS-CoV-2 spike protein, averaged by variant. (A) 3-Down state. (B) 1-Up state. PDB-Ids are listed in Table S2 (ESI†). | ||
The CSM pattern of each variant in Fig. 7 is similar to Fig. 5 above, affirming the generality of the local distortion trend. This is to be expected since each mutation alter few residues along the RBD sequence, while the CSM spectra are based on 10-residues fragments. The 1-Up spectra display high similarity to each other, teaching that all variants eventually reach the same level of distortion which is required to bind the protein to ACE2. The most striking evident is the large variability of the 3-Down state spectra that allows excellent discrimination between the variants. The different mutations lead to small structural changes of the starting conformation of the RDB domains by creating local islands of distortion with varying levels. The differences between the variants are not arbitrary – they follow the trend of variants transmissibility. The more transmissible the variant (and the latest the variant on the evolutionary path), the higher is its local distortion at the 3-Down state. As discussed above, along with the RBD migration, distortion naturally increases. Our findings suggest that if the protein experiences higher distortion at the initial state – its path to the final state becomes shorter since a larger extent of the final distortion already occurred. This may reduce the energy barrier of the overall transition, and lead to a faster transition towards the 1-Up state.
The differences between the protein variants stems from sequence mutations, and most of them are outside the RBD domain.39 Table S4 (ESI†) lists the mutations in the RBD domain for variants included in this study. Few mutations appear at a center of a distortion peak (e.g., L452R of the Delta variant, and N501Y of the Alpha, Beta and Omicron BA.1 variants), while others appear unrelated to a CSM peak (e.g., K417N for the Beta and Omicron BA.1 variants). Direct link between the mutation in the RBD and distortion peaks was not found in our data set. It is possible that mutations outside the RBD affect the local distortion of the RBD. Further research is needed in order to explore this issue more closely, e.g., by applying a shorter CSM ruler and analyzing regions beyond the RBD domain.
Distortion progress along the minimum energy path
Fallon et al.7 used MD simulation to study the RBD migration process of the WT variant.7 Their reported trajectories span 32 steps along the minimum energy path starting with the 3-Down state and up to the 1-Up state. We used these coordinates to calculate the CSM spectra of local distortion and validate our findings from the experimental data presented in Fig. 7. Spectra of selected steps from the MD analysis are presented in Fig. 8, showing increased distortion as the RBD domain opens up, particularly at the vicinity of the CSM peaks. The CSM calculations were performed with all the atoms of the side-chains, excluding the hydrogen atoms. Analysis at the backbone level produced similar results with no significant differences at this scale. The trend of the curves in Fig. 8 displays high resemblance to Fig. 7A, as more advanced steps show higher CSM levels. At step 15, which is half the way towards the 1-Up state, the local distortion approximately equals that of the final 1-Up state. Comparing Fig. 8 with Fig. 7A we postulate that: (1) the peaks of the CSM spectra are not arbitrary, and teach us on the level of openness of the RBD chain along the path to the 1-Up state. (2) The RBD domain of variants with higher distortion at the 3-Down state, has a shorter way to migrate towards the open state. Gobeil et al.9 suggested that increased transmissibility of the Omicron BA.1 spike is related to the ease of accessing the 1-Up state. Fig. 7 and 8 quantify this interpretation and provide grounds for variants comparison. The CSM spectra appears here as a predictive tool that quantifies the relation between symmetry, structure and protein function. | ||
| Fig. 8 CSM spectra of 10-residues fragments along the sequence of the RBD of the WT SARS-CoV-2 spike protein. Coordinates are based on MD analysis.7 Step 0 is the 3-Down state, and step 31 is the 1-Up state. | ||
Conclusions
The RBD migration of the SARS-CoV-2 spike protein involves a major symmetry breaking of the protein, as well as hidden local changes to the symmetry of the RBD trimer. Using continuous symmetry and chirality measures, we quantified these structural changes and provided a quantitative ground for structural comparison of different variants. Using the CCM as a three-dimensional conformational similarity descriptor, we showed that symmetry breaking during the 3-Down to 1-Up transition involves local conformational changes of specific residues and their side chains. CSM spectra that capture the local distortion with respect to C3 symmetry of 10-residues trimeric fragments along the RBD sequence were used to follow the distortion trend, compare variants and identify the residues that lead the change.The power of the approach is in its predictive nature. Analysis of both Cryo-EM and MD simulation data showed that the distortion of the 3-Down state implies on the distortion of the 1-Up state. Higher asymmetry at the 3-Down state is related with higher variant's transmissibility. Analysis of MD simulation data teaches that increased distortion appears at more advanced stages along the RBD migration process, suggesting that a higher transmissibility may stem from a shorter path between the 3-Down and 1-Up states. Along the CSM spectra, residues with extreme CSM values indicating on high distortion, are generally preserved between the two states. Their majority are located at the RDB-ACE2 binding interface, with relatively close proximity to each other that allows non-bonding interactions, indicating that they might be involved in an allosteric network of information. Further research that will extend the analysis beyond the RBD region is needed to establish this view. Our findings provide a clear and quantitative demonstration to the notion that the capacity to change is encoded in the initial structure of the protein,16 particularly in the framework of allosteric regulation. Extending this type of analysis to other protein systems can contribute to better understanding of the role of symmetry in protein structure and function.
