
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
SPICY: a method for single scan rotating frame relaxometry†
Katja
Tolkkinen
 a,
Sarah E.
Mailhiot
a,
Anne
Selent
a,
Otto
Mankinen
a,
Henning
Henschel
b,
Miika T.
Nieminen
cde,
Matti
Hanni
cde,
Anu M.
Kantola
a,
Timo
Liimatainen
cde and
Ville-Veikko
Telkki
*a
a,
Sarah E.
Mailhiot
a,
Anne
Selent
a,
Otto
Mankinen
a,
Henning
Henschel
b,
Miika T.
Nieminen
cde,
Matti
Hanni
cde,
Anu M.
Kantola
a,
Timo
Liimatainen
cde and
Ville-Veikko
Telkki
*a
aNMR Research Unit, University of Oulu, Oulu, Finland. E-mail: ville-veikko.telkki@oulu.fi
bDepartment of Medicinal Chemistry, Uppsala University, Uppsala, Sweden
cResearch Unit of Health Sciences and Technology, University of Oulu, Oulu, Finland
dDepartment of Diagnostic Radiology, Oulu University Hospital, Oulu, Finland
eMedical Research Center Oulu, University of Oulu and Oulu University Hospital, Oulu, Finland
First published on 13th April 2023
Abstract
T 1ρ is an NMR relaxation mode that is sensitive to low frequency molecular motions, making it an especially valuable tool in biomolecular research. Here, we introduce a new method, SPICY, for measuring T1ρ relaxation times. In contrast to conventional T1ρ experiments, in which the sequence is repeated many times to determine the T1ρ time, the SPICY sequence allows determination of T1ρ within a single scan, shortening the experiment time remarkably. We demonstrate the method using 1H T1ρ relaxation dispersion experiments. Additionally, we combine the sequence with spatial encoding to produce 1D images in a single scan. We show that T1ρ relaxation times obtained using the single scan approach are in good agreement with those obtained using the traditional experiments.
1. Introduction
Nuclear magnetic resonance (NMR) experiments provide versatile information on time-dependent dynamic processes of molecules. NMR relaxation experiments give information on molecular rotational motions and environments. Most often, NMR relaxation studies utilize longitudinal T1 and transverse T2 relaxation. T1 is sensitive to molecular motions with frequencies close to the Larmor frequency, ω0 = γB0, which is determined using gyromagnetic ratio γ and the strength of the static magnetic field B0.1 To probe a range of molecular motion frequencies, B0 needs to be changed,2 which is not possible with conventional NMR instruments.However, there exists a third relaxation process called T1ρ, or spin–lattice relaxation in the rotating frame, where the nuclear magnetization returns to equilibrium under the influence of continuous wave spin lock (SL) field B1.1,3,4 Since the ω1 frequency can be changed, typically between 1 and 6 kHz, T1ρ can be utilized to probe motional processes over a wide dynamic timescale.4 The T1ρ relaxation time extends between T2 and T1 as the SL field increases, and this increase of T1ρ is called T1ρ relaxation dispersion.4–7
The other common method for measuring relaxation dispersion is T2 dispersion in which the effective transverse relaxation time is measured as a function of the time between the refocusing pulses in the Carr–Purcell–Meiboom–Gill (CPMG) pulse sequence.8,9 However, in conventional NMR instruments, T2 relaxation dispersion measurements are limited to lower frequency ranges than T1ρ dispersion.4,10
Relaxation dispersion measurements have been shown to be a powerful tool for characterizing micro- and millisecond timescale biomolecular motions, such as chemical exchange and protein dynamics4–7,11–16 and biomechanical properties of objects like rat tissues17,18 and articular cartilage of various origins.19–22 Articular cartilage, which is a connective tissue possessing very specialized biomechanical properties, is composed of cartilage cells which are surrounded by an extracellular matrix, principally consisting of collagen, proteoglycans and water.23T1ρ relaxation and dispersion studies have been utilized in both NMR spectroscopy and MRI to track biochemical changes of the cartilage extracellular matrix.19–22,24–27 An additional use of T1ρ measurements is T1ρ-weighted magnetic resonance imaging (MRI).28T1ρ preparation is often paired with MRI to obtain T1ρ image contrast which provides higher resolution to some pathological changes and disease progression than only with T1 and T2 weighted imaging. For example, T1ρ is sensitive to proteoglycan loss in articular cartilage as osteoarthritis progresses and T1ρ mapping can detect early articular cartilage degradation with more sensitivity than T2 mapping.29–32 Furthermore, T1ρ has shown potential to probe diseases such as spinal disc degeneration,33,34 Alzheimer's disease,35,36 Parkinson's disease37 and ischemia.38
The conventional T1ρ pulse sequence consists of a continuous wave (CW) radiofrequency (RF) pulse followed by the spin lock period and signal acquisition.39–41 Alternatively, the T1ρ experiments can be performed using adiabatic spin locking.42–44 The conventional experiments must be repeated with incremented spin lock times, such that the total experiment time ranges from minutes to days depending on the number of repetitions. Furthermore, T1ρ dispersion experiments are more time consuming since the experiments must be repeated with several SL frequencies. Anoardo et al. have presented an off-resonance technique (SLOAFI)45 for determining T1ρ relaxation dispersion in a shorter time, but to our knowledge, no methods for acceleration of the T1ρ preparation portion of the on-resonance rotating frame relaxation experiment have been reported. To circumvent the long experiment times, several rapid T1ρ imaging methods have been developed. For example, Duvvuri et al.46 combined a fast spin echo sequence with T1ρ preparation, Li et al.47 developed a T1ρ imaging sequence based on spiral k-space readout, Liimatainen and Gröhn48 acquired T1ρ decay of one k-space line during single adiabatic pulse train, and Bothakur et al.49 introduced a pulse sequence for spin locked echo planar imaging (SLEPI). There are also accelerated T1ρ methods based on gradient echoes and steady state free precession,50–53 and reconstruction of undersampled k-space data.54–60 The development of rapid imaging methods is advantageous not only for reducing patient imaging time, but also for allowing imaging of dynamic objects like a cardiovascular system.54–57 In addition, the short experiment time of SLEPI has expanded the application of T1ρ to functional MRI.61
Here, we introduce a novel single scan method for rotating frame relaxometry. In this method, the T1ρ is produced by the SPIn lock CYcle (SPICY), in which a single RF excitation pulse is followed by a loop of spin locking and signal acquisition lobes. This allows for the determination of T1ρ within a single scan, similar to the time domain Carr–Purcell–Meiboom–Gill (CPMG)8 acquisition, which measures T2. The total scan time is from a few seconds to minutes. We perform 1H T1ρ dispersion experiments with both the conventional and the SPICY T1ρ sequences on liquid samples and protein hydrogels prepared from the main constituents of the articular cartilage extracellular matrix, and we show that the relaxation time information obtained with these methods is in good agreement. In addition, we show that the SPICY sequence can be combined with spatial encoding to produce 1D images in a single scan.
