
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Optical frequency comb-based measurements and the revisited assignment of high-resolution spectra of CH2Br2 in the 2960 to 3120 cm−1 region†
Ibrahim
Sadiek
 *ab,
Adrian
Hjältén
a,
Frances C.
Roberts
c,
Julia H.
Lehman
d and
Aleksandra
Foltynowicz
a
*ab,
Adrian
Hjältén
a,
Frances C.
Roberts
c,
Julia H.
Lehman
d and
Aleksandra
Foltynowicz
a
aDepartment of Physics, Umeå University, Umeå, Sweden. E-mail: aleksandra.foltynowicz@umu.se
bLeibniz Institute for Plasma Science and Technology, Greifswald, Germany. E-mail: ibrahim.sadiek@inp-greifswald.de
cSchool of Chemistry, University of Leeds, UK
dSchool of Chemistry, University of Birmingham, UK. E-mail: j.lehman@bham.ac.uk
First published on 14th February 2023
Abstract
Brominated organic compounds are toxic ocean-derived trace gases that affect the oxidation capacity of the atmosphere and contribute to its bromine burden. Quantitative spectroscopic detection of these gases is limited by the lack of accurate absorption cross-section data as well as rigorous spectroscopic models. This work presents measurements of high-resolution spectra of dibromomethane, CH2Br2, from 2960 cm−1 to 3120 cm−1 by two optical frequency comb-based methods, Fourier transform spectroscopy and a spatially dispersive method based on a virtually imaged phased array. The integrated absorption cross-sections measured using the two spectrometers are in excellent agreement with each other within 4%. A revisited rovibrational assignment of the measured spectra is introduced, in which the progressions of features are attributed to hot bands rather than different isotopologues as was previously done. Overall, twelve vibrational transitions, four for each of the three isotopologues CH281Br2, CH279Br81Br, and CH279Br2, are assigned. These four vibrational transitions are attributed to the fundamental ν6 band and the nearby nν4 + ν6 − nν4 hot bands (with n = 1–3) due to the population of the low-lying ν4 mode of the Br–C–Br bending vibration at room temperature. The new simulations show very good agreement in intensities with the experiment as predicted by the Boltzmann distribution factor. The spectra of the fundamental and the hot bands show progressions of strong QKa(J) rovibrational sub-clusters. The band heads of these sub-clusters are assigned and fitted to the measured spectra, providing accurate band origins and the rotational constants for the twelve states with an average error of 0.0084 cm−1. A detailed fit of the ν6 band of the CH279Br81Br isotopologue is commenced after assigning 1808 partially resolved rovibrational lines, with the band origin, rotational, and centrifugal constants as fit parameters, resulting in an average error of 0.0011 cm−1.
1 Introduction
Dibromomethane, CH2Br2, is one of the photochemically reactive halogenated volatile organic compounds (HVOCs) which are mainly emitted from the ocean.1–5 Once emitted, HVOCs affect the oxidation capacity of the atmosphere and exhibit high ozone destruction potential. Dibromomethane and bromoform, CHBr3, are classified by the World Metrology Organization as very short-lived substances, i.e., species that have a local lifetime comparable to or shorter than the characteristic time of mixing processes in the troposphere and hence have a non-uniform tropospheric distribution.5 Despite their short local atmospheric lifetimes, 123 days and 24 days for CH2Br2 and CHBr3, respectively, they contribute to the stratospheric bromine budget due to the fast vertical transportation by tropical deep convection.5 A potential anthropogenic source of brominated VOCs is treated ballast water systems,6 where HVOCs are produced as disinfection by-products (DBPs) with much higher concentrations compared to their natural abundance.7–9 Related to this, in situ measurements are necessary to confirm the existing models of the spread of ballast water discharge at harbours,10 and to help to identify if the DBPs have an ecotoxicological effect on marine life as well as human health by monitoring the exposure limits at workplaces of harbours.So far, quantitative detection of HVOCs has been mainly based on gas chromatography coupled with a mass spectrometer or an electron capture detector (GC/MS-ECD). GC/MS-ECD techniques require considerable calibration efforts and provide only discrete measurements, thus hindering the continuous monitoring in the environment and at workplaces. Optical detection techniques have the potential to overcome these limitations. As a prerequisite to develop a sensitive spectroscopic detection scheme, high-resolution absorption spectra of these molecules have to be recorded, and accurate spectral models have to be provided.
There exist two high-resolution spectral measurements of CH2Br2 in the mid-infrared (mid-IR) region made using the continuous wave (CW) cavity ring-down spectroscopy (CRDS) technique. Brumfield et al.11,12 measured the strong CH2 wagging vibration, ν8, around 1197 cm−1 using a quantum cascade laser coupled to a supersonic expansion source, providing no information about the absorption cross-section. Another measurement was performed on the symmetric C–H vibration, ν1, around 3013 cm−1, and the asymmetric C–H vibration, ν6, around 3076 cm−1, by Sadiek and Friedrichs,13 using a mid-IR CW optical parametric oscillator coupled to an optical cavity at room temperature, with a reported absorption cross-section.
A major challenge to providing an accurate spectroscopic model for brominated molecules is the complexity of their spectra caused by the nearly equal isotopic abundance of the 79Br and 81Br atoms. Another challenge is the existence of energetically low-lying vibrational states sharing a considerable fraction of the population at room temperature; hence, several hot bands may exist and cross-interfere with nearby fundamental bands. Nevertheless, the assignment of the ν8 band was straightforward,11,12 where three progressions of rovibrational features, measured in the supersonic jet expansion, could be assigned to the three isotopologues of dibromomethane: CH279Br79Br, CH279Br81Br, and CH281Br81Br. Following the same approach, Sadiek and Friedrichs13 provided an initial simulation and assignment of the ν6 fundamental band. However, a mismatch between the measured intensities and the predicted normal abundances of the three isotopologues was observed and attributed to the strong anharmonicities in vibrations, which are common in such halogenated compounds. Apart from the seemingly justified intensity mismatch between the simulations and the measurement of the ν6 band, the estimated large isotopic shifts of the three isotopologues did not agree with the theoretical predictions of isotopic shifts for C–H stretching vibrations, which involve a nearly negligible displacement of bromine atoms. Given the larger than predicted isotopic shift reported previously13 and our very recent work on iodomethane, CH3I,14 and diiodomethane, CH2I2,15 where similar progressions of rovibrational structures were observed for monoisotopic iodine, we believe that a revisited assignment should be provided for the spectrum of CH2Br2 in the C–H stretching region.
In this work, we present two independent measurements of CH2Br2 in the 2960–3120 cm−1 range using two direct frequency comb-based spectroscopic techniques, and we introduce a revisited analysis of the measured spectra in the asymmetric C–H stretching region at around 3076 cm−1. The spectra were recorded independently using (i) Fourier transform spectroscopy (FTS), and (ii) a spatially dispersive method using a virtually imaged phased array (VIPA), in a wider range than previously available from CW-CRDS.13 Compared to CW-CRDS, direct comb spectroscopy provides a wide bandwidth in a much shorter acquisition time, which makes the measurement less susceptible to long timescale fluctuations.
