
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Surface species in direct liquid phase synthesis of dimethyl carbonate from methanol and CO2: an MCR-ALS augmented ATR-IR study†
Matteo
Signorile
 *a,
Davide
Salusso
a,
Valentina
Crocellà
a,
Maria Cristina
Paganini
a,
Silvia
Bordiga
a,
Francesca
Bonino
a and
Davide
Ferri
b
*a,
Davide
Salusso
a,
Valentina
Crocellà
a,
Maria Cristina
Paganini
a,
Silvia
Bordiga
a,
Francesca
Bonino
a and
Davide
Ferri
b
aDipartimento di Chimica, NIS e INSTM, Universita di Torino, Via P. Giuria 7, I-10125 Torino and Via G. Quarello 15/A, I-10135, Torino, Italy. E-mail: matteo.signorile@unito.it
bPaul Scherrer Institut, CH-5232, Villigen, Switzerland
First published on 17th February 2023
Abstract
The reaction mechanism of dimethyl carbonate (DMC) production over ZrO2 from CO2 and CH3OH is well-known, but the level of understanding has not improved in the last decade. Most commonly, the reaction mechanism has been explored in the gas phase, whilst DMC production occurs in the liquid phase. To overcome this contradiction, we exploited in situ ATR-IR spectroscopy to study DMC formation over ZrO2 in the liquid phase. A multiple curve resolution-alternate least square (MCR-ALS) approach was applied to spectra collected during the CO2/CH3OH interaction with the catalyst surface, leading to the identification of five pure components with their respective concentration profiles. CO2 and CH3OH activation to carbonates and methoxide species was found to strongly depend on the reaction temperature. Low temperature prevents methanol dissociation leaving a catalyst covered with stable carbonates, whilst higher temperature decreases the stability of the carbonates and enhances the formation of methoxides. A reaction path involving the methoxide/carbonate interaction at the surface was observed at low temperature (≤50 °C). We propose that a different reaction path, independent of carbonate formation and involving the direct CO2/methoxide interplay, occurs at 70 °C.
1. Introduction
Human activities have exponentially increased across the last two centuries, implying the uncontrolled rise of emission of greenhouse gases.1,2 The most immediate threat arises from CO2: despite its lower greenhouse potential compared with more threatening molecules (e.g. CH4, N2O or CFCs), its orders of magnitude are more abundant than others.3 For this reason, mitigation of CO2 emissions is mandatory in view of contrasting the present climate crisis. Strategies accounting for CO2 capture and storage (CCS) have been proposed on a short-mid term time lapse to alleviate the issue.4,5 Nonetheless, from the perspective of a sustainable industrial economy, a change of paradigm is required: CO2 should become a feedstock rather than a waste in order to re-introduce it in a closed-loop anthropogenic carbon cycle with zero net emissions. Such a strategy requires the development of CO2 capture and utilization (CCU) technologies.6–13Many possible CCU strategies have been proposed, which are ideally divided into two main families:7 (i) methods exploiting the (partial) reduction of CO2 in the presence of H2;13–16 and (ii) methods leaving the oxidation state of the C atom unaltered, but aiming at the functionalization of the CO2 molecule.12 The former class is undoubtedly an attractive opportunity, also from the perspective of establishing an anthropogenic chemical carbon cycle as forecasted by Olah et al.17 Nevertheless, the issues in provisioning H2 produced without CO2 emission (the so called “green hydrogen”) make this strategy not easily applicable from a short term perspective because further development in the H2 production market is a mandatory prerequisite to its full exploitation.10 In this framework, the H2-free chemistry offered by CO2 functionalization strategies could represent an immediate boost to CCU. Among the many possible processes that can be exploited for CO2 functionalization, its conversion to organic carbonates upon reaction with alcohols is an attractive option.12,18,19 In particular, the simplest carbonate, dimethyl carbonate (DMC, synthesized by reaction with methanol), is an interesting product with increasing demand, due to its growing market share as a fuel additive, mild methylating agent and Li-ion battery electrolyte.18,20–25 The current methods adopted for DMC production are based on toxic reagents and/or environmentally unfriendly procedures.26 The major limitation toward a real-scale application of synthesis of DMC from CO2 and alcohols is the lack of efficient catalysts featuring high conversion levels and long lifetime. Many homogeneous and heterogeneous catalysts have been proposed so far, but reaction yields rarely overcome 10% in the absence of a sacrificial dehydration agent.26,27 Thereby, the rational development of new catalysts based on the understanding of the existing systems is of utmost importance. Their low cost, negligible toxicity and easy separation from the reaction environment make simple metal oxides optimal protype catalysts. In particular, ZrO2 and CeO2 showed excellent selectivity (close to 100%), despite the poor yield in DMC (typically lower than 5%).26 A thorough characterization of the catalytic processes occurring at the surface of these oxides could drive the accurate design of a novel generation of catalysts through a better understanding of the elementary reaction steps involved in the synthesis of DMC. Mechanistic studies involving CeO228–30 are generally more common than for ZrO2, where the surface chemistry of CO2 and methanol is analysed from a gas phase perspective.31–34 It is worth noting that DMC synthesis over oxides is usually carried out in the liquid phase,22 so the effect of working in a condensed phase is not accounted for in these pioneering studies.