2. Experimental
2.1. Samples
Three liquid samples, A, B and C, were used. The samples were made by dissolving CuSO4·5H2O in water (10% H2O, 90% D2O), with the sample A containing 0.013 wt% and sample B containing 0.04 wt% CuSO4. The solutions were placed in 5 mm NMR tubes. Sample C represents a two-compartment sample of liquids A and B made by placing the 0.04 wt% solution to the bottom of a 5 mm tube inside which a 3 mm tube containing the 0.13 wt% solution was placed. The tubes were placed in the RF coil in such a way that the liquid B covered the bottom half and liquids A and B the top half of the coil. The illustration of the samples is shown in Fig. 3f.A hydrogel sample containing 40 mg g−1 collagen type II, 10 mg g−1 chondroitin sulfate and phosphate-buffered saline (PBS) was used. 10 mL of collagen solution (rat tail, ∼6 mg mL−1 in 0.01 M acetic acid, Merck KGaA, Darmstadt, Germany) was added to a sample tube together with 0.15 mL 10× PBS. Stock solution of CS (50–80 mg mL−1 CS powder from bovine trachea, Merck KGaA, Darmstadt, Germany in double distilled water) was added to the collagen solution and the mixture was diluted with double distilled water to give a total volume of 6 mL. The sample was thoroughly mixed and pH adjusted to basic (pH: 8–9) using 1 M sodium hydroxide (NaOH) and 0.5 M hydrochloric acid (HCl). The sample tube was placed for 45–60 min in a water bath at 37 °C to solidify the mixture into a gel. The solid was removed from the sample tube, transferred into a plastic cell culture dish, and dried at 40 °C in a laminar flow oven until the mass of the sample was reduced to 1.50 ± 0.03 g. Finally, the sample was placed in a 5 mm NMR tube in the middle of the RF coil.
2.2. Pulse sequence design
The conventional T1ρ pulse sequence39 (Fig. 1a) consists of a 90° excitation pulse, two CW spin lock pulses separated by a 180° pulse, and an acquisition period in which the free induction decay (FID) is recorded. To encode for T1ρ, the sequence is repeated with incremented SL pulse durations. Contrary to the conventional T1ρ pulse sequence, the SPICY pulse sequence (Fig. 1b) utilizes a loop structure which repeats the spin locking of constant length and signal acquisition blocks n times during one scan. To accomplish this, the acquisition period acquires the signal intensity of the spin echo. While in the conventional experiment T1ρ is determined from the FID signal, in the SPICY, the T1ρ fit is obtained from the echo intensities corresponding to different echo numbers. The 1D imaging versions of the conventional T1ρ and the SPICY sequences are shown in Fig. 1c and d. In the SPICY imaging sequence, the third 180° pulse and the dephasing gradient block are needed to return to the center of the k-space before the beginning of the next loop. An 8-step phase cycle is used in the SPICY sequences (see the ESI†).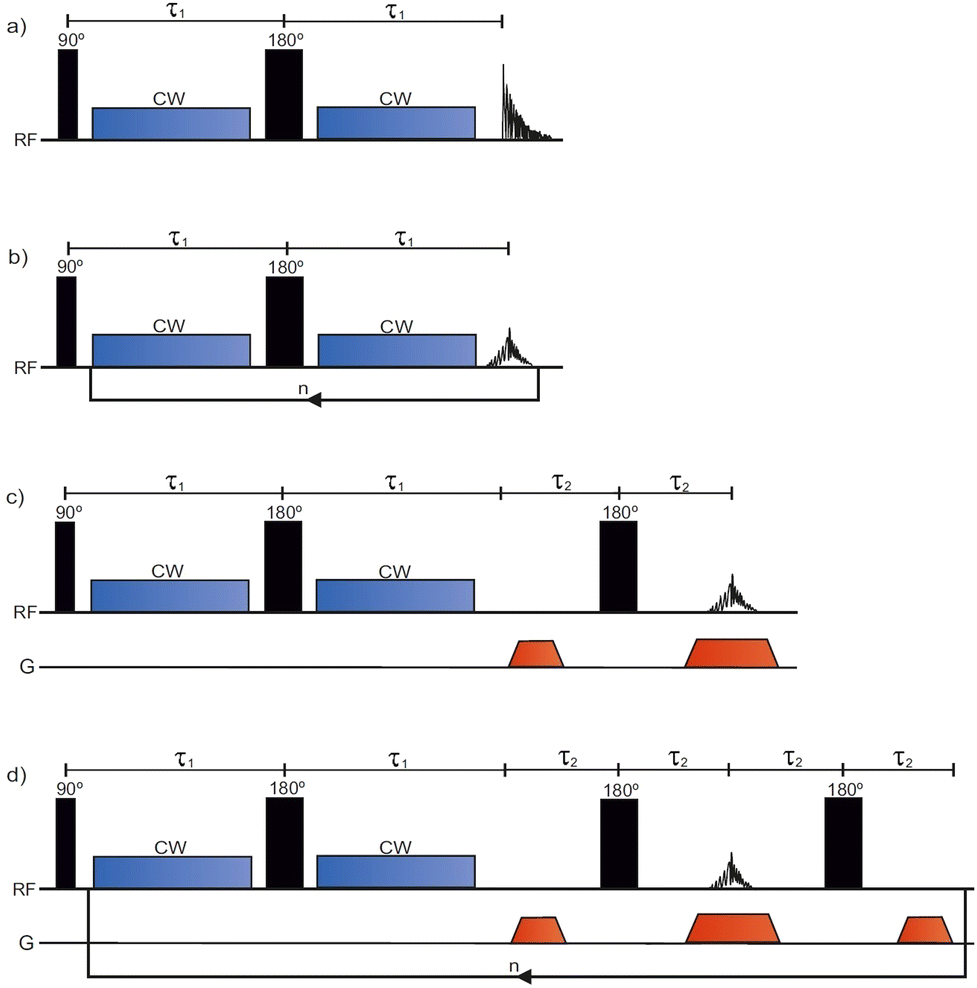 | ||