In the revisited spectral analysis, we attribute the sharp rovibrational QKa(J) transitions to the nearby nν4 + ν6 − nν4 hot bands, where the integer n = 1–3 is the initial vibrational level, as a result of the population of the low-lying Br–C–Br bending vibrational state, ν4, located at 171.1 ± 0.2 cm−1.16 The new spectral analysis is supported by ab initio quantum chemical calculations at different model chemistries.
2 Experimental and computational details
2.1 Experimental methods
Two separate comb-based setups were used to record the high-resolution spectra of CH2Br2 in the fundamental C–H stretching region in the range from 2960 cm−1 to 3120 cm−1. The FTS measurements were performed at Umeå University, Sweden, and the VIPA measurements were conducted at the University of Leeds, United Kingdom. Schematics of the two setups are shown in Fig. 1(a and b). FTS has previously been used to measure the line positions and intensities of the v4 band of CH3I,14 and the resulting line list was included in the newest edition of the HITRAN database,17 while the VIPA spectrometer was used to measure the absorption cross-section of CH2I2.15 | ||
| Fig. 1 Schematic depictions of the experimental setups: (a) the Fourier transform spectrometer and (b) the dispersive VIPA-based spectrometer. | ||
The mid-IR beam was passed through a Herriott multipass absorption cell (Aerodyne AMAC-76LW) with a path length of 7600 cm. The analytical grade CH2Br2 sample (Acros organics – 99%) was introduced in an Ace glass tube connected to the gas supply system and was brought into the Herriott cell by its vapour pressure. All measurements were conducted at room temperature. Prior to introducing CH2Br2 into the cell, the headspace was evacuated through a bypass valve. Afterwards, the cell was filled using a needle valve to the desired pressure, measured using a pressure transducer (CERAVAC CTR 101 N) with a resolution of 10 μbar.
After the absorption cell, the beam was coupled to a home-built fast-scanning FTS with an auto-balancing HgCdTe detector and a nominal resolution matched to frep. A beam of a narrow-linewidth CW diode laser with a wavelength of 1563 nm propagating on a path parallel to the comb beam in the FTS was used for calibrating the optical path difference. The sub-nominal resolution data analysis approach was used,19,20 where the comb modes were stepped across the absorption spectrum by tuning the comb frep and the spectra acquired at different steps were then interleaved. Spectra were recorded at 13 frep steps separated by 15 Hz, corresponding to an 11 MHz step in the optical domain. At each step, 200 interferograms were recorded, resulting in a total acquisition time of 2 h for all steps. Afterwards, the spectra recorded at the different frep steps were normalized to a background spectrum recorded using the absorption cell evacuated at the start of the measurement.
To correct the baseline fluctuations resulting from drifts in the emission envelope of the comb, we used the spectrum recorded at the first frep step, which was the closest in time to the background measurement, as a model of the overall contour of the CH2Br2 bands. We fit this spectrum to the normalized spectra at other frep steps together with a baseline consisting of a 5th order polynomial and slowly varying sine terms. The fitted baselines were then subtracted before interleaving the spectra.
To obtain the correct frequency scale, the value of the CW reference laser wavelength, λref, used in the analysis is usually optimized to minimize the instrumental line shape (ILS) distortions of individual lines.20 Such optimization could not be performed for the CH2Br2 spectrum due to the lack of fully resolved absorption lines. Instead, the weak absorption lines of H2O, originating most likely from the air leakage into the vial, that were separated from the CH2Br2 bands, were used for optimization. These lines were too broad to be affected by the ILS, so instead we fit Voigt profiles to them and varied λref to match their positions to those listed in the HITRAN database.17 We took the final λref value as the mean of the optima found for five different H2O lines. From the spread in the optima (max–min) and from the shifts of the lines for a given change in λref, we estimate that the optimization process contributes a frequency scale uncertainty of 1.6 MHz. Summing this value in quadrature with the maximum HITRAN line center uncertainty of the targeted H2O lines (∼3 MHz) yields a final uncertainty of 3.4 MHz of the FTS frequency scale.
The mid-IR frequency comb laser beam was passed through a Herriott multipass cell containing a known amount of vapour produced from the liquid sample of CH2Br2 (Sigma Aldrich, 99%). The Herriott cell path length of 1130 ± 30 cm was calibrated using a known concentration of CH4 (BOC, certified mixture of 4.85% CH4 in N2). The liquid sample of CH2Br2 was placed in a vacuum-tight sample container, and a flow of buffer gas was used to remove air impurities before the vapour pressure of CH2Br2 was allowed to equilibrate. A throttled vacuum pump in combination with a mass flow controller (Tylan FC-280) operating at a flow rate of 2.7 ± 0.3 standard cm3 min−1 was used to control the pressure of CH2Br2 in the multipass cell. The spectra were acquired with a pure sample of CH2Br2 at 0.70 ± 0.03 mbar, where the pressure and its uncertainty are the average and standard deviation of the pressure in the flow cell before, during, and after the measurements (MKS 722B Baratron capacitance manometer).
After passing through the Herriott cell, the laser beam was fibre coupled to the cross-dispersive VIPA spectrometer, where the beam was spatially dispersed into a two dimensional (2D) array using a VIPA etalon (Light Machinery, 12 GHz free spectral range),22 and a diffraction grating (Laser Components, 450 lines mm−1). This 2D array was imaged onto an InSb infrared camera (Infratec, ImageIR 8320, 512 × 640 pixels, 15 micron pitch), where the individual comb modes were clearly visible.
For the CH2Br2 spectrum spanning from 2922 cm−1 to 3123 cm−1, a total of 12 different diffraction grating positions (referred to as separate spectral windows) at a frep of 250 MHz were used. For each acquisition, where the sample cell contained either CH2Br2 (signal) or pure N2 (background), 2000 individual images were averaged together to give one image for the analysis, with camera parameters of a 50 μs integration time and a 125 Hz imaging rate. The images were analysed in pairs, where the signal and its corresponding background image were analysed using a home-written MATLAB code that records the intensity of each comb mode, orders the modes in ascending frequency wavenumber, and then utilises the Beer–Lambert law to determine the absorption spectrum. Each separate spectral window (image) was measured four times and averaged together, yielding an acquisition time of 2 minutes per spectral window. Finally, the adjacent windows were stitched together using overlapping data points, yielding the full spectrum.