In this work, we aim at characterizing the surface species involved in the direct reaction of CO2 with methanol over a high surface area ZrO2. In the attempt to study the catalytic process under representative environmental conditions, we present an ATR-IR study of the solid–liquid interface. We further tried to disclose the effect of temperature on the equilibria between the observed surface species, whose IR fingerprints were disentangled using the multiple curve resolution–alternate least square (MCR-ALS) algorithm. The competition between surface carbonates and methoxides will be discussed in view of its potential effect on DMC production.
2. Experimental
2.1. Materials
ZrO2 was synthesized according to the literature.35 This specific synthetic method provides a final material in a 83![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :17 mixture of the monoclinic and tetragonal polymorphs, respectively (see the XRD pattern in Fig. S2, ESI†) with a BET specific surface area of 73 m2 g−1 (N2 adsorption isotherm at 77 K in Fig. S3, ESI†). The cyclohexane (VWR, >99.8%) solvent was dried overnight over 4A molecular sieves prior to use. Methanol (VWR, >99.9% HPLC), dimethyl carbonate (DMC, Sigma-Aldrich, >99.0%), and CO2 (PANGAS, 99.995%) were used as received, without further purification.
:17 mixture of the monoclinic and tetragonal polymorphs, respectively (see the XRD pattern in Fig. S2, ESI†) with a BET specific surface area of 73 m2 g−1 (N2 adsorption isotherm at 77 K in Fig. S3, ESI†). The cyclohexane (VWR, >99.8%) solvent was dried overnight over 4A molecular sieves prior to use. Methanol (VWR, >99.9% HPLC), dimethyl carbonate (DMC, Sigma-Aldrich, >99.0%), and CO2 (PANGAS, 99.995%) were used as received, without further purification.
2.2. Methods
The powder X-ray diffraction (PXRD) pattern of the sample was collected with a PW3050/60 X'Pert PRO MPD diffractometer (PANalytical) in the Bragg–Brentano geometry in the 10 to 90° 2θ range at a step size of 0.0167° and an integration time of 40 s using a Cu Kα1,2 X-ray source. The Rietveld refinement method implemented in the FullProf software package was used to extract the relative abundance of monoclinic and tetragonal ZrO2 polymorphs.36The specific surface area (SSA) was determined by applying the Brunauer–Emmett–Teller (BET) method in the 0.05–0.3 p/p0 range to the adsorption isotherm of N2 at 77 K obtained with a Micromeritics ASAP 2020 physisorption analyser.
Infrared spectra were collected with a Bruker Vertex 70 Fourier transform spectrometer, equipped with a mercury cadmium telluride detector operated at the liquid nitrogen temperature. Each spectrum consisted of an average of 32 scans (64 for the background spectrum). IR spectra during adsorption of CO2/dimethyl carbonate from the gas phase were measured in the transmission mode (2 cm−1 resolution) with a home-made cell equipped with KBr windows suitable for thermal treatments under a controlled atmosphere. The ZrO2 sample (ca. 10 mg) was pressed into a self-supporting pellet and inserted in a gold envelope. Prior to measurements the pellet underwent the following activation protocol: (i) heating (5 °C min−1) under vacuum (residual pressure < 10−3 mbar) to 400 °C, (ii) holding for 30 min under an O2 atmosphere (100 mbar) and (iii) cooling to room temperature (RT) under vacuum. A detailed description of the gas phase experiments is given in the ESI.†
In situ IR experiments were conducted in the attenuated total reflection (ATR) mode using a commercial horizontal ATR mirror unit and cell (Bruker), allowing circulation of a liquid phase over the internal reflection element. Circulation was controlled through a peristaltic pump operated at 0.5 mL min−1 constant flow (Fig. S4, ESI†). In a typical experiment, ZrO2 was deposited from a water suspension (15 mg per 0.5 ml of MilliQ H2O) on the prismatic ZnSe single crystal internal reflection element (72 × 10 × 6 mm, 45°, Specac) and allowed to dry at room temperature overnight. A fresh deposition was performed prior to each experiment. The sample was pre-treated at 70 °C under cyclohexane for 60 min to favour the removal of weakly interacting surface species. After cooling to the target temperature (if required), the background spectrum was collected and the experiment started. In a typical run, a 0.2 M solution of methanol in cyclohexane was mixed in 1:1 volume ratio with CO2-saturated cyclohexane. In contrast to standard reaction conditions for DMC synthesis where pure methanol is used, the use of a dilute methanol solution has been selected in order to obtain a sufficiently intense signal from alcohol in the ATR spectra, while avoiding signal saturation that could occur in pure methanol and obscuring surface processes and related reaction intermediates. Such a reactive mixture was dosed onto the sample for 60 min. Spectra were collected continuously (1 spectrum every 20 s, 4 cm−1 resolution): a typical dataset consisted of approximately 180 spectra. This procedure was repeated at four different temperatures: 10 °C, 30 °C, 50 °C and 70 °C. Such temperatures are lower than usually adopted in catalytic tests (typically, 100–150 °C and under a pressure in the order of MPa), as a consequence of the use of cyclohexane (boiling point ca. 80 °C) as the working solvent at ambient pressure. The experimental strategy is graphically summarized in Fig. 1.