| Fig. 1 The pulse sequences for (a) conventional T1ρ, (b) SPICY, (c) conventional 1D T1ρ imaging and (d) SPICY 1D imaging. | ||
2.3. Experiments
The T1ρ experiments were performed on a spectrometer with 600 MHz 1H operating frequency equipped with a 5 mm QXI probe with a gradient strength of 0.6 T m−1 and TopSpin 3.6.2 software (Avance III, Bruker Biospin, MA, USA). The probe temperature was set to 290 K during the experiments. In all the experiments, the 90° excitation pulse had a length of P1 ≈ 9.5 μs, and the 180° pulse had P2 ≈ 19 μs. The relaxation delay in all the experiments was 15 s. In all T1ρ experiments, the SL-pulses were applied on-resonance. In the conventional T1ρ experiments (Fig. 1a and c), the SL-pulse length was incremented between 30 and 480 ms in 16 steps. In the non-imaging SPICY measurements (Fig. 1b) of the water samples, the SL pulse duration was 30 ms and the number of echoes n was 16. The signal at the center of the echo was acquired for 50 μs with 12 points and the total SL interruption time during a single loop including all the SL gaps was 130 μs. In the SPICY measurements of hydrogels, the SL pulse duration was 40 ms, the number of echoes was 12, the signal acquisition time was 25 μs, the number of points was 6 and the total SL interruption time was 100 μs. In the SPICY imaging sequence (Fig. 1d), the signal at the center of the echo was acquired for 0.82 ms with 512 points and the total SL interruption time was 8 ms. In both imaging sequences, the read gradient length was 0.82 ms, the gradient ramp time was 100 μs, and the gradient strength was 0.3 T m−1. The length of the dephasing gradients was half of the read gradients. The non-imaging sequences were performed with SL frequencies of 2–11 kHz and the imaging experiments with 7 kHz. The number of scans was 16 for the water samples and 32 for the hydrogel sample. The experiment time of one scan was 4 min 12 s for the conventional and 16 s for the SPICY sequences.2.4. Data analysis
To find T1ρ relaxation times, the equation was fitted to the experimentally obtained signal decay. In the analysis of the SPICY data, the signal was summed across all the points acquired for each echo to perform T1ρ fitting while the area under the spectra was used in the conventional T1ρ analysis. The 1D images were obtained by Fourier transform. The data analysis was done using MATLAB (Mathworks, R2020b, Natick, MA, USA).
was fitted to the experimentally obtained signal decay. In the analysis of the SPICY data, the signal was summed across all the points acquired for each echo to perform T1ρ fitting while the area under the spectra was used in the conventional T1ρ analysis. The 1D images were obtained by Fourier transform. The data analysis was done using MATLAB (Mathworks, R2020b, Natick, MA, USA).