Methods
Protein selection and filtering
A set of 63 structures of the SARS-CoV-2-spike protein measured by cryo-EM experiments and without ligands were downloaded from the RCSB-PDB.32 These included the WT and four past and present variants of concern: Alpha, Beta, Delta and Omicron BA.1. Proteins with 10 or more missing residues on a single RBD chain were excluded. These criteria left us with 42 proteins, each representing one of the known conformations (3-Down, 1-Up). For the remaining proteins we calculated the CSM with respect to C3 symmetry using a running ruler of 10 residues, and the CCM per residue for all three chains. All atoms were included in these calculations. We performed a Wilcoxon test on of all pairs of proteins with the same variant and conformation, to test whether the CSM distributions of the samples were equal. When a pair of proteins was identified as statistically identical (p > 0.05) we kept only the protein with the better resolution. This process left us with 33 proteins divided to conformers and variants specified in Tables S2 and S3 (ESI†). CSM and CCM calculations were performed on selected chains and residues. The pdbtools package40 was used for coordinates' selection. Secondary structure was extracted with the Biopython pakage.41The choice of 10 residues for the ruler size is empirical. Bonjack-Shterengartz and Avnir29 recommended to use a ruler of 10 to identify a hinge (which is a flexible and less symmetric region in a protein) for systems were nothing is known about its location. In a different study, we showed that the tendency of amino acids with long or polar side chains to distort the protein is larger compared to small and non polar residues, since the conformational freedom of the side-chains may cause distortion even if the backbone symmetry is high.31 This may cause the spectra calculated with a smaller ruler to be more noisy and more difficult to analyze. On the other hand, a large ruler tends to flatten most of the fluctuations and loses important information. After testing different ruler sizes on a small set of proteins (with 1, 3, 5, 7, 10, 15 and 20 residues), we found that for the Sars-CoV-2 spike protein, a ruler of 10 residues is a reasonable compromise, that averages over residues identity effects on the one hand while retaining important information regarding the level of local symmetry on the other hand.
Setting a unified sequence
In several variants, mutations appear in the form of missing residues (e.g., residues H69-V70 and Y144 of the Alpha variant). In order to maintain a consecutive sequence, several researchers renumbered the residues that follow such mutations, in order to eliminate sequence gaps. This renumbering changed the sequence numbers that define the RBD domain, making it difficult to compare different proteins on a unified sequence. Prior to our analysis we reverted this renumbering such that the RBD domains for all the proteins in our data were between P330 and P527. It should be noted that mutations that create gaps are generally found in the NTD domain, and do not exist in the RBD.Coordinates of MD analysis
MD simulation of the free energy path of the 3-Down to 1-Up RBD transition for the WT spike protein were reported by Fallon et al.7 The details of the simulation are specified in the original paper and are briefly summarized here. The simulation was performed with Amber v20,42 and relied on two cryo-EM measurements of the protein representing the 3-Down (PDB-ID: 6VXX8) and the 1-Up (PDB-ID: 6VSB,43 chain A) states. Amino acids substitutions were converted back to the WT sequence, and both structures were cleaved at the furin site (R685|S686) of the S1/S2 interface, to mimic the expected state in situ. Sequence gaps were corrected based on several other measurements, particularly the crystallographic structure of the spike-ACE2 complex (PDB-ID: 6M0J44) which was used to replace missing coordinates of atoms at the RBD domain. Water and glycan molecules were added. After several equilibration steps, production simulation of each system (3-Down and 1-Up) were conducted for about 0.3 μs each, at 310 K and constant NPT, with a 4 fs time step.7We performed a symmetry analysis for 32 structures (including the initial and final states) taken from the above simultation,7 representing snapshots of the protein structure along the free energy path of the RBD opening process. Prior to the analysis, Glycan and solvent molecules were excluded, and the residues were renumbered to match the sequence numbers of the experimental data included in our study. CSM and CCM calculations were performed without the Hydrogen atoms since these do not exist in the experimental data.