For a given spectral window, the starting comb mode number was determined by acquiring a spectrum of CH4 and comparing it to the HITRAN database.17 The accuracy of the frequency axis in the HITRAN CH4 reference spectrum is much better than the spacing between the comb teeth (given by the repetition rate of the laser). Thus, assuming the repetition rate of the comb is well known since it is referenced to a Rb frequency standard, the starting mode number, and therefore the remaining ascending integer mode numbers in the image, can be determined without any uncertainty. However, it should be noted that the spectra are affected by instrumental broadening caused by the fact that the comb modes are not fully resolved by the VIPA. This results in a Lorentzian ILS function with a full-width-at-half-maximum (FWHM) of 0.0142 cm−1.15,21
2.2 Computational methods
The optimized molecular structure and the anharmonic frequency calculations of the three isotopologues, CH281Br81Br, CH279Br81Br, and CH279Br79Br, were performed using the Gaussian 09 package,23 with different model chemistries. The density functional theory (DFT), including the three-parameter hybrid density functional (B3LYP), the long-range corrected wB97X functional of Chai and Head-Gordon,24 and the M06 hybrid meta-exchange correlation functional of Truhlar and Zhao25 were used together with the post-Hartree Fock, second-order Möller–Plesset perturbation (MP2). The 6-311++G(d,p) split-valence basis set and the Karlsruhe basis set of valence quadruple-zeta with two sets of polarization functions (def2-QZVPP) were used. The anharmonic rotational constants of the vibrational excited states needed for the semi-empirical simulations of the twelve vibrational bands of the three isotopologues were obtained by applying the second order vibrational perturbative (VPT2) quantum chemistry assisted approach, as implemented in Gaussian 09 and demonstrated elsewhere.13 The spectral simulations and fitting were performed using the PGOPHER software from C. Western.26 The PGOPHER input file of the revisited simulations is given in the ESI† (S1).3 Results and discussion
3.1 High-resolution absorption cross-sections
Fig. 2 shows the high-resolution absorption cross-section of CH2Br2 measured in the region from 2960 cm−1 to 3120 cm−1 using the two independent setups: (i) the comb-based FTS setup (red) at 0.47 ± 0.01 mbar and (ii) the comb-based VIPA setup (black) at 0.70 ± 0.03 mbar. The previous high-resolution measurement using the CW-CRDS (blue), covering the region from 2955 cm−1 to 3086 cm−1,13 and a low resolution Fourier transform infrared (FT-IR) spectrum (green) adopted from the PNNL database27 are included for comparison. The spectrum is dominated by the fundamental symmetric, ν1, and asymmetric, ν6, C–H stretching vibrational bands around 3013 cm−1 and 3076 cm−1, respectively. Panels (b–d) show three zoomed-in windows at the band centre and at the wings of the asymmetric C–H stretching vibrations, showing at least three progressions of the rovibrational structure in each, as indicated by the dotted vertical lines. Panel (d) shows no CRDS measurements as it was reported up to 3086 cm−1. | ||
| Fig. 2 (a) Absorption cross-section of CH2Br2 measured in this work using the FTS (red) and the VIPA (black) setups, together with the previously reported spectra based on the CRDS measurement (blue) and the low-resolution FT-IR spectrum from the PNNL database (green). Panels (b–d) show zoomed-in windows at different parts of the asymmetric C–H stretching band, with vertical lines indicating the progressions discussed in the text. There is no reported CRDS spectrum exceeding 3086 cm−1. | ||
It should be noted that the CRDS data showed a systematic offset at a frequency of ∼60 MHz (0.002 cm−1) with respect to the FTS spectrum, which could be partially attributed to the reported accuracy of ±0.0009 cm−1 or ±27 MHz of the wavemeter used in the CRDS experiment.13 This offset cannot be observed in the VIPA data as the spectrum was broadened by the ILS function.
The noise on the baseline of the two comb-based measurements is 6 × 10−23 cm2 mol−1 for the FTS data and 5 × 10−24 cm2 mol−1 for the VIPA data. Expressed in terms of the minimum absorption coefficient, αmin, it is 7 × 10−7 cm−1 for the FTS spectrum, and 9 × 10−8 cm−1 for the VIPA spectrum. This yields a noise equivalent absorption sensitivity, NEAS = αmin × t1/2, of 6 × 10−5 cm−1 Hz−1/2 for the FTS and 1 × 10−6 cm−1 Hz−1/2 for the VIPA, where the measurement time t was 2 hours for the entire interleaved FTS spectrum, and 2 min for one spectral VIPA window. To reflect the multiplexed advantage of comb-based measurements, we also report the figure of merit, FoM = NEAS × M−1/2, where M is the number of spectral elements in the spectrum, equal to 5 × 105, for the entire interleaved FTS spectrum and 4.1 × 103 for one spectral VIPA window. The FoM is 8.6 × 10−8 cm−1 Hz−1/2 per spectral element for the FTS and 1.5 × 10−8 cm−1 Hz−1/2 per spectral element for the VIPA. While it is not straightforward to directly compare the sensitivities of the two different measurement setups, the VIPA sensitivity (in terms of NEAS and FoM) is expected to be better by a factor of M1/2 than the FTS assuming equal laser power incident on the camera/detector, equal integration time, number of spectral elements and interaction length.28
In order to assess the uncertainty of the absorption cross-section measured using the FTS, we checked the linearity of the absorption spectra measured at different partial pressures of CH2Br2. Fig. 3 shows the integrated absorption coefficient, 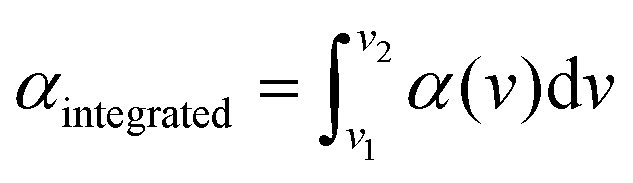 , evaluated between 2960 cm−1 and 3120 cm−1 as a function of the partial pressure of CH2Br2. Two sets of measurements (labelled A and B) were performed with pure CH2Br2 by increasing and decreasing the cell pressure and acquiring a spectrum at a 125 MHz sample point spacing. The baseline in these spectra was corrected with the same routine used for the interleaving process. The error bars on the partial pressure correspond to the resolution of the pressure sensor of 0.01 mbar, while the y-scale relative uncertainties were calculated from the inverse of the signal-to-noise ratio of the measurements, with the noise estimated on the baseline to the side of the band. The dotted line is a linear fit to the data weighted by the uncertainties. The standard deviation of the scatter of the residuals of this fit yields a 4% relative error in the absorption cross-section. This uncertainty stems mostly from uncertainties in the CH2Br2 concentration (e.g., leakage into the sample vials, adsorption onto cell walls and tubes, and sample impurities), and possible errors in the baseline correction.
