
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA study of the thermodynamics and mechanisms of the atmospherically relevant reaction dimethyl sulphide (DMS) with atomic chlorine (Cl) in the absence and presence of water, using electronic structure methods†
Lydia
Rhyman
 *ab,
Edmond P. F.
Lee
c,
Ponnadurai
Ramasami
*ab and
John M.
Dyke
*c
*ab,
Edmond P. F.
Lee
c,
Ponnadurai
Ramasami
*ab and
John M.
Dyke
*c
aComputational Chemistry Group, Department of Chemistry, Faculty of Science, University of Mauritius, Réduit, 80837, Mauritius. E-mail: lyd.rhyman@gmail.com; ramchemi@intnet.mu
bCentre For Natural Product Research, Department of Chemical Sciences, University of Johannesburg, Doornfontein, Johannesburg, 2028, South Africa
cSchool of Chemistry, University of Southampton, Highfield, Southampton, SO17 1BJ, UK. E-mail: jmdyke@soton.ac.uk
First published on 18th January 2023
Abstract
The thermodynamics and mechanisms of the atmospherically relevant reaction dimethyl sulphide (DMS) + atomic chlorine (Cl) were investigated in the absence and presence of a single water molecule, using electronic structure methods. Stationary points on each reaction surface were located using density functional theory (DFT) with the M06-2X functional with aug-cc-pVDZ (aVDZ) and aug-cc-pVTZ (aVTZ) basis sets. Then fixed point calculations were carried out using the UM06-2X/aVTZ optimised stationary point geometries, with aug-cc-pVnZ basis sets (n = T and Q), using the coupled cluster method [CCSD(T)], as well as the domain-based local pair natural orbitals coupled cluster [DLPNO-UCCSD(T)] approach. Four reaction channels are possible, formation of (A) CH3SCH2 + HCl, (B) CH3S + CH3Cl, (C) CH3SCl + CH3, and (C′) CH3S(Cl)CH3. The results show that, in the absence of water, channels A and C′ are the dominant channels. In the presence of water, the calculations show that the reaction mechanisms for A and C formation change significantly. Channel A occurs via submerged TSs and is expected to be rapid. Channel B occurs via TSs which present significant energy barriers indicating that this channel is not significant in the presence of water relative to CH3SCH2 + HCl and DMS·Cl adduct formation, as is the case in the absence of water. Channel C was not considered as it is endothermic in the absence of water. In the presence of water, pathways which proceed via (a) DMS·H2O + Cl, (b) Cl·H2O + DMS and (c) DMS·Cl + H2O were considered. It was found that under tropospheric conditions, reactions via pathway (b) are of minor importance relative to those that proceed via pathways (a) and (c). This study has shown that water changes the mechanisms of the DMS + Cl reactions significantly but the presence of water is not expected to affect the overall reaction rate coefficient under atmospheric conditions as the DMS + Cl reaction has a rate coefficient at room temperature close to the collisional limit.
1. Introduction
The sulfur cycle in the earth's atmosphere has been the subject of intense investigation in recent years because of the need to assess the contribution of anthropogenically produced sulfur to acid rain, visibility reduction, aerosol production and climate modification.1–4 Roughly half of the global flux of sulfur in the atmosphere is thought to be from natural sources, with a significant fraction of all natural sulfur arising from dimethyl sulphide (DMS, CH3SCH3) emitted from the oceans.5–7 DMS is produced by biodegradation of oceanic phytoplankton in the upper layers of the oceans, initiated by ultraviolet radiation from the sun, as well as from anthropogenic activities. Subsequent oxidation of DMS (to SO2) leads to aerosol production and cloud formation. As a result, DMS plays an important role in climate regulation.1,2 In the atmosphere, the main oxidants of DMS are thought to the OH radical during the day and the NO3 radical at night. However, oxidation of DMS in the troposphere appears to be more rapid than would be expected solely from reaction with OH and NO3, and other DMS removal reactions involving atomic and molecular halogens and halogenated radicals, such as Cl, Cl2 and BrO, have therefore been proposed.8–12This paper investigates the energetics and mechanism of the DMS + Cl reaction as well as the effect of water, a major atmospheric constituent, on this reaction. The chlorine atom is a key atmospheric oxidant,13 which can be produced via photolysis of chlorine-containing species generated in sea salt aerosols,14 and surface reactions of gaseous hydrogen chloride and nitrogen oxides followed by photolysis of the ClNO and ClNO2 produced.15 Also, rate coefficients for reactions of volatile organic compounds (VOCs) with Cl atoms are generally 10–100 times larger than those of the corresponding OH-reactions.16 The estimated day-time global average Cl atom concentration in the marine boundary layer (MBL) is 103 atom cm−3 whereas that of OH radical is 106 molecule cm−3,14,17 although peak Cl atom concentrations have been measured in the region (104–106 atoms cm−3), giving [Cl]:[OH] ratios up to 103 higher than usual.14 All these considerations suggest that oxidation reactions of VOCs by Cl atoms can be important in the troposphere.18,19
The DMS + Cl reaction has been studied experimentally and theoretically, and based on these reports,20–30 four reaction channels have been proposed, as shown in Table 1. This table compares experimentally derived reaction enthalpies (ΔHϕf,298), obtained from available heats of formation of the reagents and products,20 with values obtained in this work from DLPNO-UCCSD(T) calculations. As can be seen good agreement is obtained between the experimentally derived and computed values for the four reaction channels.
There have been several laboratory kinetics studies on DMS + Cl at different temperatures and pressures.21–25 In the work of Stickel et al.,21 the rate coefficient was found to decrease with temperature and increase with pressure and they showed, using time resolved diode laser spectroscopic detection of HCl, that the H-atom abstraction reaction (channel A) dominates at low pressure whereas adduct formation (channel C′) and H-atom abstraction (channel A) contribute approximately equally at atmospheric pressure. The adduct, CH3S(Cl)CH3, formed in the DMS + Cl reaction, has been detected and studied, notably by electron impact mass spectrometry,24 cavity-ring down spectroscopy25 and UV-visible absorption spectroscopy.26
The DMS + Cl reaction has also been the subject of a number of theoretical studies,25,27–30 where reaction enthalpies (ΔHϕf,298) have been computed. A summary of the computed values, and their expected reliability, is given in Table S1 in the ESI† section.
Water is the third most abundant species in the troposphere behind only N2 and O2 with concentrations of up to 1018 molecules cm−3. Recently, it has been demonstrated that even a single water molecule can increase the rate coefficient of a reaction. Water can form complexes with the reagents, products and transition states in a reaction and lower their energies, and in this way, the activation barrier for a reaction may be reduced.31–33 However, complex formation is a process that occurs with a large reduction of entropy. Therefore, a decrease of an enthalpy barrier by one water molecule does not necessarily lead to an enhancement of the rate of reaction as the free energy, relative to the reagents, of the transition state on the reaction surface determines the value of the reaction rate coefficient.