Continuous symmetry measures
The CSM, S(G), represents the minimal distance of a molecular structure from a structure of the same set of atoms and bonds that belongs to the point group G. The scale of the measure is continuous between 0 and 100, where 0 represents perfect symmetry of the original structure and 100 is obtained in the extreme case where the nearest symmetric structure collapses to the center of mass. The methodology is based on using the original structure as a starting point, and systematically searching for a structure with the same connectivity map, but with the desired symmetry. The final structure is the one for which the square sum of these distances is minimal. Mathematically, it is defined as: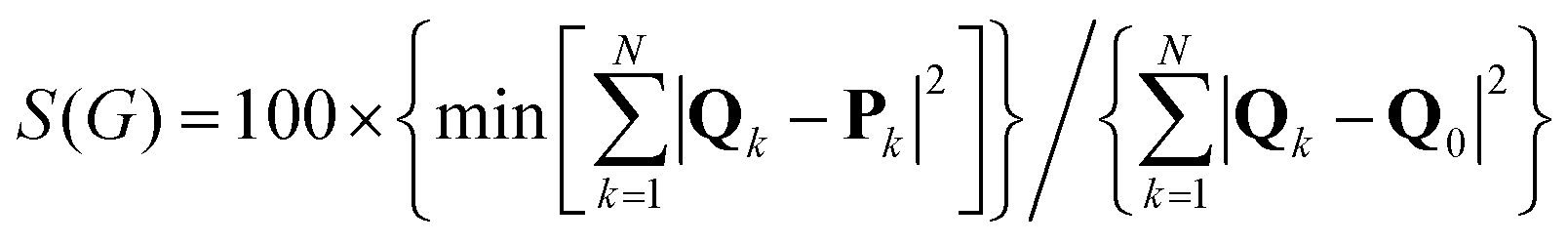 | (1) |
| CCM = min[S(Sn)] n = 1, 2, 4, 6… | (2) |
The main challenge in CSM calculation is finding the closest symmetric structure that serves as the reference structure in eqn (1). An exact algorithm has been recently developed for small-to medium-sized molecules,21 in which only structure-preserving permutations are scanned to find the closest symmetric structure. In the case of proteins, this algorithm is not applicable due to the enormous number of possible permutations, and an approximate calculation is applied.23 This algorithm uses the Hungarian algorithm45 to efficiently solve the assignment problem and find the correct permutation. It utilizes the sequence of the peptides to reduce the size of equivalence groups of atoms, and compels the code to preserve both the sequence and the peptide structure. In this study, the approximated algorithm was used. The atoms of the backbone and the side chains were included in all CSM and CCM calculations. Hydrogen atoms are generally absent in experimental measurements of proteins, and were therefore excluded.
Accuracy considerations
CSM calculations rely on the accuracy of the coordinates at hand. In the case of the SARS-CoV-2 spike proteins, experimental data were based on Cryo-EM measurements with resolution in the range of 3–4 Å. In many of these measurements, high values of the B-factor are reported. In our set of 33 proteins, the median B-factor in the RBD domain was 121 Å2 for the 3-Down proteins and 209 Å2 for the 1-Up proteins. These values raise questions regarding the reliability of the coordinates. High B-factors is a common issue for all SARS-CoV-2 Cryo-EM measurements that characterizes substantial parts of the RBD and NTD domains of the S1 subunit, for both the 3-Down and 1-Up states. Any structural analysis of these proteins that relies on these coordinates may suffer from this caveat. It has been shown that certain refinement procedures may overestimate the B-Factors in Cryo-EM measurements.46 New refinement procedures claim to improve the quality of the data,47 but the extent of this improvement for the SARS-CoV-2 spike protein is not yet clear.High B-factors do not necessarily indicate on high distortion in terms of CSM. Pervious studies showed that the CSM errors calculated based on the crystallographic B-factors in CSM calculations are negligible.33,48 No correlation between the CSM and the B-factors was found for our set of proteins. We also did not find significant differences in the shape of the CSM spectra presented in Fig. 5 and 7 between measurements with higher or lower B-factors. The CSM trend thus appear unrelated to the B-factors. We concluded that these values, although high, do not carry direct information regarding the CSM trends. The analysis presented here, based on a global descriptor of fragments of 10-residues naturally reduces the bias due to measurement errors. The fact that similar trends were obtained by analysis of several measurements of the same protein, strengthens the validity of our findings and the reliability of the final conclusions. Further support is obtained from analyzing the MD simulation by Fallon et al.,7 which was based on a high resolution measurement (PDB-ID: 6VXX, at 2.8 Å) with much smaller B factor values in the RBD domain, and a median value of 63 Å2, which is a reasonable value at this resolution.49 We note that this protein cannot be analyzed by CSM, since perfect symmetry was enforced. The shapes of the CSM spectra from the simulation files were similar to the spectra obtained from experimental data, teaching that the spectra are authentic and valid.
