
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Solvent-controlled self-assembly of Fmoc protected aliphatic amino acids†
Bharti
Koshti‡
a,
Hamish W A
Swanson‡
 b,
Basil
Wilson
a,
Vivekshinh
Kshtriya
a,
Soumick
Naskar
a,
Hanuman
Narode
a,
King Hang Aaron
Lau
b,
Tell
Tuttle
*b and
Nidhi
Gour
*a
b,
Basil
Wilson
a,
Vivekshinh
Kshtriya
a,
Soumick
Naskar
a,
Hanuman
Narode
a,
King Hang Aaron
Lau
b,
Tell
Tuttle
*b and
Nidhi
Gour
*a
aDepartment of Chemistry, Indrashil University, Kadi, Mehsana, Gujarat, India. E-mail: nidhi.gour@indrashiluniversity.edu.in
bDepartment of Chemistry, University of Strathclyde, 295 Cathedral Street, Glasgow G1 1XL, UK. E-mail: tell.tuttle@strath.ac.uk
First published on 28th March 2023
Abstract
Self-assembly of modified amino acids facilitate the formation of various structures that have unique properties and therefore serve as excellent bio-organic scaffolds for diverse applications. Self-assembly of Fmoc protected single amino acids has attracted great interest owing to their ease of synthesis and applications as functional materials. Smaller assembly units enable synthetic convenience and potentially broader adoption. Herein, we demonstrate the ability to control the morphologies resulting from self-assembly of Fmoc modified aliphatic single amino acids (Fmoc-SAAs) namely, Alanine, Valine, Leucine, Isoleucine, and Proline. Controlled morphological transitions were observed through solvent variation and the mechanism that allows this control was investigated using coarse-grained molecular dynamics simulations. These show that FmocA can form well defined crystalline structures through uniform parallel Fmoc stacking and the optimization of ion concentrations, which is not observed for the other Fmoc-SAAs. We demonstrate that Fmoc protected aliphatic single amino acids are novel scaffolds for the design of distinct micro/nanostructures through a bottom-up approach that can be tuned by control of the environmental parameters.
Introduction
Elucidating the link between discrete molecular construction and resultant self-assembled nanostructures is the first step towards the de novo design of functional materials and doing so would be highly beneficial in manifold contexts. Self-assembling materials can find diverse applications from antimicrobial materials1,2 to emulsifiers for food, cosmetic, and biomedical industries.3,4 Given the inherent biological interactions within these application areas, a wide variety of small biomimetic assembling molecules are the focus of this research, including peptides and peptide amphiphiles,5–7 heterochiral peptides,8,9 peptoids,10,11 and single amino acids (SAAs). The assembly of SAAs is of particular interest for many reasons, including their ease of synthesis,12 functional diversity,13 stability,14 and biocompatibility.15 SAAs with sidechains presenting aromatic and other functional groups assemble into a variety of morphologies such as fibres,16 micelles,17 and tubes14,18 to highlight a few. Self-assembly of short amino acids has also drawn attention due to its implications in the pathogenesis of in-born errors of metabolisms (IEMs) relating the aetiology of IEMs to amyloid associated diseases.19A popular strategy to promote amphiphilicity and consequently induce the aqueous assembly of SAAs and other short oligopeptides is the functionalisation of the N-terminus with an N-(9-fluorenylmethoxycarbonyl) Fmoc group.20–22 In the study of assembling Fmoc modified peptidic molecules, there has been a particular focus on the characterisation of di- and tripeptides.23 Most notably that of Fmoc-FF which is a particularly well described assembly system.20,24 In contrast work surrounding the assembly forming properties of Fmoc-SAAs is disjointed15,25–28 with many of these studies focusing on self-assembly either by co-assembly or conjugation with other functional groups as opposed to the native self-assembly properties of the individual amino-acid residue.29,30 It is surprising that this class of molecules have not received more attention, as Fmoc-SAAs present an appealing alternative to more complex peptide based assembling materials: their small size means that extensive synthetic procedures can be avoided in their preparation which is both cost effective and more environmentally friendly. Furthermore, the antimicrobial properties of Fmoc modified amino acid short peptides have been reported, highlighting a avenue to application to this concern.31–34 A well-established methodology by which to control/induce molecular assembly is through the dilution of a stock solution with a more polar solvent, such as water.11,14 In related work many small simple molecular precursors have been shown to form assembled hydro/organogels35 The small size of Fmoc-SAAs means that chemical expectations can be subverted, for example Kundu et al. have shown that the dipeptide Fmoc-KK can assembly in a variety of different organic solvents36 as can Fmoc-K in DMSO: water solutions as demonstrated by Sato et al.37 With these perspectives in mind, we decided to undertake a systematic survey of the assembly characteristics of N-(9-fluorenylmethoxycarbonyl) modified single aliphatic L-amino acids namely: L-alanine (FmocA), L-valine (FmocV), L-leucine (FmocL), L-isoleucine (FmocI), L-proline (FmocP) (Fig. 1) under varied concentration, pH, temperature, and solvent conditions. A variety of optical microscopy methods (bright-field, phase contrast, and fluorescence) and field-emission scanning electron microscopy (FE-SEM) to characterise assemblies under these varied conditions. In particular we make use of fluorescence labelling, with fluorescein and rhodamine B dye. This was done to assess the formation of aggregates and self-assembled structures while in solution, through the incorporation of dyes into organic domains present within assemblies, as has been reported elsewhere.38–40 Moreover, by focusing on the Fmoc modified aliphatic amino acids the contributions of both aromatic and aliphatic hydrophobicity to assembly may be better understood. To this end we also undertook co-incubation experiments with tannic acid, urea and solution state 1H-NMR experiments have also been performed. Further the mechanism of self-assembly was further characterised assessed using coarse-grained molecular dynamics (CG-MD) methods. In this report we show that despite their small size, a wide range of self-assembled morphologies comparable to those formed by larger peptidic molecules, may be obtained ranging from crystalline structures, rods, spheres, and fibres. The ability to control this behaviour through environmental controls is of interest for varied materials applications. Lastly, we can show how in mixed organic/aqueous solvent conditions the formation of ordered structures is dependent on the steric effects of aliphatic sidechains.
