Understanding photochemical pathways of laser-induced metal ion reduction through byproduct analysis†
Received
4th January 2023
, Accepted 9th June 2023
First published on 29th June 2023
Abstract
Laser-induced reduction of metal ions is attracting increasing attention as a sustainable route to ligand-free metal nanoparticles. In this work, we investigate the photochemical reactions involved in reduction of Ag+ and [AuCl4]− upon interaction with lasers with nanosecond and femtosecond pulse duration, using strong-field ionization mass spectrometry and spectroscopic assays to identify stable molecular byproducts. Whereas Ag+ in aqueous isopropyl alcohol (IPA) is reduced through plasma-mediated mechanisms upon femtosecond laser excitation, low-fluence nanosecond laser excitation induces electron transfer from IPA to Ag+. Both nanosecond and femtosecond laser excitation of aqueous [AuCl4]− produce reactive chlorine species by Au–Cl bond homolysis. Formation of numerous volatile products by IPA decomposition during both femtosecond and nanosecond laser excitation of [AuCl4]− is attributed to enhanced optical breakdown by the Au nanoparticle products of [AuCl4]− reduction. These mechanistic insights can inform the design of laser synthesis procedures to improve control over metal nanoparticle properties and enhance byproduct yields.
1. Introduction
Photochemical reduction of metal salts has been explored for decades as a sustainable method to produce metal nanoparticles without toxic chemical reducing agents.1–6 Light sources from the infrared through γ-ray spectral regions initiate distinct chemical reaction pathways that reduce metal ions.1 Ionizing radiation directly dissociates solvent molecules to provide strong reducing agents such as solvated electrons and organic radicals.2,3 Ultraviolet (UV) photons reduce metal ions through electronic excitation of a photosensitizer to produce organic radicals or direct excitation and dissociation of metal ion complexes.4,5 Intense pulsed lasers initiate reaction pathways observed with both ionizing and UV radiation to reduce metal ions in what is called laser reduction in liquid (LRL).6
LRL typically involves focusing intense laser pulses into a liquid medium, which ionizes and dissociates solvent molecules to produce a localized plasma containing reactive chemical species that reduce metal ions. In aqueous solution, the primary reactive species are the same as those observed in radiolysis: hydrated electrons (eaq−), hydroxyl radicals (OH˙), hydrogen radicals (H˙) and hydrogen peroxide (H2O2).7–11 LRL has been widely used to produce Au nanoparticles (NPs) from aqueous solutions of the tetrachloroaurate complex [AuCl4]−7–20 and Ag NPs from aqueous solutions of the salts AgNO3 or AgClO4.21–26 For [AuCl4]−, Au3+ reduction is initiated by cleavage of an Au–Cl bond, whereas reduction of an Ag salt involves direct electron capture by free Ag+ ions in solution.
Under LRL conditions that form plasma, hydrated electrons and H2O2 contribute to the reduction of [AuCl4]− into Au atoms and clusters through the reactions10
| | | [Au3+Cl4]− + 3eaq− → Au0+ 4Cl− | (1) |
| | 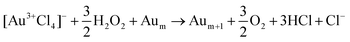 | (2) |
We recently reported that LRL using 532 nm, 8 ns laser pulses at low fluences below the plasma formation threshold can also reduce [AuCl
4]
− to Au NPs.
19 To account for the observed [AuCl
4]
− reduction without the aid of reactive plasma species, we proposed that the reduction to Au atoms proceeds through direct photolysis of Au–Cl bonds by mechanisms that had previously been proposed for photochemical reduction by UV photons,
27,28| | 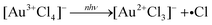 | (3) |
| | | 2[Au2+Cl3]− → [Au3+Cl4]− + [Au1+Cl2]− | (4) |
| | 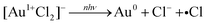 | (5) |
Under low-fluence nanosecond LRL at 532 nm,
reaction (3) was proposed to be initiated by laser-induced heating based on simulations showing that transient temperatures of 580 K, near the spinodal temperature of water, were sustained for about 100 μs after irradiation with a single 8 ns laser pulse.
19
A recent report by Liu et al. confirmed that reaction (3) initiates UV photolysis of aqueous [AuCl4]− through X-ray transient absorption spectroscopy measurements.29 They found that excitation at 263 nm formed [Au2+Cl3]− within the instrument response function of 79 ps, indicating rapid Au–Cl bond homolysis following electronic excitation of [AuCl4]−. In contrast, upon excitation at 800 nm (the wavelength used in femtosecond LRL studies7–20), the [Au2+Cl3]− did not appear until 5 ns after the initial pulse. The authors surmised that this delayed Cl loss resulted from the time required for a hydrated electron to diffuse to an [AuCl4]− molecule, indicating that the initial reduction step in LRL with near-infrared femtosecond lasers is
| | | [Au3+Cl4]− + eaq− → [Au2+Cl3]− +Cl−, | (6) |
which is consistent with the general mechanism in
reaction (1).
Whereas aqueous [AuCl4]− can easily be reduced due to its labile Au–Cl bonds and high reduction potential (E0 = +1.002 V for reaction (1)30), efficient photoreduction of Ag+ typically requires an auxiliary polymer reducing agent,31 photosensitizer,32 or aliphatic alcohol.33–35 LRL experiments with both nanosecond and femtosecond pulsed lasers indicate that reduction of aqueous Ag+ to produce stable Ag NPs only occurs in the presence of a capping agent,21–23 or a radical scavenger.24–26 The requirement of a radical scavenger for Ag NP formation by LRL is attributed to the facile back-oxidation of Ag0 atoms by OH˙ radicals present in laser plasma.24,25 LRL of Ag+ with 355 nm nanosecond pulses at a fluence sufficient for two-photon ionization of water but not plasma generation was found to produce stable Ag NPs in the presence of surfactants.22 However, it remains unknown whether two-photon excitation of Ag+ in the presence of alcohols using visible wavelengths can initiate Ag+ reduction at fluences below the plasma threshold.
To unravel the photochemical reaction pathways associated with [AuCl4]− and Ag+ reduction, we consider LRL under two different excitation conditions: high-density plasma from tightly focused femtosecond laser pulses and nonionizing low-fluence nanosecond laser excitation. For both conditions, we characterize stable molecular byproducts of metal ion reduction in water and aqueous solutions of isopropyl alcohol (IPA). Strong-field ionization mass spectrometry36 is used to analyze the composition of the headspace gas after LRL because the more common method of gas chromatography-mass spectrometry (GC-MS)37–40 is not suitable for aqueous solutions due to water-induced degradation of GC columns.41 Combined with spectroscopic assays for oxidized byproducts, we both verify the pathways in reactions (1)–(5) and identify new pathways involving activation of IPA by metal ions and the metal nanoparticle products. These insights into metal ion reduction pathways can lead to better control of the properties of metal nanoparticles produced by LRL.
2. Experimental methods
2.1 Materials
Potassium tetrachloroaurate(III) (KAuCl4, Strem Chemicals); silver perchlorate (AgClO4), titanium dioxide (Sigma Aldrich); potassium permanganate, sodium acetate (Alfa Aesar); N,N-diethyl-p-phenylenediamine sulfate (DPD, ACROS Organics); sulfuric acid, hydrogen peroxide, sodium oxalate, potassium hydroxide, potassium chloride, isopropyl alcohol, and acetic acid (Fisher Scientific) were used as received.
2.2 Sample preparation
All solutions were prepared using Millipore Ultrapure water with resistivity 18.2 M Ω cm−1 at 25 °C as the solvent. All working solutions were freshly prepared before laser processing. The pH of working solutions containing metal ions was adjusted to 5.6 ± 0.3 with KOH. For headspace gas analysis, working solutions contained 1 mM KAuCl4 or AgClO4 in water or 100 mM aqueous IPA, along with solutions containing no metal ions as controls. For samples used in DPD and titanium sulfate assays, working solutions contained 0.1 mM aqueous [AuCl4]−. Samples of water were also irradiated as controls. The lower metal ion concentration for spectroscopic assays was used to ensure that absorbances of OD < 2.0 were obtained.
2.3 Laser processing
The experimental setups for processing with laser pulses of nanosecond19 and femtosecond10 duration have been described previously. Briefly, nanosecond laser processing was conducted with a Nd:YAG laser system (Lab 170–30, Spectra Physics) operated at the second harmonic, producing 532 nm, 8 ns, pulses at a 30 Hz repetition rate was set to a pulse energy of 100 mJ. A loosely focused geometry was used where laser beam was down-collimated to a diameter of 7 mm before interaction with the precursor solutions. Femtosecond laser processing was conducted using a commercial titanium-sapphire chirped-pulse amplifier (Astrella, Coherent, Inc., Santa Clara, CA, USA), delivering 30 fs pulses, with the bandwidth centered at 800 nm and a repetition rate of 1 kHz. A tight-focusing geometry with large numerical aperture producing a measured focal beam waist of approximately 6.5 μm10 was used with pulse energy of 1 mJ, producing a peak intensity of approximately 2 × 1016 W cm−2 (ignoring losses). Experimental working solutions were processed with the nanosecond or femtosecond laser in 3 mL batches in a sealed cuvette for 60 minutes for headspace gas analysis. The cuvette containing the working solution was first evacuated so that the mass spectrum before laser processing could be measured (see Section 2.4).
