
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Gas phase models of hydride transfer from divalent alkaline earth metals to CO2 and CH2O†
Christian
Sant Gjermestad
 ab,
Mauritz Johan
Ryding
b and
Einar
Uggerud
*b
ab,
Mauritz Johan
Ryding
b and
Einar
Uggerud
*b
aDepartment of Chemistry, University of Bergen, Allégaten 41, NO-5007 Bergen, Norway
bDepartment of Chemistry and Hylleraas Centre for Quantum Molecular Sciences, University of Oslo, PO Box 1033, Blindern, NO-0135 Oslo, Norway. E-mail: einar.uggerud@kjemi.uio.no
First published on 29th May 2023
Abstract
The reactivity of HMg+, HMgCl, and HMgCl2− in hydride transfer reactions with CO2 and CH2O were studied by means of the reverse reactions—decarboxylation of HCO2MgCln+/0/− and deformylation of CH3OMgCln+/0/− (n = 0–2)—by a combination of quantum chemical computations and mass spectrometry experiments. HCO2Mg+, HCO2MgCl and HCO2MgCl2− display similar energetics for unimolecular carbon dioxide loss; for CH3OMg+, CH3OMgCl and CH3OMgCl2−, formaldehyde loss is more favourable for the cationic species than for the anionic one, with the neutral species found in-between. Despite very similar overall thermochemistry for each of the charge states of the CO2 and CH2O systems, the intermediate reaction barriers are higher for the CO2 eliminations due to a more complex and demanding reaction mechanism. Exothermic ligand exchange is observed for CH3OMg+ reacting with CO2, forming HCO2Mg+ and CH2O. Both the thermochemistry and the presence of intermediate energy barriers slightly disfavour this type of ligand exchange for CH3OMgCl and CH3OMgCl2−. It was also found that CH3OMg+ reacts readily with H2O to eliminate H2, whereas quantum chemical computations predict that the corresponding neutral and anionic species suffer unfavourable reaction thermochemistry. A corresponding reaction for the magnesium formate compounds was not observed. Quantum chemical computations were performed to investigate periodic trends in reactivity. The energetic requirements for decarboxylation of HCO2M+, HCO2MCl and HCO2MCl2− increase in the order M = Be, Mg, Ca, Sr and Ba; the only exception is the cationic Be species for which the reaction is more endothermic than for the corresponding Mg species. For deformylation of CH3OM+, CH3OMCl and CH3OMCl2− the trends are more irregular and less pronounced than for the decarboxylation reactions; however, the Be species was found to have higher reaction energies than the Mg species for all the methoxy-compounds irrespective of charge state. The effect on decarboxylation and deformylation upon replacing Cl with F, Br or I was found to be minimal for all aforementioned species. The consequences of these observations on the reverse reactions, hydride transfer to CO2 and CH2O, are discussed, and the effects of systematic structural changes on reactivity are rationalized on the basis of thermochemical cycles and well-known periodic trends.
Introduction
Main group metal hydrides, MHn, and metal hydride anions, MHn+1−, are powerful reducing agents that find much use in organic chemistry.1 Well-known examples are B2H6 and AlH4−. Within this group of compounds, we also find MgH2, which is a stable solid that releases hydrogen upon heating. Since this process is reversible, magnesium hydrides are promising lightweight hydrogen storage materials.2,3 Mechanically or chemically activated MgH2 has also been suggested as a potential anode material for use in Li-ion batteries due to their good hydride transfer properties.4,5 While MgH2 itself is poorly soluble in organic solvents, derivatives of the type HMgX (X = NR2, OR, halide, Cp, N-heterocyclic carbenes)—in analogy with the closely related Grignard reagent—often exist in the form of dimers or higher oligomers in such solvents.6 These derivatives are effective and selective reducing agents, in particular towards the carbonyl group,2,7–9 but also towards the CO2 molecule.10–12 A β-diketiminato magnesium hydride has even been demonstrated to reductively couple two CO molecules, giving an ene–diolate complex as product.13 The same complex has also been found to be reactive towards double and triple carbon–carbon bonds.14Because the above-mentioned magnesium hydride derivatives (HMgX) exist as oligomers in solution, they are exceedingly difficult to isolate and characterise. Therefore, in order to study the basic reaction mechanism for hydride reduction, isolating the monomeric species in the gas phase appears to be a particularly attractive strategy. In this respect, OHair and co-workers were pioneers by being able to synthesize and isolate gas phase HMgX2− by mass spectrometry.15 These anions were made by decarboxylation of the corresponding formate complex, HCO2MgX2−, upon collision-induced dissociation (CID). It was also observed that the formate complex could be recreated in the reaction between HMgX2− and formic acid in the gas phase, upon dehydrogenation:
| HMgCl2− + HCO2H → HCO2MgCl2− + H2 | (1) |
It should also be mentioned that more recently, Attygalle and co-workers, using a similar technique, reported the formation of a plethora of metal hydride anions, including AlH4−.16
In this work, we aim to increase our understanding of the reactions of HMgX species (X = halide) with carbonyl compounds, in particular carbon dioxide and formaldehyde. For practical reasons, the reactions studied will be the reverse of the hydride transfer, i.e., the decarboxylation and deformylation of HCO2MgXn+/0/− and CH3OMgXn+/0/−, respectively (n = 0–2). The essential thermochemical and kinetic information is obtained by quantum chemical computations supplemented by mass spectrometry experiments. Additional work included are possible ligand exchange reactions with CO2 and H2O. In addition, quantum chemical computations were performed on the effects of substituting the other alkaline earth metals for Mg.
Methods
Experimental details
The experiments were conducted using a QTOF 2 mass spectrometer (Micromass/Waters) with a quadrupole—time-of-flight geometry, electrospray ionisation (ESI) source and microchannel plate detectors. A collision cell with hexapole ion guide is situated in-between the quadrupole and the TOF unit; a modified collision gas inlet, fitted with a leak-valve, allows greater precision in setting the collision gas pressure compared to the original instrument. The setup has been described in detail previously.17–20The ions studied, CH3OMgCl2−, CH3OMg+, HCO2MgCl2− and HCO2Mg+, were produced by ESI using millimolar solutions of MgCl2, with addition of HCO2H for the formation of the formate-containing ions. The solvents were either methanol or methanol–water 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1. For specific concentrations, additives and solvent ratios, please see the ESI;† the source conditions used to generate the ions can also be found in the ESI.† All reagents were obtained from commercial suppliers (Sigma-Aldrich, VWR Chemicals) and utilized without further purification.
:1. For specific concentrations, additives and solvent ratios, please see the ESI;† the source conditions used to generate the ions can also be found in the ESI.† All reagents were obtained from commercial suppliers (Sigma-Aldrich, VWR Chemicals) and utilized without further purification.