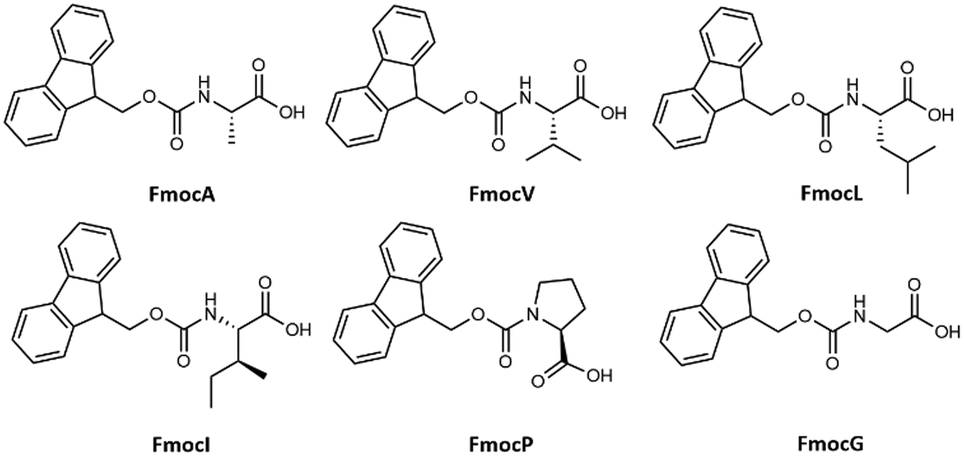 | ||
| Fig. 1 Chemical structure of Fmoc protected aliphatic single amino acids studied for their self-assembly. | ||
Results and discussion
Aqueous solution study
The aliphatic sidechains of alanine, valine, leucine, isoleucine, and proline are non-polar as such their interaction with polar solvents is not favoured and this hydrophobicity drives their coalescence. At close range, aliphatic dispersion interactions will make coalescence enthalpically favoured.41 Modification of these SAAs with Fmoc will increase the entropic penalty of solvation, further driving coalescence. Enthalpic and entropic gains will also be made through π–π stacking interactions of Fmoc, which have the potential to impart order within the hydrophobic domain that can subsequently become crystalline.23 Our experiments began with preparation of 20 mM stock solutions in 50% water: methanol of each amino acid. These were then diluted to 3 mM and 8 mM using deionized water. Optical microscopy and FE-SEM analysis of FmocA (3 mM) revealed a unique crystalline structure reminiscent of a Catharanthus flower with angular hexagonal ‘petals’ (Fig. 2). This structure is regular and uniform and there is no morphological difference between the 3 mM and 8 mM systems (Fig. 2, Fig. S4 and S5, ESI†). Fluorescence microscopy suggests these structures could bind with the fluorescein dye and green fluorescence was observed (Fig. S4 and S5, ESI†). No morphological transition or rearrangement occurred after heating at 70 °C for 30 minutes (Fig. 2) indicating that this crystalline form is thermodynamically stable. Crystallinity is further confirmed by phase contrast microscopy, in which the structure appears light with respect to the background (Fig. 2 and Fig. S6, ESI†).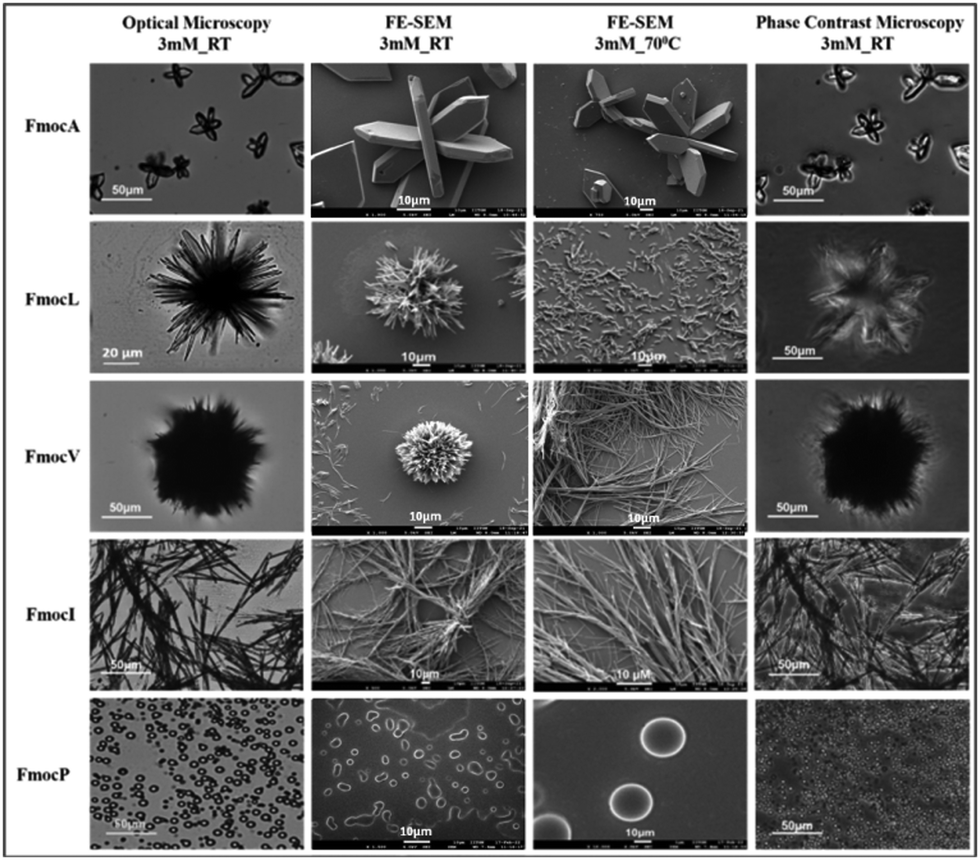 | ||
| Fig. 2 Microscopy images of 3 mM Fmoc-SAAs studied by different techniques i.e., Optical microscopy, FE SEM, and Phase contrast microscopy. | ||
FmocV on the other hand assembled into fibrous ball like structures at 3 mM which changed to fibrillar assemblies at 8 mM. Concentration dependent self-assembly studies of FmocV suggest fibrous balls merge with each other and gradually change to fibres (Fig. S7–S10, ESI†). In contrast, FmocL form star-like flowers with petals composed of needles at 3 mM (Fig. 2, Fig. S11 and S12, ESI†). Phase contrast microscopy suggests these structures are semi-crystalline, and the flowers appear to be arranged from rod-like morphologies. The structural isomer FmocI assembled into conventional fibrous structures under all conditions at 3–8 mM concentration which was surprising (Fig. 2, Fig. S13 and S14, ESI†). Therefore, from the microscopy analysis, it was evident that aliphatic Fmoc-SAAs namely FmocA, FmocV, FmocL and FmocI self-assembled into unique and distinct structures. The structure of Fmoc-SAAs just differs in the length of carbon chain attached it. Moreover, FmocL and FmocI are structural isomers and the different morphologies observed suggest a crucial role of branching of carbon chain in determining the structure. FmocI is more hydrophobic than FmocL which may promote the formation of more uniform fibrillar morphologies. Further we also studied the self-assembly of the more unusual amino acid, FmocP, which assembled to conventional spherical morphologies at 3 and 8 mM concentration both before and after heating (Fig. 2 and Fig. S15, ESI†). FmocP has a compact, closed cyclic ring in the side chain which might impart it relatively “more hydrophilicity” than suggested by the length of its hydrophobic aliphatic side chain; hence it may assemble to vesicle like structures in the polar aqueous environment rather that forming fibres as observed for other Fmoc-SAAs. Indeed, from the phase contrast microscopy it appears only FmocA and FmocL self-assembled structures have some crystalline characteristics while FmocV, FmocI, and FmocP assembled into amorphous morphologies as they appeared dark in contrast to the background. After annealing at 70 °C for 30 minutes the morphologies deviate structurally for some Fmoc-SAAs. The temperature of 70 °C was chosen for heating since it was an optimal temperature wherein enhanced aggregation can be achieved without altering the composition of aqueous solvent mixture and chemical stability of Fmoc-SAAs. In the case of FmocV a network of fibres is formed since the temperature rise might lead to enhanced aggregation driving merging of fibrous ball like structures (Fig. 2). Intriguingly, in contrast to FmocV, FmocL (3 mM) disintegrates into small rods which are crystalline, as indicated by the phase contrast microscopy; it may be the case that the extra thermal energy herein enables a structural rearrangement to occur for this sequence (Fig. 2). In case of other Fmoc-SAAs the effect of annealing is not so prominent, and the structures remained robust both before and after heating (Fig. 2).