2.4 Strong-field ionization mass spectrometry
Mass spectrometry (MS) of headspace gas was conducted using 1300 nm, 20 fs pulses for ionization as described previously.36 Briefly, a portion of the 800 nm titanium-sapphire output was converted to 1300 nm in an optical parametric amplifier and focused to a peak intensity of 1014 W cm−2 into the extraction region of a linear time-of-flight mass spectrometer (Jordan TOF, Grass Valley, CA). A sealed quartz cuvette containing working solution was attached to the ultrahigh vacuum chamber (base pressure 3 × 10−9 Torr) and evacuated through a variable effusive leak valve. Once evacuated, the valve was adjusted to produce a working pressure of 3 × 10−7 torr consisting of headspace gases. Mass spectra were recorded by averaging over 100![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 laser shots using a 1 GHz digital oscilloscope (WaveRunner 610Zi, Teledyne LeCroy). Mass spectra were normalized either to the area under the H2O+ ion signal at m/z 18 (for samples without IPA) or to IPA+ peak at m/z 60 (for samples containing IPA).
000 laser shots using a 1 GHz digital oscilloscope (WaveRunner 610Zi, Teledyne LeCroy). Mass spectra were normalized either to the area under the H2O+ ion signal at m/z 18 (for samples without IPA) or to IPA+ peak at m/z 60 (for samples containing IPA).
2.5 Spectroscopic assays
Free chlorine species (Cl2, ClOH, ClO−) were detected using the standard assay based on N,N-diethyl-p-phenylenediamine (DPD).42 Details of DPD oxidation by free chlorine from the initial dication, dpd2+, to the semiquinoid radical, dpd2+, are given in the ESI.† The dpd2+ exhibits strong absorbance at 551 nm that is used to quantify oxidizing species based on a calibration curve (ESI,† Fig. S1). Detection of H2O2 was performed using an adapted spectroscopic assay from ref. 43 based on the formation of pertitanic acid from titanium sulfate, Ti(SO4)2, and described in our previous work.10 The absorbance of pertitanic acid at 407 nm was used to quantify H2O2 concentration based on a calibration curve (ESI,† Fig. S2).
The general procedure for both spectroscopic assays was as follows. 25 mM KCl was added to laser-processed samples to precipitate any Au NPs that were formed, then samples were centrifuged using a Fisher Scientific accuSpin Micro17 microcentrifuge for 30 min at 13000 rpm. Following centrifugation, 2 mL of supernatant liquid was collected. All types of samples were processed following the above procedure (even those that produced no NPs) to eliminate any artifacts of sample processing. For DPD assay, acetate buffer (200 μL) was added to adjust the sample pH to ∼5. Then, DPD reagent (100 μL) was added right and the absorbance at 551 nm was measured using a commercial UV-vis spectrophotometer (Agilent 8453). For titanium sulfate assay, 400 μL of titanium(IV) sulfate (25 mM) was added to the cuvette and the absorbance of pertitanic acid at 407 nm measured.
3. Results and discussion
Chemical byproducts of laser-induced Ag+ and [AuCl4]− reduction were measured in two solutions that have been widely employed in LRL synthesis of Ag and Au NPs: metal ions in water8–10,12–15,19–23 and in water/IPA.11,17,18,21,26 MS analysis was performed on the headspace gas from working solutions in a sealed cuvette both before and after laser processing. The MS before laser processing was recorded to determine the species initially present so that any new products formed by laser processing could easily be identified. To corroborate reaction pathways suggested by MS analysis, spectroscopic assays using DPD and titanium sulfate were conducted for selected reaction conditions. We first report the products and reaction pathways induced by laser processing of water and aqueous IPA in the absence of metal ions in Section 3.1. Analysis of additional reaction pathways activated by the presence of Ag+ and [AuCl4]− is then presented in Sections 3.2 and 3.3, respectively. A summary of reaction pathways and further discussion is provided in Section 3.4.
3.1 Laser-induced reactions in water and aqueous IPA
Femtosecond laser processing of water is known to produce H2 and H2O2 from the optical breakdown plasma.7,10,44,45 MS analysis of laser-processed water confirmed H2 production using femtosecond laser pulses, but no H2 was observed after nanosecond laser processing (ESI,† Fig. S3). H2O2 was detected by titanium sulfate assay only after femtosecond laser processing (ESI,† Fig. S4). These results indicate that ionization and decomposition of water does not occur for nanosecond laser processing under the low-fluence conditions used.
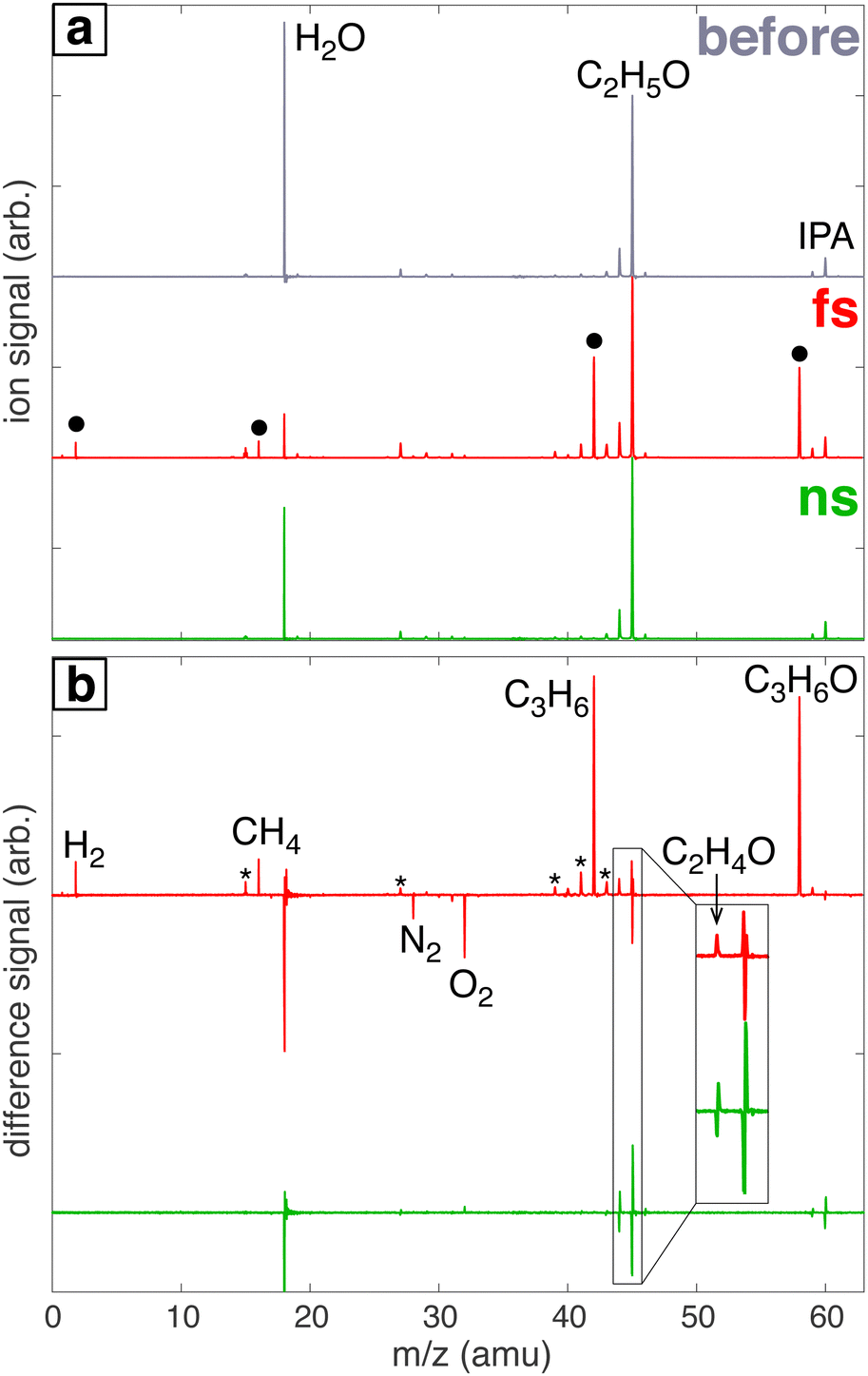
Fig. 1a shows a representative mass spectrum of 100 mM aqueous IPA before laser processing (‘before’, grey) and spectra of aqueous IPA after processing with the femtosecond laser (‘fs’, red) and nanosecond laser (‘ns’, green). The ‘before’ spectrum contains prominent peaks at m/z 60 (IPA parent ion), 45 (C2H5O fragment of IPA), 44 (C2H4O fragment of IPA), and 18 (H2O). The IPA fragments at m/z 45 and 44 are also observed in electron impact mass spectra from the NIST spectral database.46 Whereas processing with the nanosecond laser results in a spectrum with the same peaks as the ‘before’ spectrum, processing with the femtosecond laser produces four new peaks indicated by the black circles in Fig. 1a. To more easily visualize these spectral changes, the ‘before’ spectrum was subtracted off from the spectra after laser processing to produce the difference spectra shown in Fig. 1b. New species in the spectrum processed with the femtosecond laser (red) at m/z 2, 16, 42, 44, and 58 are assigned to H2, CH4, C3H6 (propene), C2H4O (acetaldehyde), and C3H6O (acetone). The peaks marked with a * are fragmentation products of acetone and propene.46 Although m/z 44 was observed in the ‘before’ spectrum as an IPA fragmentation product, the positive difference peak after femtosecond processing (inset, Fig. 1b) suggests that a small amount of acetaldehyde was produced. In contrast, the m/z 44 feature after nanosecond laser processing contains negative and positive components due to slight shifts in the peak location between the spectra taken before and after laser processing. Peak shapes consisting of negative and positive components are observed in difference spectra for all species present in the ‘before’ spectrum due to this peak shifting (ESI,† Fig. S5 illustrates the shift of the m/z 45 peak).
