
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceIn silico capture and activation of methane with light atom molecules†
Stefan
Mebs
 *a and
Jens
Beckmann
b
*a and
Jens
Beckmann
b
aInstitut für Experimentalphysik, Freie Universität Berlin, Arnimallee 14, 14195, Berlin, Germany. E-mail: stefan.mebs@fu-berlin.de
bInstitut für Anorganische Chemie und Kristallographie, Universität Bremen, Leobener Straße 7, 28359, Bremen, Germany
First published on 26th January 2023
Abstract
Methane (CH4) can be captured in silico with a light atom molecule containing only C, H, Si, O, and B atoms, respectively. A tripodal peri-substituted ligand system was employed, namely, [(5-Ph2B-xan-4-)3Si]H (1, xan = xanthene), which after hydride abstraction (1+) carries four Lewis acidic sites within the cationic cage structure. In a previous study, this system was shown to be able to capture noble gas atoms He–Kr (Mebs & Beckmann 2022). In the corresponding methane complex, 1+CH4, a polarized Si+⋯CH4 contact of 2.289 Å as well as series of (H3)CH⋯O/CPh hydrogen bonds enforce spatial CH4 fixation (the molecule obeys C3-symmetry) and slight activation. A trigonal-pyramidal Si–CHeq3–Hax local geometry is thereby approached with Hax–C–Heq angles decreased to 103.7°. All attempts to replace the Lews acidic –BPh2 fragments in 1 with basic –PR2 (R = Ph, tBu) fragments indeed increased intra-molecular hydrogen bonding between host molecule and CH4, and thus caused stronger activation of the latter, however ultimately resulted in the formation of energetically favorable quenched structures with short P–Si contacts, making CH4 binding hard to achieve. The electronic situation of two hypothetic methane complexes, 1+CH4 and [(5-tBu2P-xan-4-)3SiCH4]+ (2+CH4), was determined by a set of calculated real-space bonding indicators (RSBIs) including the Atoms-In-Molecules (AIM), non-covalent interactions index (NCI), and electron localizability indicator (ELI-D) methods, highlighting crucial differences in the level of activation. The proposed ligand systems serve as blueprints for a more general structural design with adjustable trigonal ligand systems in which central atom, spacer fragment, and functional peri-partner can be varied to facilitate different chemical tasks.
Introduction
Methane is the main component in natural gas, a key molecule in chemical synthesis and the second most important greenhouse gas, thus of global relevance for modern society. Moreover, it is a highly inert molecule, which resists trapping and its use as source for value added products requires energy consumptive conversion into syngas (CO + H2) at high temperatures.1 Of particular industrial relevance is the separation of CH4 from CO2 and N2. Besides cryogenic distillation, different adsorbent-based techniques are applied.2 In a large-scale computational study, the potential of liquid solvents or zeolite structures to capture methane was addressed, but with limited success.3 Experimental studies employed metal–organic frameworks (MOFs) to selectively bind CO2 over CH4 (enriching the latter)4 or adsorb CH4 under high pressure or recently even under ambient conditions.5,6 Methane indeed is trapped in enormous quantities as methane hydrate (also methane clathrate, methane ice, fire ice,…) within water crystal structures in the shallow maritime geosphere and permafrost, and as such may have a massive effect on the world climate when the average temperature rises further.7 To date, however, light-atom organic molecular guest–host complexes with CH4 are yet unknown, to the best of our knowledge, and no X-ray diffraction study of such a complex was published.8,9 Indirect proof of CH4-complex formation with noble transition metals such as Rh were given by different kinds of spectroscopy, such as NMR.10 A metal-free activation of methane by a borenium complex was recently published, but the procedure requires 60 bar of CH4 and 110 °C to give reasonable yields.11We describe here a computational study on the first two molecular transition-metal free methane complexes, one of which potentially could be formed under ambient conditions. In our conceptual DFT screening approach we previously found that a tripodal peri-system ligand design with a central silyl cation as central linker, and suitable combinations of spacer fragments, such as acenaphthene or xanthene, with Lewis acidic and/or basic peri-, such as –BPh2 or –PPh2 groups, which serve as secondary functional groups, allows to successfully address important chemical tasks such as N2 or CO2 activation and capture of noble gases.12–15 In the course of fixing and activation of methane we exploit the C3-symmetry of particular tailor-made ligand systems, which guarantees an ideal embedment of the tetrahedral guest molecule within the active site (or void) of the surrounding host molecule. The first ligand system, [(5-Ph2B-xan-4-)3Si]H (1, xan = xanthene), contains four Lewis acidic sites and itself is actually also capable to energy efficiently fix noble gases.13 The second ligand system, [(5-tBu2P-xan-4-)3Si]H (2), combines the Lewis acidic silyl cation in the center with three Lewis basic –PPh2 groups in the side-arms, which results in considerably stronger fixation and activation of CH4, actually facilitating its subsequent deprotonation, but unfortunately suffers from premature quenching. In our screening approach, we could exclude several other spacer molecules, such as naphthalene (dperi = 2.5 Å), acenaphtene (dperi = 2.7 Å), biphenylene (dperi = 3.9 Å), and dibenzofuran (dperi = 5.4 Å) to provide suitable hosts for methane. Xanthene apparently has a proper peri-distance (dperi = 4.9 Å), offers a hydrogen bond donor (the O-atoms), and is flexible to accommodate for structural changes associated with CH4-uptake. Moreover, xanthene is a common organic molecule, derivatives of which are widely used in chemical industry, e.g. for dyes. A potential synthesis route of compound 1 is given in Scheme S1 (ESI†).