Mixed solvent studies
To assess the relative importance of different noncovalent interactions in the assemblies observed, 3 mM of each Fmoc-SAA was dissolved in tetrahydrofuran (THF) which is a compatible solvent for all aliphatic Fmoc-SAAs. To induce aggregation/assembly, water was then added in increasing proportions. For FmocA it was observed that as the water concentration was increased fibre like structures were formed at room temperature in the range of 10% water to 90% water in the water: THF system. These fibre-like morphologies appear flexible and curved at low water concentrations. However, they gain linearity and become needle like as the water concentration is increased (Fig. 3). Clearly, the hydrophobic Fmoc group is entropically driven to minimize its water contacts in solvent systems with a higher water composition; a crystalline arrangement appears to offer optimal minimisation.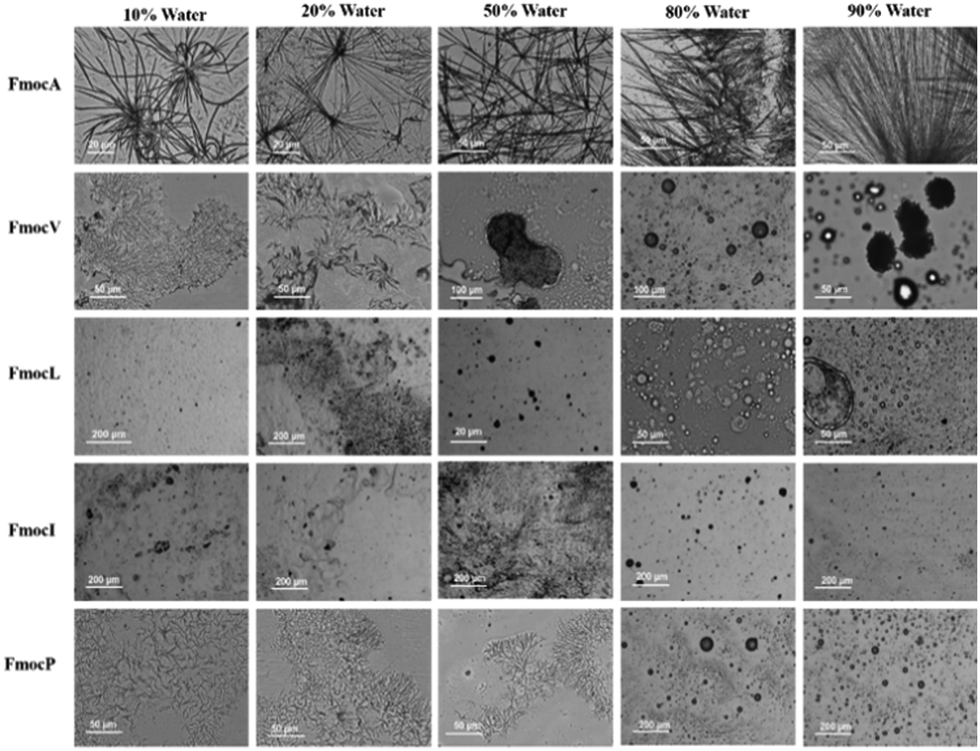 | ||
| Fig. 3 Since no morphological transition was seen from 3 mM to 8 mM in the THF: water systems a 3 mM concentration was taken as standard. Optical microscopy images of increasing percentage of water in THF at room temperature. | ||
When the THF composition is higher, the Fmoc residues are likely to be less solvophobic and so the assemblies qualitatively appear to be less crystalline and more amorphous in nature. In contrast to FmocA, the water: THF studies of FmocV, FmocL and FmocI yielded no structures, except for some sphere-like aggregates which generally began to form at 80% Water: THF (Fig. 3). In the case of FmocP the same trend was observed. However, as the percentage of water in THF system was increased, self-assembly is induced and at 10% Water: THF small fibres were obtained (Fig. 3). Interestingly, these fibres disappear after addition of more water, possibly due to the comparatively more hydrophilic nature of proline. As the percentage of water is increased to above 80% Water: THF spherical aggregates as observed in Fig. 2 again appear. Notably, spheres are the most thermodynamically stable due to the minimum surface area to volume ratio they enable. Clearly the assembly pathway of FmocA is distinct from that of the other aliphatic Fmoc-SAAs, both in mixed solvent and in aqueous assembly. We also performed mixed solvent studies with methanol and water (Fig. S44, ESI† for details). It was observed that similar distinctions between assembled morphologies for different Fmoc-SAAs formed, reminiscent of those in the aqueous solution study at high water contents (Fig. 2), which implies that the driving forces for assembly are consistent for both aprotic and protic solvent systems. To further probe the role of hydrogen bonding and π–π stacking in the self-assembled structure formation we co-incubated Fmoc-SAAs with urea and tannic acid. Urea is a well-known hydrogen bond breaker,42 while tannic acid is a generic amyloid inhibitor known to disrupt π–π stacking.43 Hence, any morphological transition or disruption occurring due to co-incubation of urea or tannic acids can provide insights into the crucial role of π–π stackings and hydrogen bonding in the self-assembled structures. We observed disruption of self-assemblies formed by all Fmoc-SAAs on co-incubation with tannic acid while with urea the assemblies remain unaffected (Fig. S18–S31, ESI†). Hence, hydrogen bonding is of relatively less importance to these assemblies than the π–π stacking of the Fmoc group. Further, the pH dependent studies of Fmoc modified aliphatic single amino acids suggest that the assemblies are formed only in neutral and acidic condition while in basic condition the assemblies were not formed (Fig. S32–S37, ESI†) indicating electrostatics are also important to the Fmoc-SAA assemblies. XRD analysis on FmocA self-assembled structures further confirmed their crystalline morphologies as the diffraction peaks appeared prominent unlike FmocI where the peaks appeared broad indicating its soft nature (Fig. S39, ESI† for details). The critical role of π–π stacking of the Fmoc groups is further supported by the up-field shift in the aromatic protons observed via NMR when a drop of D2O or increasing % of water was added (see Fig. S40–S43, ESI† for details). Clearly, this feature is dominant in the generation of assemblies and yet clear morphological differences still arise between the Fmoc-SAA's.