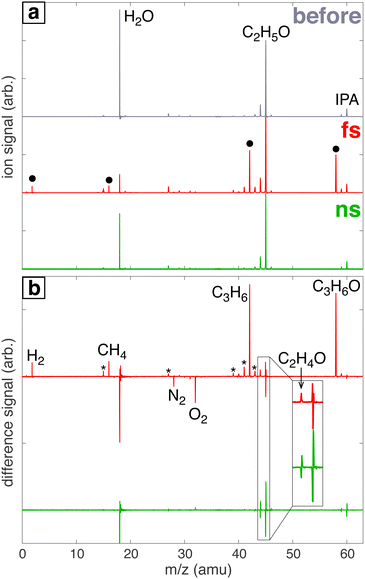 |
| | Fig. 1 (a) Mass spectra of headspace gas samples of water/IPA solution before (grey) and after processing with femtosecond (red) or nanosecond (green) laser. (b) Difference mass spectra after laser processing with new species indicated. Peaks with a * denote fragmentation products of acetone and propene. The inset magnifies the region of m/z 44–45 to show the formation of C2H4O after femtosecond laser processing. | |
The products H2, CH4, acetaldehyde, and acetone have been previously observed in γ radiolysis47 and 185 nm UV photolysis48 of IPA. It is expected that femtosecond laser processing of aqueous IPA would produce similar products as these previous studies because both irradiation methods produce solvated electrons, hydroxyl radicals (OH˙), and hydrogen radicals (H˙) in water.2,6 In particular, the reactions that produce acetone during femtosecond laser processing are likely initiated by OH˙ or H˙ radicals according to the proposed reactions in γ radiolysis studies,2
| | | (CH3)2CHOH + OH˙ → (CH3)2C˙OH + H2O | (7) |
| | | (CH3)2CHOH + H˙ → (CH3)2C˙OH + H2 | (8) |
Abstraction of a hydrogen atom from the resulting ketyl radical, (CH
3)
2C˙OH, with OH˙ or H˙ then produces acetone,
| | | (CH3)2C˙OH + OH˙ → (CH3)2CO + H2O | (9) |
| | | (CH3)2C˙OH + H˙ → (CH3)2CO + H2 | (10) |
In contrast to acetone, the formation of propene (
m/
z 42) during femtosecond laser processing is unexpected because both γ radiolysis and UV photolysis report <1% propene yield compared to H
2, CH
4, and acetone.
47,48 IPA can react with H˙ and OH˙ or 2H˙ to produce propene through the reactions
| | | (CH3)2CHOH + OH˙ + H˙ → C3H6 + 2H2O | (11) |
| | | (CH3)2CHOH + 2H˙ → C3H6 + H2 + H2O | (12) |
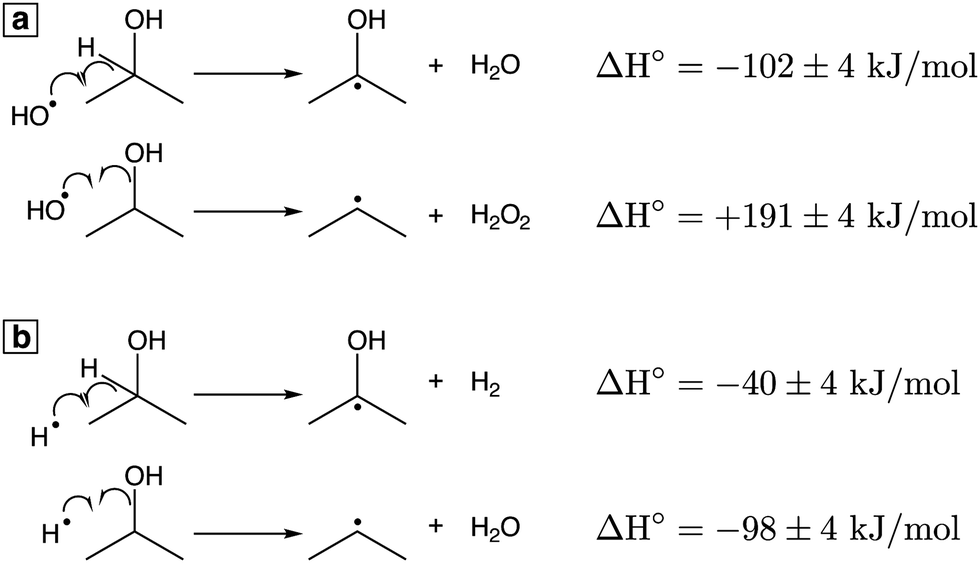
To understand the competition between acetone and propene formation, we consider the possible reactions of IPA with OH˙ (
Fig. 2a) and H˙ (
Fig. 2b). All thermochemical values are taken from ref.
49. Both OH˙ and H˙ can abstract the hydrogen on the carbon
α to the OH group to produce the ketyl radical (Δ
fH° = −96 ± 4 kJ mol
−1), which is the precursor to acetone formation (
reactions (9) and (10)). Formation of propene requires cleavage of the C–O bond to produce the less stable isopropyl radical (Δ
fH° = + 89 ± 4 kJ mol
−1); abstraction of an additional H atom then forms propene. The highly exothermic abstraction of the
α hydrogen by OH˙ (Δ
H°= −102 ± 4 kJ mol
−1,
Fig. 2(a)) is consistent with radiolysis studies indicating that 89% of OH˙ reaction with IPA occurs by
α hydrogen abstraction.
50 This favorable reaction enthalpy explains why acetone is a major product of IPA oxidation. In contrast, formation of the isopropyl radical by reaction with OH˙ is endothermic (Δ
H° = +191 ± 4 kJ mol
−1,
Fig. 2a), but the same reaction with H˙ is exothermic (Δ
H° = −98 ± 4 kJ mol
−1,
Fig. 2b). Because abstraction of the
α hydrogen by H˙ is less exothermic (Δ
H° = −40 ± 4 kJ mol
−1,
Fig. 2b) than C–O bond cleavage, the formation of propene likely proceeds through H˙ attack on IPA to produce the isopropyl radical, as previously proposed in gas-phase Ar plasma studies of IPA.
51
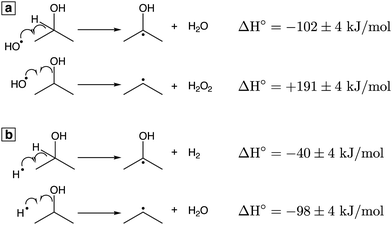 |
| | Fig. 2 Reactions of IPA with OH˙ (a) and H˙ (b) with ΔH° values at 298.15 K.49 | |
The low propene yield in radiolysis despite an exothermic ΔH° for attack of H˙ on IPA to produce the isopropyl radical implies the existence of a high kinetic barrier to this reaction, likely due to the energy required to break the C–O bond. To rationalize the apparent ease of overcoming this barrier, we consider a key difference between femtosecond laser processing and γ radiolysis or UV photolysis. Specifically, focusing intense laser pulses in liquid media under optical breakdown conditions generates laser-driven shock waves that can reach transient pressures exceeding 1 GPa52 and cavitation bubbles where gas-phase reactions can occur.39 These conditions have been proposed to induce mechanical C–C bond formation reactions in liquid alkanes.53 Hence, laser-induced shock waves can open up additional reaction pathways in femtosecond laser processing that are inaccessible in radiolysis. For propene formation from IPA, laser-induced shock waves could facilitate C–O bond cleavage through the reaction shown in Fig. 2b or overcome the modest endothermic barrier to direct dehydrogenation of IPA to propene49
| | | C3H8O → C3H6 + H2O ΔH° = 50 ± 2 kJ mol−1 | (13) |
In either case, the high propene yield observed in
Fig. 1 indicates femtosecond laser processing can overcome kinetic barriers present under radiolytic conditions.
The experiments in this section have established the baseline solvent reactions that occur in water and IPA/water solution under femtosecond laser processing and confirmed that no reactions occur under low-fluence nanosecond laser processing. With this knowledge, we turn to investigations of how added metal ions open up additional reaction pathways for both nanosecond and femtosecond laser processing.
3.2 Laser-induced reduction of Ag+
LRL of Ag+ in water does not produce stable Ag NPs.21–26 Although Ag+ is reduced by hydrated electrons at diffusion-limited rates34 to produce Ag0 atoms,OH˙ radicals in laser plasma rapidly back-oxidize Ag024,25| | | Ag0 + OH˙ → Ag+ + OH− | (15) |
In femtosecond laser processing, no net reduction of Ag+ was observed at concentrations below 300 mM.23 Although nanosecond laser processing at sufficiently high fluence to ionize water can accomplish net reduction of Ag+ to Ag0, these studies reported unstable large Ag NPs that rapidly precipitated out of solution.21,22 Consistent with these prior studies, we observed no Ag NP formation under nanosecond or femtosecond laser processing in water. MS analysis of the headspace gases found identical results as found for pure water: H2 was produced during femtosecond laser processing and no new products were formed during nanosecond laser processing (ESI,† Fig. S6). Results from the titanium sulfate assay confirmed that Ag0 is back-oxidized through reaction (15): the H2O2 yield in the presence of 0.1 mM Ag+ was reduced by 50% compared to the yield obtained in pure water at the same femtosecond laser processing conditions (ESI,† Fig. S7).