DFT calculations
Structural optimizations were conducted for all compounds by density functional theory (DFT) at the B3PW91/6-31 + G*16,17 level of theory using Gaussian1618 at the curta super-computer system of the Freie Universität Berlin. London dispersion was modelled using Grimme's GD3BJ parameters.19 The COSMO solvation model was applied to mimic the dichloromethane environment.20 Normal mode (or frequency) analysis proved the relaxed geometries of all but one relevant states to be local minima on the potential energy hypersurface and provided ΔG values. A weak negative frequency of −21.6 cm−1, which is a torsion motion on one tert-butyl group, remained for 2+CH3 (see below), despite efforts to reoptimize the structure. Potential energy scans (PES) with fixed Si⋯C(H4) distances have been conducted for both CH4-adducts (1+CH4, 2+CH4) in order to understand the “entry behavior” of CH4 into the hosts voids and to quantify it energetically, however, due to complex potential energy profiles (e.g. rotation motion of CH4 against the host molecules), the optimizations mostly didn’t fully converge even after long calculation times, so they were stopped when energetic changes didn’t exceed 1 kJ mol−1 over several steps. The transition state (2+TS) relevant for the CH4 deprotonation step was calculated using the QST3-algorythm of Gaussian16. It shows a strong negative band of −714.4 cm−1, which corresponds to the migration of the abstracted proton from C- to P-atom, i.e. H3C⋯H⋯PR3. As a consequence of the complexity of the structural analysis, some energetic trends are discussed by means of ΔE (e.g. structural isomerism), others in ΔG (e.g. reaction steps). The electronic structure of the CH4-adducts, the transition state towards methane deprotonation, and of the reaction product (2+CH3) was extracted from the calculated electron and electron pair densities employing the Atoms-In-Molecules – AIM21–23 (topological analysis of the electron density with AIM200024), Electron-Localizability-Indicator – ELI-D25 (bonding and lone-pair basin analysis with DGRID-5-1;26 grid step size: 0.05 a.u.), and non-covalent interactions index – NCI27 (intra-molecular contact patches with NCIplot;28 grid step size: 0.07 a.u.) toolkits. Analyses of the reduced density gradient, s(r) = [1/2(3π2)1/3]|∇ρ|/ρ4/3, according to the NCI method is used to visualize non-covalent bonding aspects. An estimation of different non-covalent contact types according to steric/repulsive (λ2 > 0, red-colored), van der Waals-like (λ2 ≈ 0, green-colored), and attractive (λ2 < 0, blue-colored) is facilitated by mapping the ED times the sign of the second eigenvalue of the Hessian (sign(λ2)ρ) on the iso-surfaces of s(r). For ELI-D figures, additional grids of 0.15 a.u. step size were computed. Structures are displayed with GaussView, bond paths are displayed with AIM2000, NCI and ELI-D figures are displayed with MolIso.29Results and discussion
In order to capture and activate methane with a light-atom host molecule of the here proposed type a number of criteria have to be fulfilled, which we stepwise address (the molecular design should of course be rational, i.e. the ligand system should be principally accessible by synthetic chemistry): 1. the pre-active neutral state as well as the charged/activated state of the ligand system are likely prone to conformational isomerism and the void-forming conformer should be either the lowest in energy or at least energetically accessible; 2. Hydride abstraction and formation of the methane complex should be exergonic or at least only slightly endergonic to provide a stable compound, which is potentially accessible by structural determination techniques like X-ray diffraction; 3. While methane enters the host, the latter needs to change its conformation temporarily to give access to the void, which shouldn’t be too energy consumptive, i.e. a low CH4 entry-barrier is demanded.Compound 1: we investigate first the capability of ligand system [(5-Ph2B-xan-4-)3Si]H (1, xan = xanthene) to form a suitable activated host molecule after hydride abstraction (1+) and to give access to a stable CH4-complex (1+CH4), see Fig. 1, 2, Fig. S1 and S2 (ESI†) as well as Tables S1 and S2 (ESI†). Its criteria represent as follows:
 | ||
| Fig. 1 Structures of 1+ (a), 1+CH4 (b), 2+C3 (c), 2+CH4 (d), 2+TS (e), 2+CH3 (f). | ||
 | ||
| Fig. 2 Relative energies (ΔE, dark blue) of structural isomers (e.g. exo-, dead-, and endo-variants of the neutral starting state 1, or methane complex of 1+ with the CH4-fragment at different Si⋯CH4 distances, which serve as rough estimate for transition states for CH4 complex formation) and relative enthalpies (ΔG, light blue) of different electronic states. The energy of the endo-conformer of 1 was set to zero. ΔG of 1+CH4 is corrected for BSSE and 1-atm-to-1-M conversion. | ||