 | ||
| Fig. 1 Schematic of the temperature/concentration profiles adopted in the in situ ATR-IR experiments. | ||
The adsorption/desorption of pure reagents/products, i.e. dosed separately on the sample, was also studied. Pure reagents were dosed onto the fresh sample until spectral invariance was achieved, and then desorption was performed by flowing pure cyclohexane for over 1800 s.
2.3. Data analysis by MCR-ALS
Multiple curve resolution is a chemometric technique aimed at describing a complex spectral dataset by decomposing it in a given number of “pure” spectra and their concentrations at each point in the dataset. The optimal number of pure (principal) components (PC) was selected via principal component analysis (PCA) by visual analysis of the ‘Scree plots’ (PC and eigenvalues as a function of PC number). Briefly, the algorithm is intended to solve the following bilinear problem:| D = C·ST + E |
Though featuring the positive aspects of decoupling the experimental dataset in pure spectra and their concentration (that make the chemical evolution of a complex system directly accessible), MCR-ALS is affected by intrinsic limitations that must be kept in mind while its outcomes are interpreted. In particular, linear dependencies among pure components, distinguished chemical species with similar kinetic behaviour, signal dependent noise, background effects, and ambiguities in the resolution (i.e. non-unicity of the solution) may lead to the derivation of misleading conclusions.37 These drawbacks can be mitigated by imposing constraints to the algorithm, by including the spectra of known pure species to the dataset and/or by pre-processing data to remove background/to filter noise. In the specific case of IR spectroscopy, deviations from the bilinear behaviour assumed in the decomposition can be expected, i.e. chemical components can affect each other (by causing shifts of the bands, their broadening, etc.). This issue can affect the spectra when concentration-dependent shifts occur, e.g. in the case of lateral coupling of oscillators taking place among molecules adsorbed over extended surfaces.38
MCR-ALS data analysis was performed using the Matlab® based MCR-ALS 2.0 toolbox by Jaumot and coworkers.39 Multiple MCR-ALS runs were performed on each dataset by testing different numbers of PCs and by verifying the effective purity of the obtained spectra. Non-negativity for both spectra and concentrations (fnnls algorithm) was imposed during MCR-ALS runs.
3. Results and discussion
3.1. Adsorption of pure reagents/products
The in situ ATR-IR spectra related to the adsorption of methanol (0.2 M in cyclohexane) over ZrO2 at 30 °C are presented in Fig. 2a. | ||
| Fig. 2 In situ ATR-IR spectra collected during (a) adsorption of methanol on ZrO2 (from a 0.2 M solution of methanol in cyclohexane, time evolution, over 1000 s, from black to red) at 30 °C and (b) its desorption (by exposure to cyclohexane, time evolution, over 1800 s, from red to blue). (c) Concentration profiles as a function of time (the vertical grey line represents the switch from the methanol solution to cyclohexane). (d) Corresponding pure component spectra obtained by MCR-ALS (the ATR-IR spectrum of the bare 0.2 M solution of methanol in cyclohexane is shown as a dotted curve). (e) Molecular structures of species associated with components 2 (weakly physisorbed methanol) and 3 (surface methoxide species); component 1 is liquid phase methanol. | ||
As the sample was contacted with the solution, a sharp band centred at 1035 cm−1 appeared and intensified, which was accompanied by other signals in the range up to 1600 cm−1. Between 1100 and 1200 cm−1, a finer structure composed of two sharp features at 1095 and 1150 cm−1 built up at longer contact times, i.e. when the intensity of other bands was stable. During desorption (Fig. 2b), the bands at 1035, 1095, 1400 and 1600 cm−1 were quickly depleted, while the band at 1150 cm−1 remained substantially unaltered. At late desorption stages, a component at 1050 cm−1 was also observed that was previously overshadowed by the intense peak at 1035 cm−1. Since the temporal evolution of the experimental data suggested the existence of multiple spectral components and taking advantage of the significant variance of the whole adsorption–desorption dataset, MCR-ALS was applied. Principal component analysis (PCA) allowed the identification of three components. The spectra of the pure components and the time dependence of their concentration obtained from MCR-ALS are reported in Fig. 2c and d, respectively. It should be remembered here that the concentration in MCR-ALS is not intended in the strict chemical sense, rather it represents the weight of each pure component at a given time. True concentration and intensity weights correlate through the extinction coefficients of the vibrational modes associated with the pure spectra, that are unluckily unknown in most of the cases.