3. Results and discussion
To determine the accuracy of the single scan acquisition method, the T1ρ relaxation times recorded using the SPICY method were compared to the T1ρ relaxation times recorded using the two-dimensional conventional acquisition scheme (Fig. 2). The T1ρ values were measured for three aqueous solutions A, B and C with variable SL fields in the range of 2–11 kHz (Fig. 2a). The T2 values were measured for all samples with SPICY sequence by turning off the CW pulses. All samples show stable T1ρ values over the whole frequency range, and the difference between conventional and SPICY T1ρ values is less than 2% for all three aqueous solutions, which indicates that the SPICY sequence is precise and works under different conditions.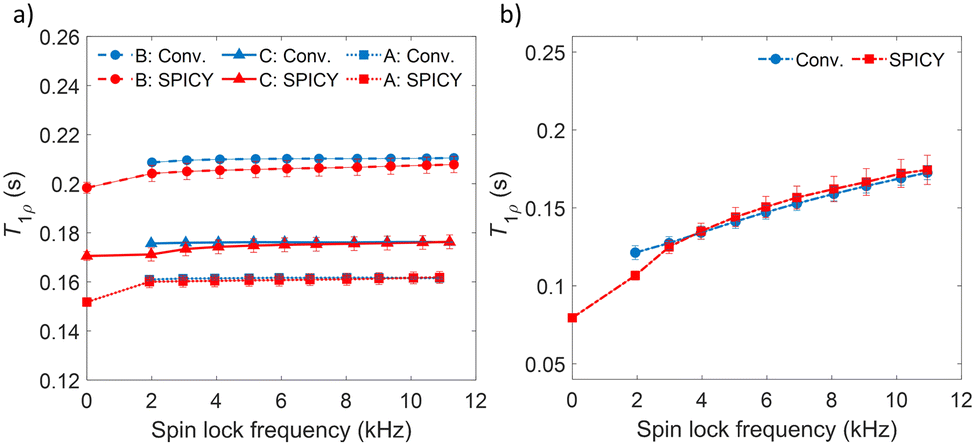 | ||
| Fig. 2 (a) T1ρ measured with different spin lock frequencies for the water samples A, B and C. (b) T1ρ dispersion measured for the hydrogel sample. Blue color indicates the conventional T1ρ and red SPICY. The T1ρ value measured with 0 kHz SL frequency is the T2 of the sample. | ||
To test the applicability of the SPICY sequence on a biochemically more relevant sample, T1ρ dispersion was measured for the hydrogel sample containing collagen and chondroitin sulfate which acts as a model of the cartilage extracellular matrix (Fig. 2b). The SL frequency was varied between 2–12 kHz leading the T1ρ values to increase between 0.12–0.17 s. The difference between the SPICY and reference T1ρ values is only 1–2% showing that the SPICY method is applicable to T1ρ dispersion studies of more complicated samples as well. The SPICY sequence was further tested with imaging such that 1D read gradients were used to record 1D signal intensity data (Fig. 1d and 3). The 1D images of the samples A and C measured with the conventional and the SPICY readouts are shown in Fig. 3a–d. The image profiles obtained with both methods are consistent. The T1ρ profiles measured for all three samples are shown in Fig. 3e. The T1ρ values obtained with SPICY imaging are about 8% smaller than the values measured with the conventional imaging sequence. The small difference is expected to arise predominantly from the signal decay caused by molecular diffusion in the presence of gradients, because the SPICY imaging experiment includes more gradient pulses due to the additional rephasing gradient and loop structure.62 The difference can be minimized by minimizing the gradient strength and length. Both methods give stable profiles, however the T1ρ values measured with SPICY show decreased T1ρ values at the edges of the coil. This is a consequence of a decrease of the B1 field close to the edges of the coil and much higher number of 180° pulses in the SL gaps during the SPICY experiment. One of the major differences between the principles of SPICY and conventional sequences is the T2 decay occurring during the SL gaps during the SPICY experiment. For that reason, the delays between the SL pulses and the signal acquisition period should be set as short as possible. Especially samples with strong dispersion can be very sensitive to non-locked periods, which is why T2 relaxation should be considered when performing the SPICY measurements. In the SPICY imaging sequence, which includes the longer acquisition period due to the imaging gradients, the T2 decay affects the signal more than in the non-imaging experiments. The T2 effect can be reduced either by shortening the non-locked periods or by reducing the number of echoes and increasing the SL pulse length. Since the acquisition time of the echo signal should be short, the SPICY sequence cannot be used to resolve spectral lines by Fourier transform.