, evaluated between 2960 cm−1 and 3120 cm−1 as a function of the partial pressure of CH2Br2. Two sets of measurements (labelled A and B) were performed with pure CH2Br2 by increasing and decreasing the cell pressure and acquiring a spectrum at a 125 MHz sample point spacing. The baseline in these spectra was corrected with the same routine used for the interleaving process. The error bars on the partial pressure correspond to the resolution of the pressure sensor of 0.01 mbar, while the y-scale relative uncertainties were calculated from the inverse of the signal-to-noise ratio of the measurements, with the noise estimated on the baseline to the side of the band. The dotted line is a linear fit to the data weighted by the uncertainties. The standard deviation of the scatter of the residuals of this fit yields a 4% relative error in the absorption cross-section. This uncertainty stems mostly from uncertainties in the CH2Br2 concentration (e.g., leakage into the sample vials, adsorption onto cell walls and tubes, and sample impurities), and possible errors in the baseline correction.
 | ||
| Fig. 3 The integrated absorption coefficient, αintegrated, in the range from 2960 cm−1 to 3120 cm−1 as a function of the CH2Br2 partial pressure measured using the FTS. The red and blue markers belong to two measurement sets A and B and the dotted black line is a linear slope fitted to the data. The blue circle and black star correspond to the FTS measurement at 0.47 mbar and VIPA measurement at 0.70 mbar, respectively, as shown in Fig. 2. | ||
The integrated absorption coefficient measured using the VIPA spectrometer is shown by the black star in Fig. 3, lining up nicely with the linear fit to the FTS data. The relative uncertainty in the measurement of the absorption cross-section using the VIPA method is a combination of the 5% error in the CH2Br2 concentration (from the pressure uncertainty) and 2.7% error in the Herriott cell path length. The combined relative uncertainty in the absorption cross-section measured using the VIPA spectrometer is thus 5.7%.
The integrated absorption cross-sections evaluated between 2956 cm−1 and 3086 cm−1, the common range for the four data sets shown in Fig. 2, is 50 ± 2 cm for the FTS, 52 ± 3 cm for the VIPA, 44 ± 1 cm for the CW-CRDS, and 50 ± 2 cm for the PNNL spectrum. The integrated absorption cross-sections from the two comb-based measurements and PNNL agree within 4%, which is within the combined uncertainty of 7.7% of the three methods. The CW-CRDS cross-section is within ∼10%, which is slightly higher than the 7.6% combined uncertainty of the two comb-based methods and the CRDS measurement (estimated to 3% from the reported signal-to-noise ratio and sample impurities13). This discrepancy in the absorption-cross section between the two comb-based methods and CRDS can be attributed to possible saturation issues in the CRDS measurement, particularly in the sharp rovibrational features of the ν6 band around 3076 cm−1 or background subtraction issues.
The absorption cross-sections measured using the two comb-based techniques are provided in the ESI† (S2).
3.2 Isotopic shift
Isotopic substitution is expected to affect all the vibrational motions of a molecule, resulting in a shift in their vibrational frequencies. The magnitude of the isotopic shift strongly depends on the involvement of the substituted isotope in the vibrational motion. Fig. 4 shows the molecular structure of CH2Br2, including the displacement vectors (blue) of the involved atoms, and the direction of the dipole moment change (yellow) of the Br–C–Br bending, ν4, the CH2 wagging, ν8, the symmetric C–H stretching, ν1, and the asymmetric C–H stretching, ν6, normal modes. Here, the principal inertial axes a, b, and c correspond to the rotational constants A, B, and C and the y-, z-, and x-axes.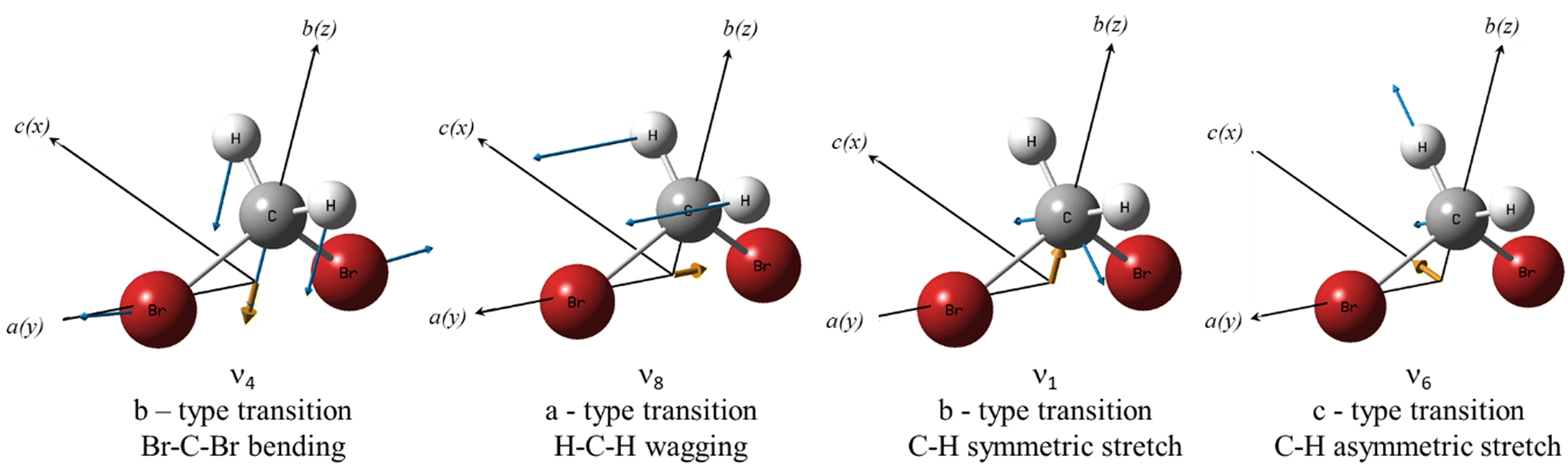 | ||
| Fig. 4 Molecular structure of CH2Br2. The blue and yellow arrows represent the displacement vectors and the direction of the transition dipole moment, respectively, for the ν4, ν8, ν1, and ν6 vibrational bands. | ||
It is anticipated that a larger isotopic shift would be observed for vibrations involving C–Br motion than vibrations involving C–H motion only. Therefore, the isotopic shift of the ν4 band, involving Br–C–Br bending, is predicted to be much larger than that of both the ν1 and ν6 vibrations. In addition, the ν8 mode is expected to involve a larger isotopic shift than the ν6 and ν1 vibrations as the CH2 wagging motion involves larger displacement within the Br–C–Br plane.
The ab initio calculations of the anharmonic frequencies of the ν4, ν8, ν1, and ν6 vibrational modes for CH281Br2 and CH279Br2 isotopologues at different levels of theory together with the available experimental data are presented in the ESI† (S3). The difference in calculated frequencies for light and heavy isotopologues, i.e., ν(CH279Br2)–ν(CH281Br2), represents the theoretically predicted frequency shift due to a decrease of two mass units. As shown in this table, the different model chemistries predict the same trend of isotopic shift for different vibrations, with almost negligible shift for the ν6 and ν1 vibrations. The M06 and the wB97X functionals seem to produce negative values for the isotopic shifts of the ν6 and ν1 vibrations; however, the predicted shift is less than the accuracy of these model chemistries.