The spectra of the three pure components can be classified according to the time evolution of the corresponding concentrations. Components 1 and 2 were characterized by a fast increase as methanol was dosed onto ZrO2, then they slightly decreased at longer exposure times. The evolution of component 3 was slower compared to that of the previous ones and required an induction time of ca. 150 seconds to be observed before reaching a plateau in the late adsorption stages. During desorption, components 1 and 2 rapidly decreased, the former confirming the complete desorption of the associated species. Conversely, the concentration of component 3 steeply increased as the methanol feed was interrupted. Such kinetic behaviour suggests that components 1 and 2 are related to species whose interaction with the ZrO2 surface is weak or negligible, whereas component 3 is representative of a strongly interacting surface species, most probably chemisorbed. Furthermore, component 3 formation occurred when the competition with other surface species diminished.
A further piece of information arises from the analysis of the spectra of the pure components. The spectrum of component 1 shows a perfect match with the experimental spectrum of the methanol solution,40 which is characterized by the intense band at 1020 cm−1 (ν(C–O) mode) and other secondary features at ca. 1400 cm−1 (δ(CH3) modes) and 1600 cm−1 (not strictly related to methanol, most probably ascribable to the δ(OH) mode from water traces present as impurity). The assignment of component 1 to liquid phase methanol is corroborated by its fast evolution, suggesting that no adsorption/desorption process is involved. The spectrum of component 2 is qualitatively similar to that of component 1, but it shows a significant shift toward higher frequencies of the ν(C–O) band maximum by 15 cm−1 compared to the liquid phase value. This shift can be related to the weak physisorption (as suggested by the quick desorption rate, testifying a low interaction energy) of methanol with some surface sites of ZrO2. A similar behaviour was reported for methanol adsorption on MgO, where the shift of the signal to higher wavenumbers was attributed to undissociated methanol interacting with the surface Lewis sites through the lone pair located on its oxygen atom and eventually through the hydrogen atom as the H-bond donor.41,42 We can reasonably ascribe the spectrum of component 2 to this type of molecular adduct. Finally, the spectrum of component 3 shows a peculiar fingerprint in the ν(C–O) region, ascribable to the formation of surface methoxy groups from the dissociative chemisorption of methanol. The component at 1150 cm−1 is related to surface linear methoxy groups coordinated to exposed Zr4+.43,44 The other intense feature at 1050 cm−1 is assigned to bridged methoxy groups involving two surface Zr4+ atoms.43,45 The 1095 cm−1 band, partly retained at the end of desorption, has not been identified previously. A similar feature, at 1100 cm−1, was assigned to the C–O stretching of methoxy groups perturbed by neighbouring OH groups formed by methanol chemisorption.43 The small shift from the maximum position reported in gas phase experiments can be ascribed to the perturbation induced by the liquid environment, i.e. the presence of a solvent. This assignment is fully supported by the kinetic data of Fig. 2d. Indeed, component 3, showing an initial induction time and a slow growth, is certainly representative of species formed through an activated process, characterized by a superior spectroscopic stability (slower desorption rate compared with the other species).
The three components identified by MCR-ALS for methanol adsorption were considered as the starting point for the analysis of experiments under reaction conditions (i.e. with methanol/CO2 co-feeding at different temperatures, vide infra section 3.2).
In a second experiment (Fig. 3), we also characterized the adsorption of CO2 on ZrO2.