A number of examples have been identified where a single water molecule increases the reaction rate coefficient.31–37 For example, a single water molecule lowers the barrier of the reaction SO3 + H2O → H2SO4 by over 25 kcal mol−1.34,35 It is important to note, however, that it is possible for water to give rise to a decrease of rate coefficient, as has been demonstrated for the OH + CH4 reaction.38 In this case, a calculation of the free energy of activation in the absence and presence of water shows that the free energy of activation is more positive in the presence of water than in the absence of water, giving rise to a reduction of rate coefficient in the presence of water.
In related work, the reaction DMS + OH, in the absence and presence of water, has been investigated theoretically at the MP2/cc-pVTZ//B3LYP/aVTZ level by Jorgensen and Kjaergaard.39 It was found that for the H-abstraction channel, the presence of water reduces the reaction barrier height and hence it was expected that this would increase the reaction rate coefficient. However, it was concluded that water is only likely to have a small effect on the overall rate coefficient because the DMS + OH reaction is moderately fast in the absence of water (room temperature rate coefficient 6.5 × 10−12 cm3 molecule−1 s−1),40 and in the troposphere, the concentrations of the hydrates DMS·H2O and OH·H2O were calculated to be small compared to the concentrations of DMS and OH, respectively.39
We have previously studied the reaction of DMS with molecular chlorine (DMS + Cl2) using UV photoelectron spectroscopy (PES), infrared matrix isolation spectroscopy and electronic structure calculations.41–43 It was found that this reaction proceeds through the formation of a covalent reaction intermediate ((CH3)2SCl2), in which sulfur is four coordinate. This then decomposes into the final products monochlorodimethylsulfide (CH3SCH2Cl) and hydrogen chloride (HCl). Also, using PES as the detection technique, the room temperature rate coefficient of DMS + Cl2 has been measured as (3.4 ± 0.7) × 10−14 cm3 molecule−1 s−1,41 four orders of magnitude lower than the DMS + Cl room temperature rate coefficient. An objective of our studies on DMS reactions of atmospheric importance is to investigate the effect of water on these reactions, notably for DMS + Cl and DMS + Cl2 to investigate if the reaction mechanisms and energetics are changed significantly in the presence of water.
The aim of this present work, therefore, is to study the reaction DMS + Cl in the absence and presence of water to investigate the mechanism and to determine if the energies of the transition states relative to the reagents are changed significantly when water is present. The DMS + Cl experimentally determined rate coefficient at room temperature and atmospheric pressure, (3.15 ± 0.31) × 10−10 cm3 molecule−1 s−1,21 is close to the collisional limit and therefore, water is unlikely to enhance the overall rate coefficient. Nevertheless, it was felt essential to study the DMS + Cl reaction with and without water to see if there are changes in mechanism, and relative energies of the stationary points on the reaction surface, prior to the study of DMS + Cl2 in the absence and presence of water. This necessitated calculations on DMS + Cl at a higher level than those performed by Resende and Almeida27 in order to obtain more reliable relative energies and structures for the stationary points on the reaction surface.
Although the DMS + Cl reaction is studied in this work in the presence of one water molecule, it is recognised that water can be present in the marine boundary layer as droplets and aerosol particles (as well as dimers and trimers) and understanding their formation is of great importance. Potential surfaces for reactions on and within these particles may well be significantly different from their gas-phase counterparts.32,44,45
2. Computational details
2.1 DMS + Cl
Electronic structure calculations were carried out to optimise the geometries of the reactants, reactant complexes, transition state structures (TSs), product complexes and products. The M06-2X functional was used with aug-cc-pVDZ (aVDZ) and aug-cc-pVTZ (aVTZ)46 basis sets. These computations were performed in the spin unrestricted formalism. The M06-2X functional was chosen because it has been shown to perform particularly well for TS structures and reaction barrier heights in benchmark studies.47–50 Harmonic vibrational frequencies were calculated at each level of theory to verify the nature of the stationary points (minima and TSs), and to provide the zero-point energy (ZPE) and the thermodynamic contributions to the enthalpy and free energy changes. Intrinsic reaction coordinate (IRC) calculations were also performed to ensure that a given TS connects with the desired minima.51,52Fixed point calculations were carried out using the UM06-2X/aVTZ optimised stationary point geometries, with the aug-cc-pVnZ basis sets (n = T and Q), using the coupled cluster method [CCSD(T)].53–55 The methods used for these UCCSD(T) computations were, therefore, UCCSD(T)/aVTZ//UM06-2X/aVTZ and UCCSD(T)/aVQZ//UM06-2X/aVTZ. The total energy values obtained with these UCCSD(T)/aVnZ//UM06-2X/aVTZ (n = T and Q) calculations were extrapolated to the complete basis set (CBS) limit. The extrapolation scheme employed the two parameter formula (eqn (1)).56,57
| E(x) = ECBS + Ax−3 | (1) |
Further single point calculations were carried out with the UM06-2X/aVTZ optimised geometries using the domain-based local pair natural orbitals coupled cluster (DLPNO-UCCSD(T)) approach.58–62 These were DLPNO-UCCSD(T)/aVnZ//UM06-2X/aVTZ (n = T and Q) calculations, with total energies extrapolated to the CBS limit using eqn (1). This method employs localised orbitals and obtains the correlation energy as a sum over the correlation energies of electron pairs. It recovers a large part of the CCSD(T) correlation energy at low computational cost.