The quadrupole was used as a mass filter in order to isolate the reactant ions of interest; the selected ions then entered the collision cell, where CID occurred using Ar as the collision gas. The resulting products and surviving reactant ions were then analysed and detected in the TOF unit. The primary products of the CID, as well as being the products of interest, were HMg+ (for the positive parent ions) and HMgCl2− (for the negative parent ions).
For each parent ion, a measurement series comprised 20 measurements at various centre-of-mass (CoM) collision-energies in the approximate range of 50–600 kJ mol−1 (depending upon the ion in question). Data were related to an internal reference in order to ensure comparability between different measurements. Appearance curves were obtained by plotting the phenomenological cross section for formation of HMg+ or HMgCl2− against the CoM collision energy. Five appearance curves were recorded for each ion using different collision gas pressures between 1 × 10−4 mbar and 5 × 10−4 mbar.
The appearance curves were analysed using the L-CID software, developed in the group of Chen.21 L-CID produces a fit to an experimental appearance curve with the main output being the threshold energy, i.e., the collision energy necessary to induce fragmentation (accounting for thermal shift, kinetic shift, Doppler broadening etc.). The software has previously been used by us to successfully analyse data from the experimental setup in use here;22 a more in depth description of the software, and considerations for its use with the QTOF 2 instrument can be found in that paper. It should be noted that L-CID assumes an instrumental configuration in which the ions are thermalized to the same temperature as the collision gas prior to collision, which is not the case for our setup; as such, a systematic error in the threshold energies is to be expected (depending upon the internal energy distribution of the ions). Furthermore, in order to circumvent the need to know the vibrational frequencies explicitly, L-CID utilizes a single effective frequency, an approximation requiring the ion to be of sufficient size. The ions studied here—with as few as five atoms—are smaller than the smallest test case used in the development of L-CID, the latter being toluene with 15 atoms. Regardless, the obtained threshold energies appear reasonable and internally consistent when compared to the appearance curves from which they were extracted.
Using the threshold energies from five different collision gas pressures, an extrapolation to zero pressure was done in order to compensate for the apparent lowering of a threshold energy caused by the ion experiencing multiple collisions with the gas molecules.
Each experiment was carried out at least twice in order to verify repeatability. To ensure no bias towards the detection of ions with different abundancies, the ability of the mass spectrometer to faithfully reproduce the expected isotope distribution of magnesium chloride clusters was tested before each experiment.
In comparing the results from different experiments, it appears that the greatest uncertainty arising from the process outlined above comes from the extrapolation of threshold energies to zero collision gas pressure. We estimate the final threshold energies to be semi-quantitative in nature and reproducible within ±10–20 kJ mol−1 between experiments.
Computational details
The computational results presented in this work were obtained using the Gaussian-4 (G4) method as implemented in Gaussian 09/16.23 This is a thermochemical method which utilizes several levels of theory and is estimated to be accurate to within 10–15 kJ mol−1.24 Selected G4 geometries were reoptimized using CCSD(T)/aug-cc-pVTZ, MP2 and a handful of DFT functionals, as presented in Table S3 of the ESI.† CCSD(T)/aug-cc-pVTZ computations were performed in NWChem.25 All minima and transition states were checked for the correct number of imaginary frequencies. Transition states were subjected to an intrinsic reaction coordinate (IRC) computation in order to determine the connected minima. Options selected for the computations can be found in the ESI.†Results and discussion
Hydride affinities and complexation energies
Before discussing the individual hydride transfer reactions, it is necessary to establish the thermochemistry. In particular, this requires knowledge of the hydride affinities (HA) and complexation energies of the key species involved: CO2, CH2O and the relevant Mg(II) complexes. This information was obtained from the quantum chemical computations, using the G4 scheme, and is presented in Fig. 1. The computed values of HA(CO2) = 254 kJ mol−1 and HA(CH2O) = 197 kJ mol−1 are higher than the literature estimates of 216 ± 10 kJ mol−1 and 170 ± 4 kJ mol−1, respectively.26,27,28 The literature values were based on Born–Haber cycles obtained from experimentally determined electron affinities and gas phase enthalpies-of-formation for the species involved. The substantial deviation between the computed and the experimental data is very likely due to the inadequacy of the G4 method in describing the naked hydrogen anion.29 As a result, the electron affinity of H is underestimated by a factor of two, which indicates the magnitude of the error in predicting the above hydride affinities. Besides this, the rest of the computations presented here should be void of this shortcoming and comply with the documented “chemical accuracy” of G4 of approximately ± 10 kJ mol−1. | ||
| Fig. 1 Energy diagrams based on the quantum chemical computations (G4, 0 K) indicating the key reaction thermochemistry (in kJ mol−1) for hydride transfer from the Mg(II) species to (a) CO2 and (b) CH2O. The ionic species have been investigated experimentally, see below, while the neutral species could only be studied computationally. | ||
As noted above, the gas phase HA of carbon dioxide is higher than that of formaldehyde; however, the gas phase HA of the Mg(II) complexes are significantly higher than the HA of either of the organic substrates. For example, the hydride transfer from HMgCl2− to CO2 forming MgCl2 and HCO2− is a reaction that is endothermic by (380–254) kJ mol−1 = 126 kJ mol−1. This may at first sight appear counterintuitive, since main group metal hydrides are used as reducing agents in organic chemistry. However, it is important to note that the driving force for the reaction is not necessarily the reduction itself, but the complexation in the resulting product. The possibility to form strong bonds between the oxophilic magnesium centre and the formate or methoxide anions results in reactions that are overall exothermic, as seen in reactions (2)–(7).