Mechanism of assembly
Intrigued by these results, we turned to molecular dynamics to interrogate the self-assembly mechanism of FmocA versus the other aliphatic Fmoc-SAAs. Specifically, we chose to use coarse grained molecular dynamics (CG-MD) using the MARTINI force field (version 2.1.) which enables access to the kinds of timescales and systems sizes required to assess this process.44 We used an Fmoc model developed in our lab which has been used to successfully reproduce a variety of experimentally observable properties of other assembling Fmoc short peptide conjugates.45 We performed simulations with C-termini fully deprotonated, as would be expected at neutral pH, and with the C-termini fully protonated. This was done on the basis that pKa shifts are known to happen in similar Fmoc assembly systems22,24 and that non-aqueous organic solvents, in this case THF, can also cause substantial changes the pKa of titratable groups.46 Given the ambiguity it was deemed most appropriate to study both possibilities. To ensure system neutrality, sodium ions were added to the charged assembly simulations. In this study 1600 molecules were simulated for 1 μs at 0, 20, 50 and 80% water. Each THF molecule was represented by a single bead (with 5 heavy atoms per bead), the bead type was Na and is closely related to that reported by Patti et al.47 For solvent systems containing water, 10% were antifreeze water beads, the inclusion of which is necessary when using Martini version 2.1. which may irreversibly freeze if a high degree of order emerges, such as crystalline regions within the system.48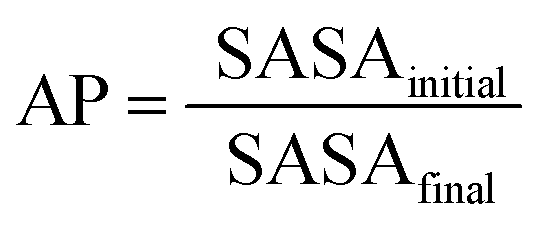 | (1) |
The first parameter we chose to measure for these systems was the Aggregation Propensity (AP); which corresponds to the ratio of Solvent Accessible Surface Area (SASA) at the beginning of the simulation in the non-aggregated state (SASAinitial) verses this same property in the final frame of the simulation (SASAfinal). As it depends on only two time-points it may be considered a static measurement (eqn (1)). If the surface area decreases over the course of the trajectory due to aggregation, then the AP score will be greater than 1.0. The larger this number, the greater a molecules preference for coalescence/assembly. This metric, and variations of it, have been used effectively in previous studies for the directed discovery of peptide-based materials.49–51 Firstly, the AP metric indicates that the assembly of FmocA is distinct from that of the other Fmoc-SAAs, with a higher AP score at 50% water content for both charged and neutral C-termini, though the assembly of FmocV and FmocP yields a slightly higher AP score at 80% water in the neutral case (Fig. 4). Inspection of the trajectories immediately revealed the cause of the increased AP score for FmocA; which forms highly crystalline structures, with parallel packed Fmoc groups which over the course of the simulation form initially as small platelets and consequently combine into extended structures (Fig. 5b). In contrast the other sequences form amorphous aggregates with only small regions of ordered stacking (FmocV, Fig. 5b). In the charged systems, such an arrangement by FmocA is supported by ions which adopt regular and uniform positions along the parallel stacks. However, this same crystallisation is observed in the neutral state when the C-termini are assumed to be neutrally charged. The persistence of this structuring irrespective of charge implies that the small steric profile of FmocA, compared to the other aliphatic sidechains is central to the formation of well-defined crystalline assemblies.
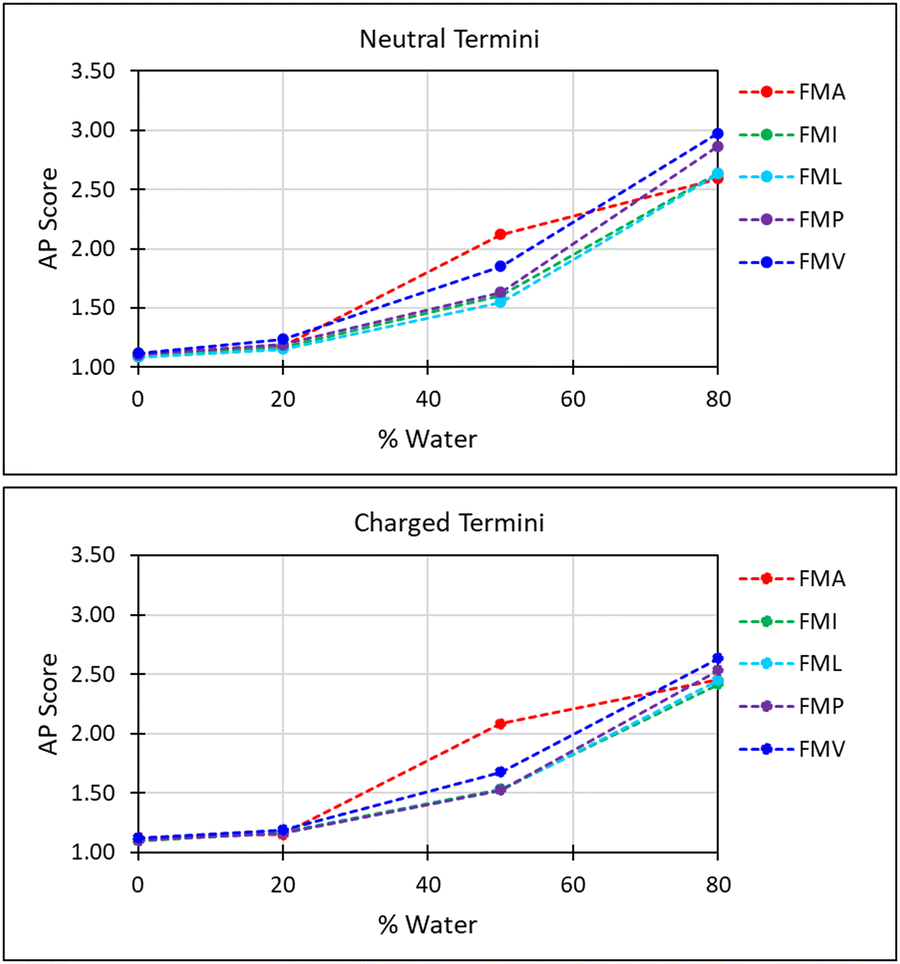 | ||
| Fig. 4 Aggregation propensity (AP) score with respect to water fraction; FmocA and FmocV exhibit distinct behaviour. The low AP scores for FmocA in high THF concentrations are attributed to the slower rate of assembly in these more compatible solvent conditions. | ||
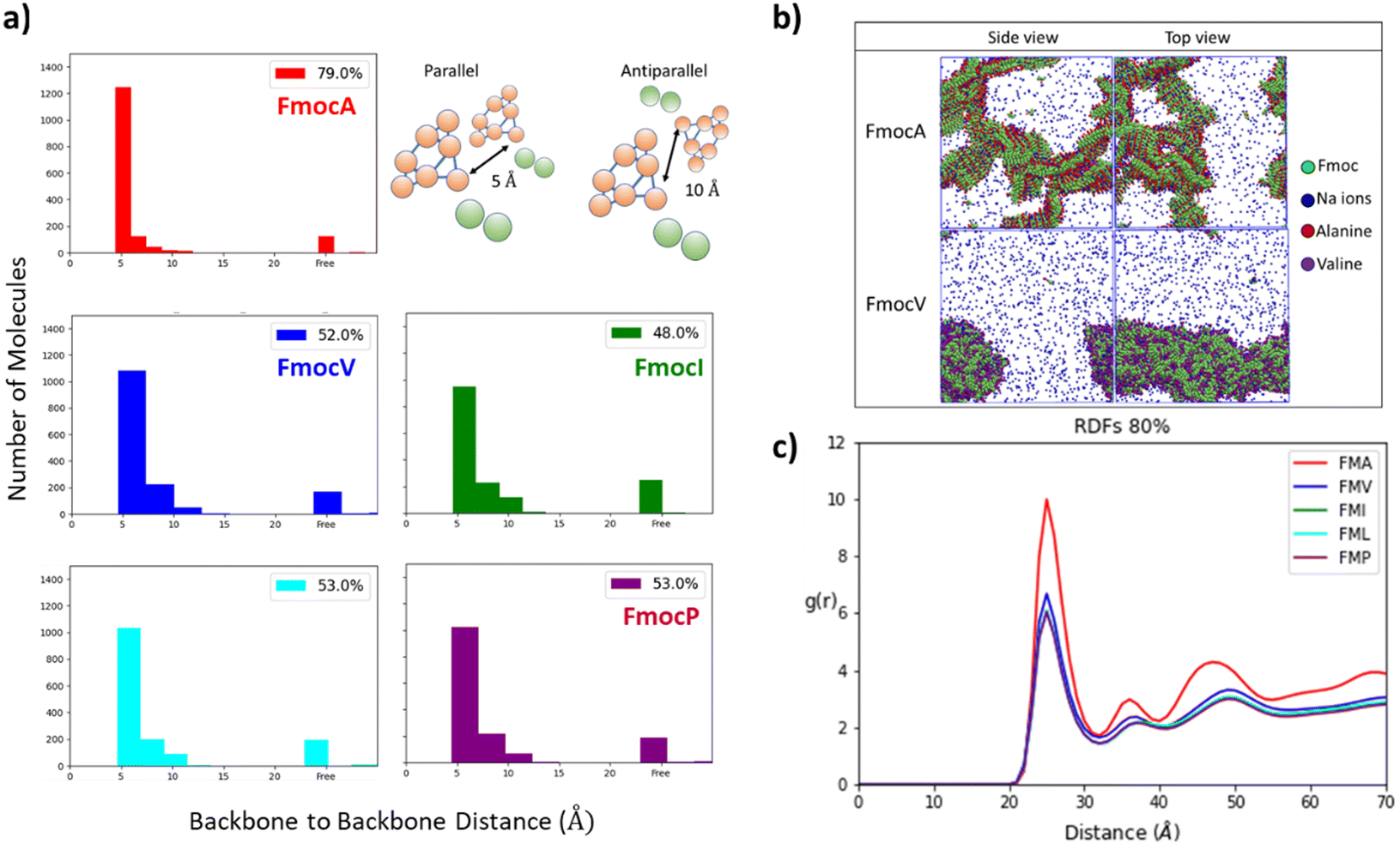 | ||
| Fig. 5 (a) The number of parallel stacked Fmoc groups in 80% water is highest for charged FmocA. For other aliphatic sequences, a range of spacings <10 Å are found to be present in the assemblies. We hypothesise that steric and dispersive effects of the aliphatic sidechains disrupt homogenous stacking in these cases. (b) Assembly structures of FmocA and FmocV in 80% water after 1 μs. It is evident that more ions are solvated in FmocV than in FmocA; as these are more readily incorporated into the crystalline assembly of FmocA, as supported by radial distribution function analysis (RDF), (c) ion–molecule Radial Distribution Function (RDF) evaluated for all charged C-termini molecules in 80% solvent compositions. Clearly the substantially larger RDF for FmocA is higher than all other aliphatic Fmoc amino acid derivatives. It is expected that this greater ion density supports crystallisation. | ||