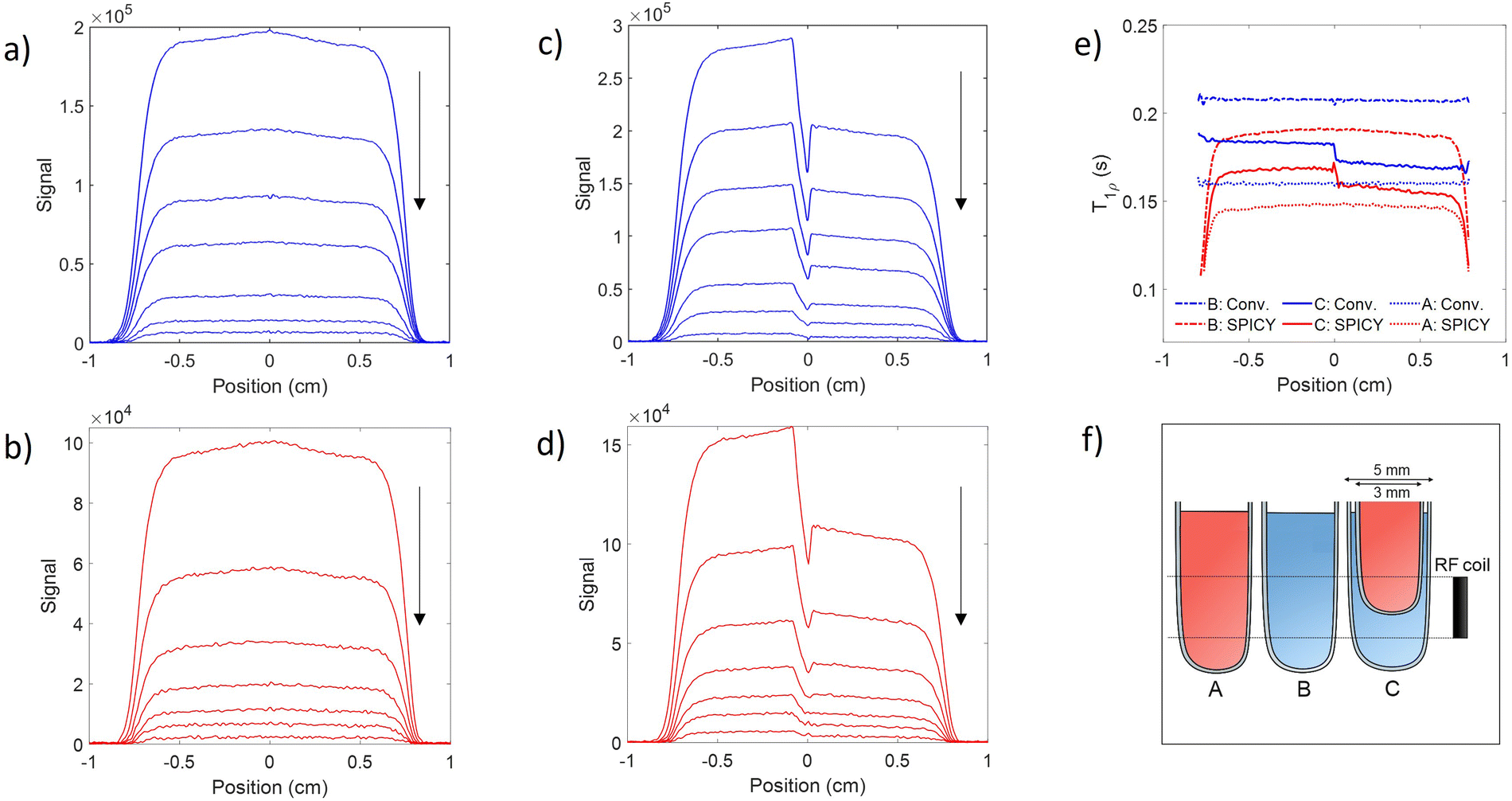 | ||
| Fig. 3 (a–d) 1D MR images of the samples A (left; a and b) and C (right; c and d) measured with the conventional (blue; a and c) and the SPICY T1ρ (red; b and d) sequences. The arrow indicates the direction of increasing spin lock time (a and c) or echo number (b and d). (e) T1ρ profiles of samples A (dotted line), B (dashed-dotted line) and C (solid line) measured with the conventional (blue) and the SPICY sequence (red). (f) Illustration of samples A, B and C with respect to the RF coil. | ||
The SPICY method has a lot of potential for future applications in both spectroscopy and imaging. The SPICY method could be applied to accelerate the T1ρ relaxation dispersion studies of biomacromolecules and to determine, for example, the rates of chemical exchange or conformational changes.4–7,11–22 We showed dispersion measurements with SL fields of 2–11 kHz, however, a wider range of SL fields could be applicable. The SPICY sequence could be further developed for accelerating multidimensional T1ρ imaging by adding additional spatial encoding dimensions, for example, in the same way as in the RARE imaging.63 Because SPICY is much faster and the spin locking does not produce more energy deposition compared to the conventional method, it could have a lot of potential in clinical MRI. The higher number of 180° pulses may cause more heating compared to the standard method, however, the problem could be avoided by replacing the image readout part by gradient echo. The T1ρ values of the SPICY imaging remain slightly smaller than the reference values due to the higher number of gradient pulses and longer SL gap during signal acquisition, but it must be noted that in clinical imaging, the contrast between tissues is generally more important than the quantitative T1ρ values. Also, the decreased T1ρ values at the edges of the RF coil can be managed by placing the imaged object in the middle of the sensitive area of the coil. In the same way as the SLEPI sequence,49,60 SPICY could be applied to imaging of dynamic processes. It might be possible to use adiabatic spin locking in the SPICY sequence.42–44 Because the SPICY allows signal readout in a similar manner to the CPMG,8 it could be added to multidimensional NMR or Laplace NMR experiments64 to correlate T1ρ with the other relaxation or diffusion parameters. Furthermore, the 1D imaging version of SPICY could replace the CPMG readout part in multidimensional ultrafast NMR65 or Laplace NMR (LNMR) experiments.62,66–69 The single scan approach also significantly facilitates the use of nuclear spin hyperpolarization techniques70–73 to boost the sensitivity of the experiments by several orders of magnitude. For example, recently Qi et al.74 boosted T2 relaxation dispersion measurements by dissolution dynamic nuclear polarization (dDNP) to study protein–ligand binding. Similarly, SPICY could be exploited in hyperpolarized studies as well.