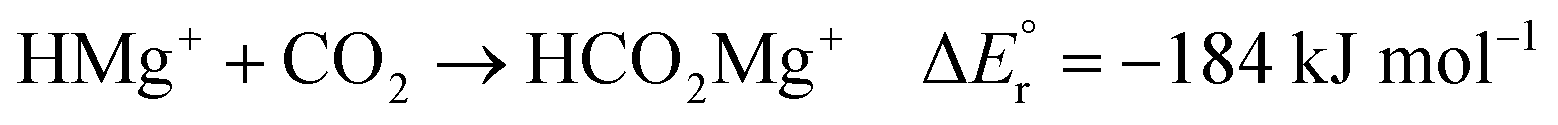 | (2) |
 | (3) |
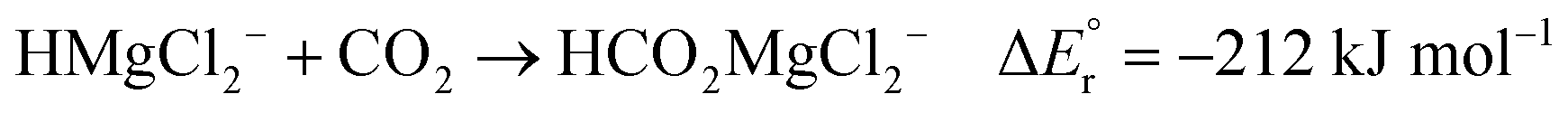 | (4) |
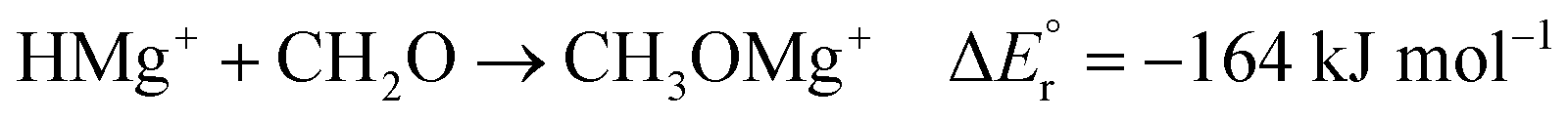 | (5) |
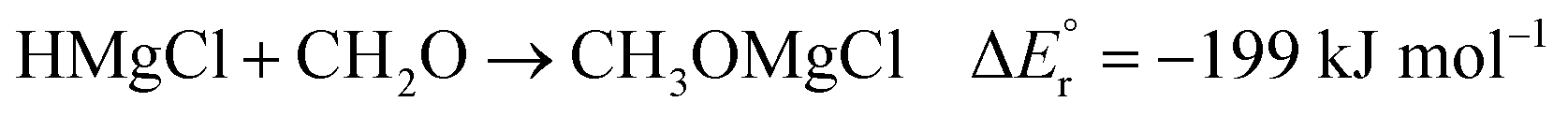 | (6) |
 | (7) |
Collision-induced dissociation of HCO2Mg+ and CH3OMg+
Each of the ions HCO2Mg+ and CH3OMg+ were produced by electrospray ionisation and mass selected before being subject to energy variable CID (see the Methods section). At the fairly low collision energies used here, the primary processes are, respectively, decarboxylation and deformylation:| HCO2Mg+ → HMg+ + CO2 | (8) |
| CH3OMg+ → HMg+ + CH2O | (9) |
An example of experimentally recorded appearance curves for reactions (8) and (9) is shown in Fig. 2, collected at a collision gas pressure PAr = 5 × 10−4 mbar. Using the L-CID software,21 it is possible to estimate the threshold energies producing the curves to be 163 kJ mol−1 and 104 kJ mol−1, respectively.
 | ||
| Fig. 2 Appearance curves for HMg+ upon energy variable CID (PAr = 5 × 10−4 mbar) of HCO2Mg+ (open circles) and CH3OMg+ (closed squares). | ||
To compensate for the CID results being affected by ions experiencing multiple collisions, threshold energies from five different collision gas pressures were extrapolated to produce a threshold energy at PAr = 0 mbar (all threshold values and extrapolation parameters can be found in the ESI†). The resulting values were 198 kJ mol−1 for HCO2Mg+ and 136 kJ mol−1 for CH3OMg+.
In addition to HMg+, some minor products were also detected upon CID of the reactant ions. For CH3OMg+, loss of elements corresponding to CH3O•, giving rise to Mg+, was observed above approximately 300 kJ mol−1 (CoM). For HCO2Mg+, we observed small amounts of MgOH+, corresponding to loss of CO. However, MgOH+ could potentially be formed from the primary product MgH+ reacting with restgas H2O and eliminating H2; our computations show this reaction to be exothermic at −81 kJ mol−1.
Collision-induced dissociation of HCO2MgCl2− and CH3OMgCl2−
For the anions, HCO2MgCl2− and CH3OMgCl2−, formally derived from each of the two cations discussed above by the addition of two chloride ions, we also observe decarboxylation/deformylation upon CID at low collision energies:| HCO2MgCl2− → HMgCl2− + CO2 | (10) |
| CH3OMgCl2−→ HMgCl2− + CH2O | (11) |
Appearance curves for formation of HMgCl2− from HCO2MgCl2− and CH3OMgCl2− are shown in Fig. 3 for the collision gas pressure 5 × 10−4 mbar; the associated threshold energies are 136 and 132 kJ mol−1, respectively. Upon extrapolation to zero collision gas pressure, the threshold energies for reactions (10) and (11) become 189 and 181 kJ mol−1, respectively.
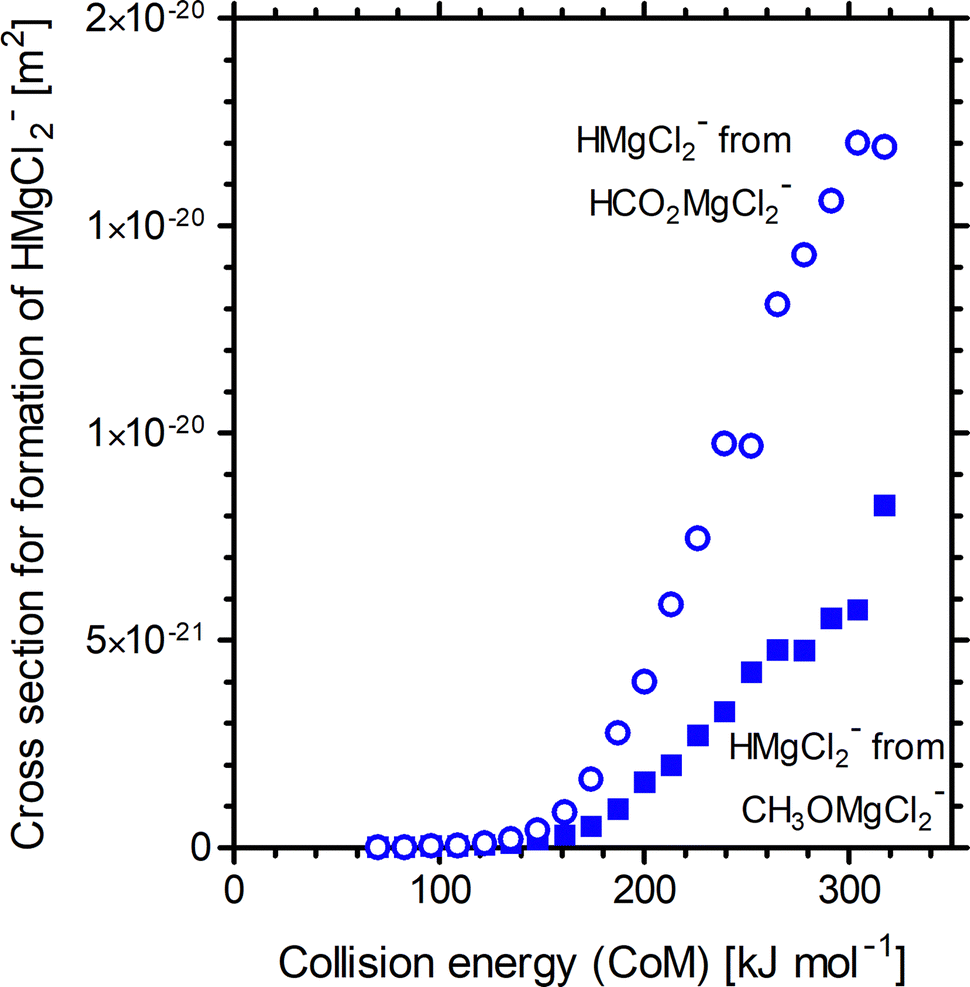 | ||
| Fig. 3 Appearance curves for HMgCl2− upon energy variable CID (PAr = 5 × 10−4 mbar) of HCO2MgCl2− (open circles) and CH3OMgCl2− (closed squares). | ||
For the anionic precursors, formation of Cl− is observed as a secondary reaction-channel in the CID experiments. For HCO2MgCl2− the abundance of Cl− was too small to get a good estimate on the threshold energy for its formation. For CH3OMgCl2−, the abundance of Cl− is lower, albeit comparable, to that of HMgCl2− and the threshold energy (at zero collision gas pressure) could be estimated to 200 kJ mol−1. In comparing Fig. 2 and 3, we see that for the cations, the methoxide precursor produces a greater abundance of the magnesium hydride product compared to the formate precursor, whereas for the anions, the relationship is reversed. The fact that, for CH3OMgCl2−, formation of HMgCl2− is in competition with an easily accessible second reaction channel is the likely explanation as to why. Regardless, the second reaction channel has been explicitly accounted for in the L-CID analysis and should not impact the estimated threshold energies for formation of HMgCl2−.