Data availability
The CSM code is freely available through our GitHub page at: https://continuous-symmetry.github.io/CSM-OUI/. CSM and CCM calculations can also be performed online through the CoSyM website at: https://csm.ouproj.org.il.Author contributions
Conceptualization and supervision was performed by ITA. Data curation and calculations were performed by YS. Both authors contribute to the formal analysis. The manuscript was written through contributions of both authors. All authors have given approval to the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank Sagiv Barhoom (the Open University of Israel) for his help in programming. The research was supported by The Open University of Israel's Research Fund, grants 102128, 102558, and 511711.References
- A. G. Wrobel, D. J. Benton, C. Roustan, A. Borg, S. Hussain, S. R. Martin, P. B. Rosenthal, J. J. Skehel and S. J. Gamblin, Nat. Commun., 2022, 13, 1–7 Search PubMed.
- J. Zhang, Y. Cai, T. Xiao, J. Lu, H. Peng, S. M. Sterling, R. M. Walsh, S. Rits-Volloch, H. Zhu, A. N. Woosley, W. Yang, P. Sliz and B. Chen, Science, 2021, 372, 525–530 CrossRef CAS PubMed.
- A. Winger and T. Caspari, Viruses, 2021, 13, 1–15 CrossRef PubMed.
- A. Telenti, E. B. Hodcroft and D. L. Robertson, Cold Spring Harbor Perspect. Med., 2022, 12, a041390 CrossRef CAS PubMed.
- Y. Cai, J. Zhang, T. Xiao, C. L. Lavine, S. Rawson, H. Peng, H. Zhu, K. Anand, P. Tong, A. Gautam, S. Lu, S. M. Sterling, R. M. Walsh, S. Rits-Volloch, J. Lu, D. R. Wesemann, W. Yang, M. S. Seaman and B. Chen, Science, 2021, 373, 642–648 CrossRef CAS PubMed.
- S. M.-C. Gobeil, J. Katarzyna, M. Shana, M. Katayoun, P. Robert, S. Victoria, M. F. Kopp, M. Kartik, L. Dapeng, W. Kevin, K. O. Saunders, R. J. Edwards, B. Korber, B. F. Haynes, R. Henderson and A. Priyamvada, Science, 2021, 373, eabi6226 CrossRef CAS PubMed.
- L. Fallon, K. A. A. Belfon, L. Raguette, Y. Wang, D. Stepanenko, A. Cuomo, J. Guerra, S. Budhan, S. Varghese, C. P. Corbo, R. C. Rizzo and C. Simmerling, J. Am. Chem. Soc., 2021, 143, 11349–11360 CrossRef CAS PubMed.
- A. C. Walls, Y. J. Park, M. A. Tortorici, A. Wall, A. T. McGuire and D. Veesler, Cell, 2020, 181, 281–292 CrossRef CAS PubMed.
- S. M. C. Gobeil, R. Henderson, V. Stalls, K. Janowska, X. Huang, A. May, M. Speakman, E. Beaudoin, K. Manne, D. Li, R. Parks, M. Barr, M. Deyton, M. Martin, K. Mansouri, R. J. Edwards, A. Eaton, D. C. Montefiori, G. D. Sempowski, K. O. Saunders, K. Wiehe, W. Williams, B. Korber, B. F. Haynes and P. Acharya, Mol. Cell, 2022, 82, 2050–2068.e6 CrossRef CAS PubMed.
- P. V. Raghuvamsi, N. K. Tulsian, F. Samsudin, X. Qian, K. Purushotorman, G. Yue, M. M. Kozma, W. Y. Hwa, J. Lescar, P. J. Bond, P. A. MacAry and G. S. Anand, eLife, 2021, 10, e63646 CrossRef CAS PubMed.