2.2 DMS + Cl + H2O
The calculations performed for this reaction were similar to those carried out for DMS + Cl. Geometry optimizations were carried out with the M06-2X functional using aug-cc-pVTZ (aVTZ) basis sets. Then, as in the DMS + Cl case, fixed point UCCSD(T) and DLPNO-UCCSD(T) calculations were performed, using the UM06-2X/aVTZ optimised stationary point geometries, with the aug-cc-pVnZ (n= T and Q) basis sets. Extrapolations were then carried out to obtain the CBS total energies using eqn (1).All electronic structure computations were carried out using Gaussian 0963 and Gaussian 1664 running on SEAGrid.65–68 The DLPNO-UCCSD(T) single point calculations were performed using the ORCA package.69,70 Transition state theory (TST) and TST including Wigner tunneling correction (TST + W) rate coefficients were calculated using the KisTheIP program.71
3. Results and discussion
3.1 The DMS + Cl reaction
Schematic energy profiles for the DMS + Cl reaction channels obtained in this work at the UM06-2X/aVTZ level are shown in Fig. 1, and the computed relative energies, enthalpies (ΔHϕf,298K) and free energies (ΔGϕf,298K) of the stationary points relative to the reagents are shown in Table 2 and Table S4 (ESI†). The agreement between the UCCSD(T) and DLPNO-CCSD(T) values in these tables is good with the difference between the UCCSD(T) and DLPNO-UCCSD(T) results for a given basis set (aVTZ or aVQZ) being small (<1 kcal mol−1) with the DLPNO values being slightly more positive than the UCCSD(T) values. On comparing the extrapolated UCCSD(T)/CBS results with the DLPNO-UCCSD(T)/CBS values for all the stationary points, a difference of <1 kcal mol−1 (ranging between 0.1 to 0.8 kcal mol−1) can be seen. This is in good agreement with a previous report where it was found that for hydrogen abstraction reactions the DLPNO-CCSD(T) results compare satisfactorily with CCSD(T) results within an uncertainty of 0.5 kcal mol−1.72 | ||
| Fig. 1 Energy profiles for the reaction of DMS + Cl using the UM06-2X/aVTZ and DLPNO-UCCSD(T)/CBS//UM06-2X/aVTZ methods. The UM06-2X/aVTZ relative electronic energies (ΔE) including ZPE are reported in the figure, with the DLPNO-UCCSD(T)/CBS//UM06-2X/aVTZ values shown in brackets; values are in kcal mol−1). Adduct 1 is the reactant complex of channel C. Formation of Adduct 1 from DMS + Cl is referred to as channel C′ in the text. | ||
| UM06-2X/aVTZ | UCCSD(T)/CBS//UM06-2X/aVTZ | DLPNO-UCCSD(T)/CBS//UM06-2X/aVTZ | |
|---|---|---|---|
| a The thermal correction to the enthalpy obtained using the UM06-2X/aVTZ method were added to the UCCSD(T) single point energy (ZPE is included in the electronic energies). b The structure of RC1 was obtained from its respective IRC and the energetic parameters were obtained by single point calculations. | |||
| Channel A | |||
| RC1b | −10.6 | −8.2 | −7.4 |
| (−10.9) | (−8.5) | (−7.7) | |
| −4.5 | −2.1 | −1.3 | |
| TS1 | −11.5 | −9.7 | −8.9 |
| (−12.0) | (−10.2) | (−9.4) | |
| −5.2 | −3.3 | −2.6 | |
| MS1 | −12.9 | −14.4 | −13.7 |
| (−12.7) | (−14.1) | (−13.5) | |
| −7.6 | −9.1 | −8.5 | |
| CH3SCH2 + HCl | −9.1 | −11.1 | −10.8 |
| (−8.2) | (−10.2) | (−10.0) | |
| −11.5 | −13.5 | −13.2 | |
| Channel B | |||
| RC2 | −0.5 | −0.8 | −0.6 |
| (−0.4) | (−0.7) | (−0.5) | |
| 4.2 | 4.0 | 4.2 | |
| TS2 | 15.5 | 18.0 | 18.3 |
| (15.4) | (17.9) | (18.2) | |
| 20.8 | 23.3 | 23.6 | |
| MS2 | −11.7 | −10.5 | −10.2 |
| (−11.4) | (−10.1) | (−9.8) | |
| −7.8 | −6.6 | −6.3 | |
| CH3S + CH2Cl | −11.1 | −10.1 | −10.0 |
| (−11.2) | (−10.2) | (−10.1) | |
| −14.2 | −13.2 | −13.1 | |
| Channel C, C′ | |||
| Adduct 1 | −22.7 | −20.9 | −20.3 |
| (−23.1) | (−21.2) | (−20.7) | |
| −16.2 | −14.4 | −13.8 | |
| TS3 | 7.2 | 8.0 | 8.8 |
| (7.1) | (7.9) | (8.7) | |
| 13.4 | 14.2 | 15.0 | |
| MS3 | 4.3 | 5.5 | 6.0 |
| (5.1) | (6.3) | (6.9) | |
| 8.9 | 10.1 | 10.6 | |
| CH3SCl + CH3 | 5.6 | 5.4 | 5.6 |
| (6.4) | (6.2) | (6.3) | |
| 2.8 | 2.6 | 2.8 | |
Fig. 1 shows that channel A (hydrogen abstraction) and channel C′ (formation of adduct 1) are likely to be the dominant channels at tropospheric temperatures. This is in good agreement with available experimental evidence21–26 which shows that channel A dominates at low pressure but at higher pressures channel C′ becomes competitive with channel A. It was noted earlier that adduct 1 [(CH3)2SCl] has been detected by electron impact mass spectrometry,24 cavity-ring down laser spectroscopy25 and UV-visible absorption spectroscopy.26 Channel B, which produces CH3S and CH3Cl, is the most exothermic. However, it has a high transition state (TS2; at a relative energy of +18.3 kcal mol−1 in Fig. 1) between the reagents and product complex (MS2) and therefore, the rate coefficient of this pathway at temperatures relevant to the troposphere (200–300 K) will be very low. Channel C, which produces CH3SCl + CH3, also has a relatively high TS (TS3) of +8.8 kcal mol−1.
The standard reaction enthalpies of channels A, B, C and C′ were computed by Resende and Almeida in ref. 27 at the UQCISD(T)/DZP//UMP2/DZP level as −2.5, −8.5, +13.1 and −12.7 kcal mol−1, respectively. (see Table S1, ESI†). We repeated these calculations at the same level and obtained values of −3.0, −7.5, 20.9 and −11.4 kcal mol−1. Improving the basis set from DZP to aVDZ gave values of −6.0, −9.4, 12.7 and −12.3 kcal mol−1 (UQCISD(T)/aVDZ//UMP2/aVDZ values) (see Table S3, ESI†). These latter values are in better agreement with the experimentally derived values of (−9.4 ± 1.5), (−10.1 ± 0.8), (8.0 ± 1.7) and ∼−20.0 kcal mol−1. Our M06-2X/aVDZ values are −7.0, −10.4, 9.1 and −23.6 kcal mol−1 and for channels A, and C′, respectively, where we carried out UCCSD(T)/CBS//UM06-2X/aVDZ calculations, we obtained −9.8 and −21.1 kcal mol−1 for these channels (see Table S3, ESI†) in good agreement with the expected values of (−9.4 ± 1.5), and ∼−20.0 kcal mol−1. Our M06-2X/aVTZ values for channels A, B, C and C′ are −8.2, −11.2, +6.4 and −23.1 kcal mol−1, respectively, and our UCCSD(T)/CBS//UM06-2X/aVTZ values for A and C′ are −10.2 and −21.2 kcal mol−1 (Table 2). A comparison of the potential energy profiles obtained by Resende and Almeida27 with those obtained in this work is given in the ESI† section.