Computational results for decarboxylation of magnesium formate complexes
The potential energy diagrams for decarboxylation of HCO2Mg+, HCO2MgCl and HCO2MgCl2− are displayed in Fig. 4. It can be seen that the three analogous reactions have very similar transition state energies and show a slight increase in the overall reaction energy with increasing number of chloride ligands. The computed transition state energies and reaction energies are so similar that it becomes impossible to decide which of the two reaction steps determines the threshold energy for product formation. | ||
| Fig. 4 Potential energy diagram for loss of CO2 from HCO2Mg+ (orange), HCO2MgCl (black) and HCO2MgCl2− (blue). Based on the quantum chemical computations (G4, 0 K) with relative energies given in kJ mol−1. The reverse reactions equal a hydride transfer coupled with formate complex formation. | ||
There are some distinct differences between the species. On the reactant side, for the cation, a shallow energy minimum is found, featuring the formate entity bound in a monodentate fashion (1′A). No such energy minimum could be located for the neutral or the anion. To which extent this difference is of importance to the overall reaction dynamics is, however, unclear. In any instance, and irrespective of charge state, one of the two Mg–O bonds of the bidentate formate complex must be fully broken in order to reach the transition state of the actual hydrate transfer (TS1A, TS1B or TS1C). The energetic cost of this is approximately the same in all three cases, and consequently, the threshold energy is nearly the same. However, the differences in the binding energies of the product complexes are much more significant. In the cation complex, 2A, there is strong interaction between the cationic magnesium centre, HMg+, and one of the electron rich oxygen atoms of CO2. In contrast, in the anion complex, 2C, there is only a very weak interaction between HMgCl2− and CO2. Not surprisingly, the situation for the neutral product complex, 2B, is in-between those of the cationic and the anionic complexes.
Fig. 5 shows the most important binding parameters of the three transition state structures. While the transition state energies are similar, the geometries differ, in particular for the formyl moiety. The geometry is the most reactant-like for the cation in that it has the shortest C–H bond length of the three; the transition state TS1A can therefore be considered to be earlier than TS1B and TS1C with respect to the reaction coordinate 1 → 2; this is consistent with 2A being the most strongly bonded of the product complexes (Hammond's postulate). Thus, the gradual increase in the C–H bond length and the O–C–O bond angle of TS1A, TS1B and TS1C can be said to reflect the trend in the complex energies of 2A, 2B and 2C.
 | ||
| Fig. 5 Transition state geometries for decarboxylation of HCO2Mg+ (TS1A), HCO2MgCl (TS1B) and HCO2MgCl2− (TS1C). Bond lengths are given in pm and bond angles in degrees. | ||
Computational results for deformylation of magnesium methoxide complexes
For the methoxy complexes, the variation in reaction energies for the deformylation reactions, shown in Fig. 6, is slightly more pronounced than for the decarboxylation reactions discussed above (Fig. 4). It is worth noting that while the reaction energies show only a small charge dependence, the energies of the transition states (TS2) and ion–neutral complexes (5) are more strongly dependent upon charge. In any case, for all three charge states (+, 0 and −), the transition state energy is lower than the product energy and the energetic threshold of the reaction is determined by the overall reaction endothermicity. | ||
| Fig. 6 Potential energy diagram for loss of CH2O from CH3OMg+ (orange), CH3OMgCl (black) and CH3OMgCl2− (blue). Based on the quantum chemical computations (G4, 0 K) with relative energies given in kJ mol−1. The reverse reactions equal a hydride transfer coupled with methoxy complex formation. | ||
In contrast to the decarboxylation reactions, for the deformylation reactions the route from the reactant towards the transition state is a direct hydride transfer without the need to first break an additional Mg–O bond; as a result of this, the assumptions inherent to the Hammond postulate are fulfilled. Therefore, we expect increasing endothermicity of the elementary reaction step 4 → 5 to correlate with a higher barrier; we also expect the position of the transition state to be further along the reaction path (i.e. the geometry of the transition state to be less like the reactant). Inspection of Fig. 6 and the transition state geometries in Fig. 7 reveals this to be the case.
 | ||
| Fig. 7 Transition state geometries for decarboxylation of CH3OMg+ (TS2A), CH3OMgCl (TS2B) and CH3OMgCl2− (TS2C). Bond lengths are given in pm and bond angles in degrees. | ||
Comparison of experimental and computational results
The experimental threshold energies—198 kJ mol−1 for loss of carbon dioxide from HCO2Mg+ and 136 kJ mol−1 for loss of formaldehyde from CH3OMg+—can be compared to the rate determining energies from the G4 (0 K) computations. For the former reaction, the relevant computed energy is 196 kJ mol−1 (corresponding to the height of TS1A); for the latter reaction, the computational threshold energy is 164 kJ mol−1 (the overall reaction energy of 4A → 6A).For loss of carbon dioxide from HCO2MgCl2−, and loss of formaldehyde from CH3OMgCl2−, the experimental threshold energies are 189 and 181 kJ mol−1, respectively; the corresponding values from our computations (G4, 0 K) are 212 and 211 kJ mol−1, respectively. The G4 data for reaction (10) are in good qualitative accord with the B3LYP/6-31+G(d) model of Khairallah and O'Hair.15
Typically, G4 energies are accurate within ±10–15 kJ mol−1.24 Our experimental threshold energies are reproducible within ±10–20 kJ mol−1 (±2 estimated standard deviations), with the extrapolation to zero pressure constituting the highest uncertainty factor. In this respect, the experimental and computed data are not significantly different, although a nominal difference of up to 30 kJ mol−1 may appear high. It is also relevant that DFT methods appear to give lower estimates for reaction energies, by 10–20 kJ mol−1 (see ESI†), compared to the explicitly correlated method used here.