1. The neutral starting compound 1 potentially exists in three different conformational isomers, two of which are void-forming after hydride abstraction (“endo”- or “exo”-variant of 1; both providing 1+active, see below), one of which is not (“dead”-variant). The exo-conformation (1exo, Fig. S1a, ESI†) would principally be preferred as starting material, as the Si–H section is located at the outer-sphere of the ligand system and is thus easy accessible by hydride abstraction agents, however, this conformation is 103 kJ mol−1 higher in energy (ΔE) compared to its endo-counterpart (1endo, Fig. S1c, ESI†). Advantageously, the unfavored dead-conformation (1dead, Fig. S1b, ESI†) is also 35 kJ mol−1 higher in energy compared to “endo”, making the latter the likely main product in ligand synthesis. Accordingly, 1endo is labeled as 1 in the following. The charged compound, 1+, principally also exists in two conformational isomers, one of which is void-forming (1+active, Fig. S1d, ESI†), one of which is not (1+dead, Fig. S1e, ESI,† 17.1 kJ mol−1 lower in energy than 1+active), but since hydride abstraction from 1endo causes formation of 1+active, the three Si–Cxan bonds now exhibit partial double-bond character, and rotation along this bond would be accompanied with a significant rotational energy barrier, it can be excluded at this stage. Accordingly, 1+active is labeled as 1+ in the following.
2. The reaction enthalpy (ΔG) of the hydride abstraction step to form the active state 1+ from 1 is calculated to be slightly endergonic (ΔG(1→1+) = 31.5 kJ mol−1, Fig. 1a and 2), which is assumed to be feasible under common reaction conditions (e.g. slightly elaborated temperatures). The reaction enthalpy of the subsequent CH4-complex formation step to form 1+CH4 (Fig. 1b and 2) from 1+ is almost enthalpy-neutral (ΔG(1+→1+CH4) = 4.2 kJ mol−1). This value already includes correction for basis-set superposition error (BSSE = 4.0 kJ mol−1) and conversion from 1 atm to 1 M condition (7.9 kJ mol−1).
3. By keeping the Si⋯CCH4 distance fixed to 5, 7.5, 10, and 12.5 Å (restrained optimizations, Fig. S2, ESI†), the energy barrier (ΔE) for gaseous CH4 to enter 1+ was estimated to be only about 2 kJ mol−1 (at d(Si⋯CCH4) = 7.5 Å), so energetic costs induced by bending of the host molecule to form 1+CH4 are considered negligible. When CH4 further approaches the Si center, 57.2 kJ mol−1 are released, which in return means the exit barrier of CH4 in 1+CH4 is equally large. In other words, once formed, 1+CH4 should form a stable complex.
In summary, controllable rotational isomerism of the starting material, medium to small positive reaction enthalpies of hydride abstraction and CH4 complex formation, and a negligible CH4 entry barrier into the void of the host, but a considerable CH4 exit barrier, render formation of 1+CH4 feasible with moderate synthetic efforts. 1+CH4 would then be the first stable methane complex employing a light-atom molecule. In order to account for the level of theory (particular the lack of polarization functions on the H-atoms), single-point calculations were performed at the B3PW91/6-311 + G(2df,p) level using the structural coordinates optimized at the lower B3PW91/6-31 + G* level, see Table S1 (ESI†). Since the geometries were not optimized at the higher level, the results may be considered as semi-quantitative, but it should be noted that the overall trends found at the lower level are fully conserved, and that the two crucial steps of hydride abstraction and CH4-complex formation become more favorable at the higher level by 5–13 kJ mol−1, which gives further confidence into our results.
Compound 2: a considerably more complex and less controllable situation, however, is unfortunately obtained for the [(5-tBu2P-xan-4-)3Si]H (2) analogue, see Fig. 1, and Fig. S3–S6 as well as Tables S1 and S2 (ESI†). Its criteria represent as follows:
1. As in 1, three rotational isomers are relevant for the neutral starting state of 2, which show equal relative energies (2endo is 91 kJ mol−1 lower in energy than 2exo and 49 kJ mol−1 lower in energy than 2dead, Fig. S3a–c, ESI†). Accordingly, 2endo is labeled as 2 in the following. But in contrast to 1+, which forms only one relevant structural isomer, 2+ diverges in at least three different isomers, one obeying C3-symmetry (2+C3, void-forming, Fig. S3d, ESI†) with three Si⋯P distances of 4.608 Å, an asymmetric one being 6.1 kJ mol−1 lower in energy (2+C1, potentially void-forming, Fig. S3e, ESI†) with Si⋯P distances of 4.530, 4.614, and 4.727 Å, as well as a quenched state being 46.9 kJ mol−1 lower in energy (2+quench, not void-forming, Fig. S3f, ESI†) exhibiting a very short Si–P distance of 2.470 Å, likely preventing CH4 uptake. 2+dead (Fig. S3g, ESI†) was also calculated, and is even 75.5 kJ mol−1 lower in energy than 2+C3, but considered inaccessible, thus negligible.