UM06-2X/aVTZ structures for stationary points obtained in this work on the potential energy surface of channels A and C′ including selected geometrical parameters, are shown in Fig. 2. A discussion of the geometrical parameters of these stationary points, shown in Fig. 1 and 2, is given in the ESI† section.
 | ||
| Fig. 2 UM06-2X/aVTZ optimised geometries of RC1 (obtained from IRC), TS1, MS1, CH3SCH2, HCl and adduct 1. The optimised geometries of the other stationary points in Fig. 1 are shown in Fig. S3 (ESI†). i.e. RC2, TS2, MS2, CH3S, CH3Cl, TS3, MS3, CH3SCl and CH3. These geometries and geometrical parameters are discussed in the ESI† section. | ||
3.2 The DMS + Cl + H2O reactions


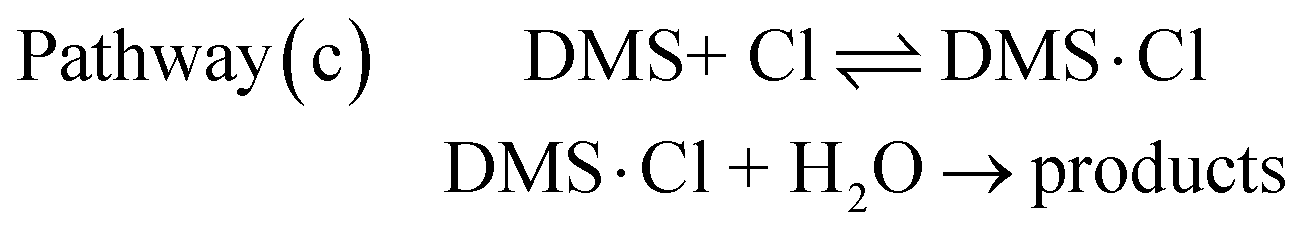
The complexes DMS·H2O, Cl·H2O and DMS·Cl and their reactions were investigated computationally to estimate which pathway is likely to be dominant under typical conditions in the marine boundary layer (see computed UM06-2X/aVDZ and UM06-2X/aVTZ structures in Fig. 3 and Fig. S4, ESI†).
 | ||
| Fig. 3 UM06-2X/aVTZ optimised geometries for DMS·H2O, Cl·H2O and DMS·Cl. | ||
The DMS·H2O complex has several configurations depending on the orientation of the water molecule. The most stable DMS·H2O complex obtained using the UM06-2X/aVDZ and UM06-2X/aVTZ methods is displayed in Fig. 3 and Fig. S4 (ESI†) (which also shows computed DMS·Cl and Cl·H2O structures). In the optimised geometry of the DMS·H2O complex, water is located with its O atom in the Cs plane where the S atom lies, with its H atoms out of the Cs plane, with the S–H bond distance of 2.411 Å computed with both methods. These values are in agreement with reported data obtained using the B3LYP/6-311G(d,p) (S–H: 2.487 Å)67 and MP2/cc-pVTZ (S–H: 2.418 Å)39 methods. The minimum energy UM06-2X/aVDZ and UM06-2X/aVTZ structures computed for the reaction between Cl and water is also shown in Fig. S4 (ESI†) where the Cl–O bond distance is 2.577 Å and 2.599 Å, respectively. The computed UM06-2X/aVDZ stabilisation energies (without ZPE) of DMS·H2O and Cl·H2O are −6.3 and −5.6 kcal mol−1, respectively. These stabilisation energies are slightly more positive at the UM06-2X/aVTZ level: −5.9 kcal mol−1 for DMS·H2O and −5.0 kcal mol−1 for Cl·H2O (Table 2).
The equilibrium constants (Keq) at 298 K for the formation of DMS·H2O, Cl·H2O and DMS·Cl (pathways (a), (b) and (c)) were calculated and are listed in Table 3. They were calculated using equation (2):
| Keq = exp(−ΔGϕf,298K/RT) | (2) |
| Pathway | (a) | (a) | (a) | (b) | (b) | (b) | (c) | (c) | (c) |
|---|---|---|---|---|---|---|---|---|---|
| ΔEs | ΔGϕf,298K | K eq | ΔEs | ΔGϕf,298K | K eq | ΔEs | ΔGϕf,298K | K eq | |
| UB3LYP/aVTZ | −4.0 | 3.53 | 2.6 × 10−3 | −6.2 | −0.10 | 1.2 | −23.3 | −16.1 | 6.1 × 1011 |
| UMP2/aVDZ | −6.5 | 1.48 | 8.3 × 10−2 | −3.0 | 2.13 | 2.7 × 10−2 | −20.7 | −13.1 | 3.9 × 109 |
| UM06-2X/aVDZ | −6.3 | 3.26 | 4.0 × 10−3 | −5.6 | 0.48 | 4.5 × 10−1 | −24.0 | −16.8 | 2.2 × 1012 |
| UM06-2X/aVTZ | −5.9 | 2.54 | 1.4 × 10−2 | −5.0 | 0.55 | 4.0 × 10−1 | −23.5 | −16.2 | 8.0 × 1011 |
| UCCSD(T)/CBS//UM06-2X/aVDZ | −5.2 | 3.16 | 4.8 × 10−3 | −3.6 | 2.01 | 4.5 × 10−1 | −21.5 | −14.4 | 3.5 × 1010 |
| UCCSD(T)/CBS//UM06-2X/aVTZ | −5.3 | 3.13 | 5.1 × 10−3 | −5.3 | 2.01 | 4.4 × 10−1 | −21.6 | −14.4 | 3.7 × 1010 |
The concentration of DMS in the atmosphere is about 200 ppt at sunrise and 120 ppt by late afternoon.73 Thus, as the total number of density of air at 1 atm and 298 K is 2.46 × 1019 molecules cm−3, the afternoon concentration of DMS (120 ppt) is expected to be ∼2.95 × 109 molecules cm−3. The estimated day-time global average concentration of Cl is 103 atom cm−3 and the concentration of water is 7.38 × 1017 molecules cm−3 in the marine boundary layer with 100% humidity.39 These concentrations were used with the Keq values in Table 3 to estimate the [DMS·H2O]/[Cl·H2O], [DMS·H2O]/[DMS·Cl] and [DMS·Cl]/[Cl·H2O] ratios in the troposphere and these are shown in Table 4.
| UM06-2X/aVDZ | UM06-2X/aVTZ | UCCSD(T)/CBS//UM06-2X/aVDZ | UCCSD(T)/CBS//UM06-2X/aVTZ | |
|---|---|---|---|---|
| [DMS·H2O]/[Cl·H2O] | 2.6 × 104 | 1.0 × 105 | 3.2 × 104 | 3.4 × 104 |
| [DMS·H2O]/[DMS·Cl] | 1.3 | 1.3 × 101 | 1.0 × 102 | 1.0 × 102 |
| [DMS·Cl]/[Cl·H2O] | 2.0 × 104 | 8.0 × 103 | 3.1 × 102 | 3.4 × 102 |
On the basis of the calculated concentration ratios using the UM06-2X/aVDZ and UM06-2X/aVTZ methods, it can be concluded that [DMS·H2O] and [DMS·Cl] are significantly greater than [Cl·H2O], indicating that pathways via [Cl·H2O] (Pathway (b)) are not important relative to those via DMS·H2O (Pathway (a)) and DMS·Cl (Pathway (c)). Also, improved relative energies and ΔGϕf,298K values for pathways (a), (b) and (c) obtained via fixed point UCCSD(T) calculations extrapolated to the CBS limit lead to the same overall conclusion i.e. based on these approximate calculations it is expected that [DMS·H2O] > [DMS·Cl] » [Cl·H2O], and, therefore, reactions that proceed via [Cl·H2O] + DMS are not significant.