These results align with the experimental findings for these mixed solvent systems (Fig. 3). Where only small aggregates are observed in 50% THF: water systems for all systems except FmocA. For solvent systems with 80% water the same pattern is observed, with all species except FmocA forming amorphous aggregates. Lastly, it is curious that the AP at 20% water is low for FmocA given that assemblies were observed in this solvent environment experimentally (Fig. 3). This is attributed to the short duration of the simulation, on the basis that the rate at which assembly occurs may be reduced by the improved solvent compatibility in this system. To assess the structuring present within the assemblies we measured the degree of parallel/antiparallel Fmoc alignment between adjacent molecules. In our methodology, molecules are deemed to be parallel stacked if the distance between ‘backbone’ (BB) Fmoc beads is <6 Å while packing is antiparallel when this distance is ∼10 Å. If the distance between the centre of geometry of a given Fmoc group and its next closest neighbour >7 Å then it is considered to not be in the stack and thus is deemed ‘free’ (Fig. 5a). This analysis revealed that the degree of parallel Fmoc alignment is substantially higher in the last frame of the 1 μs simulation for FmocA in 80% water compared to all other aliphatic Fmoc-SAAs both in the charged and neutral state (82% are within the stack with neutral termini and 79% for the charged termini) (Fig. 5 and Fig. S3, ESI†). In previous work the fluorenyl spacing distance in Fmoc assemblies has been evaluated as being ∼3.6–5.0 Å using atomistic molecular dynamics and wide-angle X-ray scattering (WAXS) measurements.52,53 We find our estimated spacing for the crystalline stacking in FmocA, ∼5 Å, to be in good agreement with these results particularly given the reduced level of molecular detail. In contrast to FmocA, for all other systems in which amorphous assemblies arise, in 80% water only ∼50% of molecules have BB–BB distances less than 6 Å apart (Fig. 5a). By this method it is not possible to evaluate the sizes of stacked domains distributed throughout the assemblies; however clearly the prevalence of well-defined fluorenyl arrangements is reduced in all cases other than FmocA and we attribute this to the presence of the aliphatic sidechains which disrupt extended π–π stacking based structuring. The efficient parallel packing of Fmoc residues, in which π–π interactions is optimised, and the solvent accessible surface area is minimised, can only be achieved by drawing FmocA molecules close together. A thermodynamic hindrance to this is the increased proximity of charged C-termini which electrostatically repel each other, and this effect can only be overcome by adequate screening by cationic ions. Alanine has a very small methyl sidechain, which in the Martini representation is subsumed into the backbone bead. All other aliphatic amino acids have a sidechain, which is represented by a second hydrophobic bead. Upon aggregation, FmocA will enable a larger number of ions to come into proximity as opposed to the other aliphatic amino acids, for which the non-polar sidechain will ‘frustrate’ ion organisation. To assess this hypothesis, we measured the radial distribution function (RDF) of sodium ions with respect to Fmoc-SAAs over the course of the 1 μs trajectory (Fig. 5c) in the 80% water simulations. In all systems the first sodium solvation shell, occurs at 2.5 Å and is substantially larger for FmocA than for the other compounds. This supports the hypothesis that crystallinity only emerges for FmocA as it does not hinder counter ion screening and therefore enables close and highly ordered packing, which in turn enables crystallinity to arise. Notably, ionic support for Fmoc protected amino acids has been reported elsewhere, so this observation is not without precedent.54 We also performed these simulations with neutral termini and found the same stacking occurs for FmocA and its absence for all other systems. The emergence of this structuring irrespective of C-termini state implies that the sidechain steric effects is of greatest significant for the FmocSAA assembly structure.