- H. M. Dokainish, S. Re, T. Mori, C. Kobayashi, J. Jung and Y. Sugita, eLife, 2022, 11, e75720 CrossRef CAS PubMed.
- G. Verkhivker, Int. J. Mol. Sci., 2022, 23, 2172 CrossRef CAS PubMed.
- D. Mannar, J. W. Saville, Z. Sun, X. Zhu, M. M. Marti, S. S. Srivastava, A. M. Berezuk, S. Zhou, K. S. Tuttle, M. D. Sobolewski, A. Kim, B. R. Treat, P. M. Da Silva Castanha, J. L. Jacobs, S. M. Barratt-Boyes, J. W. Mellors, D. S. Dimitrov, W. Li and S. Subramaniam, Nat. Commun., 2022, 13, 1–12 Search PubMed.
- R. A. Mansbach, S. Chakraborty, K. Nguyen, D. C. Montefiori, B. Korber and S. Gnanakaran, Sci. Adv., 2022, 7, eabf3671 CrossRef PubMed.
- S. M. C. Gobeil, K. Janowska, S. McDowell, K. Mansouri, R. Parks, K. Manne, V. Stalls, M. F. Kopp, R. Henderson, R. J. Edwards, B. F. Haynes and P. Acharya, Cell Rep., 2021, 34, 108630 CrossRef CAS PubMed.
- D. Thirumalai, C. Hyeon, P. I. I. Zhuravlev and G. H. H. Lorimer, Chem. Rev., 2019, 119, 6788–6821 CrossRef CAS PubMed.
- E. Papaleo, G. Saladino, M. Lambrughi, K. Lindorff-Larsen, F. L. Gervasio and R. Nussinov, Chem. Rev., 2016, 116, 6391–6423 CrossRef CAS PubMed.
- H. Zabrodsky, S. Peleg and D. Avnir, J. Am. Chem. Soc., 1992, 114, 7843–7851 CrossRef CAS.
- M. Pinsky, C. Dryzun, D. Casanova, P. Alemany and D. Avnir, J. Comput. Chem., 2008, 29, 2712–2721 CrossRef CAS PubMed.
- C. Dryzun, A. Zait and D. Avnir, J. Comput. Chem., 2011, 32, 2526–2538 CrossRef CAS PubMed.
- G. Alon and I. Tuvi-Arad, J. Math. Chem., 2018, 56, 193–212 CrossRef CAS.
- H. Zabrodsky and D. Avnir, J. Am. Chem. Soc., 1995, 117, 462–473 CrossRef CAS.
- I. Tuvi-Arad and G. Alon, J. Cheminform., 2019, 11, 39 CrossRef PubMed.
- P. Alemany, D. Casanova, S. Alvarez, C. Dryzun and D. Avnir, in Reviews in Computational Chemistry, ed. A. L. Parrill and K. B. Lipkowitz, 2017, vol. 30, pp. 289–352 Search PubMed.
- I. Tuvi-Arad and D. Avnir, J. Math. Chem., 2010, 47, 1274–1286 CrossRef CAS.
- I. Tuvi-Arad and D. Avnir, Chem. – Eur. J., 2012, 18, 10014–10020 CrossRef CAS PubMed.
- A. W. A. W. Kaspi-Kaneti, J. Barroso, G. Merino, D. Avnir, I. Garzon, I. Tuvi-Arad, I. L. I. L. Garzón and I. Tuvi-Arad, J. Org. Chem., 2020, 85, 15415–15421 CrossRef CAS PubMed.
- I. Tuvi-Arad, T. Rozgonyi and A. Stirling, J. Phys. Chem. A, 2013, 117, 12726–12733 CrossRef CAS PubMed.
- M. Bonjack-Shterengartz and D. Avnir, PLoS One, 2017, 12, e0180030 CrossRef PubMed.
- H. Wang, D. Avnir and I. Tuvi-Arad, Biochemistry, 2018, 57, 6395–6403 CrossRef CAS PubMed.
- Y. Shalit and I. Tuvi-Arad, PLoS One, 2020, 15, e0235863 CrossRef CAS PubMed.
- H. M. Berman, J. Westbrook, Z. Feng, G. Gilliland, T. N. Bhat, H. Weissig, I. N. Shindyalov and P. E. Bourne, Nucleic Acids Res., 2000, 28, 235–242 CrossRef CAS PubMed.