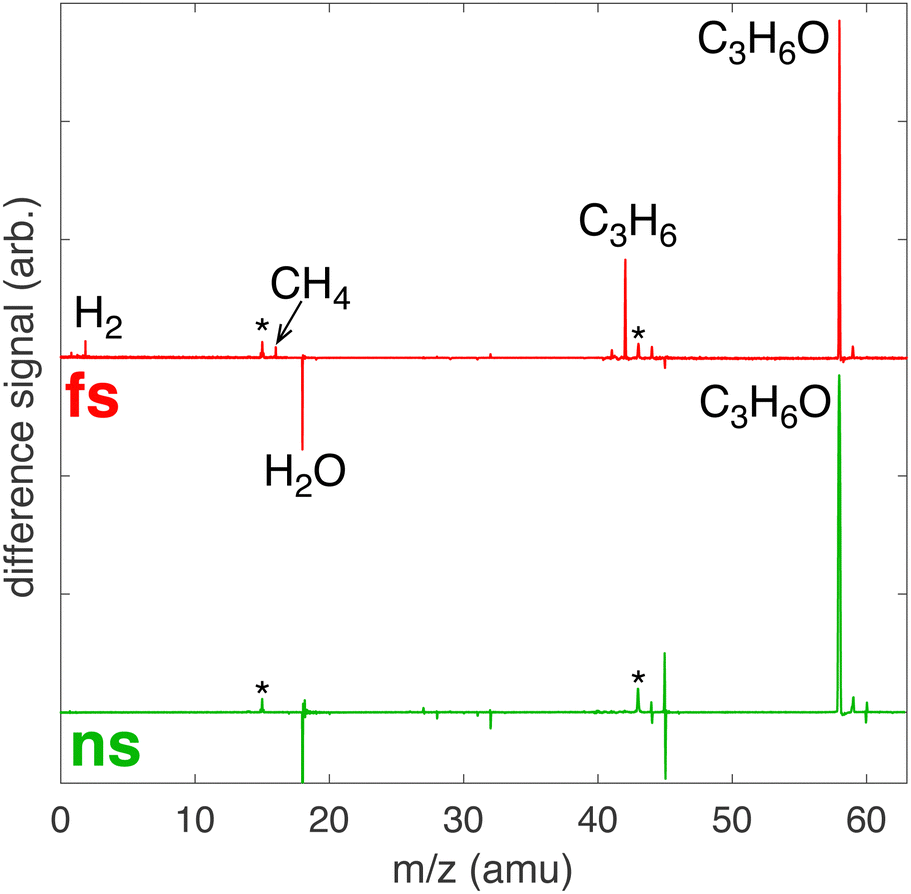
Hydroxyl radical scavengers including ammonia24,25 and IPA26 suppress reaction (15), enabling production of Ag NPs by femtosecond LRL. In high-fluence nanosecond LRL, the addition of IPA was reported to lower the Ag+ reduction rate by 25% due to its scavenging behavior.21 MS analysis of the headspace gas was performed after laser processing of 1 mM Ag+ in 100 mM aqueous IPA (raw spectra shown in ESI,† Fig. S8). The resulting difference spectra in Fig. 3 indicate that the same reaction products are formed as in femtosecond laser processing of aqueous IPA (cf., Fig. 1), whereas only acetone is formed during nanosecond laser processing. Oxidation of IPA to acetone during nanosecond laser processing is accompanied by partial conversion of Ag+ to Ag NPs, observed in the absorbance spectrum of the laser-processed solution (ESI,† Fig. S9). These reactions do not appear to involve OH˙ radicals because no H2O2 was detected from nanosecond laser processing (ESI,† Fig. S10).
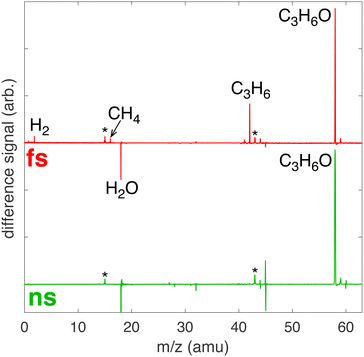 |
| | Fig. 3 Difference MS for femtosecond (red) and nanosecond (green) laser processing of 1 mM Ag+ in 100 mM aqueous IPA with major products indicated. Peaks with a * denote fragmentation products of acetone. | |
The observed formation of acetone as a byproduct of Ag+ reduction during nanosecond laser processing suggests that the reaction mechanism proceeds by electron transfer from an IPA molecule to Ag+ within a solvent cage, as proposed by Hada et al. for Ag+ reduction by 254 nm UV photolysis,33
| | | Ag+ + (CH3)2CHOH + H2O → Ag0 + H3O+ + (CH3)2C˙OH | (16) |
Reaction (16) produces a ketyl radical that can further be oxidized to acetone. Because no H
2O
2 was detected, it is unlikely that the ketyl radical oxidizes to acetone by OH˙
viareaction (9). Instead, the ketyl radical can reduce an additional Ag
+ ion to produce acetone. It has been proposed that the ketyl radical directly reduces Ag
+via electron transfer through the reaction
4,32,33| | | Ag+ + (CH3)2C˙OH + H2O → Ag0 + H3O+ + (CH3)2CO | (17) |
However, the standard oxidation potential
E0 = +1.8 V for the ketyl radical is not strong enough to reduce aqueous Ag
+ because
E0 (Ag
+/Ag
0) = −1.8 V according to Henglein
et al.34,35 Instead, they proposed that the ketyl radical reduces the Ag
2+ cluster that rapidly forms from Ag
0via the reaction
Because
E0 (Ag
2+/Ag
2) = −0.44 V,
34 the reduction of Ag
2+ by the ketyl radical to produce acetone
| | | Ag2+ + (CH3)2C˙OH + H2O → Ag20 + H3O+ + (CH3)2CO | (19) |
is thermodynamically favorable. Hence, we expect that
reaction (19) produces the acetone observed in our experiments.
The photochemical reactions (16) through (19) were initiated by 254 nm (4.9 eV) photons in ref. 33. For the same reactions to occur with 532 nm (2.3 eV) photons from the nanosecond laser, absorption of two photons by the solvent cage containing Ag+ and IPA must occur. The peak intensity of ∼3 × 108 W cm−2 in our experiments19 is likely sufficient for 2-photon absorption. Nevertheless, our observation that Ag+ reduction proceeds in the presence of IPA but not in pure water suggests that the energy required to initiate reaction (16) is lower than that required for electron transfer from a water molecule to Ag+ in a solvent cage also proposed by Hada et al.33
| | | Ag+ + 2H2O → Ag0 + H3O+ + OH˙ | (20) |
Because
reaction (20) is initiated with 254 nm photons, we can conclude that the energy required for
reaction (16) with IPA is likely less than 4.6 eV (two 532 nm photons) and the energy required for
reaction (20) is above 4.6 eV but less than 4.9 eV.
In contrast to the electron transfer mechanism that produces only acetone as a byproduct for nanosecond laser excitation, femtosecond LRL of Ag+ appears to proceed primarily by plasma-mediated reactions involving solvated electrons and IPA scavenging of OH˙ as previously proposed24,26 because no additional byproducts beyond those of IPA decomposition were observed in Fig. 3. We cannot rule out the participation of the electron-transfer mechanism in reactions (16) through (19) since higher quantities of acetone relative to other products are observed from femtosecond LRL in Fig. 3. However, experimental constraints preclude quantitative analysis of products using strong-field ionization mass spectrometry, so we cannot conclude that significantly more acetone is formed from IPA when Ag+ is present.
3.3 Laser-induced reduction of [AuCl4]−
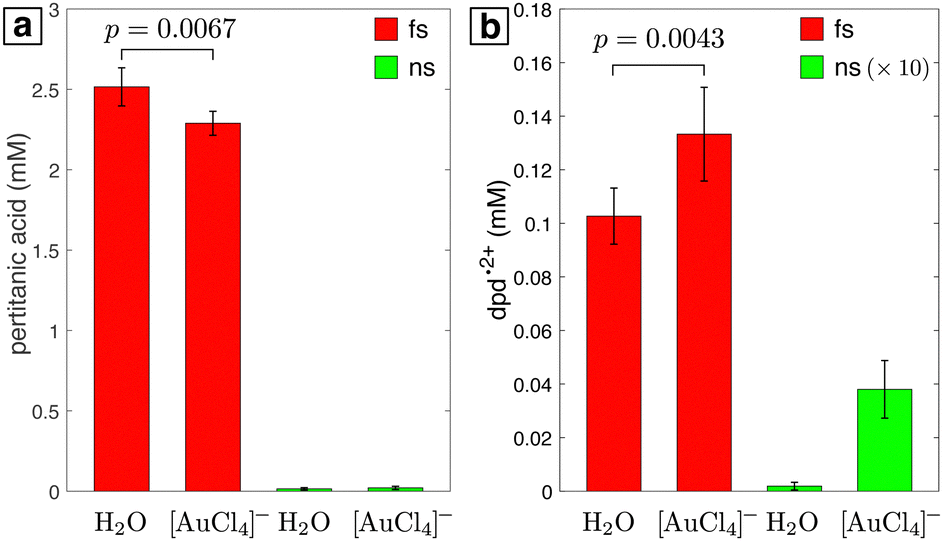
Whereas both femtosecond and nanosecond LRL require IPA for net reduction of Ag+, [AuCl4]− is easily converted to Au NPs in water.7–10,13–15,18–20 MS analysis of 1 mM [AuCl4]− in water (ESI,† Fig. S11) showed that H2 was produced with femtosecond laser processing, as for pure water and aqueous Ag+. No H2 was formed by nanosecond laser processing. Neither condition produced Cl2 or other volatile chlorine species that might be expected if Cl˙ radicals are formed in the Au3+ reduction pathways described in reactions (3) through (5) in the Introduction. To determine whether Cl˙ radicals are formed, we used the DPD assay (Section 2.5) to quantify the presence of stable free chlorine species (ClOH, ClO−). The titanium sulfate assay was also performed for solutions subject to the same laser processing conditions to isolate the contributions of H2O2, which can oxidize DPD.54 Laser processing was performed for 10 minutes with the femtosecond laser and 60 minutes with the nanosecond laser to ensure that 100% of the [AuCl4]− was converted to Au NPs (ESI,† Fig. S12) because [AuCl4]− also oxidizes DPD.