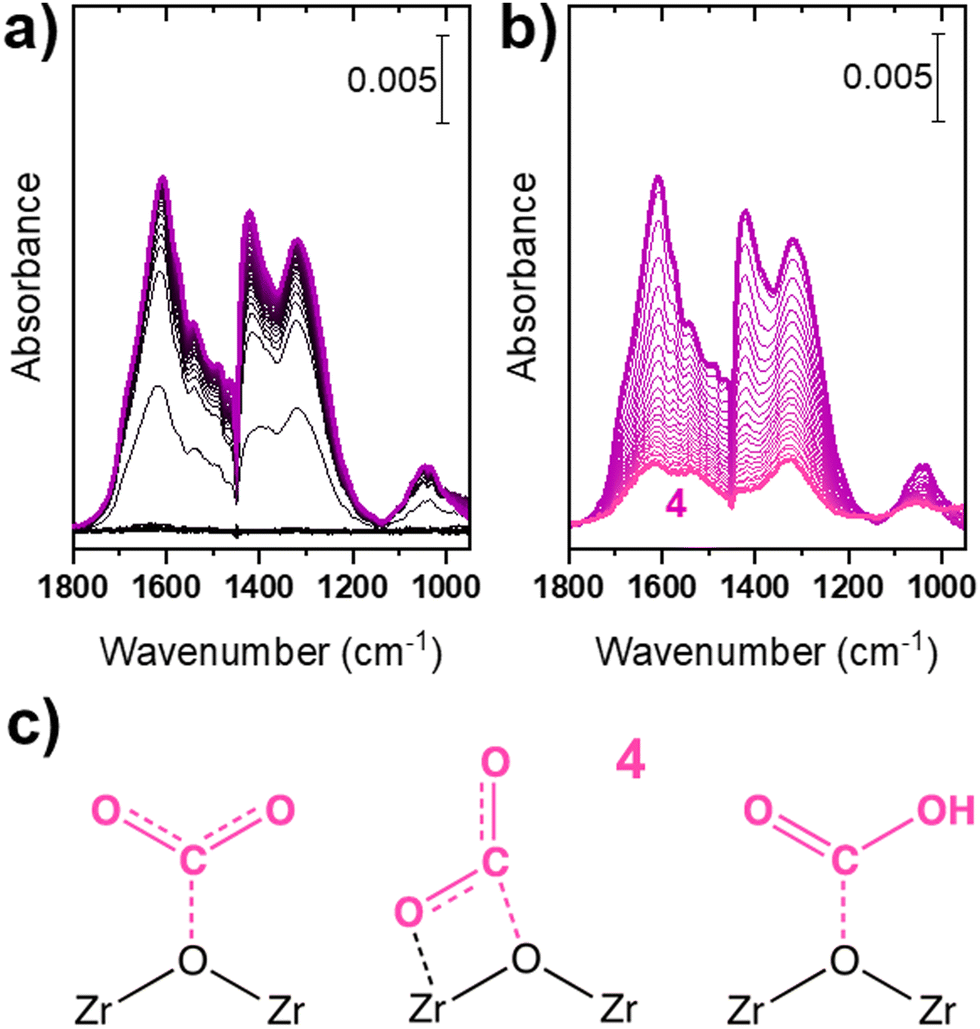 | ||
| Fig. 3 In situ ATR-IR spectra collected during (a) adsorption of CO2 on ZrO2 (from CO2-saturated cyclohexane, time evolution, over 1800 s, from black to violet) and (b) desorption (cyclohexane, time evolution, over 1800 s, from violet to pink) at 30 °C. (c) Molecular structures of species associated with component 4 (from left to right: surface monodentate carbonate-like, bidentate carbonate-like and bicarbonate-like species). | ||
Upon exposure to CO2-saturated cyclohexane (Fig. 3a), several signals appeared in the spectral region typical of the vibrational modes of adsorbed carbonate-like species. The intensity increase continued with time and at the maximum coverage a clear pattern of four bands at 1610, 1420 (uncertain because of a close negative signal due to poor compensation of the solvent phase), 1315 and 1040 cm−1 was visible. Compared to the other bands, the one at 1420 cm−1 continued to intensify also at the longest contact time, suggesting that more than a single surface carbonate-like species is present. The same behaviour is confirmed during desorption (Fig. 3b), where the band at 1420 cm−1 showed complete reversibility: this signal, typical of the bicarbonates-like ν(CO)sym mode, is clearly observed in the gas-phase experiment (see Fig. S5 and comments herein, ESI†) and its intensity is covariant with typical modes of bicarbonates. Conversely, the bands at 1610, 1315 and 1040 cm−1, together with an additional component at 1530 cm−1 evident at lower CO2 coverage, decreased during desorption but stabilized at the end of the experiment, finally yielding the spectrum labelled as 4 in Fig. 3b. These signals can be assigned to various families of carbonate-like species, most probably adsorbed mono- and bi-dentate carbonates.33,46,47 A more precise assignment is not trivial due to the liquid phase environment, indeed a very heterogenous population of surfaces sites is expected as compared to gas-phase activated samples. As a further confirmation, in contrast to the spectra of surface bicarbonates adsorbed on ZrO2 collected in the gas phase (Fig. S5, ESI†), the sharp component assigned to the δ(OH) mode (ca. 1225 cm−1)46 is not observed. This can be specifically ascribed to the liquid-phase conditions, especially to the presence of a solvent, which could interact with the bicarbonate proton and alter its IR signatures. It is otherwise possible that the bands assigned to bicarbonates are actually related to other types of carbonates, differing from those typically observed in gas phase studies, used here as references for spectral assignment.
Since at least two spectral components seem to be involved, MCR-ALS analysis was attempted. Unluckily, the procedure did not converge to meaningful spectra, which is reasonable for species like surface carbonates, because a structural variation implies minor influence on their spectral fingerprints. Accordingly, in the analysis of reactivity experiments presented hereafter (Section 3.2), we considered that surface carbonates contribute as a single component and are thus represented by the spectrum labelled as 4 in Fig. 3, without any relevant contribution of bicarbonates because most relevant bands of bicarbonates (1420, 1210 cm−1) are absent in the spectra.