Computational results for other ligands and metals
Additional computations were performed in order to investigate how different charge states, ligands and metals impact the overall reaction energy and transition states of the reactions presented in Fig. 4 and 6. In order to include also the heavier elements, these computations were done using B3LYP/LANL2DZ. This compromise clearly affects the quality of computed energies, and we note that this rather rudimentary basis set give both higher reaction energies and higher transition state energies compared to the more accurate G4 data—compare values from Tables 1 and 2 (M = Mg and X = Cl) to the results in Fig. 4 and 6. On the other hand, the charge dependence (corresponding to the number of anionic ligands) is the same, and we therefore assume that also the periodic trends to be discussed below are correctly reproduced using B3LYP/LANL2DZ. , for decarboxylation and deformylation of magnesium formate and -methoxy complexes, upon substituting F, Br or I for Cl. Energies in kJ mol−1 obtained with B3LYP/LANL2DZ. Note that there is no separate entry for HCO2Mg+ since there is no halide involved
, for decarboxylation and deformylation of magnesium formate and -methoxy complexes, upon substituting F, Br or I for Cl. Energies in kJ mol−1 obtained with B3LYP/LANL2DZ. Note that there is no separate entry for HCO2Mg+ since there is no halide involved
| Ligand (X) | HCO2MgX → CO2 + HMgX | HCO2MgX2− → CO2 + HMgX2− | CH3OMgX → CH2O + HMgX | CH3OMgX2− → CH2O + HMgX2− | ||||
|---|---|---|---|---|---|---|---|---|
| E(TS) |
|
E(TS) | ΔEr° | E(TS) |
|
E(TS) |
|
|
| F | 204 | 216 | 234 | 261 | 197 | 233 | 226 | 249 |
| Cl | 204 | 215 | 225 | 246 | 200 | 237 | 231 | 245 |
| Br | 204 | 214 | 222 | 238 | 202 | 239 | 229 | 242 |
| I | 205 | 214 | 220 | 234 | 204 | 241 | 227 | 240 |



 , for decarboxylation of HCO2M+, HCO2MCl and HCO2MCl2−. M = Be, Mg, Ca, Sr and Ba. Energies in kJ mol−1 obtained with B3LYP/LANL2DZ
, for decarboxylation of HCO2M+, HCO2MCl and HCO2MCl2−. M = Be, Mg, Ca, Sr and Ba. Energies in kJ mol−1 obtained with B3LYP/LANL2DZ
| Metal (M) | HCO2M+ → CO2 + HM+ | HCO2MCl → CO2 + HMCl | HCO2MCl2− → CO2 + HMCl2− | |||
|---|---|---|---|---|---|---|
| E(TS) |
|
E(TS) |
|
E(TS) |
|
|
| Be | 187 | 215 | 185 | 172 | 205 | 224 |
| Mg | 201 | 195 | 204 | 215 | 225 | 246 |
| Ca | 208 | 240 | 220 | 257 | 251 | 281 |
| Sr | 216 | 252 | 231 | 279 | 261 | 292 |
| Ba | 229 | 274 | Not found | 298 | 273 | 306 |
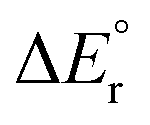


Table 1 shows how the nature of the halide ligand (X) affects the overall reaction energy, and the energy of the transition state, upon decarboxylation of HCO2MgXn+/0/− and deformylation of CH3OMgXn+/0/− (n = 1, 2). For comparison, in these computations, the decarboxylation of HCO2Mg+ have E(TS) = 201 kJ mol−1 and  , while the deformylation of CH3OMg+ have E(TS) = 151 kJ mol−1 and
, while the deformylation of CH3OMg+ have E(TS) = 151 kJ mol−1 and  . From the G4-computations (Fig. 4 and 6) we saw that both the reaction energy and the transition state energy tends to increase with the number of Cl-ligands on Mg; the exception is the transition state for decarboxylation of HCO2Mg+ which is higher, not lower, than the transition states of the neutral and anionic species. It appears from the data in Table 1 that the general trend holds true also upon substituting F, Br or I for Cl; that is, the reaction energy and transition state energy increases with increasing number of ligands. It is quite interesting to see that, for the neutral formate- and methoxy complexes, the transition state energies and reaction energies are surprisingly similar for all four halides in Table 1. At variance, for n = 2 (the anions), both the transition state energies and the reaction energies tend to decrease going down in the group.
. From the G4-computations (Fig. 4 and 6) we saw that both the reaction energy and the transition state energy tends to increase with the number of Cl-ligands on Mg; the exception is the transition state for decarboxylation of HCO2Mg+ which is higher, not lower, than the transition states of the neutral and anionic species. It appears from the data in Table 1 that the general trend holds true also upon substituting F, Br or I for Cl; that is, the reaction energy and transition state energy increases with increasing number of ligands. It is quite interesting to see that, for the neutral formate- and methoxy complexes, the transition state energies and reaction energies are surprisingly similar for all four halides in Table 1. At variance, for n = 2 (the anions), both the transition state energies and the reaction energies tend to decrease going down in the group.
Table 2 shows the effect of different alkaline earth metals on the decarboxylation reactions at various charge states of the HCO2MCln+/0/− complexes. The clear trend is that decarboxylation becomes less and less favourable going down in the group of the alkaline earth metals (Be, Mg, Ca, Sr and Ba); both the transition state energies and the reaction energies are affected in the same way, with exception of the reaction energy for decarboxylation of HCO2Be+ which is actually larger than that of HCO2Mg+. Some insight into the observed trend can be found by decomposing the reaction into five steps, with their associated energies (IE = ionisation energy, EA = electron affinity, PA = proton affinity), as follows:
| HCO2M+ → HCO2− + M2+ −ΔEcomplex | (12) |
| H− → H+ + 2e− EA(H) + IE(H) | (13) |
| HCO2− → H− + CO2 HA(CO2) | (14) |
| M + H+ → HM+ −PA(M) | (15) |
| M2+ + 2e− → M −(IE1(M) + IE2(M)) | (16) |
| HCO2M+ → HM+ + CO2 | (17) |
 | (18) |
Since the contributions from reactions (13) and (14) are constant in the series, the variation in the overall reaction energy, 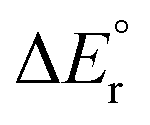 , is governed by the contribution from the complexation energy (reaction (12)) minus the combined contributions from the metal's proton affinity and two ionisation energies (reactions (15) and (16)). These contributions show clear trends when going down the periodic table: both the complexation energy and the ionisation energy decrease in magnitude while the proton affinity increases in magnitude (Table 3). As noted above,
, is governed by the contribution from the complexation energy (reaction (12)) minus the combined contributions from the metal's proton affinity and two ionisation energies (reactions (15) and (16)). These contributions show clear trends when going down the periodic table: both the complexation energy and the ionisation energy decrease in magnitude while the proton affinity increases in magnitude (Table 3). As noted above, 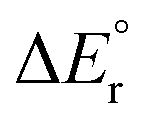 is found to increase with increasing atomic number of the metal, with the exception of beryllium in the cationic complex; this irregularity can be traced to the very high complexation energy of Be2+.