2. The reaction enthalpy (ΔG) of the hydride abstraction step to form the active state 2+C3 from 2 is calculated to be almost enthalpy-neutral (ΔG(2→2+C3) = 7.0 kJ mol−1, Fig. 1c), even better values are of course expected if 2+C1 or 2+quench are considered as product (not shown). The reaction enthalpy of the subsequent CH4-complex formation step to form 2+CH4 (Fig. 1d) from 2+C3 is also almost enthalpy-neutral (ΔG(2+C3→2+CH4) = 7.2 kJ mol−1); taking 2+quench as reference, a much less favorable value is obtained (ΔG(2+q→2+CH4) = 35.5 kJ mol−1). These values again include correction for basis-set superposition error (BSSE = 5.6 kJ mol−1) and conversion from 1 atm to 1 M condition (7.9 kJ mol−1).
3. By keeping the Si⋯CCH4 distance fixed to 2.5, 3.0, 3.5, 4.0, 4.5, and 5.0 Å (restrained optimizations, Fig. S4, ESI†), the energy barrier (ΔE) for gaseous CH4 to enter 2+C3 was estimated to be about 90 kJ mol−1 (at d(Si⋯CCH4) = 4.5 Å, Fig. S4f, ESI†), so energetic costs induced by bending of the host molecule to form 2+CH4 are significant, as 2+ preferably would form a short Si–P bond (2+quench) and CH4 would remain at the outer side of the ligand system (2+CH4(exo), Fig. S4h, ESI†). Once 2+CH4 would be formed, however, it would experience a large exit barrier (72 kJ mol−1), and since CH4 is considerably activated in 2+CH4, it would preferably be deprotonated than leave, see below, as well as Fig. S6 (ESI†) for more details of the energy scheme.
In summary, unfavorable structural isomerism of the activated material, a significantly positive reaction enthalpy of CH4 complex formation starting from the quenched active state, as well as a considerable CH4 entry barrier into the void of the host, render formation of 2+CH4 feasible only with elaborated synthetic efforts, such as purging of the neutral ligand system 1 with pressurized methane via hydride abstraction, if at all. It remains to be seen if variation of the spacer fragment or the peri-partner can solve this issue, and DFT-work is ongoing in this direction.
In 1+CH4, the methane molecule is only slightly activated. To the contrast, it is significantly more strongly bound and notably more activated in 2+CH4 and requires only 17 kJ mol−1 to be deprotonated by the host molecule; the transfer of the proton to the non-bonding electron pair of an adjacent P atom thereby releases considerable 182 kJ mol−1. As the bound CH4 would need a higher amount of energy to leave the void as gaseous CH4 again (exit barrier), being trapped once by 2+ is likely an irreversible process for methane.