By comparing the calculated values with the experiment, the isotopic shift of the ν8 band reported by Brumfield et al.11 agrees very well with the ab initio calculations, particularly at the B3LYP, M06, and MP2 model chemistries. However, the value of the shift of the ν6 band reported by Sadiek and Friedrichs13 is more than two orders of magnitude larger than the theory predictions. Therefore, a new analysis of the high-resolution spectrum of CH2Br2 in the asymmetric C–H stretching region should be provided using the predicted small isotopic shift, as well as the population of the low-lying ν4 vibration, as presented here. For CH2Br2, population in the ν4 vibration at 171.1 cm−1 results in a progression of nν4 + ν6 − nν4 hot bands, with n = 1–3. A similar effect was recently observed for CH3I14 and CH2I2,15 where the measured spectra showed progressions of rovibrational features near their fundamental bands. For CH3I, the fundamental C–H stretching vibration, ν4, around 3080 cm−1 contains a nearby progression which was attributed to the ν3 + ν4–ν3 hot band with an intensity equal to ∼7.6% of the fundamental ν4 band, as predicted by the Boltzmann distribution factor. Similarly, the CH2I2 spectrum shows congested progressions of rovibrational spectral features, which were explained by the population of the low lying ν4 vibrational state of the I–C–I bend motion, resulting in the progression of nν4 + ν6 − nν4 hot bands, where the integer n = 1–5 is the initial vibrational level. The fact that halogenated hydrocarbons, particularly CH2I2, CH2Br2, and CF2I2, have very low-lying vibrations resulting in a large density of vibrational states. For this reason, they have been used as model systems for studying the intramolecular quantum dynamics and rovibrational energy redistribution.29,30
3.3 Spectral simulations and analysis
The spectral analysis in our study is limited to the asymmetric C–H stretching region because (i) the sharp, more clearly resolved rovibrational features of the ν6 band hold the potential for being selected for future monitoring applications and enable spectral fitting procedures to be undertaken and (ii) the previously reported assignment turned out to be incorrect. Using the entire measured ν6 band, rather than the truncated band previously reported from CRDS measurements, the revisited assignment of these spectral features takes into account the almost 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 normal abundances of the two stable isotopes of bromine and the fact that the low-lying ν4 band at 171.1 cm−1 is populated at room temperature. At room temperature, assuming the Boltzmann distribution of the population, a series of nν4 + ν6 − nν4 hot bands, with n = 1 to 3, are predicted to be visible nearby the fundamental ν6 band, with population ratios of 0.43, 0.18, and 0.08 relative to the ν6 band, respectively.
:1 normal abundances of the two stable isotopes of bromine and the fact that the low-lying ν4 band at 171.1 cm−1 is populated at room temperature. At room temperature, assuming the Boltzmann distribution of the population, a series of nν4 + ν6 − nν4 hot bands, with n = 1 to 3, are predicted to be visible nearby the fundamental ν6 band, with population ratios of 0.43, 0.18, and 0.08 relative to the ν6 band, respectively.
In this section, a semi-empirical approach is adopted for simulating the spectra of the twelve vibrational states, relying on the ab initio calculations and the ground state parameters. The semi-empirical simulations are then used as a starting point for fitting the band heads of the sharp rovibrational features to the measured spectrum. Finally, a detailed line fit is introduced for the dominant ν6 band of the CH279Br81Br isotopologue.
For the fundamental ν6 band of CH2Br2, the M06/6-311++G(d,p) level of theory has shown to give effective rotational constants of the vibrational exited states that best reproduce the experimental spectrum.13 For the ν6 and ν1 fundamental bands, and nearby hot bands of CH2I2,15 the wB97X/def-2QZVPP level of theory was tested to provide very good semi-empirical excited vibrational state rotational constants. The wB97X/def-2QZVPP level of theory is particularly attractive with respect to the computation time and validity for halogenated molecules.32 Nevertheless, several model chemistries were used in the current work to estimate the isotopic shift and the Δ values of the anharmonic rotational constants upon vibrational excitation. The values of the calculated anharmonic rotational constants and the estimated Δ values of the ν4 and ν6 bands used in the simulations of the twelve vibrational states are given in the ESI† (S4). The evaluated semi-empirical rotational constants for the twelve vibrational states of the three isotopologues of CH2Br2 are also given in the ESI† (S5). Using these semi-empirical values of the rotational constants for the twelve vibrational states, i.e., four for each isotopologue, the major P/RQKa(J) clusters of rovibrational transitions could be reproduced, providing a very good initial match with the experiment for further fitting to the experimental spectrum.
Fig. 5(a) shows the spectrum recorded (black) in the range from 3040–3110 cm−1 using the VIPA setup with pure CH2Br2 vapour at 0.70 mbar together with the overall simulations (blue, inverted for clarity) of the three isotopologues. For each isotopologue, the simulation includes the fundamental ν6 band, and three hot bands, nν4 + ν6 − nν4 with n = 1–3. As shown in Fig. 5(b), four progressions of transitions could be clearly identified in this spectrum, as indicated by the vertical dotted lines around 3071 cm−1. The simulations also involve the Lorentzian contribution that accounts for the ILS function (FWHM 0.0142 cm−1) and the Gaussian contribution that accounts for the Doppler broadening (FWHM 0.0029 cm−1). Note that the experimental data used here are from low pressure CH2Br2 vapour, so the Lorentzian contribution is not impacted by pressure broadening. As shown in this figure, the spectra exhibit progressions of strong clusters of QKa(J) transitions typical for c-type bands. These clusters are separated by approximately 2(A − ![[B with combining macron]](https://www.rsc.org/images/entities/i_char_0042_0304.gif) ), where = (B + C)/2.
), where = (B + C)/2.