is found to increase with increasing atomic number of the metal, with the exception of beryllium in the cationic complex; this irregularity can be traced to the very high complexation energy of Be2+.
 for the decarboxylation of HCO2M+ with M = Be, Mg, Ca, Sr and Ba (reactions (12)–(18)). Energies in kJ mol−1 obtained with B3LYP/LANL2DZ
for the decarboxylation of HCO2M+ with M = Be, Mg, Ca, Sr and Ba (reactions (12)–(18)). Energies in kJ mol−1 obtained with B3LYP/LANL2DZ
| Be | Mg | Ca | Sr | Ba | |
|---|---|---|---|---|---|
| ΔEcomplex (M(II) + HCO2− → HCO2M+) | −1965 | −1487 | −1209 | −1126 | −1042 |
| IE1(M) + IE2(M) (M(0) → M(II)) | 2671 | 2153 | 1757 | 1634 | 1481 |
| PA(M(0) + H+ → HM+) | 710 | 769 | 843 | 870 | 917 |
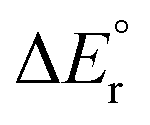
|
215 | 196 | 240 | 252 | 274 |
It is evident from Table 3 that the complexation energy for formate to M2+ decreases in magnitude with M = Be, Mg, Ca, Sr, Ba. In addition, it seen in Fig. 1a that the complexation energy of formate to Mg significantly decreases in magnitude upon addition of one or two chloride ligands to the metal: ΔEcomplex = −338 kJ mol−1 for the latter case. We must therefore consider the possibility that for HCO2MCl2−, loss of formate can become competitive with the decarboxylation reaction when going down the alkaline earth metal group. However, computed reaction energies for loss of HCO2− from HCO2MCl2− with M = Be, Mg, Ca, Sr and Ba shows this not to be the case (see ESI,† Table S6).
The effect of different alkaline earth metals on the deformylation reactions of CH3OMCln+/0/− is shown in Table 4 (M = Be, Mg, Ca, Sr and Ba, n = 0–2). Also in this case we observe increasing reaction energies when going down the group of alkaline earth metals, with the exception of beryllium, which now show a higher reaction energy for all three charge states. However, the increase in reaction energy is less pronounced compared to the decarboxylation reactions in Table 2.
 , for deformylation of CH3OM+, CH3OMCl and CH3OMCl2−. M = Be, Mg, Ca, Sr and Ba. Energies in kJ mol−1 obtained with B3LYP/LANL2DZ
, for deformylation of CH3OM+, CH3OMCl and CH3OMCl2−. M = Be, Mg, Ca, Sr and Ba. Energies in kJ mol−1 obtained with B3LYP/LANL2DZ
| Metal (M) | CH3OM+ → CH2O + HM+ | CH3OMCl → CH2O + HMCl | CH3OMCl2− → CH2O + HMCl2− | |||
|---|---|---|---|---|---|---|
| E(TS) |
|
E(TS) |
|
E(TS) |
|
|
| Be | 149 | 330 | 277 | 290 | 272 | 265 |
| Mg | 151 | 221 | 200 | 237 | 231 | 245 |
| Ca | 145 | 235 | 174 | 238 | 208 | 248 |
| Sr | 143 | 231 | 172 | 247 | 203 | 249 |
| Ba | 155 | 248 | 181 | 259 | 206 | 256 |

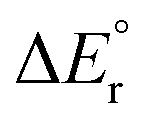

If one compares the overall reaction energies for decarboxylation of HCO2M+ (Table 2) and deformylation of CH3OM+ (Table 4), it can be seen that for M = Be and Mg, decarboxylation is easier by 115 and 26 kJ mol−1, respectively. However, for Ca, Sr and Ba, deformylation is the more facile reaction by 5, 21 and 26 kJ mol−1, respectively. In terms of Lewis hard/soft acid/base theory, we note that the Lewis acid, M, becomes increasingly soft when going down the group; consequently, the magnitude of the complexation energy decreases more quickly for the harder of the Lewis bases, CH3O− (Table S7 in ESI†), compared to the softer of the Lewis bases, HCO2−.
Ligand exchange reactions
The reactions occurring when CH3OMg+ and CH3OMgCl2− collide with CO2 were investigated, briefly, in mass spectrometry experiments. The anion was found to have an efficient complexation reaction wherein it simply adds CO2 (ESI,† Fig. S1) and likely forms a stable carbonate-type intermediate (7C′, Fig. 9). For the cation, a ligand exchange reaction was observed instead:| CH3OMg+ + CO2 → HCO2Mg+ + CH2O | (19) |
 | ||
| Fig. 8 Mass spectra observed in isotopic reactions: (a) for CH3OMg+ (m/z 55) + CO2 (ECoM = 46 kJ mol−1, PCO2 = 2.94 × 10−4 mbar), (b) for CH3OMg+ + 13CO2 (ECoM = 48 kJ mol−1, P13CO2 = 2.62 × 10−4 mbar), and (c) for CD3OMg+ (m/z 58) + CO2 (ECoM = 48 kJ mol−1, PCO2 = 2.62 × 10−4 mbar). Note that the intensity of the reactant peaks have been scaled down by a factor of 50. | ||
To rationalize the outcome of these observations, we surveyed the potential energy surfaces using the G4 method. The potential energy curves for CH3OMg+, CH3OMgCl and CH3OMgCl2− reacting with CO2 is shown in Fig. 9. For CH3OMg+ we find a transition state geometry (TS3A, −3 kJ mol−1) involving a carbon-to-carbon hydride transfer, analogous to the Meerwein–Ponndorf–Verley mechanism.30–32 This energy barrier—just below the energy of the reactants—explains the low reactivity observed in the measurement. Overall, the reaction is slightly exothermic with the products HCO2Mg+ + CH2O at −20 kJ mol−1 relative to the reactants. The reactions of the neutral and the anion, CH3OMgCl and CH3OMgCl2−, are almost thermoneutral; however, their transition states, TS3B and TS3C, give rise to substantial energy barriers of 55 and 51 kJ mol−1, respectively, which explains why the latter reaction was not observed in our experiments. In all three reactions, the rate determining step is found to be the hydride transfer from the leaving formaldehyde unit to the entering CO2 unit. The reactant side intermediate for the neutral, 7B, has a covalently bonded CO2 group and is structurally different from the complexes 7A and 7C. In order for the neutral reaction to reach TS3B, one C–O bond must first be broken before the hydride transfer can occur; this provides the rationale as to why the neutral reaction has a higher barrier (relative to the reactant side intermediate) than the anion reaction, even though the product complex of the former (8B) is lower in energy.