Real-space bonding indicator analysis: despite the fact that formation of 2+CH4 apparently is not a trivial task, we were interested in the bonding situation of both methane complexes, which were thus analyzed in detail computationally using a suitable set of real-space bonding indicators (RSBI), providing insight into strengths and nature of primary and secondary atom–atom interactions within these complex molecular systems (Fig. 3 and 4 and Table 1).
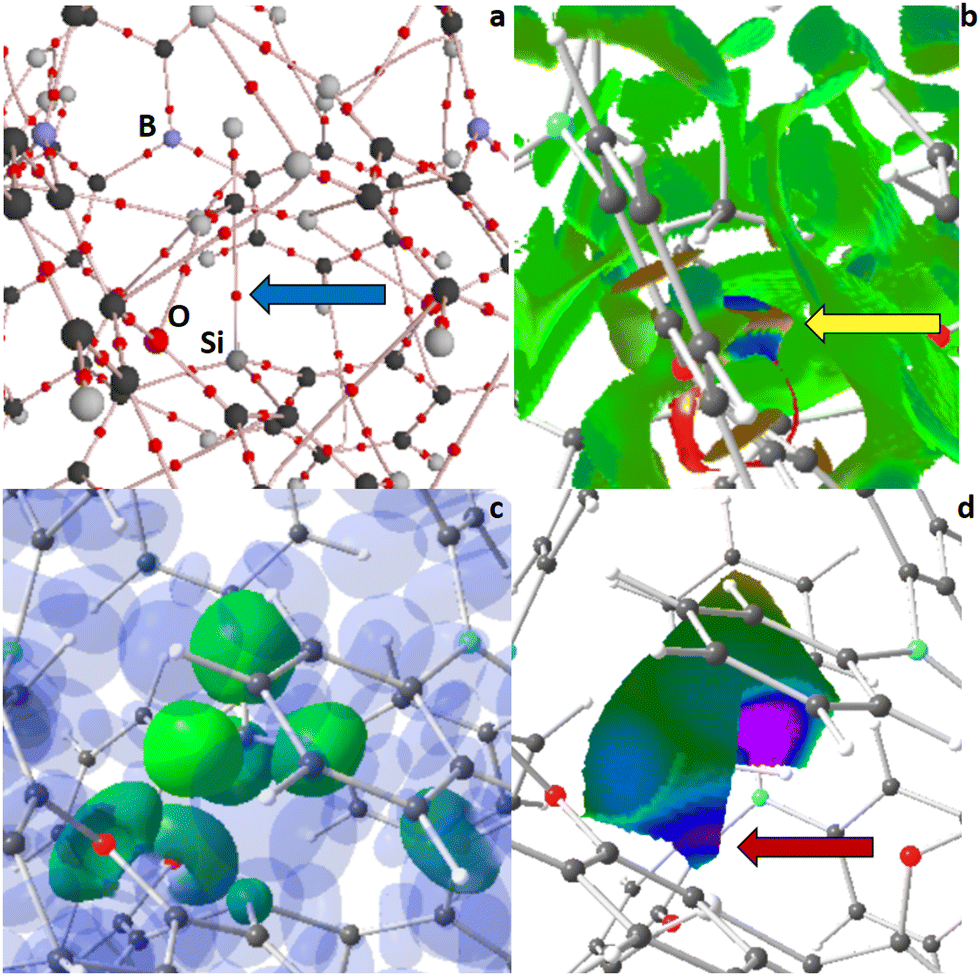 | ||
| Fig. 3 RSBI analysis of 1+CH4 (a) AIM bond paths motif, (b) NCI iso-surface at s(r) = 0.5, (c) ELI-D localization domain representation at an iso-value of 1.4, (d) ELI-D distribution mapped on two HCH4 ELI-D basins (equatorial and axial). Color code atoms: hydrogen – light gray, carbon – medium gray, oxygen – medium red, phosphor – dark or pale red, boron – light blue. | ||
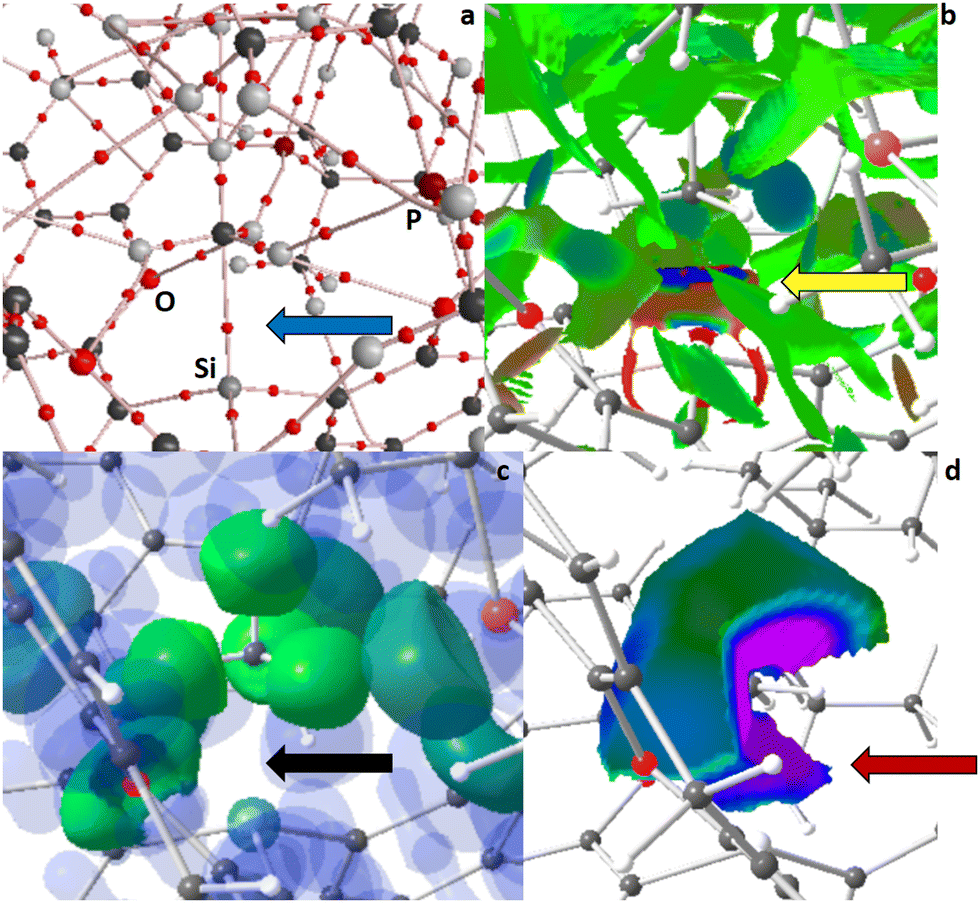 | ||
| Fig. 4 RSBI analysis of 2+CH4 (a) AIM bond paths motif, (b) NCI iso-surface at s(r) = 0.5, (c) ELI-D localization domain representation at an iso-value of 1.4, (d) ELI-D distribution mapped on two HCH4 ELI-D basins (equatorial and axial). Color code atoms: hydrogen – light gray, carbon – medium gray, oxygen – medium red, phosphor – dark or pale red. | ||
| Model | d C–H1 [Å] | d C–H2 [Å] | d C–H3 [Å] | d C–H4 [Å] | H1–C–H4 [°] | H2–C–H4 [°] | H3–C–H4 [°] |
|---|---|---|---|---|---|---|---|
| H1–3 are equatorial, H4 is axial; ρ(r)bcp: ED at the bcp, ∇2ρ(r)bcp: laplacian, ε: bond ellipticity d1: distance atom(1)-bcp, G/ρ(r)bcp, H/ρ(r)bcp: kinetic and total energy density over ρ(r)bcp ratios, QAIM: AIM fragmental and atomic charges, NELI, VELI: electron populations and volumes the ELI-D basin, γELI: ELI-D value at the attractor position, RJI: Raub–Jansen Index; QAIMC = −0.12 e for gaseous CH4. | |||||||
| 1+CH4 | 1.099 | 1.099 | 103.7 | ||||
| 2+CH4 | 1.116 | 1.096 | 97.8 | ||||
| 2+TS | 1.091 | 1.125 | 1.254 | 1.095 | 114.2 | 92.5 | 89.3 |
| 2+CH3 | 1.091 | 1.091 | 2.687 | 1.090 | 108.0 | 107.2 | |
| Model | d Si–C [Å] | d 1/d | ρ(r)bcp [eÅ−3] | ∇2ρ(r)bcp [eÅ−5] | ε | G/ρ(r)bcp [a.u.] | H/ρ(r)bcp [a.u.] |
|---|---|---|---|---|---|---|---|
| 1+CH4 | 2.289 | 0.78 | 0.27 | 0.9 | 0.00 | 0.56 | −0.34 |
| 2+CH4 | 2.081 | 0.74 | 0.41 | 2.5 | 0.00 | 0.86 | −0.43 |
| 2+TS | 2.010 | 0.73 | 0.55 | 3.2 | 0.15 | 0.93 | −0.52 |
| 2+CH3 | 1.878 | 0.72 | 0.80 | 6.1 | 0.01 | 1.13 | −0.60 |
| Model | Q AIMlig [e] | Q AIMCHn [e] | Q AIMC [e] | N ELI [e] | V ELI [Å3] | γ ELI | RJI [%] |
|---|---|---|---|---|---|---|---|
| 1+CH4 | 0.95 | 0.06 | −0.43 | ||||
| 2+CH4 | 1.02 | −0.02 | −0.72 | 0.38 | 1.0 | 1.36 | 72.0 |
| 2+TS | 1.11 | −0.11 | −0.87 | 1.07 | 2.9 | 1.64 | 81.4 |
| 2+CH3 | 1.69 | −0.67 | −0.80 | 2.00 | 5.9 | 2.02 | 85.2 |