As has already been outlined, in the absence of water, channel A (production of CH3SCH2 + HCl) and channel B (production of CH3S + CH3Cl) are both exothermic whereas channel C (production of CH3SCl + CH3) is endothermic. Computed relative electronic energies (ΔE, kcal mol−1), relative enthalpies (ΔHϕf,298K, kcal mol−1) and relative free energies (ΔGϕf,298K, kcal mol−1) for the channels A and B are given in Tables 5 and 6, Tables S5 and S6 (ESI†) respectively. Fig. 1 shows that channel A proceeds via a submerged TS (TS1) whereas channel B has a significant energy barrier in the forward direction via TS2. In the presence of water, the mechanisms of both these reactions are changed significantly as can be seen on comparing Fig. 1 with Fig. 4 and 5 (for channel A) and Fig. 1 with Fig. 6 and 7 (for channel B). For channel A in the presence of water, the reaction still occurs via submerged TSs with the submerged TS being lower via DMS·H2O + Cl (Fig. 4, pathway (a)) than via DMS·Cl + H2O (Fig. 5, pathway (c)). The lowest TS is TS1.H2O-B in Fig. 4. However, in the presence of water channel B still has significant energy barriers in the forward direction (with TSs of TS2·H2O-A, Fig. 6 and TS2·H2O-B, Fig. 7). It can be seen that water does not give rise to a significant lowering of the activation energy barrier for channel B such that channel B becomes competitive with channel A. Channel A and complex formation (channel C′) are still the dominant reaction channels in the presence of water. More detail on the comparison of Fig. 1 with Fig. 4 and 5 (for Channel A) and Fig. 1 with Fig. 6 and 7 (for Channel B) is given in the ESI† section. The structures of the TSs in Fig. 4–7 are shown in Fig. 8.
| UM06-2X/aVTZ | UCCSD(T)/CBS//UM06-2X/aVTZ | DLPNO-UCCSD(T)/CBS//UM06-2X/aVTZ | |
|---|---|---|---|
| a The thermal correction to the enthalpy obtained using the UM06-2X/aVTZ method was added to the UCCSD(T) single point energy (ZPE is included in the electronic energies). b Reliable value could not be obtained; calculations were very lengthy involving several re-starts, thus extrapolation to the CBS limit could not be performed. However, a reliable DLPNO-UCCSD(T)/CBS//UM06-2X/aVTZ value was obtained. | |||
| Pathway (a) | |||
| DMS·H2O + Cl | −4.4 | −3.8 | −3.7 |
| (−4.5) | (−3.9) | (−3.8) | |
| 2.5 | 3.1 | 3.2 | |
| DMS·H2O + Cl → DMS·H2O·Cl-1 | −26.0 | −23.5 | −22.8 |
| (−26.2) | (−23.7) | (−23.0) | |
| −11.8 | −9.3 | −8.6 | |
| TS1·H2O-A | −7.7 | —b | −2.6 |
| (−8.2) | (−3.1) | ||
| 6.3 | 11.4 | ||
| TS1·H2O-B | −14.1 | −12.0 | −11.1 |
| (−14.7) | (−12.5) | (−11.7) | |
| 0.0 | 2.2 | 3.0 | |
| MS1·H2O-1 | −15.8 | −16.6 | −15.8 |
| (−15.4) | (−16.2) | (−15.4) | |
| −3.6 | −4.4 | −3.6 | |
| Pathway(c) | |||
| DMS·Cl + H2O | −22.7 | −20.9 | −20.3 |
| (−23.1) | (−21.2) | (−20.7) | |
| −16.2 | −14.4 | −13.8 | |
| DMS·Cl + H2O → DMS·H2O·Cl-2 | −29.9 | −27.3 | −26.5 |
| (−30.5) | (−27.9) | (−27.1) | |
| −15.1 | −12.5 | −11.7 | |
| TS1·H2O-C | −4.7 | −1.1 | 0.3 |
| (−6.4) | (−2.7) | (−1.4) | |
| 11.2 | 14.8 | 16.1 | |
| MS1·H2O-2 | −18.1 | −18.7 | −17.9 |
| (−18.3) | (−18.9) | (−18.1) | |
| −4.1 | −4.7 | −3.9 | |
| CH3SCH2·H2O + HCl | −12.4 | −13.5 | −12.9 |
| (−11.9) | (−13.0) | (−12.4) | |
| −7.1 | −8.2 | −7.6 | |
| CH3SCH2 + HCl·H2O | −13.0 | −15.0 | −14.6 |
| (−12.7) | (−14.7) | (−14.4) | |
| −9.7 | −11.7 | −11.4 | |
| CH3SCH2 + HCl + H2O | −9.1 | −11.1 | −10.8 |
| (−8.2) | (−10.2) | (−10.0) | |
| −11.5 | −13.5 | −13.2 | |
| UM06-2X/aVTZ | UCCSD(T)/CBS//UM06-2X/aVTZ | DLPNO-UCCSD(T)/CBS//UM06-2X/aVTZ | |
|---|---|---|---|
| a The thermal correction to the enthalpy obtained using the UM06-2X/aVTZ method was added to the UCCSD(T) single point energy (ZPE is included in the electronic energies). | |||
| Pathway (a) | |||
| DMS·H2O + Cl | −4.4 | −3.8 | −3.7 |
| (−4.5) | (−3.9) | (−3.8) | |
| 2.5 | 3.1 | 3.2 | |
| DMS·H2O + Cl → DMS·H2O·Cl-3 | −15.5 | −12.1 | −10.8 |
| (−16.1) | (−12.6) | (−11.4) | |
| −1.6 | 1.9 | 3.1 | |
| TS2·H2O-A | 9.8 | 12.9 | 13.6 |
| (9.3) | (12.4) | (13.2) | |
| 23.8 | 26.9 | 27.7 | |
| MS2·H2O-1 | −18.4 | −14.8 | −14.0 |
| (−18.3) | (−14.8) | (−14.0) | |
| −5.4 | −1.9 | −1.1 | |
| Pathway (c) | |||
| DMS·Cl + H2O | −22.7 | −20.9 | −20.3 |
| (−23.1) | (−21.2) | (−20.7) | |
| −16.2 | −14.4 | −13.8 | |
| DMS·Cl + H2O → DMS·Cl·H2O-4 | −29.5 | −26.9 | −26.0 |
| (−30.0) | (−27.4) | (−26.7) | |
| −14.9 | −12.2 | −11.4 | |
| TS2·H2O-B | 32.1 | 34.6 | 35.7 |
| (31.7) | (34,2) | (35.3) | |
| 46.2 | 48.7 | 49.8 | |
| MS2·H2O-2 | −18.8 | −15.4 | −14.8 |
| (−18.7) | (−15.4) | (−14.7) | |
| −6.1 | −2.7 | −2.1 | |
| DMS·Cl + H2O → DMS·Cl·H2O-2 | −29.9 | −27.3 | −26.5 |
| (−30.5) | (−27.9) | (−27.1) | |
| −15.1 | −12.5 | −11.7 | |
| TS2·H2O-C | 31.7 | 34.1 | 35.2 |
| (31.2) | (33.7) | (34.7) | |
| 46.0 | 48.5 | 49.5 | |
| MS2·H2O-3 | −17.6 | −14.9 | −14.4 |
| (−17.3) | (−14.6) | (−14.0) | |
| −5.6 | −2.9 | −2.3 | |
| CH3S + CH3Cl·H2O | −13.8 | −12.4 | −12.2 |
| (−13.9) | (−12.5) | (−12.3) | |
| −10.9 | −9.5 | −9.3 | |
| CH3S·H2O + CH3Cl | −15.0 | −13.4 | −13.1 |
| (−15.2) | (−13.6) | (−13.4) | |
| −10.9 | −9.3 | −9.0 | |
| CH3S + CH3Cl + H2O | −11.1 | −10.1 | −10.0 |
| (−11.2) | (−10.2) | (−10.1) | |
| −14.2 | −13.2 | −13.1 | |
 | ||
| Fig. 4 Channel A, pathway (a). Energy profile for the reaction of DMS·H2O + Cl using the UM06-2X/aVTZ and DLPNO-UCCSD(T)/CBS//UM06-2X/aVTZ (within brackets) methods. The relative electronic energies (ΔE) including ZPE are reported in kcal mol−1. | ||