Experimental
Materials and methods
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, 1:3 and 1:5. A fixed concentration of Fmoc protected amino acid as 3 mM was taken. A 20 μL solution of this was drop casted on a clean glass slide and visualized in Leica DM2500 upright fluorescent microscope under bright field mode.
:1, 1:3 and 1:5 against modified amino acids. The images were visualized under Leica DM2500 upright fluorescent microscope under bright filed with different magnification.
000 solvent beads with varied compositions, selected to leverage the fact that one Martini water bead is equivalent to four water atoms while one THF bead represents only one molecule. For example, in a 50% water: THF solution, there are 4.49 moles of H2O to 1 mole of THF present. If the total number of solvent beads is conserved (80000) then to obtain this same ratio, 37680 THF beads (1 molecule per bead) and 42320 water beads (4 molecules per bead) would be required. These compositions are detailed in Table S3 (ESI†). 10% of the water component were antifreeze beads to prevent freezing of water as crystalline domains emerge which is a recognized issue with Martini force field version 2.1.55 Simulations were performed both with neutral and charged C-termini, for the latter case an additional 1600 sodium ions were added to neutralize the charge of the system. All systems were build using Gromacs version 2020.7.56 Visualization of simulation results was done using Visual Molecular Dynamics (VMD)57 and analysis was done using the python package MDAnalysis.58
000 steps. Following this an NPT equilibration (250000 steps with a 20 fs timestep) was performed for 5 ns using a Berendsen pressure coupling (compressibility = 4.5 × 10−5 bar−1 and pressure = 1 atm) with velocity rescaling at a temperature of 298.15 K with a time constant for coupling of 1 ps. For the production simulation, a timestep of 25 fs was used for a total of 40000000 steps for 1 μs, an NPT ensemble was used using isotropic Parrinello-Rahman pressure coupling (compressibility = 4.5 × 10−5 bar−1, pressure = 1 atm and a time constant for pressure coupling of 12 ps) and again velocity rescaling was used with a reference temperature of 298.15 K. Both electrostatic and Lennard Jones (LJ) potentials were shifted to a straight cutoff at 11 Å as consistent with Martini simulation methodology for dealing with non-bonded interactions.59 The LINCS algorithm was used for constraints within the Fmoc ring model.60
:1, 1:3 and 1:5. A fixed concentration of Fmoc protected amino acid as 3 mM was taken. A 20 μL solution of this was drop casted on a clean glass slide and visualized in Leica DM2500 upright fluorescent microscope under bright field mode.
:1, 1:3 and 1:5 against modified amino acids. The images were visualized under Leica DM2500 upright fluorescent microscope under bright filed with different magnification.
000 solvent beads with varied compositions, selected to leverage the fact that one Martini water bead is equivalent to four water atoms while one THF bead represents only one molecule. For example, in a 50% water: THF solution, there are 4.49 moles of H2O to 1 mole of THF present. If the total number of solvent beads is conserved (80000) then to obtain this same ratio, 37680 THF beads (1 molecule per bead) and 42320 water beads (4 molecules per bead) would be required. These compositions are detailed in Table S3 (ESI†). 10% of the water component were antifreeze beads to prevent freezing of water as crystalline domains emerge which is a recognized issue with Martini force field version 2.1.55 Simulations were performed both with neutral and charged C-termini, for the latter case an additional 1600 sodium ions were added to neutralize the charge of the system. All systems were build using Gromacs version 2020.7.56 Visualization of simulation results was done using Visual Molecular Dynamics (VMD)57 and analysis was done using the python package MDAnalysis.58
000 steps. Following this an NPT equilibration (250000 steps with a 20 fs timestep) was performed for 5 ns using a Berendsen pressure coupling (compressibility = 4.5 × 10−5 bar−1 and pressure = 1 atm) with velocity rescaling at a temperature of 298.15 K with a time constant for coupling of 1 ps. For the production simulation, a timestep of 25 fs was used for a total of 40000000 steps for 1 μs, an NPT ensemble was used using isotropic Parrinello-Rahman pressure coupling (compressibility = 4.5 × 10−5 bar−1, pressure = 1 atm and a time constant for pressure coupling of 12 ps) and again velocity rescaling was used with a reference temperature of 298.15 K. Both electrostatic and Lennard Jones (LJ) potentials were shifted to a straight cutoff at 11 Å as consistent with Martini simulation methodology for dealing with non-bonded interactions.59 The LINCS algorithm was used for constraints within the Fmoc ring model.60
Conclusions
We report for the first time a comparative study of the assembly characteristics of all Fmoc protected aliphatic single amino acids. As anticipated, given that hydrophobicity is enhanced via Fmoc protection, all molecules assemble in high water content systems with diverse morphologies. However, FmocA exhibits distinct behaviour with respect to the other sequences as it yields flower like structures formed by the assembly of hexagonal crystalline morphologies. Unlike FmocA other amino acids assemble to amorphous or semicrystalline morphologies. The importance of the Fmoc stacking to assembly was confirmed via urea and tannic acid co-incubation; in which only the latter did not yield assemblies, indicating that pi–pi stacking caused due to presence of Fmoc group is the central non-covalent interaction supporting assembly. Furthermore, the up-field shift in the aromatic protons observed via NMR cements this finding. To rationalise these interesting deviations in assembly, despite the uniform assembly drivers in Fmoc-SAAs, we turned to coarse grained molecular dynamics. In doing so it was found that FmocA is unique in its ability to (i) optimize ion and sidechains arrangements and (ii) adopt uniform parallel Fmoc stacking in a manner not found for the other Fmoc aliphatic amino acids. We attribute this to the small steric profile of the methyl sidechain in alanine compared to the relatively bulky sidechains present in all other molecules, in which favourable dispersion interactions may result in more amorphous structure formation.Author contributions
All experimental work was done by BK, NG and the NG group. HS and TT completed all computational work and drafted the manuscript.Data availability
All computational data described in this publication are openly available from the University of Strathclyde KnowledgeBase at https://doi.org/10.15129/35e73840-1374-43e6-ad7a-b7986ca89226.Conflicts of interest
There is no conflict of interest to declare.Acknowledgements
NG, BK, BS VK and SN greatly acknowledge support from SERB research grant (EMR/2016/003186) and SERB SPG/2021/000521 for funding and fellowships. VK thanks to ICMR for the senior research fellowship No (45/13/2020-/BIO/BMS). BK thanks SHODH for funding and Indrashil University for infrastructure support. NG greatly acknowledges the support of IIT Gandhinagar Central Instrumentation Facility (IITGn-CIF) for the help provided in FE-SEM analysis. HWAS thanks the Carnegie Trust for funding. Computational results were obtained using the EPSRC-funded ARCHIE-WeSt High-Performance Computer (http://www.archie-west.ac.uk; EPSRC grant no. EP/K000586/1).References
- Z. Azoulay, P. Aibinder, A. Gancz, J. Moran-Gilad, S. Navon-Venezia and H. Rapaport, Acta Biomater., 2021, 125, 231–241 CrossRef CAS PubMed.