The quantified yields of pertitanic acid and dpd2+ after laser processing of water and 0.1 mM [AuCl4]− are shown in Fig. 4. Tabulated yields can be found in the ESI,† Table S1. Pertitanic acid is only observed upon femtosecond laser processing (Fig. 4a), with a 9% lower yield in 0.1 mM [AuCl4]− than in water. The lower yield is due to H2O2 consumption through the autocatalytic reduction of [AuCl4]− in the presence of Au NPs, as reported previously.7,8,10 In contrast, the dpd2+ yield after femtosecond laser processing is 29% higher in [AuCl4]− than in water (Fig. 4b). Because a decrease in dpd2+ yield in [AuCl4]− would be expected if H2O2 were the only oxidant present, the statistically significant increase in dpd2+ (p = 0.0043 by two-tailed t-test performed in MATLAB) indicates presence of free chlorine species in addition to H2O2. The presence of oxidized dpd2+ after nanosecond laser processing of [AuCl4]− (Fig. 4b) indicates that free chlorine species are generated under these conditions as well. Since no H2O2 is formed, this result confirms that direct Au–Cl bond homolysis drives [AuCl4]− reduction in nanosecond laser processing, consistent with the mechanism proposed in our previous work.19 The formation of free chlorine species during femtosecond laser processing is surprising because [AuCl4]− reduction is widely thought to proceed by reaction with hydrated electrons instead of Au–Cl bond homolysis.9–11 This issue will be further discussed in Section 3.4.
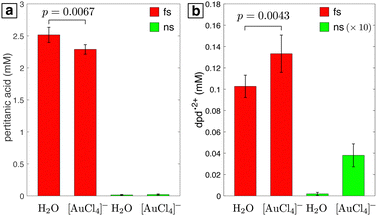 |
| | Fig. 4 Quantified yields of pertitanic acid (a) and dpd2+ (b) in samples of water and 0.1 mM [AuCl4]− after processing with femtosecond (red) or nanosecond (green) laser. Values for the nanosecond laser in (b) are magnified by a factor of 10 for clarity. Brackets between the femtosecond samples indicate p values obtained from the two-tailed t-test. | |
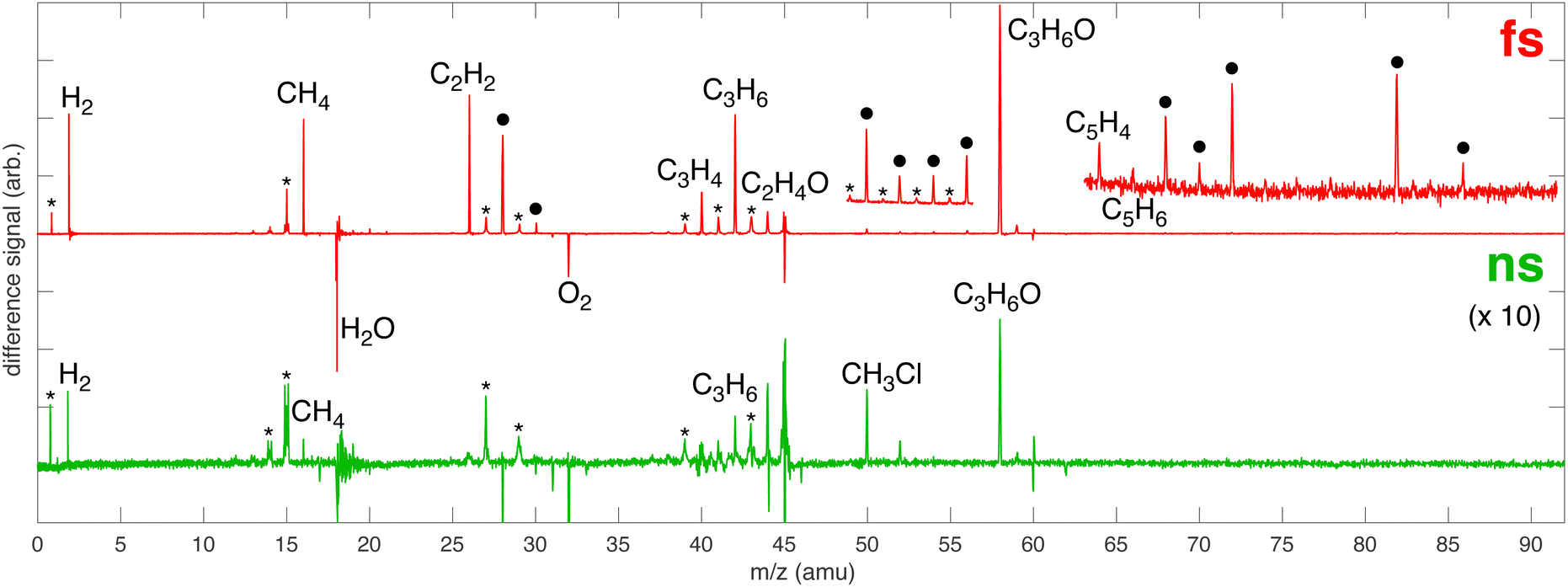
MS analysis of the volatile reaction products of [AuCl4]− in IPA/water indicates that addition of Au substantially enhances the decomposition of IPA in both femtosecond and nanosecond laser processing. A plethora of new products are observed with both femtosecond and nanosecond laser processing in Fig. 5, enumerated in Table 1. The enhanced formation of chemical products from IPA decomposition likely arises from the presence of the Au NPs that are formed by LRL of [AuCl4]−.
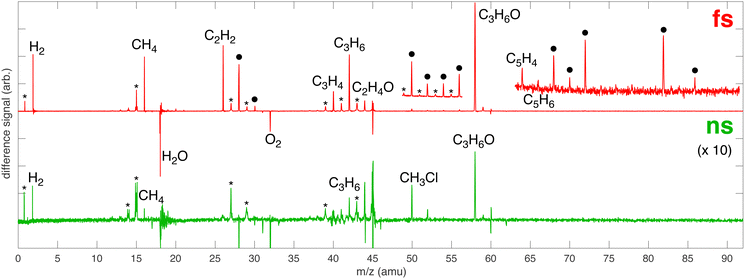 |
| | Fig. 5 Difference MS for femtosecond (red) and nanosecond (green) laser processing of 1 mM [AuCl4]− in 100 mM aqueous IPA with major products indicated. The ● denotes that multiple species can contribute to the peak and the * indicates a peak due to fragmentation. The nanosecond spectrum is magnified by a factor of 10 to clearly show all products. Insets magnify the regions m/z 49–57 and 63–91 in the femtosecond spectrum to show low-yield products. | |
Table 1 Peak assignments for new species in Fig. 5 based on data from NIST Standard Reference Database 69.46 The ‘x’ denotes the presence of the species
|
m/z |
Assignment |
fs |
ns |
|
Also observed with added Ag+ (Fig. 3).
|
| 2 |
H2 |
x
|
x
|
| 16 |
CH4 |
x
|
x
|
| 26 |
C2H2 |
x
|
|
| 28 |
C2H4, CO |
x
|
|
| 30 |
C2H6, CH2O |
x
|
|
| 40 |
C3H4 |
x
|
|
| 42 |
C3H6 |
x
|
x
|
| 44 |
C2H4O |
x
|
|
| 50 |
CH335Cl, C4H2 |
x
|
x
|
| 52 |
CH337Cl, C4H4 |
x
|
x
|
| 54 |
C4H6, C3H2O |
x
|
|
| 56 |
C4H8, C3H4O |
x
|
|
| 58 |
C3H6O |
x
|
x
|
| 64 |
C5H4 |
x
|
|
| 66 |
C5H6 |
x
|
|
| 68 |
C5H8, C4H4O |
x
|
|
| 70 |
C5H10, C4H6O |
x
|
|
| 72 |
C5H12, C4H8O, C3H4O2 |
x
|
|
| 82 |
C6H10, C5H6O |
x
|
|
| 86 |
C6H14, C5H10O, C4H6O2 |
x
|
|
Au NPs are known to enhance optical breakdown of water and alcohols during nanosecond laser processing.37–39 Low-fluence nanosecond laser processing of IPA and Au NPs was reported to produce acetone, acetaldehyde, and methanol as major products.38 In another study, nanosecond laser ablation of an Au target in ethylene glycol was found to produce H2, CH4, and CO as major products.39 The nanosecond sample shows no clear evidence of acetaldehyde due to the peak shape at m/z 44 and we cannot conclude whether methanol and CO formed due to contamination from O2 at m/z 32 and N2 at m/z 28. Nevertheless, the formation of acetone, CH4, and H2 supports the assertion that the Au NPs formed by LRL of [AuCl4]− facilitate optical breakdown under low-fluence nanosecond laser processing because neither CH4 nor H2 were observed in Fig. 1 or 3.
Femtosecond laser excitation at 800 nm is known to efficiently form and excite plasma in the vicinity of Au NPs.55 The resulting enhancement of the laser-induced shock waves can facilitate C–C coupling reactions, as proposed previously.53 The MS peaks in the range of m/z 64–86 in Fig. 5 contain four or more carbon atoms (Table 1), suggesting that enhanced plasma reactivity imparted by the Au NPs induces C–C bond formation in our experiments. Excess plasma energy imparted by Au NPs can also account for the formation of the alkynes C2H2 (m/z 26), C3H4 (m/z 40), C4H2 (m/z 50), and C5H4 (m/z 64). The alkynes acetylene (C2H2, 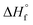 = 54.6 ± 0.1 kJ mol−1) and propyne (C3H4,
= 54.6 ± 0.1 kJ mol−1) and propyne (C3H4, 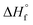 = 44.3 ± 0.2 kJ mol−1) have substantially higher heats of formation than the corresponding alkenes ethylene (C2H4,
= 44.3 ± 0.2 kJ mol−1) have substantially higher heats of formation than the corresponding alkenes ethylene (C2H4, 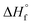 = 12.5 ± 0.1 kJ mol−1) and propene (C3H6,
= 12.5 ± 0.1 kJ mol−1) and propene (C3H6, 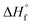 = 4.6 ± 0.3 kJ mol−1).49 The formation of alkynes from femtosecond laser processing only in the presence of Au NPs suggests that the enhanced plasma energy they provide opens up additional high-energy reaction pathways.