 | ||
| Fig. 5 Channel A, pathway (c). Energy profile for the reaction of DMS·Cl + H2O using the UM06-2X/aVTZ and DLPNO-UCCSD(T)/CBS//UM06-2X/aVTZ (within brackets) methods. The relative electronic energies (ΔE) including ZPE are reported in kcal mol−1. Note DMS·H2O·Cl-2 also occurs in Fig. 7. | ||
 | ||
| Fig. 6 Channel B, pathway (a). Energy profile for the reaction of DMS·H2O + Cl using the UM06-2X/aVTZ and DLPNO-UCCSD(T)/CBS//UM06-2X/aVTZ (within brackets) methods. The relative electronic energies (ΔE) including ZPE are reported in kcal mol−1. | ||
 | ||
| Fig. 7 Channel B. pathway (c). Energy profile for the reaction of DMS·Cl + H2O using the UM06-2X/aVTZ and DLPNO-UCCSD(T)/CBS//UM06-2X/aVTZ (within brackets) methods. The relative electronic energies (ΔE) including ZPE are reported in kcal mol−1. | ||
 | ||
| Fig. 8 UM06-2X/aVTZ optimised geometries of TS1·H2O-A, TS1·H2O-B, TS1·H2O-C, TS2·H2O-A, TS2·H2O-B and TS2·H2O-C. | ||
The possibility of forming products from a reaction intermediate through an internal rearrangement was considered by taking the reaction intermediate DMS·H2O·Cl-2, as an example. A stepwise reaction sequence of the type
| DMS·H2O·Cl-2 → DMS·OH·HCl → DMS·OH + HCl → CH3SCH2 + H2O + HCl | (3) |
The results of the TST and TST + W rate coefficient calculations carried out for DMS + Cl and DMS + Cl in the presence of water are shown in Table S7 (ESI†). As expected, for DMS + Cl in the absence of water the pathway with the highest rate coefficient at 298 K is channel A via TS1 (see Fig. 1). For DMS + Cl in the presence of water, reactions that proceed via DMS·Cl + H2O (Fig. 5 and 7) have rate coefficients which are lower. For DMS·H2O + Cl channels (Fig. 4 and 6), the only pathway which has a rate coefficient comparable to that of the DMS + Cl reaction is the one which goes via channel A via TS1·H2O-B (Fig. 4). The pathway through channel A via TS1·H2O-A has a rate coefficient which is approximately four orders of magnitude lower (see Table S7, ESI† and Fig. 4). The computed equilibrium constant (Keq) for formation of DMS·H2O of 5.1 × 10−3 (Table 3, UCCSD(T)/CBS//UM06-2X/aVTZ value) and the partial pressure 7.38 × 1017 molecules cm−3 of H2O (100% humidity value) were used to calculate, [DMS·H2O]/[DMS], to be 1.5 × 10−4. This is consistent with the Keq and [DMS·H2O]/[DMS] values calculated at 298 K of 6.1 × 10−3 and 2.0 × 10−4 respectively, obtained at the MP2/cc-pVTZ//B3LYP/aVTZ level in ref. 39. Therefore, because of the low [DMS·H2O], even if the route via DMS·H2O + Cl pathway (a) via channel A through TS1·H2O-B has a room temperature rate coefficient comparable to that of the reaction in the absence of water, DMS + Cl in the absence of water will still be the dominant reaction via channel A via TS1 even under high humidity levels. An equivalent calculation for [DMS·Cl]/[DMS] gives this ratio as 1.5 × 10−6, with Keq = 3.7 × 1010 for pathway (c) (Table 3) and [Cl] = 103 cm−3. Clearly water changes the mechanisms of the DMS + Cl reactions significantly but the presence of water is not expected to change the dominant reaction or overall reaction rate coefficient under atmospheric conditions. Nevertheless, this study will be an important precursor in an equivalent study of the reaction of DMS with Cl2 in the presence of water.
4. Conclusions
The atmospherically important reaction DMS + Cl, and the effect of water on this reaction, have been investigated using electronic structure methods with the M06-2X density functional. In the absence of water, hydrogen atom abstraction from DMS to give CH3SCH2 + HCl, and Cl addition to DMS to form the adduct DMS·Cl, are found to be the dominant reaction channels. These channels are both exothermic (reaction enthalpies, ΔHϕf,298, = −10.0 and −20.7 kcal mol−1 at the DLPNO-UCCSD(T)/CBS//UM06-2X/aVTZ level). Another channel leading to the formation of CH3S + CH3Cl is also found to be exothermic (ΔHϕf,298 = −10.1 kcal mol−1 at the DLPNO-UCCSD(T)/CBS//UM06-2X/aVTZ level) but it has a high reaction barrier in the forward direction (TS1 = 18.3 kcal mol−1 at the DLPNO-UCCSD(T)/CBS//UM06-2X/aVTZ level). The only other reaction channel, production of CH3SCl + CH3, is endothermic and is not important under atmospheric conditions.In the presence of water, reactions which proceed via (a) DMS·H2O + Cl, (b) Cl·H2O + DMS and (c) DMS·Cl + H2O were considered. However, it was found that, under atmospheric conditions, reactions via pathway (b) are of minor importance relative to those that proceed via pathways (a) and (c). The reaction mechanisms for formation of CH3SCH2 + HCl, CH3S + CH3Cl, and DMS·Cl are altered significantly in the presence of water. The CH3SCH2 + HCl channel occurs via submerged TSs and is expected to be rapid. The CH3S + CH3Cl channel occurs via TSs which present significant energy barriers (13.6 kcal mol−1via DMS·H2O + Cl and 35.7 kcal mol−1via DMS·Cl + H2O at the DLPNO-UCCSD(T)/CBS//UM06-2X/aVTZ level) indicating that this channel is not significant in the presence of water relative to CH3SCH2 + HCl production, and DMS·Cl adduct formation, as is the case in the absence of water. The hydrated adduct DMS·Cl has a different minimum energy structure when formed from DMS·H2O + Cl or from DMS·Cl + H2O. TST calculations show that formation of CH3SCH2 + HCl via pathway (a) is faster than via pathway (c).