 | ||
| Fig. 9 Potential energy curves for the reaction of CO2 with CH3OMg+ (orange), CH3OMgCl (black) and CH3OMgCl2− (blue). Based on the quantum chemical computations (G4, 0 K) with relative energies given in kJ mol−1. Dashed lines indicate a reaction step which is presumed to have a non-existent or negligible barrier. | ||
In Fig. 8a, there is a small peak at m/z 71 corresponding to the product CH3O2Mg+. The formation of this product from a reaction with CO2 would necessitate the transfer of an oxygen atom to the reactant ion:
| CH3OMg+ + CO2 → CH3O2Mg+ + CO | (20) |
| CH3OMg+ + H2O → CH3O2Mg+ + H2 | (21) |
Isotope labelling of the carbon atom in carbon dioxide will not affect the mass of the product in reaction (20), nor would it affect a reaction of CH3OMg+ with rest gas H2O (reaction (21)); in agreement, no shift in the position of the peak can be seen in Fig. 8b. In contrast, Fig. 8c shows a shift in peak position by m/z +2 when starting from the isotopically labelled reactant CD3OMg+. The increase in product mass by just two mass units rules out reaction (20), while for reaction (21) it would mean that the leaving hydrogen molecule comprises one hydrogen atom from CD3OMg+ and one from H2O.
The reaction between CH3OMg+ and H2O was investigated computationally (G4); the potential energy curve, along with complementary results for CH3OMgCl and CH3OMgCl2−, is shown in Fig. 10. For the cation, we see a submerged barrier at −36 kJ mol−1 and the overall reaction is exothermic at −141 kJ mol−1. In comparing to the energetics of the CO2/CH2O ligand exchange reaction in Fig. 9 (barrier of −3 kJ mol−1 and reaction energy of −20 kJ mol−1), we see a clear indication that CH3OMg+ should indeed react more readily with H2O (reaction (21)) than with CO2 (reaction (19)). Fig. 10 also shows that reaction with water is highly unfavourable for the anion, CH3OMgCl2−, with the transition state TS4C at 110 kJ mol−1 relative to the reactants and an overall endothermic reaction. The reaction of CH3OMgCl—although nearly thermoneutral—should also be unlikely due to the sizeable barrier of TS4B at 72 kJ mol−1.
 | ||
| Fig. 10 Potential energy curves for the reaction of H2O with CH3OMg+ (orange), CH3OMgCl (black) and CH3OMgCl2− (green). Based on the quantum chemical computations (G4, 0K) with relative energies given in kJ mol−1. Dashed lines indicate a reaction step which is presumed to have a non-existent or negligible barrier. | ||
The three potential energy curves in Fig. 10 are displaced nearly parallel to each other, as determined by the overall thermochemistry; this reflects the similar reaction mechanisms of the species. For all three reactions, the preparation for H2 elimination involves the H2O entity providing a proton and the methoxy entity providing a hydride. It is evident that the cationic route is favoured due to activation by the positive charge, giving rise to the TS of lowest energy. The computationally derived reaction mechanisms thus confirm the validity of reaction (21) with regards to the isotope shifts observed upon exchanging CH3OMg+ with CD3OMg+ (Fig. 8c), i.e., each of the hydrogens in the leaving H2 originate from a different reactant. It should be noted that the corresponding reaction with methanol has been observed by Khairallah and O'Hair: the isolated HMgCl2− was found to react readily with the methanol used as electrospray solvent.15
For the sake of completeness, we performed measurements on the reaction between HCO2Mg+ and H2O (Fig. S4 in ESI†). The analogue of reaction (21) would be
| HCO2Mg+ + H2O → CHO3Mg+ + H2 | (22) |
Conclusion
In this study we have analysed a series of decarboxylation and deformylation reactions by mass spectrometry and quantum chemical computations, finding consistency between experimental observation and theoretical model. The results have provided us with a number of valuable insights into the reverse reactions, i.e., the hydride transfer from selected magnesium hydride species to the prototypical carbonyl compounds, CO2 and CH2O, and the subsequent complexation between the oxophilic Mg(II) and the newly-formed anionic ligand, coordinated via the oxygen atom(s). It should be noted that there is a key difference in the reaction mechanisms: the hydride transfer to CO2 involves a shift of the ligand, from a monodentate coordination to the metal in the pre-reaction complex, to a bidentate coordination upon formation of the product complex, which is not required in the CH2O case. Consequently, the transition states of the former reaction are likely more destabilized.In comparing the reaction energies and barriers for the three magnesium hydride species HMg+, HMgCl and HMgCl2− reacting with CO2, we find that the number of chloride ligands (or equivalently overall electric charge) has a minimal impact, although there is a slight tendency for the anion to be more reactive than the neutral (Fig. 4). For HMg+, HMgCl and HMgCl2− reacting with CH2O, the variation is somewhat larger with the neutral and the anion having more exothermic reactions compared to the cation, by 35–47 kJ mol−1 (Fig. 6); on the other hand, the reaction barrier is more submerged for the cation (−67 kJ mol−1) compared to the neutral (−33 kJ mol−1) and the anion (−16 kJ mol−1). The trends appear to hold true also when substituting other halides (F, Br and I) for Cl.
For the hydride transfer from HMg+, HMgCl or HMgCl2− to either CO2 or CH2O, we find that the reaction energies are surprisingly similar between the two substrate molecules; for instance, for the cation the difference is 20 kJ mol−1 and for the anion it is essentially nil. This is despite the fact that carbon dioxide has a HA that is 57 kJ mol−1 higher than that of formaldehyde. This means that the so-formed CH3O− ligand binds stronger to the magnesium centre compared to HCO2− by an amount that almost compensates for the difference in HA. However, in considering not just Mg, but also other alkaline earth metals, we find that the binding strength between the metal and the methoxy ligand, relative to that between the metal and the formate ligand, decreases for the series M = Be, Mg, Ca, Sr and Ba. It was also found that the hydride transfer reactions of HM+, HMCl and HMCl2− become increasingly more exothermic when descending group 2: Mg, Ca, Sr, Ba. However, the case of M = Be is exceptional in that it often results in more exothermic reactions compared to the other metals, mostly owing to the strong oxophilicity of Be; the effect is more pronounced for the cation, and more pronounced for the reaction with formaldehyde.