Finally, the interaction of the reaction product (DMC) with ZrO2 was studied, to evaluate the stability of the final product on the catalyst surface (Fig. 4).
 | ||
| Fig. 4 In situ ATR-IR spectrum of DMC adsorbed on ZrO2 (from a 0.1 M solution of DMC in cyclohexane, after 30 min of contact, solid curve) at 30 °C. The spectrum of the bare 0.1 M solution of DMC in cyclohexane is reported for the sake of comparison (dotted line). (b) Molecular structure of DMC weakly interacting with surface acid sites. | ||
The DMC-surface interaction appears to be weak and non-specific. The vibrational modes of DMC were only slightly affected by the adsorption on ZrO2 and its spectrum closely resembles that of the solution of DMC in cyclohexane. The ν(C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) mode was the most affected one and exhibited multiple low-energy shoulders (1725 and 1740 cm−1) of the principal band at 1760 cm−1. In analogy with the behaviour observed for other carbonyl-containing molecules (e.g. acetone48), these minor spectral features can be ascribed to the interaction of the weakly basic carbonyl moiety with Lewis sites exposed at the ZrO2 surface (i.e. Zr4+ sites with different local environments). The weak interaction of adsorbed DMC with the ZrO2 surface is testified by the labile nature of these spectral components, which rapidly disappear during desorption (spectra not shown for the sake of brevity).
O) mode was the most affected one and exhibited multiple low-energy shoulders (1725 and 1740 cm−1) of the principal band at 1760 cm−1. In analogy with the behaviour observed for other carbonyl-containing molecules (e.g. acetone48), these minor spectral features can be ascribed to the interaction of the weakly basic carbonyl moiety with Lewis sites exposed at the ZrO2 surface (i.e. Zr4+ sites with different local environments). The weak interaction of adsorbed DMC with the ZrO2 surface is testified by the labile nature of these spectral components, which rapidly disappear during desorption (spectra not shown for the sake of brevity).
It is noteworthy that the decomposition of DMC to form methoxy and monomethyl carbonate (MMC) surface species was not observed under the experimental conditions adopted here. Conversely, DMC dissociation occurred very quickly at 25 °C when dosed on ZrO2 from the gas phase (Fig. S6, ESI†).33 Possibly, the solvent stabilizes DMC or more likely surface sites are not available in the liquid phase experiment due to the limitations of the activation procedure, i.e. maximum 70 °C in cyclohexane compared to the gas phase experiment (Fig. S6, ESI†), where ZrO2 was activated under vacuum at 400 °C.
3.2. Reactivity of mixed methanol-CO2
Fig. 5 shows the in situ ATR-IR spectra collected during the simultaneous interaction of methanol and CO2 on ZrO2 at different temperatures.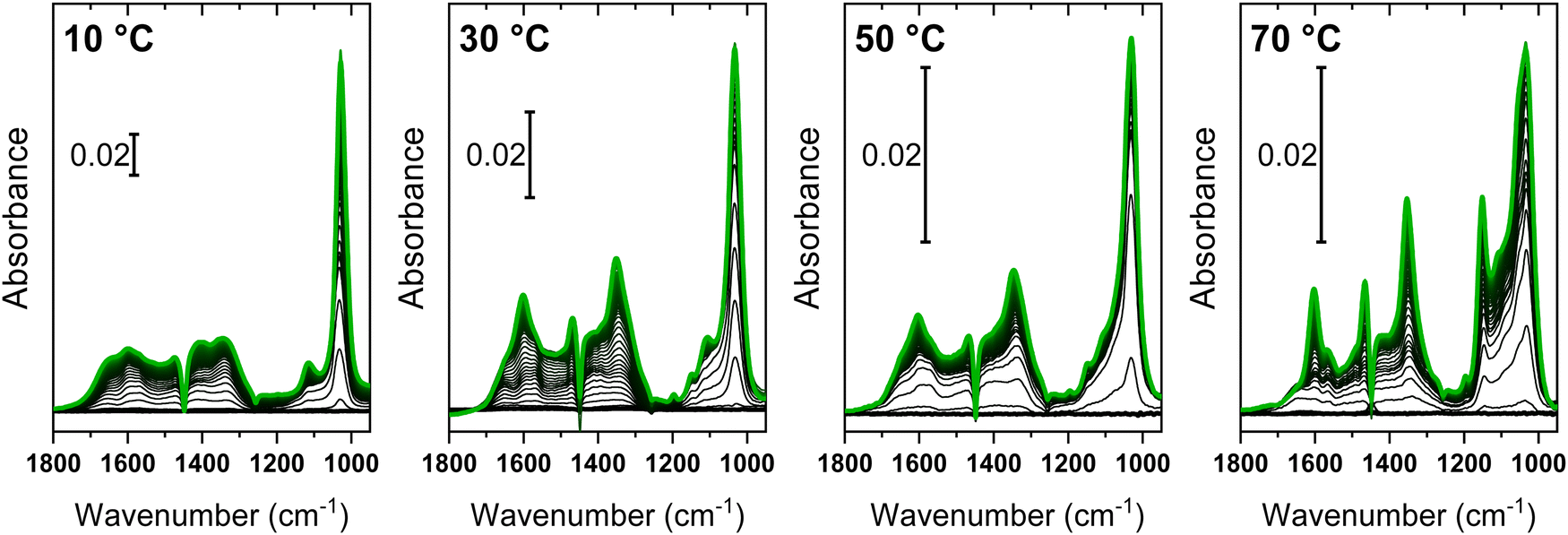 | ||
| Fig. 5 In situ ATR-IR spectra collected during the co-feeding of methanol and CO2 (from a CO2 saturated 0.1 M solution of methanol in cyclohexane) on ZrO2 at different temperatures. Spectra were recorded for 1 h (time evolution from black to green). | ||
At first glance, all the datasets present similar spectroscopic features, but these are characterized by different relative intensities depending on the reaction temperature. The absolute intensity decrease as the temperature increases is a consequence of a lowering of the amount of adsorbed species (in particular of weakly bound ones). Thermal effects related to the change of the refractive index of the ZnSe internal reflection element with temperature also contribute. The temperature increase causes a rise in the refractive index and thus a decrease in the sampling depth probed by the evanescent wave.49 At each temperature, the dominating feature was the sharp band in the 1020–1050 cm−1 range, previously ascribed to methanol related species, weakly interacting with the surface sites of ZrO2. A significant difference can be observed