= 4.6 ± 0.3 kJ mol−1).49 The formation of alkynes from femtosecond laser processing only in the presence of Au NPs suggests that the enhanced plasma energy they provide opens up additional high-energy reaction pathways.
Peaks at m/z 50 and 52 are observed in both the nanosecond and femtosecond samples in Fig. 5. These species are assigned to CH3Cl with35 Cl and37 Cl, respectively, in the nanosecond sample because the ratio of the integrated signals of m/z 50/52 is 3.05, closely matching the natural chlorine 35/37 isotopic ratio of 3.12. Moreover, no other alkynes were observed in the nanosecond sample, so the formation of C4H2 and C4H4 is highly unlikely. In contrast, the m/z 50/52 ratio in the femtosecond sample is 2.14, which indicates that these signals cannot exclusively be attributed to CH3Cl; instead at least some portion comes from C4H2 and C4H4, consistent with the observation of other alkynes. We note that the resolution of our MS instrument is not sufficient to resolve the small mass differences between C4H2 and CH335Cl, so it is not possible to definitively conclude that CH3Cl was formed during femtosecond processing. Nevertheless, the observation of H2 and reactive chlorine species from femtosecond processing suggest that CH3Cl can easily be formed under plasma conditions.
CH3Cl has been observed as a product of photochemical reactions involving Au NPs, [AuCl4]−, and IPA catalyzed by 488 nm light.56 The authors proposed that photoexcitation of the surface plasmon resonance (SPR) of the Au NP results in oxidation of IPA to acetone. Subsequent chlorine transfer from [AuCl4]− to acetone caused C–C bond cleavage to form CH3Cl and the acetyl radical, which went on to form acetaldehyde. This mechanism could also be operative under our nanosecond laser processing conditions because the 532 nm laser wavelength is resonant with the Au NP SPR frequency. Additionally, Cl˙ radicals formed by homolytic Au–Cl bond cleavage could form CH3Cl from both IPA and acetone through the reactions
| | | Cl˙ + (CH3)2CHOH → CH3Cl + CH3CHOH˙ | (21) |
| | | Cl˙ + (CH3)2CO → CH3Cl + CH3CO˙ | (22) |
According to the active thermochemical tables (ATcT) database,
57reaction (21) has Δ
H° = 12.51 ± 0.54 kJ mol
−1 and
reaction (22) is barely endothermic with Δ
H° = 2.94 ± 0.4 kJ mol
−1. Because previous gas-phase studies have identified acetone as the major product of Cl˙ reaction with IPA
58 and the barrier to
reaction (22) is lower, it is likely that the CH
3Cl is predominantly formed from reaction of Cl˙ with acetone. For femtosecond processing, it is also possible that CH
3Cl is formed by direct recombination of Cl˙ and CH
3˙ present in the plasma.
3.4 Discussion
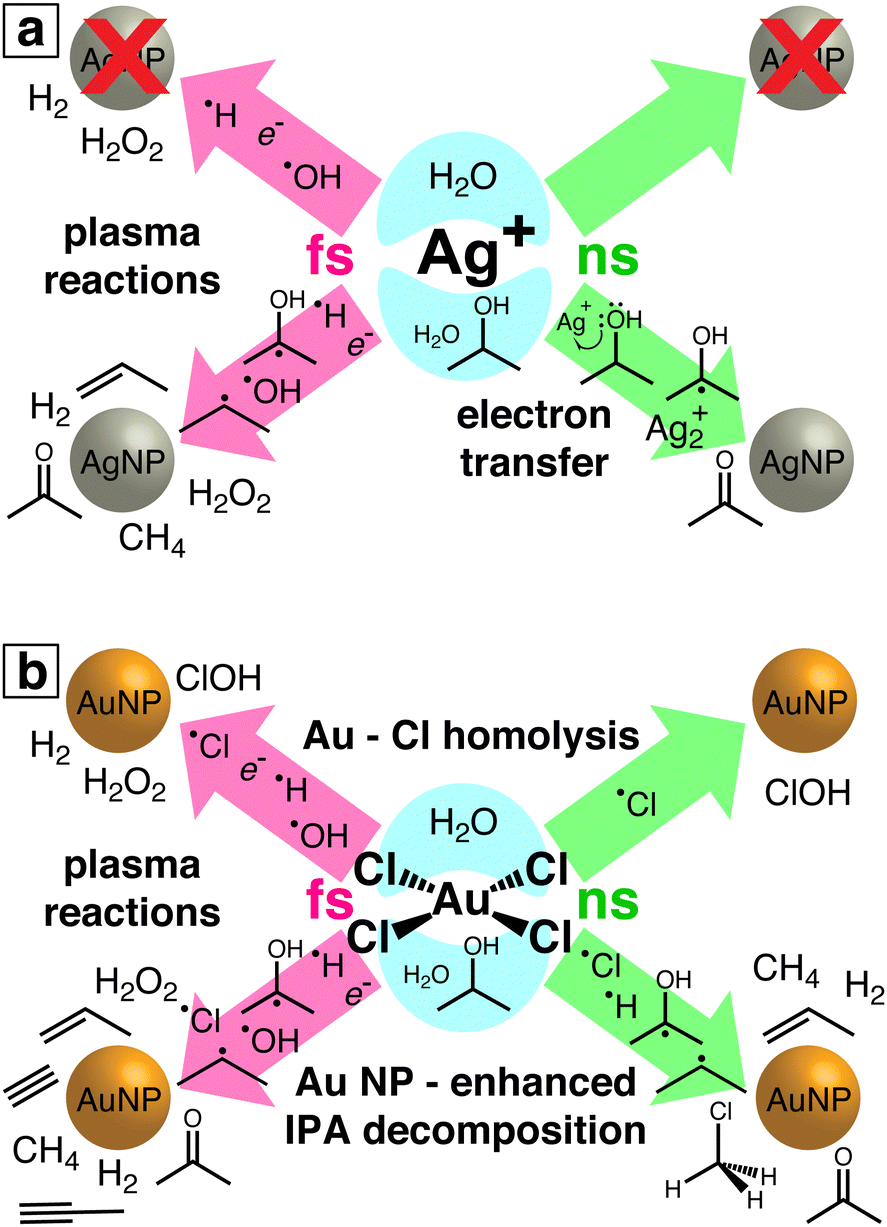
The results from this work both identify a new pathway for two-photon laser reduction of Ag+ by electron-transfer from IPA and reveal more complex reaction pathways involving [AuCl4]− and Au NPs than observed in previous LRL studies. For the first time, chemical byproducts of laser-induced metal ion reduction in the presence of an organic species have been experimentally characterized. Although previous studies have detected H2O27,8,10,16,18–20 and H213 as byproducts of femtosecond laser [AuCl4]− reduction in aqueous solution, the formation of free chlorine species has not previously been observed in any LRL study. Fig. 6 summarizes the reaction pathways of Ag+ (Fig. 6(a)) and [AuCl4]− (Fig. 6b) observed in our experiments. Detected reaction products and putative intermediate species for each reaction condition (femtosecond or nanosecond laser excitation; water or water/IPA solution) are indicated. In the remainder of this section, we discuss the four main classes of reaction pathways identified in Fig. 6: electron transfer, Au–Cl homolysis, plasma reactions, and Au NP-enhanced IPA decomposition.
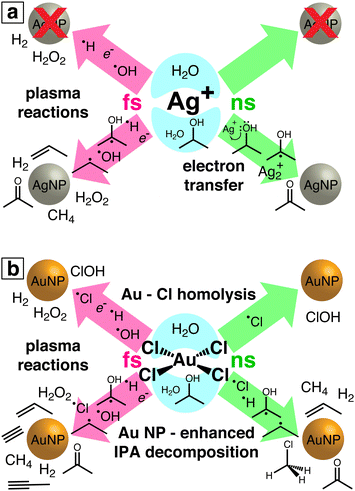 |
| | Fig. 6 Schematic of reaction pathways observed in laser-induced reduction of Ag+ (a) and [AuCl4]− (b). Detected reaction products and putative reaction intermediates are indicated for each reaction condition: femtosecond (pink) or nanosecond (green) laser excitation; water (top) or water/IPA (bottom). | |
Electron transfer from IPA to Ag+ to produce acetone by non-ionizing nanosecond laser excitation presents a new LRL mechanism for Ag+, distinct from previous nanosecond LRL reports in which water ionization reduced Ag+.21,22 Although photoinduced electron transfer from IPA to Ag+ under UV excitation was proposed decades ago,33 this work represents the first time that this mechanism was observed using visible light to induce two-photon excitation. It is possible that similar electron transfer mechanisms also operate in the numerous reported Ag NP syntheses using visible light and organic media to reduce Ag+.5
The observation of free chlorine from nanosecond LRL of [AuCl4]− (Fig. 4) provides direct evidence for the Au–Cl homolysis pathways (reactions (3)–(5)) proposed for nanosecond LRL in our previous work.19 The lack of H2O2 formation in nanosecond LRL is also consistent with the observation from X-ray transient absorption spectroscopy that [AuCl4]− reduction proceeds primarily through these bond homolysis pathways upon UV excitation.29 The detection of free chlorine in femtosecond LRL is more surprising because numerous femtosecond LRL studies have established the primacy of plasma reactions (vide infra). Moreover, delayed formation of [Au2+Cl3]− after 800 nm femtosecond excitation observed in X-ray transient absorption spectroscopy confirmed that hydrated electrons initiate [AuCl4]− reduction.29 Although a small contribution of direct Au–Cl homolysis in [AuCl4]− is possible in femtosecond LRL based on the reaction kinetics,8 it is also possible that the free chlorine species arise by Cl˙ loss from the clusters [Au2Cl2]2− and [Au4Cl4]4−. These clusters were present at times ranging from milliseconds to hundreds of seconds after 800 nm femtosecond laser excitation according to X-ray transient absorption spectroscopy.29 Excitation of these clusters by a subsequent femtosecond laser pulse could release Cl˙. Although the present measurements cannot determine whether free chlorine is released from [AuCl4]− or small Au0 clusters, its effects should be taken into account for both nanosecond and femtosecond LRL of other chlorine-containing precursor complexes such as [PtCl4]2− and [PdCl4]2−.