- V. Castelletto, C. J. C. Edwards-Gayle, I. W. Hamley, G. Barrett, J. Seitsonen, J. Ruokolainen, L. R. de Mello and E. R. da Silva, Chem. Commun., 2020, 56, 615–618 RSC.
- G. G. Scott, P. J. McKnight, T. Tuttle and R. V. Ulijn, Adv. Mater., 2016, 28, 1381–1386 CrossRef CAS PubMed.
- G. G. Scott, T. Boerner, M. E. Leser, T. J. Wooster and T. Tuttle, Front. Chem., 2022, 10, 822868 CrossRef CAS PubMed.
- Y. M. Abul-Haija, G. G. Scott, J. K. Sahoo, T. Tuttle and R. V. Ulijn, Chem. Commun., 2017, 53, 9562–9565 RSC.
- Y. Y. Xie, X. T. Qin, J. X. Zhang, M. Y. Sun, F. P. Wang, M. M. Huang, S. R. Jia, W. Qi, Y. F. Wang and C. Zhong, J. Colloid Interface Sci., 2022, 622, 135–146 CrossRef CAS PubMed.
- H. H. Xing, A. Rodger, J. Comer, A. S. Picco, C. Huck-Iriart, E. L. Ezell and M. Conda-Sheridan, ACS Appl. Bio Mater., 2022, 5, 4599–4610 CrossRef CAS PubMed.
- S. Kralj, O. Bellotto, E. Parisi, A. M. Garcia, D. Iglesias, S. Semeraro, C. Deganutti, P. D'Andrea, A. V. Vargiu, S. Geremia, R. De Zorzi and S. Marchesan, ACS Nano, 2020, 14, 16951–16961 CrossRef CAS PubMed.
- A. M. Garcia, M. Melchionna, O. Bellotto, S. Kralj, S. Semeraro, E. Parisi, D. Iglesias, P. D'Andrea, R. De Zorzi, A. V. Vargiu and S. Marchesan, ACS Nano, 2021, 15, 3015–3025 CrossRef CAS PubMed.
- V. Castelletto, J. Seitsonen, K. M. Tewari, A. Hasan, R. M. Edkins, J. Ruokolainen, L. M. Pandey, I. W. Hamley and K. H. A. Lau, ACS Macro Lett., 2020, 9, 1415–1416 CrossRef CAS PubMed.
- V. Castelletto, A. M. Chippindale, I. W. Hamley, S. Barnett, A. Hasan and K. H. A. Lau, Chem. Commun., 2019, 55, 5867–5869 RSC.
- F. Sheehan, D. Sementa, A. Jain, M. Kumar, M. Tayarani-Najjaran, D. Kroiss and R. V. Ulijn, Chem. Rev., 2021, 121, 13869–13914 CrossRef CAS PubMed.
- A. Pinazo, R. Pons, L. Perez and M. R. Infante, Ind. Eng. Chem. Res., 2011, 50, 4805–4817 CrossRef CAS.
- M. Reches and E. Gazit, Science, 2003, 300, 625–627 CrossRef CAS PubMed.
- V. Jayawarna, S. M. Richardson, A. R. Hirst, N. W. Hodson, A. Saiani, J. E. Gough and R. V. Ulijn, Acta Biomater., 2009, 5, 934–943 CrossRef CAS PubMed.
- P. Chakraborty and E. Gazit, ChemNanoMat, 2018, 4, 730–740 CrossRef CAS PubMed.
- N. Gour, P. C. Kanth, B. Koshti, V. Kshtriya, D. Shah, S. Patel, R. Agrawal-Rajput and M. K. Pandey, ACS Chem. Neurosci., 2019, 10, 1230–1239 CrossRef CAS PubMed.
- L. Adler-Abramovich, L. Vaks, O. Carny, D. Trudler, A. Magno, A. Caflisch, D. Frenkel and E. Gazit, Nat. Chem. Biol., 2012, 8, 701–706 CrossRef CAS PubMed.
- M. Ni and S. M. Zhuo, RSC Adv., 2019, 9, 844–852 RSC.
- M. Zhou, A. M. Smith, A. K. Das, N. W. Hodson, R. F. Collins, R. V. Ulijn and J. E. Gough, Biomaterials, 2009, 30, 2523–2530 CrossRef CAS PubMed.
- V. Castelletto, C. M. Moulton, G. Cheng, I. W. Hamley, M. R. Hicks, A. Rodger, D. E. Lopez-Perez, G. Revilla-Lopez and C. Aleman, Soft Matter, 2011, 7, 11405–11415 RSC.
- B. A. K. Kriebisch, A. Jussupow, A. M. Bergmann, F. Kohler, H. Dietz, V. R. I. Kaila and J. Boekhoven, J. Am. Chem. Soc., 2020, 142, 20837–20844 CrossRef CAS PubMed.
- S. Fleming and R. V. Ulijn, Chem. Soc. Rev., 2014, 43, 8150–8177 RSC.
- C. Tang, A. M. Smith, R. F. Collins, R. V. Ulijn and A. Saiani, Langmuir, 2009, 25, 9447–9453 CrossRef CAS PubMed.
- Z. M. Yang, H. W. Gu, D. G. Fu, P. Gao, J. K. Lam and B. Xu, Adv. Mater., 2004, 16, 1440–1444 CrossRef CAS.
- V. Singh, K. Snigdha, C. Singh, N. Sinha and A. K. Thakur, Soft Matter, 2015, 11, 5353–5364 RSC.
- S. Gupta, I. Singh, A. K. Sharma and P. Kumar, Front. Bioeng. Biotechnol., 2020, 8, 504 CrossRef PubMed.