upon inspection of the features related to the methoxy species, which became increasingly evident with increasing temperature. This behaviour agrees with the expected relative stability of methanol species; methoxy groups were still observed at high temperatures, as inferred by their stronger interaction with the surface. The typical broad bands of surface carbonates were well evident at all temperatures at higher energy. A triplet of sharp bands with maxima at 1600, 1465 and 1350 cm−1 appeared on top of these signals, which could be ascribed to the formation of methyl carbonates coordinated to surface sites, i.e. intermediate reaction species obtained from the reaction of methanol and CO2.33
Following the same approach used before, the datasets in Fig. 5 were analysed by MCR-ALS to obtain spectra of pure components from the experiments with the methanol-CO2 feed and the time dependence of their concentrations. Since the same spectroscopic features were observed regardless of the reaction temperature, all data were merged in a single dataset and analysed simultaneously. A critical point in the application of MCR-ALS to the dataset is the high number of expected pure components. As inferred from the dosage of pure reactants/products on ZrO2 (Section 3.1), at least three components due to methanol and methoxy groups and one to carbonates are expected. Moreover, the qualitative inspection of the spectra suggests the presence of a further component with signals at 1605, 1470 and 1350 cm−1, ascribable to surface intermediates such as MMC. Therefore, five pure components were required to obtain a chemically/spectroscopically meaningful description of the reactivity data of Fig. 5. In an unbiased MCR-ALS run with five pure components, imposed convergence threshold was hardly reached and some components were identical to each other, suggesting that they are actually not pure components. To facilitate the algorithm in the recognition of the correct spectral components, we added 50 replicas of the spectrum of each pure component obtained from the respective adsorption/desorption experiments of the pure reagents (spectra 1–4 in Fig. 2 and 3) to the dataset. This strategy ensured that the initial guess for MCR-ALS considered the expected species. However, by avoiding strictly constraining these components, this method allows (re)optimization of the estimated pure components by considering that small spectral variations could occur in the methanol-CO2 feed. An attempt to introduce the reaction product (DMC) as a sixth component was performed (Fig. S7, ESI†) and also in this case we included 50 replicas of the spectrum of adsorbed DMC as in Fig. 4. Nonetheless, inconsistent results were obtained on a chemical basis, as detailed in the ESI.† The (re)optimized spectra of the five pure components and their concentration profiles at each temperature are shown in Fig. 6.
 | ||
| Fig. 6 (a) Spectra of pure components and (b) their concentration profiles obtained from MCR-ALS for reactivity experiments carried out at different temperatures. MCR-ALS has been performed on all the datasets collected at different temperatures simultaneously. (c) Tentative molecular structures of species associated with all components, including component 5 (surface MMC intermediate); component 1 is liquid phase methanol. | ||
The spectra obtained for components 1–4 (Fig. 6a) closely resemble those obtained in the adsorption experiments involving the isolated reagents/products. Hence, they were ascribed to the same previously commented species: 1, liquid phase methanol; 2, molecular methanol adducts at the ZrO2 surface; 3, surface methoxy groups; and 4, surface carbonates. Conversely, component 5 was obtained independently from the MCR-ALS dataset. It is characterized by three principal bands at 1600, 1470 and 1350 cm−1, which match the spectrum proposed for MMC surface species,33 the key intermediate in the catalytic cycle leading to the formation of DMC over ZrO2.