Plasma reactions involving eaq−, H˙, OH˙, and H2O2 are well established as the primary reaction pathways that drive femtosecond LRL.9–11,17–20,24–26 It should be noted that the high-intensity laser excitation can also induce direct Au–Cl homolysis that contributes Cl˙ to the plasma species present during [AuCl4]− reduction, as discussed above. Numerous groups have demonstrated that the kinetics of metal ion reduction and metal NP sizes can be controlled through addition of chemical scavengers for eaq−11,17,20 and OH˙.18,24–26 Although this plasma environment is widely considered to resemble radiolysis conditions,6,11,17 the present results demonstrate that additional IPA decomposition pathways that form propene are accessible in femtosecond laser plasma that do not occur in radiolysis.47 This enhanced formation of organic byproducts during femtosecond LRL can affect the metal NP products. No organic ligands were observed on the Au NPs synthesized in water/IPA,18 but LRL of copper and silver acetylacetonate in IPA produced NPs with a carbon shell.59,60
The observed enhanced IPA decomposition when Au NPs are present opens up the possibility of driving the formation of organic products through LRL of [AuCl4]−, particularly with femtosecond excitation. In particular, our observation of numerous alkynes from IPA decomposition suggests that addition of [AuCl4]− or Au NPs may enhance the yields of polyynes (linear chains of sp-hybridized carbons) from femtosecond laser processing of organic liquids. Typically, laser processing of neat organic liquids requires hours to produce observable polyyne yields.61 Laser ablation of Au in ethanol was found to produce higher yields of polyynes,62 likely due to Au NP-enhanced plasma reactions. Hence, our results support the potential use of Au NPs to enhance the production of specific organic products by laser processing.
4. Conclusion
Photochemical reaction pathways of Ag+ and [AuCl4]− reduction during nanosecond and femtosecond laser processing in solution were determined using strong-field ionization mass spectrometry and spectroscopic assays. An electron transfer pathway from IPA to Ag+ induced by two-photon visible excitation with low-fluence nanosecond laser pulses that produces acetone as a byproduct was identified. Free chlorine species were detected after reduction of [AuCl4]− by both nanosecond and femtosecond laser pulses, indicating the formation of Cl˙ as a reaction intermediate. In addition to IPA decomposition pathways observed in radiolysis, femtosecond laser plasma also induces formation of propene, likely due to the actions of laser-induced shock waves. The presence of Au NPs was found to further enhance IPA decomposition, producing alkyne products from femtosecond laser excitation and CH3Cl from nanosecond laser excitation. Collectively, this knowledge that a plethora of complex reaction pathways occur during LRL of metal precursors can be applied to design laser processing syntheses to better control the properties of metal nanoparticles and enhance yields of organic products.
Author contributions
L. M. Frias Batista: conceptualization, methodology, software, formal analysis, investigation, writing – original draft. M. Moody: methodology, formal analysis, investigation, validation. C. Weththasingha: methodology, formal analysis, investigation, validation. E. Kaplan: methodology, formal analysis, investigation, validation. I. Faruque: formal analysis, investigation, validation. M. S. El-Shall: conceptualization, supervision, resources, funding acquisition, writing - review & editing. K. M. Tibbetts: conceptualization, supervision, resources, funding acquisition, project administration, visualization, writing – review & editing.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work was supported by the National Science Foundation through grant CHE-1900094 (MSE) and the U.S. Army Research Office through Contract W911NF-19-1-0099 (KMT). LMFB acknowledges generous financial support from an Altria Graduate Research Fellowship.
Notes and references
- K. Guo, A. Baidak and Z. Yu, J. Mater. Chem. A, 2020, 8, 23029–23058 RSC.
- J. Belloni, M. Mostafavi, H. Remita, J.-L. Marignier and A. Marie-Odile Delcourt, New J. Chem., 1998, 22, 1239–1255 RSC.
- K. Čubová and V. Čuba, Radiation Physics and Chemistry, 2020, 169, 108774 CrossRef.
- M. Sakamoto, M. Fujistuka and T. Majima, J. Photochem. Photobiol., C, 2009, 10, 33–56 CrossRef CAS.
- N. Jara, N. S. Milán, A. Rahman, L. Mouheb, D. C. Boffito, C. Jeffryes and S. A. Dahoumane, Molecules, 2021, 26, 4585 CrossRef CAS PubMed.
- L. M. Frias Batista, A. Nag, V. K. Meader and K. M. Tibbetts, Sci. China: Phys., Mech. Astron., 2022, 65, 1–45 Search PubMed.
- B. Tangeysh, K. Moore Tibbetts, J. H. Odhner, B. B. Wayland and R. J. Levis, J. Phys. Chem. C, 2013, 117, 18719–18727 CrossRef CAS.
- K. Moore Tibbetts, B. Tangeysh, J. H. Odhner and R. J. Levis, J. Phys. Chem. A, 2016, 120, 3562–3569 CrossRef CAS PubMed.
- N. Nakashima, K. Yamanaka, M. Saeki, H. Ohba, S. Taniguchi and T. Yatsuhashi, J. Photochem. Photobiol., A, 2016, 319–320, 70–77 CrossRef CAS.
- V. K. Meader, M. G. John, C. J. Rodrigues and K. M. Tibbetts, J. Phys. Chem. A, 2017, 121, 6742–6754 CrossRef CAS PubMed.
- H. Belmouaddine, M. Shi, P.-L. Karsenti, R. Meesat, L. Sanche and D. Houde, Phys. Chem. Chem. Phys., 2017, 19, 7897–7909 RSC.
- C. Zhao, S. Qu, J. Qiu and C. Zhu, J. Mater. Res., 2003, 18, 1710–1714 CrossRef CAS.
- T. Nakamura, Y. Mochidzuki and S. Sato, J. Mater. Res., 2008, 23, 968–974 CrossRef CAS.
- T. Nakamura, Y. Herbani, D. Ursescu, R. Banici, R. V. Dabu and S. Sato, AIP Adv., 2013, 3, 082101 CrossRef.
- Muttaqin, T. Nakamura and S. Sato, Appl. Phys. A: Mater. Sci. Process., 2015, 120, 881–888 CrossRef CAS.
- J. H. Odhner, K. Moore Tibbetts, B. Tangeysh, B. B. Wayland and R. J. Levis, J. Phys. Chem. C, 2014, 118, 23986–23995 CrossRef CAS.
- H. Belmouaddine, M. Shi, L. Sanche and D. Houde, Phys. Chem. Chem. Phys., 2018, 20, 23403–23413 RSC.
- L. M. Frias Batista, V. K. Meader, K. Romero, K. Kunzler, F. Kabir, A. Bullock and K. M. Tibbetts, J. Phys. Chem. B, 2019, 123, 7204–7213 CrossRef CAS PubMed.
- C. J. Rodrigues, J. A. Bobb, M. G. John, S. P. Fisenko, M. S. El-Shall and K. M. Tibbetts, Phys. Chem. Chem. Phys., 2018, 20, 28465–28475 RSC.
- K. Y. Putri, A. L. Fadli, F. A. Umaroh, Y. Herbani, C. Imawan and D. Djuhana, Radiat. Phys. Chem., 2022, 199, 110269 CrossRef CAS.
- J. P. Abid, A. W. Wark, P. F. Brevet and H. H. Girault, Chem. Commun., 2002, 792–793 RSC.
- U. Y. Qazi, S. Kajimoto and H. Fukumura, Chem. Lett., 2014, 43, 1693–1695 CrossRef.
- T. Nakamura, H. Magara, Y. Herbani and S. Sato, Appl. Phys. A: Mater. Sci. Process., 2011, 104, 1021 CrossRef CAS.
- V. K. Meader, M. G. John, L. M. Frias Batista, S. Ahsan and K. M. Tibbetts, Molecules, 2018, 23, 532 CrossRef PubMed.
- Y. Herbani, T. Nakamura and S. Sato, J. Phys. Conf. Ser., 2017, 817, 012048 CrossRef.
- C. M. Nguyen, L. M. Frias Batista, M. G. John, C. J. Rodrigues and K. M. Tibbetts, J. Phys. Chem. B, 2021, 125, 907–917 CrossRef CAS PubMed.
- S. Eustis, H.-Y. Hsu and M. A. El-Sayed, J. Phys. Chem. B, 2005, 109, 4811–4815 CrossRef CAS PubMed.
- M. Harada and S. Kizaki, Cryst. Growth Des., 2016, 16, 1200–1212 CrossRef CAS.