 | ||
| Fig. 5 (a) Absorption cross-section in the range dominated by the ν6 band measured using the VIPA spectrometer (black) together with the overall simulations (blue, inverted), involving twelve vibrational states. The different colours of the vertical dotted lines show different rovibrational progressions. (b) Zoomed-in window showing the experiment (black) and the simulation of the individual isotopologues, offset for clarity. The simulation of each of the three isotopologues contains four vibrational states indicated by the vertical dotted lines. (c) Zoomed-in window showing the experiment and the simulations of the vibrational bands for the CH279Br81Br isotopologue. | ||
| ν 0 | ν 6 | ν 4 | ν 4 + ν6 − ν4 | 2ν4 | 2ν4 + ν6 − 2ν4 | 3ν4 | 3ν4 + ν6 − 3ν4 | |
|---|---|---|---|---|---|---|---|---|
| a Ground state constants from microwave measurements of Niide et al.16 b D K was allowed to float in both the ground and excited states. | ||||||||
| Origin | 0 | 3076.7187(5) | 171.1008(3) | 3248.2287(3) | 341.6389(3) | 3419.1600(3) | 512.5325(4) | 3590.4058(8) |
| A | 0.8675192 | 0.866907(5) | 0.871208(2) | 0.870566(6) | 0.874895(3) | 0.874210(10) | 0.878501(17) | 0.878116(20) |
| B | 0.0408047 | 0.0408270(6) | 0.0407879(7) | 0.0408100(7) | 0.0407472(8) | 0.0407730(8) | 0.0407470(16) | 0.0407757(16) |
| C | 0.0392537 | 0.0392753(5) | 0.0392269(5) | 0.0392457(4) | 0.0392475(7) | 0.0392647(7) | 0.0391945(7) | 0.03921214(7) |
|
D
k × 105b |
1.271(4) | 1.277(4) | 1.240(3) | 1.239(5) | 1.240(4) | 1.224(4) | 1.240(4) | 1.225(5) |
| Num. of Ka assignments | 39 | 42 | 32 | 11 | ||||
| obs. − calc. | 0.0080 | 0.0085 | 0.0086 | 0.0086 | ||||
| ν 0 | ν 6 | ν 4 | ν 4 + ν6 − ν4 | 2ν4 | 2ν4 + ν6 − 2ν4 | 3ν4 | 3ν4 + ν6 − 3ν4 | |
|---|---|---|---|---|---|---|---|---|
| a Ground state constants from the microwave measurements of Niide et al.16 b D K was allowed to float in both the ground and excited states. | ||||||||
| Origin | 0 | 3076.7206(5) | 172.078(3) | 3249.204(3) | 343.859(13) | 3421.381(13) | 515.460(6) | 3593.360(6) |
| A | 0.8683441 | 0.867753(6) | 0.871870(5) | 0.871230(6) | 0.875430(10) | 0.874719(10) | 0.879395(17) | 0.878435(22) |
| B | 0.0413131 | 0.0413362(6) | 0.0413154(7) | 0.0413373(7) | 0.0412746(8) | 0.0413028(8) | 0.0412575(20) | 0.0413279(20) |
| C | 0.0397255 | 0.0397505(3) | 0.0397106(4) | 0.0397311(4) | 0.0397110(7) | 0.0397287(6) | 0.0396664(7) | 0.0396728(7) |
|
D
k × 105b |
1.285(4) | 1.293(5) | 1.260(2) | 1.255(2) | 1.271(3) | 1.255(4) | 1.248(8) | 1.258(9) |
| Num. of Ka assignments | 40 | 42 | 34 | 16 | ||||
| obs. − calc. | 0.0083 | 0.0086 | 0.0086 | 0.0083 | ||||
| ν 0 | ν 6 | ν 4 | ν 4 + ν6 − ν4 | 2ν4 | 2ν4 + ν6 − 2ν4 | 3ν4 | 3ν4 + ν6 − 3ν4 | |
|---|---|---|---|---|---|---|---|---|
| a Ground state constants from the microwave measurements of Niide et al.16 b D K was allowed to float in both the ground and excited states. | ||||||||
| Origin | 0 | 3076.7177(4) | 170.1369(2) | 3247.2639(2) | 340.0197(3) | 3417.5382(3) | 509.654(26) | 3587.543(18) |
| A | 0.8667564 | 0.866122(4) | 0.870371(6) | 0.869715(6) | 0.8742546(9) | 0.8735636(9) | 0.878921(23) | 0.878667(26) |
| B | 0.0402973 | 0.0403195(3) | 0.0399610(7) | 0.0399800(7) | 0.04026154(7) | 0.04028728(8) | 0.0400810(16) | 0.04011750(16) |
| C | 0.0387823 | 0.0388033(3) | 0.0384679(4) | 0.0384869(4) | 0.03873119(6) | 0.03874950(6) | 0.0387941(7) | 0.03880211(7) |
|
D
k × 105b |
1.241(5) | 1.242(5) | 1.233(2) | 1.223(2) | 1.209(3) | 1.189(3) | 1.227(2) | 1.220(4) |
| Num. of Ka assignments | 46 | 42 | 35 | 9 | ||||
| obs. − calc. | 0.00830 | 0.00830 | 0.00830 | 0.00830 | ||||
It should be highlighted here again that the observation of a third hot band, i.e., the fourth progression in the spectrum, as indicated by the vertical dotted lines in Fig. 5(b) around 3071 cm−1, although with a weak intensity, is another proof that the rovibrational progressions of the CH2Br2 spectrum are due to the population of the ν4 band and not due to the isotopic shift as previously reported,13 as there are three possible isotopologues of CH2Br2 and not four. A similar peak progression could be expected for the spectra of CH2F2 and CH2Cl2 at room temperature, just with the decreasing intensity for the hot bands due to an increased transition frequency for their ν4 bands: CH2F2 (ν4 = 528 cm−1),33 and CH2Cl2 (ν4 = 281.5 cm−1),34 compared to CH2Br2 (ν4 = 171.1 cm−1),16 and CH2I2 (ν4 = 121 cm−1).35
We note that band head fitting of the measured spectrum of the ν1 band was not possible as the symmetric stretching C–H vibration of the ν1 band results in a b-type transition with no resolved rovibrational structures that can be utilized in band head fitting as in the ν6 band. In addition, the 1:1 abundance of the Br isotopes and the predicted nν4 + ν1 − nν4 hot bands in the ν1 region are expected to add extra complexity to the assignment of the ν1 band. Therefore, the assignment of the ν1 band will be commenced in the future at low temperatures to simplify the assignment of the three isotopologues, and possibly study molecular perturbations that may exist and affect the rovibrational transitions.
 | ||
| Fig. 6 (a) Absorption cross-section measured using the FTS spectrometer (red) together with the overall simulations (blue, inverted), involving twelve vibrational states. (b and c) Zoomed-in windows around the PQ(Ka = 15) and RQ(Ka = 8) transitions at the wings of the band, showing partially resolved profiles that match with the experiment and the simulations. The different clusters of transitions correspond to different vibrational states. (d) The residuals of the least squares fit to the assigned transitions as a function of J and Ka quantum numbers. | ||
Table 4 presents the spectroscopic parameters of the ν6 band of the CH279Br81Br isotopologue obtained from the line fitting. The values of the band parameters obtained from the band head fitting approach together with the initially reported values are also included for comparison. It can be seen in this table that the band origin in the previous assignment is off by 0.4 cm−1. The residuals of the least squares fit to assigned transitions as a function of J and Ka quantum numbers are shown in Fig. 6(d). The line positions of the assigned transitions were reproduced with an average error of 0.0011 cm−1 (33 MHz). As shown in this panel, the residuals are randomly scattered around zero for transitions up to J = 75 and Ka = 13, indicating the accuracy of the reported band parameters. It should be noted, however, that there are still several peaks that were not reproduced by the spectral simulations, which could be attributed to the other remaining vibrational bands. The assignments of individual lines of the ν6 band of the other isotopologues and all the hot bands were more challenging because the spectrum is dominated by the ν6 fundamental band of the CH279Br81Br isotopologue and complicated by the normal abundance of the three isotopologues. Therefore, the reported “band head” fitting parameters in Tables 1–3 are considered as the band parameters of the remaining vibrations. The very small difference in the band origin (4 × 10−4 cm−1, within the combined fit uncertainty) and rotational constants of the ν6 band between the band head fitting and the detailed line fitting approaches, as shown in Table 4, indicate the high accuracy of the reported band parameters of the remaining eleven bands based on the band head fitting approach.