4. Conclusions
In summary, we introduced a novel single-scan method called SPICY for measuring T1ρ relaxation times. We demonstrated that the SPICY method can be successfully used to measure T1ρ dispersions with spin locks of 2–11 kHz. SPICY shortens the scan time about an order of magnitude while producing less RF energy deposition compared to the conventional T1ρ sequence. We showed that the T1ρ values obtained from the SPICY sequence are in good agreement with those of the conventional method. We also combined the SPICY with one dimensional spatial encoding and showed that the method is applicable to single scan 1D imaging as well. Overall, the single scan T1ρ method presented in this work has a lot of potential for future applications in rotating frame relaxation studies, MR imaging, ultrafast multidimensional NMR and Laplace NMR studies, as well as in hyperpolarize NMR and MRI experiments.Author contributions
K. Tolkkinen was responsible for conceptualization, methodology, validation, investigation, resources, and writing – original draft. S. Mailhiot was responsible for conceptualization, methodology, validation, investigation, and writing – review and editing. A. Selent was responsible for conceptualization, methodology, and writing – review and editing. O. Mankinen was responsible for support in the final measurements, and writing – review and editing. H. Henschel, M. Nieminen and M. Hanni were responsible for planning and preparing the hydrogel sample, and writing – review and editing. A. M. Kantola was responsible for supervision, and writing – review and editing. T. Liimatainen was responsible for supervision, and writing – review and editing. V.-V. Telkki was responsible for conceptualization, supervision, and writing – review supervision, and writing – review and editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge the financial support from the European Research Council (grant number 772110), the Academy of Finland (grant numbers 340099, 340761 and 896824), the Marie Sklodowska-Curie Actions (grant number 896824), KAUTE foundation and the University of Oulu Kvantum Institute. Part of this work was carried out with the support from the Centre for Material Analysis, the University of Oulu, Finland. The authors thank Olli-Pekka Aro for his contribution in the preparation of the protein gel sample.References
- J. Keeler, Understanding NMR spectroscopy, John Wiley & Sons, Chichester, UK, 2nd edn, 2010 Search PubMed.
- E. Anoardo, G. Galli and G. Ferrante, Appl. Magn. Reson., 2001, 20, 365–404 CrossRef CAS.
- A. G. Redfield, Phys. Rev., 1955, 98, 1787–1809 CrossRef CAS.
- A. G. Palmer and F. Massi, Chem. Rev., 2006, 106, 1700–1719 CrossRef CAS PubMed.
- A. Rangadurai, E. S. Szymaski, I. J. Kimsey, H. Shi and H. M. Al-Hashimi, Prog. Nucl. Magn. Reson. Spectrosc., 2019, 112–113, 55–102 CrossRef CAS PubMed.
- P. Neudecker, P. Lundström and L. E. Kay, Biophys. J., 2009, 96, 2045–2054 CrossRef CAS PubMed.
- D. F. Hansen, P. Vallurupalli and L. E. Kay, J. Biomol. NMR, 2008, 41, 113–120 CrossRef CAS PubMed.
- S. Meiboom and D. Gill, Rev. Sci. Instrum., 1958, 29, 688–691 CrossRef CAS.
- J. P. Loria, M. Rance and A. G. Palmer, J. Am. Chem. Soc., 1999, 121, 2331–2332 CrossRef CAS.
- J. G. Reddy, S. Pratihar, D. Ban, S. Frischkorn, S. Becker, C. Griesinger and D. Lee, J. Biomol. NMR, 2018, 70, 1–9 CrossRef CAS PubMed.
- D. M. Korzhnev, V. Y. Orekhov and L. E. Kay, J. Am. Chem. Soc., 2005, 127, 713–721 CrossRef CAS PubMed.
- F. Massi, E. Johnson, C. Wang, M. Rance and A. G. Palmer, J. Am. Chem. Soc., 2004, 126, 2247–2256 CrossRef CAS PubMed.
- A. G. Palmer, J. Magn. Reson., 2014, 241, 3–17 CrossRef CAS PubMed.
- J. G. Cobb, J. Xie and J. C. Gore, Magn. Reson. Med., 2011, 66, 1563–1571 CrossRef CAS PubMed.
- M. Marušič, J. Schlagnitweit and K. Petzold, ChemBioChem, 2019, 20, 2685–2710 CrossRef PubMed.
- P. Lundström and M. Akke, J. Biomol. NMR, 2005, 32, 163–173 CrossRef PubMed.
- S. K. Koskinen, P. T. Niemi, S. A. Kajander and M. E. Komu, Magn. Reson. Imaging, 2006, 24, 295–299 CrossRef PubMed.
- S. K. Koskinen, A. M. Virta, P. T. Niemi, S. A. Kajander and M. E. Komu, Magn. Reson. Imaging, 1999, 17(7), 1043–1047 CrossRef CAS PubMed.
- E. Rössler, C. Mattea and S. Stapf, Magn. Reson. Med., 2015, 73, 2005–2014 CrossRef PubMed.
- U. Duvvuri, A. D. Goldberg, J. K. Kranz, L. Hoang, R. Reddy, F. W. Wehrli, A. J. Wand, S. W. Englander and J. S. Leigh, Proc. Natl. Acad. Sci. U. S. A., 2001, 98, 12479–12484 CrossRef CAS PubMed.
- N. Wang and Y. Xia, J. Magn. Reson., 2011, 212, 124–132 CrossRef CAS PubMed.
- H. Elsayed, J. Karjalainen, M. J. Nissi, J. Ketola, A. W. Kajabi, V. Casula, Š. Zbýň, M. T. Nieminen and M. Hanni, Magn. Reson. Imaging, 2023, 97, 91–101 CrossRef CAS PubMed.
- A. J. Sophia Fox, A. Bedi and S. A. Rodeo, Sports Health, 2009, 1(6), 461–468 CrossRef PubMed.
- P. Wang, J. Block and J. C. Gore, Magn. Reson. Imaging, 2015, 33(1), 38–42 CrossRef CAS PubMed.
- E.-N. Salo, T. Liimatainen, S. Michaeli, S. Mangia, J. Ellermann and M. Nieminen, T1rho dispersion in constituent-specific degradation models of articular cartilage with correlation to biomechanical properties, Intl. Soc. Magn. Reson. Med., Elbourne, Australia, 2012 Search PubMed.