A zoom-in of the AIM bond topology of 1+CH4 and 2+CH4 (see ESI† for full molecule representations) unravels the formation of a Si–CH4 bond critical point (bcp) in both cases (highlighted by a blue arrow in Fig. 3a and 4a), indicating a direct chemical interaction between the silyl cationic center and the CH4 molecule. The Si–CH4 bond distance is remarkable (2.289 Å) in 1+CH4 but astonishing (2.081 Å) in 2+CH4, which is accompanied by structural deformation of CH4 towards a Si–CH3–H trigonal bipyramid, particularly in 2+CH4. Both complexes thereby retained the C3-symmetry of their parent compounds. With electron density (ED, ρ(r)bcp) values of 0.27 or 0.41 e Å−3 the Si–CH4 contacts are substantially stronger than hydrogen bonds, and the fact that both the kinetic energy density over ED ratio (G/ρ(r)bcp) as well as the total energy density over ED ratio (H/ρ(r)bcp) are considerably positive or negative, respectively, highlights the relevance of both covalent and non-covalent bonding aspects to these interactions. This is corroborated by inspection of NCI contact patches and ELI-D basins. On the one hand, NCI exhibits a blue-colored disc- or ring-shaped Si–CH4 basin (highlighted by a yellow arrow in Fig. 3b and 4b) signifying attractive non-covalent bonding aspects. On the other hand, a Si–C ELI-D bonding basin is preformed in 1+CH4, but already topologically separated and fully formed in 2+CH4 (highlighted by a red arrow in Fig. 3d and 4d), pointing towards non-negligible covalent bonding aspects of these polarized-covalent bonds. However, the Si–C ELI-D basin is small (VELI = 1.0 Å3), little populated (NELI = 0.38 e), and not fully “localized” (γELI = 1.36), so it's not (yet) visible at a common iso-surface of γ = 1.4 (highlighted by a black arrow in Fig. 4c). The NCI further shows small blueish-colored disc-shaped CH⋯O/P basins as well as extended greenish-colored flat areas, and AIM shows numerous CH⋯O/P/C/H bcps, altogether representing a plethora of secondary intramolecular interactions, such as hydrogen bonds, H⋯H contacts, and van-der-Waals interactions, which stabilize the guest within the host. Similar bonding conditions were observed in noble-gas (Ng) complexes of 1, in which 1+He and 1+Ne showed only a Si–Ng NCI basin but no corresponding ELI-D bonding basin, indicating the dominance of non-covalent bonding aspects, whereas a Si–Ng ELI-D bonding basin was additionally formed in 1+Ar and 1+Kr, indicating rising covalent bond contributions.13 CH4-complex formation is also accompanied by strongly increased internal polarization within the CH4-fragment, as well as minor charge transfer between ligand system and CH4, which is reflected in AIM atomic and fragmental charges (Table 1).