| ν 0 | ν 6 | |||
|---|---|---|---|---|
| Frequency comb spectroscopy | CRDS13 | |||
| Line fit | Band head fit | |||
| a Ground state constants based on the microwave measurements of Niide et al.16 b The value of the DK constant was fitted in the upper and lower states. | ||||
| Origin | 0 | 3076.7191(86) | 3076.7187(5) | 3077.12573(9) |
| A | 0.86751916 | 0.866887(1) | 0.866907(5) | 0.8664934(34) |
| B | 0.040804716 | 0.0408288(2) | 0.0408270(6) | 0.04082770(5) |
| C | 0.039253679 | 0.0392752(3) | 0.0392753(5) | 0.03927221(5) |
| D J × 109 | 7.75 | 7.79(5) | ||
| D JK × 107 | −3.81 | −3.81(4) | ||
|
D
K × 105b |
1.271(2) | 1.265(1) | 1.277(4) | |
| δ K × 1010 | −6.44 | −5.90(20) | ||
| δ J × 109 | −1.03 | −1.13(3) | ||
| Num. of assign. | 1808 | |||
| Error | 0.0011 | |||
4. Conclusions
We introduce frequency comb-based measurements of the absorption cross-section of dibromomethane, CH2Br2, in the range from 2960 cm−1 to 3120 cm−1 using two independent setups: Fourier transform spectroscopy and virtually imaged phased array spectroscopy. The reported cross-sections agree very well with each other and with the low-resolution FT-IR measurements of the PNNL database (within 4%) and with earlier measurements using CRDS (within 10%).We revisited the assignment of the high-resolution spectrum of CH2Br2 as the previous assignment showed more than two orders of magnitude larger isotope shift than that predicted by ab initio calculations. The new assignment takes into account the population of the low-lying ν4 mode, located at 171.1 cm−1, at room temperature, and hence the formation of progressions of nν4 + ν6 − nν4 bands, with n = 1–3 nearby the fundamental ν6 band. The overall simulations involve twelve vibrational bands, four for each isotopologue of CH2Br2: the fundamental ν6, and three nν4 + ν6 − nν4 hot bands. Each of these bands showed the formation of QKa(J) clusters of rovibrational transitions. By assigning the heads of these clusters, the rotational constants of the twelve vibrational states could be obtained with an average error (obs. − calc.) of 0.0084 cm−1. After the band head fitting, several of the partially resolved absorption profiles could be assigned to the fundamental ν6 band of the major CH279Br81Br isotopologue. The assignment of 1808 transitions allowed us to extend the fitting and extract the higher order centrifugal distortion constants, with an average error of 0.0011 cm−1. This study provides accurate absorption cross-section data as well as an accurate spectroscopic model for the three isotopologues of CH2Br2 in the asymmetric C–H stretching region, which represent the needed information to commence the optical monitoring of CH2Br2 in workplaces at the harbours and in the environment. Future measurements of CH2Br2 at low temperatures will simplify its spectrum and enable line-by-line assignment to the ν1 spectral region.
Author contributions
Ibrahim Sadiek: conceptualization, project administration, and writing – original draft preparation; Adrian Hjältén: investigation and formal analysis; Frances Roberts: investigation and formal analysis; Julia. H. Lehman: conceptualization, supervision, and funding acquisition; Aleksandra Foltynowicz: conceptualization, supervision, and funding acquisition.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The work at the University of Leeds was supported by funding from the EPSRC (Grant No. EP/R01518X/1 and EP/R513258/1). The work at Umeå University was supported by funding from the Knut and Alice Wallenberg Foundation (KAW 2015.0159 and 2020.0303) and the Swedish Research Council (2016-03593 and 2020-00238). Computational support was enabled by resources provided by the Swedish National Infrastructure for Computing (SNIC), partially funded by the Swedish Research Council through grant agreement no. 2018-05973. Some of the computational work was also undertaken on ARC3, part of the High Performance Computing facilities at the University of Leeds, UK. I. Sadiek would like to thank the German Research Foundation for financing his current position at the INP (SA 4483/1-1). The authors would like to acknowledge the help of A. Downing, and the fruitful discussions with G. Friedrichs, K. K. Lehmann, and the late C. Western.References
- J. H. Butler, D. B. King, J. M. Lobert, S. A. Montzka, S. A. Yvon-Lewis, B. D. Hall, N. J. Warwick, D. J. Mondeel, M. Aydin and J. W. Elkins, Global Biogeochem. Cycles, 2007, 21, GB1023 Search PubMed.
- L. J. Carpenter, C. E. Jones, R. M. Dunk, K. E. Hornsby and J. Woeltjen, Atmos. Chem. Phys., 2009, 9, 1805–1816 CrossRef CAS.
- H. Hepach, B. Quack, F. Ziska, S. Fuhlbrügge, E. L. Atlas, K. Krüger, I. Peeken and D. W. R. Wallace, Atmos. Chem. Phys., 2014, 14, 1255–1275 CrossRef.
- B. Quack, E. Atlas, G. Petrick and D. W. R. Wallace, J. Geophys. Res.: Atmos., 2007, 112, D09312 Search PubMed.
- S. R. S. A. Montzka, S. O’Doherty, A. Engel, A. K. Kruger, W. T. Sturges, D. Blake, M. Dorf, P. Fraser, L. Froidevaux, et al., Scientific assessment of ozone depletion: 2010, World Meteorological Organization, Geneva Switzerland, 2011 Search PubMed.
- IMO: International Convention for the Control and Management of Ships' Ballast Water and Sediments, in BWM/CONF/36, pp. 1–38.
- A. D. Shah, Z.-Q. Liu, E. Salhi, T. Höfer, B. Werschkun and U. von Gunten, Environ. Sci.: Water Res. Technol., 2015, 1, 465–480 RSC.
- R. Xue, H. Shi, Y. Ma, J. Yang, B. Hua, E. C. Inniss, C. D. Adams and T. Eichholz, Chemosphere, 2017, 189, 349–356 CrossRef CAS PubMed.
- G. Ziegler, M. Gonsior, D. J. Fisher, P. Schmitt-Kopplin and M. N. Tamburri, Environ. Sci. Technol., 2019, 53, 8006–8016 CrossRef CAS PubMed.
- J. Maas, S. Tegtmeier, B. Quack, A. Biastoch, J. V. Durgadoo, S. Rühs, S. Gollasch and M. David, Ocean Sci., 2019, 15, 891–904 CrossRef CAS.