Our experimental investigation also revealed that in the gas phase, CH3OMg+ reacts directly with CO2 by ligand switching, and CH2O is released upon formation of HCO2Mg+. The reaction occurs by direct hydride transfer between the electrophilic carbons of the two ligands in a classical Meerwein–Ponndorf–Verley-type mechanism.30–32 Both alcohols and formic acid are known to be reactive in carbon-to-carbon hydride transfers in organic chemistry.33 It was also found that CH3OMg+ appears to be highly reactive with H2O, producing CH3O2Mg+ upon release of H2. Isotope labelling in combination with quantum chemical computations revealed that the formation of the leaving hydrogen molecule uses one hydrogen from each of the reactants. The corresponding reaction was not observed for CH3OMgCl2− and our computations indicate unfavourable thermochemistry for the reaction of the anion as well as for the neutral. The barriers are, in all cases, moderate so these results do not rule out such dehydrogenation reactions in the presence of water in solution.
It should be noted that the gas-phase reactivity trends reported here are not directly transferable to the corresponding reactions in solution or on interfaces. In solution, the charged species will be strongly affected by stabilizing interactions with other molecules and ions present; despite this, the data presented here describe the inherent reactivity of the key species. In the future, it will hopefully be possible to study these reactions in solution—experimentally and computationally—in sufficient detail and with sufficient accuracy to make a meaningful comparison.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Norwegian Research Council: through Grant No. 249788. The Chemistry of CO2 Activation and Fixation; through the Hylleraas Centre for Quantum Molecular Sciences, No. 262695/F50, through the Centre of Excellence program of the Norwegian Research Council; and through the Norwegian Supercomputing Program (NOTUR) through a grant of computer time (Grant No. NN4654K). The authors thank J. Jestilä for performing the CCSD(T) computations used in the method evaluation.References
- S. Aldridge and A. J. Downs, Chem. Rev., 2001, 101, 3305–3366 CrossRef CAS PubMed.
- B. Bogdanovic, P. Bons, M. Schwickardi and K. Seevogel, Chem. Ber., 1991, 124, 1041–1050 CrossRef CAS.
- U. Eberle, M. Felderhoff and F. Schüth, Angew. Chem., Int. Ed., 2009, 48, 6608–6630 CrossRef CAS PubMed.
- S. Brutti, G. Mulas, E. Piciollo, S. Panero and P. Reale, J. Mater. Chem., 2012, 22, 14531–14537 RSC.
- R. Mohtadi and S.-I. Orimo, Nat. Rev. Mater., 2016, 2, 16091 CrossRef.
- E. N. Koukaras, A. D. Zdetsis and M. M. Sigalas, J. Am. Chem. Soc., 2012, 134, 15914–15922 CrossRef CAS PubMed.
- E. C. Ashby and A. B. Goel, Inorg. Chem., 1977, 16, 2941–2944 CrossRef CAS.
- D. Mukherjee and J. Okuda, Angew. Chem., Int. Ed., 2018, 57, 1458–1473 CrossRef CAS PubMed.
- M. Arrowsmith, M. S. Hill, D. J. MacDougall and M. F. Mahon, Angew. Chem., Int. Ed., 2009, 48, 4013–4016 CrossRef CAS PubMed.
- S. Schnitzler, T. P. Spaniol and J. Okuda, Inorg. Chem., 2016, 55, 12997–13006 CrossRef CAS PubMed.
- L. E. Lemmerz, A. Wong, G. Ménard, T. P. Spaniol and J. Okuda, Polyhedron, 2020, 178, 114331 CrossRef CAS.
- H. Schwarz, Coord. Chem. Rev., 2017, 334, 112–123 CrossRef CAS.
- M. D. Anker, M. S. Hill, J. P. Lowe and M. F. Mahon, Angew. Chem., Int. Ed., 2015, 54, 10009–10011 CrossRef CAS PubMed.
- L. Garcia, M. F. Mahon and M. S. Hill, Organometallics, 2019, 38, 3778–3785 CrossRef CAS.
- G. N. Khairallah and R. A. J. O’Hair, Int. J. Mass Spectrom., 2006, 254, 145–151 CrossRef CAS.
- Z. Zheng, J. Pavlov, Y. Wei, Y. Zhang and A. B. Attygalle, ACS Omega, 2018, 3, 3440–3452 CrossRef CAS PubMed.
- P. U. Andersson, M. J. Ryding, O. Sekiguchi and E. Uggerud, Phys. Chem. Chem. Phys., 2008, 10, 6127 RSC.
- M. J. Ryding, A. M. Jonsson, A. S. Zatula, P. U. Andersson and E. Uggerud, Atmos. Chem. Phys., 2012, 12, 2809–2822 CrossRef CAS.
- M. J. Ryding, K. Ruusuvuori, P. U. Andersson, A. S. Zatula, M. J. McGrath, T. Kurtén, I. K. Ortega, H. Vehkamäki and E. Uggerud, J. Phys. Chem. A, 2012, 116, 4902–4908 CrossRef CAS PubMed.
- A. S. Zatula, P. U. Andersson, M. J. Ryding and E. Uggerud, Phys. Chem. Chem. Phys., 2011, 13, 13287 RSC.
- S. Narancic, A. Bach and P. Chen, J. Phys. Chem. A, 2007, 111, 7006–7013 CrossRef CAS PubMed.
- J. S. Jestilä, Z. Iker, M. J. O. Ryding and E. Uggerud, Org. Biomol. Chem., 2020, 18, 9499–9510 RSC.
- G. W. T. M. J. Frisch, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery, Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revision A.02, 2009.
- L. A. Curtiss, P. C. Redfern and K. Raghavachari, J. Chem. Phys., 2007, 126, 084108 CrossRef PubMed.
- M. Valiev, E. J. Bylaska, N. Govind, K. Kowalski, T. P. Straatsma, H. J. J. Van Dam, D. Wang, J. Nieplocha, E. Apra, T. L. Windus and W. A. de Jong, Comput. Phys. Commun., 2010, 181, 1477–1489 CrossRef CAS.
- D. J. Goebbert and P. G. Wenthold, Int. J. Mass Spectrom., 2006, 257, 1–11 CrossRef CAS.
- D. B. Workman and R. R. Squires, Inorg. Chem., 1988, 27, 1846–1848 CrossRef CAS.
- R. R. Squires, in Structure/Reactivity and Thermochemistry of Ions, ed. P. Ausloos and S. G. Lias, D. Reidel Publishing Company, 1987, pp. 373–375 Search PubMed.
- B. Chan, Y. Luo and M. Kimura, J. Phys. Chem. A, 2021, 125, 835–842 CrossRef CAS PubMed.
- H. Meerwein and R. Schmidt, Justus Liebigs Ann. Chem., 1925, 444, 221–238 CrossRef CAS.
- W. Ponndorff, Angew. Chem., 1926, 39, 138–143 CrossRef.
- A. Verley, Bull. Soc. Chim. Fr., 1925, 37, 537–542 CAS.
- N. C. Deno, H. J. Peterson and G. S. Saines, Chem. Rev., 1960, 60, 7–14 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2cp05964a |
| This journal is © the Owner Societies 2023 |