This study has shown that water changes the mechanisms of the DMS + Cl reactions significantly but the presence of water is not expected to change the dominant reaction or affect the overall reaction rate coefficient under atmospheric conditions as the DMS + Cl reaction has a rate coefficient at room temperature close to the collisional limit. However, this will probably not be the case for the DMS + Cl2 reaction which has a room temperature rate coefficient which is four orders of magnitude lower than that of DMS + Cl.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work used the Extreme Science and Engineering Discovery Environment (XSEDE), which is supported by National Science Foundation grant number OCI-1053575. LR and PR would also like to acknowledge computing facilities from the Centre for High Performance Computing of South Africa. LR also acknowledges the postdoctoral fellowship from the Higher Education Commission (formerly known as Tertiary Education Commission) of Mauritius.References
- R. J. Charlson, J. E. Lovelock, M. O. Andreae and S. G. Warren, Nature, 1987, 326, 655–661 CrossRef CAS.
- S. F. Watts, Atmos. Environ., 2000, 34, 761–779 CrossRef CAS.
- T. S. Bates, B. K. Lamb, A. Guenther, J. Dignon and R. E. Stoiber, J. Atmos. Chem., 1992, 14, 315–337 CrossRef CAS.
- H. Berresheim, P. H. Wine and D. D. Davis, in Composition, Chemistry and Climate of the Atmosphere, ed., R. Nostrand, New York, 1995, pp. 251 Search PubMed.
- S. E. Schwarz, Nature, 1988, 336, 441–445 CrossRef.
- C. F. Cullis and M. M. Hirschler, Atmos. Environ., 1980, 14, 1263–1278 CrossRef CAS.
- T. S. Bates, J. D. Cline, R. H. Gammon and S. R. Kelly-Hansen, J. Geophys. Res., 1987, 92, 2930–2938 CrossRef CAS.
- M. O. Andrea and P. J. Crutzen, Science, 1997, 276, 1052–1058 CrossRef.
- R. Vogt, P. J. Crutzen and R. Sander, Nature, 1996, 383, 327–330 CrossRef CAS.
- J. Li, N. T. Tsona, S. Tang, X. Zhang and L. Du, ACS Omega, 2021, 6, 2410–2419 CrossRef CAS PubMed.
- Q. Chen, T. Sherwen, M. Evans and B. Alexander, Atmos. Chem. Phys., 2018, 18, 13617–13637 CrossRef CAS.
- W. R. Simpson, S. S. Brown, A. Saiz-Lopez, J. A. Thornton and R. von Glasow, Chem. Rev., 2015, 115, 4035–4062 CrossRef CAS PubMed.
- B. J. Finlayson-Pitts, Anal. Chem., 2010, 82, 770–776 CrossRef CAS PubMed.
- B. Finlayson-Pitts and J. R. Pitts, Chemistry of the Upper and Lower Atmosphere, Academic Press, San Diego, CA, 2000 Search PubMed.
- J. D. Raff, B. Njegic, W. L. Chang, M. S. Gordon, D. Dabdub, B. Gerber and B. J. Finlayson-Pitts, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 13647–13654 CrossRef PubMed.
- D. O. De Haan, T. Brauers, K. Oum, J. Stutz, T. Nordmeyer and B. J. Finlayson-Pitts, Int. Rev. Phys. Chem., 1999, 18, 343–385 Search PubMed.
- C. J. Young, R. A. Washenfelder, P. M. Edwards, D. D. Parrish, J. B. Gilman, W. C. Kuster, L. H. Mielke, H. D. Osthoff, C. Tsai, O. Pikelnaya, J. Stutz, P. R. Veres, J. M. Roberts, S. Griffith, S. Dusanter, P. S. Stevens, J. Flynn, N. Grossberg, B. Lefer, J. S. Holloway, J. Peischl, T. B. Ryerson, E. L. Atlas, D. R. Blake and S. S. Brown, Atmos. Chem. Phys., 2014, 14, 3427–3440 CrossRef.
- A. A. P. Pszenny, J. Moldanov, W. C. Keene, R. Sander, J. R. Maben, M. Martinez, P. J. Crutzen, D. Perner and R. G. Prinn, Atmos. Chem. Phys., 2004, 4, 147–168 CrossRef CAS.
- R. P. Wayne, Chemistry of Atmospheres, Oxford University Press, Oxford, 3rd edn, 2000 Search PubMed.
- P. J. Linstrom and W. G. Mallard, ed., NIST Chemistry WebBook, NIST Standard Reference Database Number 69, National Institute of Standards and Technology, Gaithersburg MD, 20899, http://webbook.nist.gov, (retrieved January 2, 2017).
- R. E. Stickel, J. M. Nicovich, S. Wang, Z. Zhao and P. H. Wine, J. Phys. Chem., 1992, 96, 9875–9883 CrossRef CAS.
- C. Arsene, I. Barnes, K. H. Becker and T. Benter, Int. J. Chem. Kinet., 2005, 37, 66–73 CrossRef CAS.
- D. J. Kinnison, W. Mengon and J. A. Kerr, J. Chem. Soc., Faraday Trans., 1996, 92, 369–372 RSC.
- Y. Diaz-de-Mera, A. Aranba, D. Rodriquez, R. Lopez, B. Cabanas and E. Martinez, J. Phys. Chem. A, 2002, 106, 8627–8633 CrossRef CAS.
- S. Enami, Y. Nakano, S. Hashimoto, M. Kawasaki, S. Aloiso and J. S. Francisco, J. Phys. Chem. A, 2004, 108, 7785–7789 CrossRef CAS.
- S. P. Urbanski and P. H. Wine, J. Phys. Chem. A, 1999, 103, 10935–10944 CrossRef CAS.
- S. M. Resende and W. B. De Almeida, J. Phys. Chem. A, 1997, 101, 9738–9744 CrossRef CAS.
- C. Wilson and D. M. Hirst, J. Chem. Soc., Faraday Trans., 1997, 93, 2831–2837 RSC.
- K. C. Thompson, C. E. Canosa-Mas and R. P. Wayne, Phys. Chem. Chem. Phys., 2002, 4, 4133–4139 RSC.