- B. E. Brumfield, J. T. Stewart and B. J. McCall, J. Mol. Spectrosc., 2011, 266, 57–62 CrossRef CAS.
- B. E. Brumfield, J. T. Stewart, S. L. W. Weaver, M. D. Escarra, S. S. Howard, C. F. Gmachl and B. J. McCall, Rev. Sci. Instrum., 2010, 81, 063102 CrossRef PubMed.
- I. Sadiek and G. Friedrichs, Spectrochim. Acta, Part A, 2017, 181, 180–191 CrossRef CAS PubMed.
- I. Sadiek, A. Hjältén, F. Senna Vieira, C. Lu, M. Stuhr and A. Foltynowicz, J. Quant. Spectrosc. Radiat. Transfer, 2020, 255, 107263 CrossRef CAS.
- F. C. Roberts and J. H. Lehman, J. Chem. Phys., 2022, 156, 114301 CrossRef CAS PubMed.
- Y. Niide, H. Tanaka and I. Ohkoshi, J. Mol. Spectrosc., 1990, 139, 11–29 CrossRef CAS.
- I. E. Gordon, L. S. Rothman, R. J. Hargreaves, R. Hashemi, E. V. Karlovets, F. M. Skinner, E. K. Conway, C. Hill, R. V. Kochanov, Y. Tan, P. Wcisło, A. A. Finenko, K. Nelson, P. F. Bernath, M. Birk, V. Boudon, A. Campargue, K. V. Chance, A. Coustenis, B. J. Drouin, J. M. Flaud, R. R. Gamache, J. T. Hodges, D. Jacquemart, E. J. Mlawer, A. V. Nikitin, V. I. Perevalov, M. Rotger, J. Tennyson, G. C. Toon, H. Tran, V. G. Tyuterev, E. M. Adkins, A. Baker, A. Barbe, E. Canè, A. G. Császár, A. Dudaryonok, O. Egorov, A. J. Fleisher, H. Fleurbaey, A. Foltynowicz, T. Furtenbacher, J. J. Harrison, J. M. Hartmann, V. M. Horneman, X. Huang, T. Karman, J. Karns, S. Kassi, I. Kleiner, V. Kofman, F. Kwabia-Tchana, N. N. Lavrentieva, T. J. Lee, D. A. Long, A. A. Lukashevskaya, O. M. Lyulin, V. Y. Makhnev, W. Matt, S. T. Massie, M. Melosso, S. N. Mikhailenko, D. Mondelain, H. S. P. Müller, O. V. Naumenko, A. Perrin, O. L. Polyansky, E. Raddaoui, P. L. Raston, Z. D. Reed, M. Rey, C. Richard, R. Tóbiás, I. Sadiek, D. W. Schwenke, E. Starikova, K. Sung, F. Tamassia, S. A. Tashkun, J. Vander Auwera, I. A. Vasilenko, A. A. Vigasin, G. L. Villanueva, B. Vispoel, G. Wagner, A. Yachmenev and S. N. Yurchenko, J. Quant. Spectrosc. Radiat. Transfer, 2022, 277, 107949 CrossRef CAS.
- G. Soboń, T. Martynkien, P. Mergo, L. Rutkowski and A. Foltynowicz, Opt. Lett., 2017, 42, 1748–1751 CrossRef PubMed.
- P. Maslowski, K. F. Lee, A. C. Johansson, A. Khodabakhsh, G. Kowzan, L. Rutkowski, A. A. Mills, C. Mohr, J. Jiang, M. E. Fermann and A. Foltynowicz, Phys. Rev. A, 2016, 93, 021802 CrossRef.
- L. Rutkowski, P. Masłowski, A. C. Johansson, A. Khodabakhsh and A. Foltynowicz, J. Quant. Spectrosc. Radiat. Transfer, 2018, 204, 63–73 CrossRef CAS.
- F. C. Roberts, H. J. Lewandowski, B. F. Hobson and J. H. Lehman, Mol. Phys., 2020, 118, e1733116 CrossRef.
- M. Shirasaki, Fujitsu Sci. Tech. J., 1999, 35, 113–125 Search PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 09, Revision A.02, Gaussian, Inc., Wallingford, CT, 2009 Search PubMed.
- J.-D. Chai and M. Head-Gordon, J. Chem. Phys., 2008, 128, 084106 CrossRef PubMed.
- Y. Zhao and D. G. Truhlar, Theo. Chem. Acc., 2008, 120, 215–241 CrossRef CAS.
- C. Western, University of Bristol Research Data Repository, 2018.
- S. W. Sharpe, T. J. Johnson, R. L. Sams, P. M. Chu, G. C. Rhoderick and P. A. Johnson, Appl. Spectrosc., 2004, 58, 1452–1461 CrossRef CAS PubMed.
- A. Foltynowicz, T. Ban, P. Masłowski, F. Adler and J. Ye, Phys. Rev. Lett., 2011, 107, 233002 CrossRef PubMed.
- L. R. Khundkar and A. H. Zewail, Annu. Rev. Phys. Chem., 1990, 41, 15–60 CrossRef CAS.
- P. Z. El-Khoury, S. K. Pal, A. S. Mereshchenko and A. N. Tarnovsky, Chem. Phys. Lett., 2010, 493, 61–66 CrossRef CAS.
- J. K. G. Watson, Vibrational spectra and structure: aspects of quartic and sextic centrifugal effects on rotational energy levels, Elsevier, Amsterdam, 1977 Search PubMed.
- X. Xu and D. G. Truhlar, J. Chem. Theo. Comput., 2011, 7, 2766–2779 CrossRef CAS PubMed.
- N. Tasinato, G. Regini, P. Stoppa, A. P. Charmet and A. Gambi, J. Chem. Phys., 2012, 136, 214302 CrossRef PubMed.
- Q.-H. Liua, X.-N. Li, L.-Y. Hao, C. Tan, Y.-G. Zhou, P. Chen and H. Zhu, Chin. J. Chem. Phys., 2006, 19, 15–19 CrossRef.
- T. J. Johnson, T. Masiello and S. W. Sharpe, Atmos. Chem. Phys., 2006, 6, 2581–2591 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Input PGOPHER file for the revisited simulations of CH2Br2 in the region of the asymmetric C–H stretching (see file “revisited-CH2Br2-linefit.pgo); measured absorption-cross section by the two comb-based setups; anharmonic vibrational frequencies of the CH281Br2 and CH279Br2 isotopologues calculated at different model chemistries together with the estimated isotopic shifts; anharmonic rotational constants of the three isotopologues of CH2Br2 in its vibrational ground ν(v = 0) and first excited state ν(v = 1) of ν4 and ν6; semi-empirical rotational constants used as an input for the simulations of the twelve vibrational states of CH2Br2 in the C–H stretching region. See DOI: https://doi.org/10.1039/d2cp05881b |
| This journal is © the Owner Societies 2023 |