- S. Chibh, V. Katoch, A. Kour, F. Khanam, A. S. Yadav, M. Singh, G. C. Kundu, B. Prakash and J. J. Panda, Biomater. Sci., 2021, 9, 942–959 RSC.
- I. Irwansyah, Y. Q. Li, W. X. Shi, D. P. Qi, W. R. Leow, M. B. Y. Tang, S. Z. Li and X. D. Chen, Adv. Mater., 2015, 27, 648–654 CrossRef CAS PubMed.
- K. Tao, A. Levin, L. Adler-Abramovich and E. Gazit, Chem. Soc. Rev., 2016, 45, 3935–3953 RSC.
- A. Y. Gahane, P. Ranjan, V. Singh, R. K. Sharma, N. Sinha, M. Sharma, R. Chaudhry and A. K. Thakur, Soft Matter, 2018, 14, 2234–2244 RSC.
- R. M. Burch, M. Weitzberg, N. Blok, R. Muhlhauser, D. Martin, S. G. Farmer, J. M. Bator, J. R. Connor, C. Ko, W. Kuhn, B. A. Mcmillan, M. Raynor, B. G. Shearer, C. Tiffany and D. E. Wilkins, Proc. Natl. Acad. Sci. U. S. A., 1991, 88, 355–359 CrossRef CAS PubMed.
- N. Chauhan and Y. Singh, ACS Biomater. Sci. Eng., 2020, 6, 5507–5518 CrossRef CAS PubMed.
- H. Singh, A. Gahane, V. Singh, S. Ghosh and A. Thakur, J. Antibiot., 2021, 74, 407–416 CrossRef CAS PubMed.
- H. P. R. Mangunuru, H. Yang and G. J. Wang, Chem. Commun., 2013, 49, 4489–4491 RSC.
- S. M. Hashemnejad, M. M. Huda, N. Rai and S. Kundu, ACS Omega, 2017, 2, 1864–1874 CrossRef CAS PubMed.
- N. Narang and T. Sato, Polym. J., 2021, 53, 1413–1424 CrossRef CAS.
- V. Castelletto, I. W. Hamley, J. Perez, L. Abezgauz and D. Danino, Chem. Commun., 2010, 46, 9185–9187 RSC.
- K. T. Nam, S. A. Shelby, P. H. Choi, A. B. Marciel, R. Chen, L. Tan, T. K. Chu, R. A. Mesch, B. C. Lee, M. D. Connolly, C. Kisielowski and R. N. Zuckermann, Nat. Mater., 2010, 9, 454–460 CrossRef CAS PubMed.
- S. Marchesan, L. Waddington, C. D. Easton, D. A. Winkler, L. Goodall, J. Forsythe and P. G. Hartley, Nanoscale, 2012, 4, 6752–6760 RSC.
- J. P. W. Jan, M. Schümann, A. K. Eckhardt, H. Quanz and P. R. Schreiner, J. Am. Chem. Soc., 2021, 143, 41–45 CrossRef PubMed.
- S. Funkner, M. Havenith and G. Schwaab, J. Phys. Chem. B, 2012, 116, 13374–13380 CrossRef CAS PubMed.
- K. Ono, K. Hasegawa, H. Naiki and M. Yamada, BBA, Mol. Basis Dis., 2004, 1690, 193–202 CrossRef CAS PubMed.
- L. Monticelli, S. K. Kandasamy, X. Periole, R. G. Larson, D. P. Tieleman and S. J. Marrink, J. Chem. Theory Comput., 2008, 4, 819–834 CrossRef CAS PubMed.
- A. van Teijlingen, H. W. A. Swanson, K. H. A. Lau and T. Tuttle, J. Phys. Chem. Lett., 2022, 13, 4046–4051 CrossRef CAS PubMed.
- E. Rossini, A. D. Bochevarov and E. W. Knapp, ACS Omega, 2018, 3, 1653–1662 CrossRef CAS PubMed.
- G. Campos-Villalobos, F. R. Siperstein and A. Patti, Mol. Syst. Des. Eng., 2019, 4, 186–198 RSC.
- S. J. Marrink, H. J. Risselada, S. Yefimov, D. P. Tieleman and A. H. de Vries, J. Phys. Chem. B, 2007, 111, 7812–7824 CrossRef CAS PubMed.
- P. W. J. M. Frederix, R. V. Ulijn, N. T. Hunt and T. Tuttle, J. Phys. Chem. Lett., 2011, 2, 2380–2384 CrossRef CAS PubMed.
- P. W. J. M. Frederix, G. G. Scott, Y. M. Abul-Haija, D. Kalafatovic, C. G. Pappas, N. Javid, N. T. Hunt, R. V. Ulijn and T. Tuttle, Nat. Chem., 2015, 7, 30–37 CrossRef CAS PubMed.
- A. van Teijlingen and T. Tuttle, J. Chem. Theory Comput., 2021, 17, 3221–3232 CrossRef CAS PubMed.
- H. X. Xu, A. K. Das, M. Horie, M. S. Shaik, A. M. Smith, Y. Luo, X. F. Lu, R. Collins, S. Y. Liem, A. M. Song, P. L. A. Popelier, M. L. Turner, P. Xiao, I. A. Kinloch and R. V. Ulijn, Nanoscale, 2010, 2, 960–966 RSC.
- X. J. Mu, K. M. Eckes, M. M. Nguyen, L. J. Suggs and P. Y. Ren, Biomacromolecules, 2012, 13, 3562–3571 CrossRef CAS PubMed.
- L. Chen, G. Pont, K. Morris, G. Lotze, A. Squires, L. C. Serpell and D. J. Adams, Chem. Commun., 2011, 47, 12071–12073 RSC.
- S. J. Marrink, L. Monticelli, M. N. Melo, R. Alessandri, D. P. Tieleman and P. C. T. Souza, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2022, e1620 Search PubMed.
- H. Bekker, H. J. C. Berendsen, E. J. Dijkstra, S. Achterop, R. Vondrumen, D. Vanderspoel, A. Sijbers, H. Keegstra, B. Reitsma and M. K. R. Renardus, Phys. Comput., 1993, 92, 252–256 Search PubMed.
- W. Humphrey, A. Dalke and K. Schulten, J. Mol. Graphics Modell., 1996, 14, 33–38 CrossRef CAS PubMed.
- N. Michaud-Agrawal, E. J. Denning, T. B. Woolf and O. Beckstein, J. Comput. Chem., 2011, 32, 2319–2327 CrossRef CAS PubMed.
- D. H. de Jong, S. Baoukina, H. I. Ingolfsson and S. J. Marrink, Comput. Phys. Commun., 2016, 199, 1–7 CrossRef CAS.
- B. Hess, H. Bekker, H. J. C. Berendsen and J. G. E. M. Fraaije, J. Comput. Chem., 1997, 18, 1463–1472 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2cp05938j |
| ‡ These authors contributed equally to this work. |
| This journal is © the Owner Societies 2023 |