- K. E. Keenan, T. F. Besier, J. M. Pauly, R. L. Smith, S. L. Delp, G. S. Beaupre and G. E. Gold, Cartilage, 2015, 6(2), 113–122 CrossRef CAS PubMed.
- U. Duvvuri, R. Reddy, S. D. Patel, J. H. Kaufman, J. B. Kneeland and J. S. Leigh, Magn. Reson. Med., 1997, 38(6), 863–867 CrossRef CAS PubMed.
- R. W. Brown, Y. N. Cheng, E. M. Haacke, M. R. Thompson and R. Venkatesan, Magnetic Resonance Imaging - Physical Principles and Sequence Design, John Wiley & Sons, Hoboken, New Jersey, 2nd edn, 2014 Search PubMed.
- S. V. Akella, R. R. Regatte, A. J. Gougoutas, A. Borthakur, E. M. Shapiro, J. B. Kneeland, J. S. Leigh and R. Reddy, Magn. Reson. Med., 2001, 46, 419–423 CrossRef CAS PubMed.
- U. Duvvuri, S. Kudchodkar, R. Reddy and J. S. Leigh, Osteoarthritis Cartilage, 2002, 10, 838–844 CrossRef CAS PubMed.
- R. R. Regatte, S. V. Akella, J. H. Lonner, J. B. Kneeland and R. Reddy, J. Magn. Reson. Imaging., 2006, 23, 547–553 CrossRef PubMed.
- X. Li, C. Benjamin, Ma, T. M. Link, D. D. Castillo, G. Blumenkrantz, J. Lozano, J. Carballido-Gamio, M. Ries and S. Majumdar, Osteoarthritis Cartilage, 2007, 15, 789–797 CrossRef CAS PubMed.
- M. A. Yoon, S. J. Hong, C. H. Kang, K. S. Ahn and B. H. Kim, Magn. Reson. Imaging, 2016, 34, 932–939 CrossRef PubMed.
- Y. X. Wang, F. Zhao, J. F. Griffith, G. S. Mok, J. C. Leung, A. T. Ahuja and J. Yuan, Eur. Radiol., 2013, 23, 228–234 CrossRef PubMed.
- A. Borthakur, M. Sochor, C. Davatzikos, J. Q. Trojanowski and C. M. Clark, NeuroImage, 2008, 41, 1199–1205 CrossRef PubMed.
- M. Haris, E. McArdle, M. Fenty, A. Singh, C. Davatzikos, J. Q. Trojanowski, E. R. Melhem, C. M. Clark and A. Borthakur, J. Magn. Reson. Imaging, 2009, 29, 1008–1012 CrossRef PubMed.
- I. Nestrasil, S. Michaeli, T. Liimatainen, C. E. Rydeen, C. M. Kotz, J. P. Nixon, T. Hanson and P. J. Tuite, J. Neurol., 2010, 257, 964–968 CrossRef CAS PubMed.
- M. I. Kettunen, O. Gröhn, M. Penttonen and R. A. Kauppinen, Magn. Reson. Med., 2001, 46, 565–572 CrossRef CAS PubMed.
- W. R. Witschey, A. Borthakur, M. A. Elliot, E. Mellon, S. Niyogi, D. J. Wallman, C. Wang and R. Reddy, J. Magn. Reson., 2007, 186, 75–85 CrossRef CAS PubMed.
- R. E. Sepponen, J. A. Pohjonen, J. T. Sipponen and J. I. Tanttu, J. Comput. Assisted Tomogr., 1985, 9, 1007–1011 CrossRef CAS PubMed.
- I. A. Gilani and R. Sepponen, NMR. Biomed., 2016, 29, 841–861 CrossRef PubMed.
- T. Liimatainen, D. J. Sorce, R. O'Connell, M. Garwood and S. Michaeli, Magn. Reson. Med., 2010, 64(4), 983–994 CrossRef PubMed.
- J. Rautiainen, M. J. Nissi, T. Liimatainen, W. Herzog, R. K. Korhonen and M. T. Nieminen, Osteoarthritis Cartilage, 2014, 22(10), 1444–1452 CrossRef CAS PubMed.
- N. E. Hänninen, T. Liimatainen, M. Hanni, O. Gröhn, M. T. Nieminen and M. J. Nissi, Sci. Rep., 2022, 12(1), 12155 CrossRef PubMed.
- E. Anoardo, C. Hauser and R. Kimmich, J. Magn. Reson., 2000, 142, 372–378 CrossRef CAS PubMed.
- U. Duvvuri, S. R. Charagundla, S. B. Kudchodkar, J. H. Kaufman, J. B. Kneeland, R. Rizi, J. S. Leigh and R. Reddy, Radiology, 2001, 220, 822–826 CrossRef CAS PubMed.
- X. Li, E. T. Han, D. Newitt and S. Majumdar, Conf. Proc. IEEE. Eng. Med. Biol. Soc., 2004, 2004, 1032–1035 Search PubMed.
- T. Liimatainen and O. Gröhn, A fast, quantitative T1ρ imaging method, Int. Soc. Magn. Reson. Med., Stockholm, Sweden, 2010 Search PubMed.
- A. Borthakur, J. Hulvershorn, E. Gualtieri, A. J. Wheaton, S. Charagundla, M. A. Elliott and R. Reddy, J. Magn. Reson. Imaging, 2006, 23, 591–596 CrossRef PubMed.