- M. Bonjack-Shterengartz and D. Avnir, Proteins-Structure Funct. Bioinforma., 2015, 83, 722–734 CrossRef CAS PubMed.
- I. Tuvi-Arad and A. Stirling, Isr. J. Chem., 2016, 56, 1067–1075 CrossRef CAS.
- J. Zhang, Y. Cai, C. L. Lavine, H. Peng, H. Zhu, K. Anand, P. Tong, A. Gautam, M. L. Mayer, S. Rits-Volloch, S. Wang, P. Sliz, D. R. Wesemann, W. Yang, M. S. Seaman, J. Lu, T. Xiao and B. Chen, Cell Rep., 2022, 39, 110729 CrossRef CAS PubMed.
- L. Casalino, Z. Gaieb, J. A. Goldsmith, C. K. Hjorth, A. C. Dommer, A. M. Harbison, C. A. Fogarty, E. P. Barros, B. C. Taylor, J. S. Mclellan, E. Fadda and R. E. Amaro, ACS Cent. Sci., 2020, 6, 1722–1734 CrossRef CAS PubMed.
- Z. F. Brotzakis, T. Löhr and M. Vendruscolo, Chem. Sci., 2021, 12, 9168–9175 RSC.
- W. Humphrey, A. Dalke and K. Schulten, J. Mol. Graph., 1996, 14, 33–38 CrossRef CAS PubMed.
- E. B. Hodcroft, CoVariants: SARS-CoV-2 Mutations and Variants of Interest, https://covariants.org.
- M. Harms, pdbtools, https://github.com/harmslab/pdbtools.
- P. J. A. Cock, T. Antao, J. T. Chang, B. A. Chapman, C. J. Cox, A. Dalke, I. Friedberg, T. Hamelryck, F. Kauff, B. Wilczynski and M. J. L. de Hoon, Bioinformatics, 2009, 25, 1422–1423 CrossRef CAS PubMed.
- D. A. Case, K. Belfon, I. Ben-Shalom, S. R. Brozell, D. Cerutti, T. Cheatham, V. W. D. D. T. Cruzeiro, S. Duke, G. Giambasu, M. Gilson, H. Gohlke, A. Götz, R. Harris, R. K. Izadi, S. A. Izmailov, K. Kasavajhala, A. Kovalenko, R. T. Krasny, T. Lee, S. LeGrand, P. Li, C. Lin, J. Liu, T. Luchko, H. O. Luo, V. Man, K. M. Merz, Y. Miao, G. Monard, C. A. Nguyen, F. Pan, S. Pantano, R. Qi, D. R. Roe, A. Roitberg, N. R. Sagui, S. Schott-Verdugo, J. Shen, C. Simmerling, R. M. W. Skrynnikov, J. Smith, J. Swails, R. Walker, J. Wang, L. Wilson, R. M. Wolf, Y. Xiong, Y. Xue, D. York and P. A. Kollman, Amber 2020, University of California, San Francisco, 2020 Search PubMed.
- D. Wrapp, N. Wang, K. S. Corbett, J. A. Goldsmith, C.-L. Hsieh, O. Abiona, B. S. Graham and J. S. McLellan, Science, 2020, 367, 1260–1263 CrossRef CAS PubMed.
- J. Lan, J. Ge, J. Yu, S. Shan, H. Zhou, S. Fan, Q. Zhang, X. Shi, Q. Wang, L. Zhang and X. Wang, Nature, 2020, 581, 215–220 CrossRef CAS PubMed.
- J. Munkres, J. Soc. Ind. Appl. Math., 1957, 5, 32–38 CrossRef.
- S. Kaur, J. Gomez-Blanco, A. A. Z. Khalifa, S. Adinarayanan, R. Sanchez-Garcia, D. Wrapp, J. S. McLellan, K. H. Bui and J. Vargas, Nat. Commun., 2021, 12, 1240 CrossRef CAS PubMed.
- R. Sanchez-Garcia, J. Gomez-Blanco, A. Cuervo, J. M. Carazo, C. O. S. Sorzano and J. Vargas, Commun. Biol., 2021, 4, 1–8 CrossRef PubMed.
- M. Pinsky, D. Yogev-Einot and D. Avnir, J. Comput. Chem., 2003, 24, 786–796 CrossRef CAS PubMed.
- O. Carugo, BMC Bioinf., 2018, 19, 1–9 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3cp00163f |
| This journal is © the Owner Societies 2023 |