- X. Liu, X. Zhang, D. Khakhulin, P. Su, M. Wulff, F. Baudelet, T.-C. Weng, Q. Kong and Y. Sun, J. Phys. Chem. Lett., 2022, 13, 8921–8927 CrossRef CAS PubMed.
-
P. Vanýsek, CRC Handbook of Chemistry and Physics, CRC Press, Boca Raton, FL, 1998, ch. Electrochemical Series Search PubMed.
- D. Spadaro, E. Barletta, F. Barreca, G. Currò and F. Neri, Appl. Surf. Sci., 2010, 256, 3812–3816 CrossRef CAS.
- L. Maretti, P. S. Billone, Y. Liu and J. C. Scaiano, J. Am. Chem. Soc., 2009, 131, 13972–13980 CrossRef CAS PubMed.
- H. Hada, Y. Yonezawa, A. Yoshida and A. Kurakake, J. Phys. Chem., 1976, 80, 2728–2731 CrossRef CAS.
- A. Henglein, Ber. Bunsenges. Phys. Chem., 1977, 81, 556–561 CrossRef CAS.
- R. Tausch-Treml, A. Henglein and J. Lilie, Ber. Bunsenges. Phys. Chem., 1978, 82, 1335–1343 CrossRef CAS.
- G. L. Gutsev, D. Ampadu Boateng, P. Jena and K. M. Tibbetts, J. Phys. Chem. A, 2017, 121, 8414–8424 CrossRef CAS PubMed.
- E. V. Barmina, A. V. Simakin and G. A. Shafeev, Chem. Phys. Lett., 2017, 678, 192–195 CrossRef CAS.
- A. V. Simakin, M. E. Astashev, I. V. Baimler, O. V. Uvarov, V. V. Voronov, M. V. Vedunova, M. A. Sevost'yanov, K. N. Belosludtsev and S. V. Gudkov, J. Phys. Chem. B, 2019, 123, 1869–1880 CrossRef CAS PubMed.
- M. R. Kalus, N. Bärsch, R. Streubel, E. Gökce, S. Barcikowski and B. Gökce, Phys. Chem. Chem. Phys., 2017, 19, 7112–7123 RSC.
- M.-R. Kalus, R. Lanyumba, N. Lorenzo-Parodi, M. A. Jochmann, K. Kerpen, U. Hagemann, T. C. Schmidt, S. Barcikowski and B. Gökce, Phys. Chem. Chem. Phys., 2019, 21, 18636–18651 RSC.
- C. Aeppli, M. Berg, T. B. Hofstetter, R. Kipfer and R. P. Schwarzenbach, J. Chromatogr. A, 2008, 1181, 116–124 CrossRef CAS PubMed.
-
D. Harp and C. C. Hach, Current Technology of Chlorine Analysis for Water and Wastewater, Hach Company, Loveland, CO, USA, 2003 Search PubMed.
- G. Eisenberg, Ind. Eng. Chem., Anal. Ed., 1943, 15, 327–328 CrossRef CAS.
- G. Maatz, A. Heisterkamp, H. Lubatschowski, S. Barcikowski, C. Fallnich, H. Welling and W. Ertmer, J. Opt. A, 1999, 2, 59–64 CrossRef.
- H. Kierzkowska-Pawlak, J. Tyczkowski, A. Jarota and H. Abramczyk, Appl. Energy, 2019, 247, 24–31 CrossRef CAS.
-
https://webbook.nist.gov/chemistry/. Last checked 1/17/22.
- L. Gilles and J. Sutton, J. Chim. Phys., 1970, 67, 128–138 CrossRef CAS.
-
C. Von Sonntag and H.-P. Schuchmann, Photolysis of Saturated Alcohols, Ethers, and Amines, John Wiley & Sons, Ltd, 1977, pp. 59–145 Search PubMed.
- C. F. Goldsmith, G. R. Magoon and W. H. Green, J. Phys. Chem. A, 2012, 116, 9033–9057 CrossRef CAS PubMed.
- G. E. Adams and R. L. Willson, Trans. Faraday Soc., 1969, 65, 2981–2987 RSC.
- D. C. Guerin, R. F. Fernsler and V. A. Shamamian, J. Vac. Sci. Technol., A, 2003, 21, 1724–1733 CrossRef CAS.
- T. Juhasz, G. A. Kastis, C. Suárez, Z. Bor and W. E. Bron, Lasers Surg. Med., 1996, 19, 23–31 CrossRef CAS PubMed.
- W. Ishikawa and S. Sato, Chem. Phys. Chem., 2020, 21, 2104–2111 CrossRef CAS PubMed.
- W. Liu, S. A. Andrews, M. I. Stefan and J. R. Bolton, Water Res., 2003, 37, 3697–3703 CrossRef CAS PubMed.
- R. Lachaine, É. Boulais and M. Meunier, ACS Photonics, 2014, 1, 331–336 CrossRef CAS.
- V. Mohan, E. Wu, J. Heo, A. Das and P. K. Jain, ACS Energy Lett., 2021, 6, 1980–1989 CrossRef CAS.
-
B. Ruscic and D. H. Bross, Active Thermochemical Tables (ATcT) values based on ver. 1.124 of the Thermochemical Network, Argonne National Laboratory, Lemont, Illinois 2022, https://atct.anl.gov/Thermochemical.
- T. Yamanaka, M. Kawasaki, M. D. Hurley, T. J. Wallington, W. F. Schneider and J. Bruce, Phys. Chem. Chem. Phys., 2007, 9, 4211–4217 RSC.
- A. Nag, L. M. Frias Batista and K. M. Tibbetts, Nanomaterials, 2021, 11, 814 CrossRef CAS PubMed.
- A. Nag, C. M. Nguyen and K. M. Tibbetts, Appl. Surf. Sci., 2023, 610, 155384 CrossRef CAS.
- A. Ramadhan, M. Wesolowski, T. Wakabayashi, H. Shiromaru, T. Fujino, T. Kodama, W. Duley and J. Sanderson, Carbon, 2017, 118, 680–685 CrossRef CAS.
- B. Pan, J. Xiao, J. Li, P. Liu, C. Wang and G. Yang, Sci. Adv., 2015, 1, e1500857 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Calibration curves and additional spectroscopic assay data; additional mass spectra; tabulated results of spectroscopic assays. See DOI: https://doi.org/10.1039/d3cp00052d |
|
| This journal is © the Owner Societies 2023 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence ,
Irtiza
Faruque
,
M. Samy
El-Shall
,
Irtiza
Faruque
,
M. Samy
El-Shall
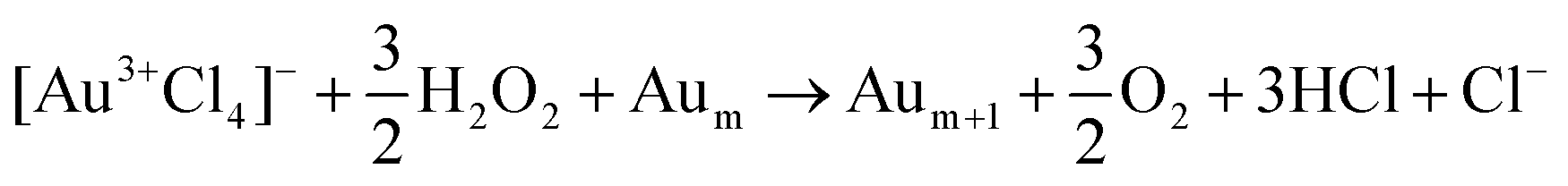
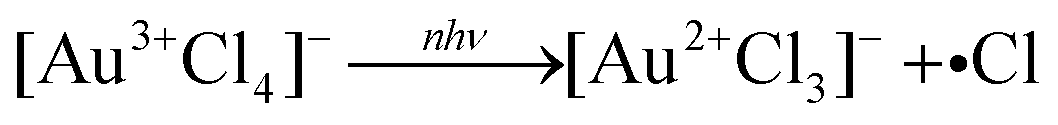
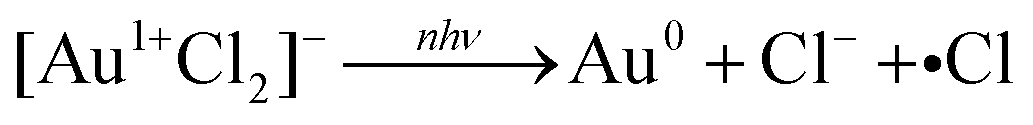
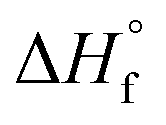 = 54.6 ± 0.1 kJ mol−1) and propyne (C3H4,
= 54.6 ± 0.1 kJ mol−1) and propyne (C3H4, 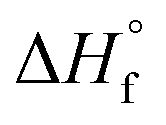 = 44.3 ± 0.2 kJ mol−1) have substantially higher heats of formation than the corresponding alkenes ethylene (C2H4,
= 44.3 ± 0.2 kJ mol−1) have substantially higher heats of formation than the corresponding alkenes ethylene (C2H4, 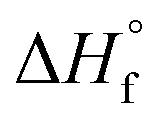 = 12.5 ± 0.1 kJ mol−1) and propene (C3H6,
= 12.5 ± 0.1 kJ mol−1) and propene (C3H6, 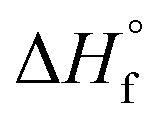 = 4.6 ± 0.3 kJ mol−1).49 The formation of alkynes from femtosecond laser processing only in the presence of Au NPs suggests that the enhanced plasma energy they provide opens up additional high-energy reaction pathways.
= 4.6 ± 0.3 kJ mol−1).49 The formation of alkynes from femtosecond laser processing only in the presence of Au NPs suggests that the enhanced plasma energy they provide opens up additional high-energy reaction pathways.