- A. Sharafi, M. V. W. Zibetti, G. Chang, M. Cloos and R. R. Regatte, NMR. Biomed., 2022, 35, e4800 CAS.
- P. Schuenke, D. Paech, C. Koehler, J. Windschuh, P. Bachert, M. E. Ladd, H. P. Schlemmer, A. Radbruch and M. Zaiss, Sci. Rep., 2017, 7, 42093 CrossRef CAS PubMed.
- W. R. Witschey, A. Borthakur, M. A. Elliott, M. Fenty, M. A. Sochor, C. Wang and R. Reddy, J. Magn. Reson. Imaging., 2008, 28, 744–754 CrossRef PubMed.
- S. Zbyn, M. J. Nissi, H. Elsayed, V. Casula, M. Hanni, T. Liimatainen and M. T. Nieminen, Accelerated T1ρ Dispersion Data Acquisition for Correlation Time Mapping of Cartilage at 3T, Proc. Intl. Soc. Mag. Reson. Med., Paris, France, 2018 Search PubMed.
- S. Kamesh Iyer, B. Moon, E. Hwuang, Y. Han, M. Solomon, H. Litt and W. R. Witschey, J. Cardiovasc. Magn. Reson., 2019, 21, 5 CrossRef PubMed.
- M. Gram, D. Gensler, P. Winter, M. Seethaler, P. A. Arias-Loza, J. Oberberger, P. M. Jakob and P. Nordbeck, MAGMA, 2022, 35, 325–340 CrossRef CAS PubMed.
- H. Qi, A. Bustin, T. Kuestner, R. Hajhosseiny, G. Cruz, K. Kunze, R. Neji, R. M. Botnar and C. Prieto, J. Cardiovasc. Magn. Reson., 2020, 22, 12 CrossRef PubMed.
- H. Qi, Z. Lv, J. Hu, J. Xu, R. Botnar, C. Prieto and P. Hu, Magn. Reson. Med., 2022, 88, 2520–2531 CrossRef PubMed.
- Y. Zhu, Q. Zhang, Q. Liu, Y. X. Wang, X. Liu, H. Zheng, D. Liang and J. Yuan, Magn. Reson. Med., 2015, 73, 263–272 CrossRef PubMed.
- M. Hanhela, A. Paajanen, M. J. Nissi and V. Kolehmainen, J. Imaging, 2022, 8, 157 CrossRef PubMed.
- H. Li, M. Yang, J. H. Kim, C. Zhang, R. Liu, P. Huang, D. Liang, X. Zhang, X. Li and L. Ying, Magn. Reson. Med., 2023, 89, 64–76 CrossRef PubMed.
- J. Hulvershorn, A. Borthakur, L. Bloy, E. E. Gualtieri, R. Reddy, J. S. Leigh and M. A. Elliott, Magn. Reson. Med., 2005, 54, 1155–1162 CrossRef PubMed.
- V.-V. Telkki, M. Urbańczyk and V. Zhivonitko, Prog. Nucl. Magn. Reson. Spectrosc., 2021, 126–127, 101–120 CrossRef CAS PubMed.
- J. Hennig, A. Nauerth and H. Friedburg, Magn. Reson. Med., 1986, 3, 823–833 CrossRef CAS PubMed.
- P. T. Callaghan, Translational Dynamics and Magnetic Resonance: Principles of Pulsed Gradient Spin Echo NMR, Oxford University Press, Oxford, 2011 Search PubMed.
- A. Tal and L. Frydman, Prog. Nucl. Magn. Reson. Spectrosc., 2010, 57, 241–292 CrossRef CAS PubMed.
- S. Ahola and V.-V. Telkki, ChemPhysChem, 2014, 15, 1687–1692 CrossRef CAS PubMed.
- S. Ahola, V. Zhivonitko, O. Mankinen, G. Zhang, A. M. Kantola, H. Y. Chen, C. Hilty, I. V. Koptyug and V.-V. Telkki, Nat. Commun., 2015, 6, 8363 CrossRef CAS PubMed.
- O. Mankinen, V. Zhivonitko, A. Selent, S. Mailhiot, S. Komulainen, N. L. Prisle, S. Ahola and V.-V. Telkki, Nat. Commun., 2020, 11, 3251 CrossRef CAS PubMed.
- S. Ullah, O. Mankinen, V. Zhivonitko and V.-V. Telkki, Phys. Chem. Chem. Phys., 2022, 24, 22109–22114 RSC.
- J. H. Ardenkjær-Larsen, B. Fridlund, A. Gram, G. Hansson, L. Hansson, M. H. Lerche, R. Servin, M. Thaning and K. Golman, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 10158 CrossRef PubMed.
- S. R. Bowers, Sensitivity enhancement utilizing parahydrogen, in Encyclopedia of Nuclear Magnetic Resonance, John Wiley & Sons, Chichester, 2007 Search PubMed.
- R. W. Adams, J. A. Aguilar, K. D. Atkinson, M. J. Cowley, P. I. Elliott, S. B. Duckett, G. G. Green, I. G. Khazal, J. López-Serrano and D. C. Williamson, Science, 2009, 323, 1708–1711 CrossRef CAS PubMed.
- B. M. Goodson, J. Magn. Reson., 2002, 155, 157–216 CrossRef CAS PubMed.
- C. Qi, Y. Wang and C. Hilty, Angew. Chem., Int. Ed., 2021, 60, 24018–24021 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Pulse sequences for the T1ρ experiments described. See DOI: https://doi.org/10.1039/d2cp05988f |
| This journal is © the Owner Societies 2023 |