The activation of methane in 2+CH4 is pronounced enough to make the transition state (2+TS) towards deprotonation only 16.7 kJ mol−1 higher in energy, as mentioned above. A nearby lone-pair of a P atom serves as acceptor for that proton. In the TS, the CH4-fragment is bent/flattened and one C–H bond is extended to 1.254 Å (Fig. 5 and Table 1). A new CH⋯P AIM bcp is formed (highlighted by a light blue arrow in Fig. 5a), the NCI exhibits a blue-colored ring-shaped CH⋯P basin (orange arrow in Fig. 5b), and the ELI-D shows strongly increased localizability on the H-basin (a so-called protonated valence basin) in direction of the P atom (purple arrow in Fig. 5d). In the process of deprotonation, the Si–C ELI-D basin becomes larger (VELI = 2.9 Å3), more populated (NELI = 1.07 e), and more localized (γELI = 1.64), making it visible at γ = 1.4. In accordance, the bond becomes shorter and the ED at the Si–CH4 bcp rises up to 0.55 e Å−1. Notably, the Si–C ELI-D basin and two equatorial H basins are topologically connected in 2+TS (black arrow in Fig. 5c), which they weren’t in 2+CH4. After the deprotonation is completed, the former lone-pair basin of the P atom (a non-bonding valence basin) has become a protonated valence-basin, which is topologically fully separated from the new R3Si+–CH3 part of the molecule (Fig. S7, ESI† and Table 1). The Si–C ELI-D basin becomes even larger (VELI = 5.9 Å3), more populated (NELI = 2.00 e), and more localized (γELI = 2.02), now resembling a “conventional” Si–C bonding basin, with even shorter bond distance and increased ED at the bcp (ρ(r)bcp = 0.80 e Å−1). The Raub–Jansen-Index (RJI) combines AIM and ELI-D and is an indicator of bond-polarity; the range spans from 50–60% for homo-polar bonds, from roughly 60–90% for polarized-covalent interactions, from 90–98% for dative and ionic bonds, to about 100% for purely van-der-Waals-like contacts. The Si–CHn (n = 4 or 3) lies in the typical range for polarized-covalent contacts, supporting the statements above, and the polarization is increasing in the series 2+CH4 → 2+TS → 2+CH3. The charge-transfer from ligand system to CHn-fragment and the internal polarization of the CHn-fragment itself are also increasing along that series (Table 1).
 | ||
| Fig. 5 RSBI analysis of 2+TS (a) AIM bond paths motif, (b) NCI iso-surface at s(r) = 0.5, (c) ELI-D localization domain representation at an iso-value of 1.4, (d) ELI-D distribution mapped on the newly formed Si–CCH4 ELI-D basin and the H basin, which is going to be transferred to the P atom. Color code atoms: hydrogen – light gray, carbon – medium gray, oxygen – medium red, phosphor – dark or pale red. | ||
Conclusion
Our computational study shows that it is possible to capture and potentially also activate methane exploiting the tripodal design and symmetry of a light-atom molecule such as [(5-Ph2B-xan-4-)3Si]H (1). In 1, which formally contains four Lewis acidic functional sites, the electron density of the B–CPh bonds serve as weak hydrogen bonding acceptor instead; attempts to further increase intramolecular hydrogen bond strengths by replacing the –BPh2 by –PtBu2 groups, however, makes synthesis of the corresponding methane complex hard to achieve due to premature quenching of the activated state. Work is ongoing to find a balanced molecular design, which captures and activates CH4 in order to make it accessible for subsequent chemical transformations.Conflicts of interest
The authors declare no conflicts of interest.Acknowledgements
The DFG is acknowledged for funding.References
- T. V. Choudhary and V. R. Choudhary, Angew. Chem., Int. Ed., 2008, 47, 1828–1847 CrossRef CAS PubMed.
- A. Alonso, J. Moral-Vico, A. A. Markeb, M. Busquets-Fite, D. Komilis, V. Puntes, A. Sanchez and X. Font, Sci. Total Environ., 2017, 595, 51–62 CrossRef CAS PubMed.
- J. Kim, A. Maiti, L. C. Lin, J. K. Stolaroff, B. Smit and R. D. Aines, Nat. Commun., 2013, 4 Search PubMed.
- Y. C. Lin, C. L. Kong, Q. J. Zhang and L. Chen, Adv. Energy Mater., 2017, 7 CAS.
- Z. Niu, X. L. Cui, T. Pham, P. C. Lan, H. B. Xing, K. A. Forrest, L. Wojtas, B. Space and S. Q. Ma, Angew. Chem., Int. Ed., 2019, 58, 10138–10141 CrossRef CAS PubMed.