- M. L. McKee, J. Phys. Chem. A, 1993, 97, 10971–10976 CrossRef CAS.
- R. J. Busek, J. S. Francisco and J. M. Anglada, Int. Rev. Phys. Chem., 2011, 30, 335–369 Search PubMed.
- V. Vaida, J. Chem. Phys., 2011, 135, 020901 CrossRef PubMed.
- V. Vaida, H. G. Kjaergaard, P. E. Hulze and D. J. Donaldson, Science, 2003, 299, 1566–1568 CrossRef CAS PubMed.
- V. Vaida, H. G. Kjaergaard and K. J. Feierabend, Int. Rev. Phys. Chem., 2003, 22, 203–219 Search PubMed.
- K. Morokuma and C. Muguruma, J. Am. Chem. Soc., 1994, 116, 10316–10317 CrossRef CAS.
- R. S. Zhu and M. C. Lin, Chem. Phys. Lett., 2002, 354, 217–226 CrossRef CAS.
- R. R. Li, J. R. A. Gorse, M. C. Sauer and S. Gordon, J. Phys. Chem., 1980, 84, 819–821 CrossRef.
- M. A. Allodi, M. E. Dunn, J. Livada, K. N. Kirschner and G. C. Shields, J. Phys. Chem. A, 2006, 110, 13283–13289 CrossRef CAS PubMed.
- S. Jorgensen and H. G. Kjaergaard, J. Phys. Chem. A, 2010, 114, 4857–4863 CrossRef PubMed.
- M. B. Williams, P. Campuzano-Jost, D. Bauer and A. J. Hynes, Chem. Phys. Lett., 2001, 344, 61–67 CrossRef CAS.
- J. M. Dyke, M. V. Ghosh, D. J. Kinnison, G. Levita, A. Morris and D. E. Shallcross, Phys. Chem. Chem. Phys., 2005, 7, 866–873 RSC.
- J. M. Dyke, M. V. Ghosh, M. Goubet, E. P. F. Lee and G. Levita, Chem. Phys., 2006, 324, 85–95 CrossRef CAS.
- S. Beccaceci, J. S. Ogden and J. M. Dyke, Phys. Chem. Chem. Phys., 2010, 12, 2075–2082 RSC.
- R. J. Buszek, J. S. Francisco and J. M. Anglada, Int. Rev. Phys. Chem., 2011, 30, 335–369 Search PubMed.
- M. A. Allodi, M. E. Dunn, J. Livada, K. N. Kirschner and G. C. Shields, J. Phys. Chem. A, 2006, 110, 13283–13289 CrossRef CAS PubMed.
- R. A. Kendall, T. H. Dunning Jr. and R. J. Harrison, J. Chem. Phys., 1992, 96, 6796–6806 CrossRef CAS.
- Y. Zhao and D. G. Truhlar, Acc. Chem. Res., 2008, 41, 157–167 CrossRef CAS PubMed.
- J. J. Zheng, Y. Zhao and D. G. Truhlar, J. Chem. Theory Comput., 2009, 5, 808–821 CrossRef CAS PubMed.
- L. Goerigk and S. Grimme, Phys. Chem. Chem. Phys., 2011, 13, 6670–6688 RSC.
- X. F. Xu, I. M. Alecu and D. G. Truhlar, J. Chem. Theory Comput., 2011, 7, 1667–1676 CrossRef CAS PubMed.
- C. Gonzalez and H. B. Schlegel, J. Chem. Phys., 1989, 90, 2154–2161 CrossRef CAS.
- C. Gonzalez and H. B. Schlegel, J. Phys. Chem., 1990, 94, 5523–5527 CrossRef CAS.
- J. Cizek, Adv. Chem. Phys., 1969, 14, 35–89 CAS.
- R. J. Bartlett, J. Phys. Chem., 1989, 93, 1697–1708 CrossRef CAS.
- J. A. Pople, R. Krishnan, H. B. Schlegel and J. S. Binkley, Int. J. Quantum Chem., 1978, 14, 545–560 CrossRef CAS.
- T. Helgaker, W. Klopper, H. Koch and J. Noga, J. Chem. Phys., 1997, 106, 9639–9646 CrossRef CAS.
- A. Halkier, T. Helgaker, P. Jørgensen, W. Klopper, H. Koch, J. Olsen and A. K. Wilson, Chem. Phys. Lett., 1998, 286, 243–252 CrossRef CAS.
- C. Riplinger and F. Neese, J. Chem. Phys., 2013, 138, 034106 CrossRef PubMed.
- C. Riplinger, B. Sandhoefer, A. Hansen and F. Neese, J. Chem. Phys., 2013, 139, 134101 CrossRef PubMed.
- C. Riplinger, P. Pinski, U. Becker, E. F. Valeev and F. Neese, J. Chem. Phys., 2016, 144, 024109 CrossRef PubMed.
- M. Saitow, U. Becker, C. Riplinger, E. F. Valeev and F. Neese, J. Chem. Phys., 2017, 146, 164105 CrossRef PubMed.
- Y. Guo, C. Riplinger, U. Becker, D. G. Liakos, Y. Minenkov, L. Cavallo and F. Neese, J. Chem. Phys., 2018, 148, 011101 CrossRef PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, T. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 09, Revision D.01, Gaussian, Inc., Wallingford CT, 2016 Search PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 16, Revision C.01, Gaussian, Inc., Wallingford CT, 2016 Search PubMed.
- S. Pamidighantam, E. Nakandala, C. Abeysinghe, S. R. Wimalasena, S. Yodage, S. Marru and M. Pierce, Int. Conf. Comput. Sci., 2016, 80, 1927–1937 Search PubMed.
- N. Shen, Y. Fan and S. Pamidighantam, J. Comput. Sci., 2014, 5, 576–589 CrossRef.
- R. Dooley, K. Milfeld, C. Guiang, S. Pamidighantam and G. Allen, J. Grid. Comput., 2006, 4, 195–208 CrossRef.
- K. Milfeld, C. Guiang, S. Pamidighantam and J. Giuliani, Proceedings of the 2005 Linux Clusters: The HPC Revolution, Apr. 2005.
- F. Neese, Mol. Sci., 2012, 2, 73–78 CrossRef CAS.
- F. Neese, F. Wennmohs, U. Becker and C. Riplinger, J. Chem. Phys., 2020, 152, 224108 CrossRef CAS PubMed.
- S. Canneaux, F. Bohr and E. Herron, J. Comput. Chem., 2014, 35, 82–93 CrossRef CAS PubMed.
- Y. Shang, H. Ning, J. Shi, H. Wang and S.-N. Luo, Phys. Chem. Chem. Phys., 2019, 21, 20857–20867 RSC.
- A. Bandy, D. C. Thornton, B. W. Blomquist, S. Chen, T. P. Wade, J. C. Ianni, G. M. Mitchell and W. Nadler, Geophys. Res. Lett., 1996, 23, 741–744 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2cp05814f |
| This journal is © the Owner Societies 2023 |