- R. B. Lin, L. B. Li, A. Alsalme and B. L. Chen, Small Struct., 2020, 1(3), 2000022 CrossRef.
- K. A. Kvenvolden, Chem. Geol., 1988, 71, 41–51 CrossRef CAS.
- M. D. Walter, P. S. White, C. K. Schauer and M. Brookhart, New J. Chem., 2011, 35, 2884–2893 RSC.
- V. D. Makhaev, Usp. Khim., 2003, 72, 287–310 Search PubMed.
- W. H. Bernskoetter, C. K. Schauer, K. I. Goldberg and M. Brookhart, Science, 2009, 326, 553–556 CrossRef CAS PubMed.
- Y. Z. Liu, W. S. Dong, Z. H. Li and H. D. Wang, Chem, 2021, 7, 1843–1851 CAS.
- S. Mebs and J. Beckmann, Phys. Chem. Chem. Phys., 2022, 24, 20953–20967 RSC.
- S. Mebs and J. Beckmann, Phys. Chem. Chem. Phys., 2022, 24, 20968–20979 RSC.
- S. Mebs, Chem. Phys. Chem., 2022, e202200621 Search PubMed.
- S. Mebs and J. Beckmann, Chem. Phys. Chem. Search PubMed , submitted.
- A. D. Becke, J. Chem. Phys., 1993, 98, 1372–1377 CrossRef CAS.
- J. P. Perdew, J. A. Chevary, S. H. Vosko, K. A. Jackson, M. R. Pederson, D. J. Singh and C. Fiolhais, Phys. Rev. B: Condens. Matter Mater. Phys., 1992, 46, 6671–6687 CrossRef CAS PubMed.
- M. J. T. Frisch, G. W. Schlegel, H. B. Scuseria, G. E. Robb, M. A. Cheeseman, J. R. Scalmani, G. Barone, V. Petersson, G. A. Nakatsuji, H. Li, X. Caricato, M. Marenich, A. V. Bloino, J. Janesko, B. G. Gomperts, R. Mennucci, B. Hratchian, H. P. Ortiz, J. V. Izmaylov, A. F. Sonnenberg, J. L. Williams-Young, D. Ding, F. Lipparini, F. Egidi, F. Goings, J. Peng, B. Petrone, A. Henderson, T. Ranasinghe, D. Zakrzewski, V. G. Gao, J. Rega, N. Zheng, G. Liang, W. Hada, M. Ehara, M. Toyota, K. Fukuda, R. Hasegawa, J. Ishida, M. Nakajima, T. Honda, Y. Kitao, O. Nakai, H. Vreven, T. Throssell, K. Montgomery, J. A. Peralta Jr., J. E. Ogliaro, F. Bearpark, M. J. Heyd, J. J. Brothers, E. N. Kudin, K. N. Staroverov, V. N. Keith, T. A. Kobayashi, R. Normand, J. Raghavachari, K. Rendell, A. P. Burant, J. C. Iyengar, S. S. Tomasi, J. Cossi, M. Millam, J. M. Klene, M. Adamo, C. Cammi, R. Ochterski, J. W. Martin, R. L. Morokuma, K. Farkas, O. Foresman, J. B. Foresman and D. J. Fox, Gaussian 09, Revision C.01, Gaussian, Inc., Wallingford CT, 2016 Search PubMed.
- S. Grimme, J. Comput. Chem., 2006, 27, 1787–1799 CrossRef CAS PubMed.
- S. Miertus, E. Scrocco and J. Tomasi, Chem. Phys., 1981, 55, 117–129 CrossRef CAS.
- R. F. W. Bader, Bull. Am. Phys. Soc., 1977, 22, 305–306 Search PubMed.
- R. F. W. Bader and T. T. Nguyendang, Adv. Quantum Chem., 1981, 14, 63–124 CrossRef CAS.
- R. F. W. Bader, Acc. Chem. Res., 1985, 18, 9–15 CrossRef CAS.
- F. Biegler-Konig, J. Schonbohm and D. Bayles, J. Comput. Chem., 2001, 22, 545–559 CrossRef.
- M. Kohout, Int. J. Quantum Chem., 2004, 97, 651–658 CrossRef CAS.
- DGRID-5.1, ed. M. Kohout, Radebeul, 2015 Search PubMed.
- E. R. Johnson, S. Keinan, P. Mori-Sanchez, J. Contreras-Garcia, A. J. Cohen and W. T. Yang, J. Am. Chem. Soc., 2010, 132, 6498–6506 CrossRef CAS PubMed.
- J. Contreras-Garcia, E. R. Johnson, S. Keinan, R. Chaudret, J. P. Piquemal, D. N. Beratan and W. T. Yang, J. Chem. Theory Comput., 2011, 7, 625–632 CrossRef CAS PubMed.
- C. B. Hübschle and P. Luger, J. Appl. Crystallogr., 2006, 39, 901–904 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2cp05821a |
| This journal is © the Owner Societies 2023 |