The optimal representation of the dataset (variance explained >99.5%) using components 1–5 revealed that the surface species involved in the reaction of methanol and CO2 over ZrO2 are the same regardless of the reaction temperature, at least within the range considered in this work. The MCR-ALS analysis unlocked access to the relative concentrations of different (surface) species, which in contrast showed a remarkable dependence on the reaction temperature (Fig. 6b). At each temperature, the dominating component (at least at early times on stream) was represented by weakly interacting methanol (2). However, its evolution changed depending on the temperature at later reaction times. In particular, at low temperatures (10 and 30 °C), only a minor fraction of methanol was able to dissociate at the ZrO2 surface by generating methoxy species (3). By increasing the temperature to 50 °C, the contribution of surface methoxy groups became comparable to that of other adsorbed species, finally becoming the dominating component at 70 °C. Surface carbonates (4) displayed the opposite behaviour, i.e. their relative concentration decreased as reaction temperature increased (remarkably at 70 °C). The concentration ratio of surface methoxy species and surface carbonates is indicative of their relative thermodynamic stability. Both methanol and CO2 interact preferentially with basic sites of the ZrO2 surface (basic O and OH), thus a competition between the two molecules is expected. Carbonates were the most stable species at low temperature (10 °C) and occupied most of available basic sites, while surface methoxy groups represented a minor fraction of the surface species. At intermediate temperatures (30–50 °C), the population of methoxy groups increased approaching that of carbonates, testifying again that the formation of these species relies on an activated process. At 70 °C the situation was reverted: with a progressive displacement of carbonates at early reaction stages and the dominant contribution of methoxy groups. The formation of the intermediate MMC (5) occurred in addition to this methoxy/carbonate equilibrium. Its formation was limited at 10 °C compared to higher temperatures, probably due to the low concentration of the precursor methoxy groups. At 30 °C, MMC formed after an induction time of ca. 300 s, that is once methoxy and carbonate groups have reached a similar concentration. Most likely, a sufficient population of these surface species is necessary to achieve their transformation toward MMC through their mutual reactivity at the surface of ZrO2, via a Langmuir–Hinshelwood mechanism.33,34 A confirmation of this observation arises from the similar behaviour observed at 50 °C, where the intermediate formed after the adsorption of the single reactants but with a negligible induction period. At 70 °C, surface carbonates are readily formed during early stages of reaction (<300 s) together with MMC but, as far as the concentration of the former decreases to zero over the following 900 s, the formation rate of MMC decreases. If the previously described Langmuir–Hinshelwood mechanism can be invoked at early reaction stages, MMC formation could take place through a different reaction mechanism after carbonates are entirely displaced and the reaction approaches a steady state. The possibility of a direct reaction of CO2 dissolved in the liquid phase with the surface methoxy species, i.e. via the Eley–Rideal type mechanism, may be considered. This type of mechanism has never been reported for ZrO2, whereas it was hypothesized in the case of CeO2.50 However, it is also possible that CO2 transiently forms carbonates at the ZrO2 surface, but these react very quickly to form MMC, so that they cannot be observed at the time resolution provided by ATR-IR.
As summarized in the reaction mechanism in Fig. 7, as the ZrO2 surface (I) is contacted with methanol and CO2 solubilizes in the working solvent, both surface carbonates and methoxide species are formed (II). Their relative ratio is strongly temperature dependent. At temperature ≤50 °C, carbonates and methoxide species react together to form MMC (III), whilst at 70 °C the reaction takes place between chemisorbed methoxide and gas-phase CO2 (III′). The formation of MMC affects the equilibria determining the population of surface species (IV). In detail, the higher the temperature, the higher the concentration of MMC at the expense of surface carbonates, which turn from the dominating surface species at 10 °C to a nearly nil concentration at 70 °C. Eventually, though not detected under the experimental conditions adopted in this study, the reaction of MMC with a second methanol molecule leads to the synthesis of DMC, which desorbs as the final reaction product. In order to achieve the product, CH3+ cations should be produced via basic dissociation of methanol, as promoted by strong surface acid sites (either Brønsted or Lewis ones) capable of abstracting an OH−. Though Brønsted acid sites are unavailable at the pristine ZrO2 surface, Lewis sites with significant strength are present in the form of coordinatively unsaturated Zr4+ cations.51 However, in order to make these sites available for reaction, pre-activation of the sample under vacuum above 250 °C is required.52 Due to the limitation on maximal temperature imposed by the working solvent (the activation cannot be performed at a temperature higher than 70 °C at ambient pressure), the strong Lewis acid sites cannot be liberated, thus the productivity of DMC under our experimental conditions is very limited.
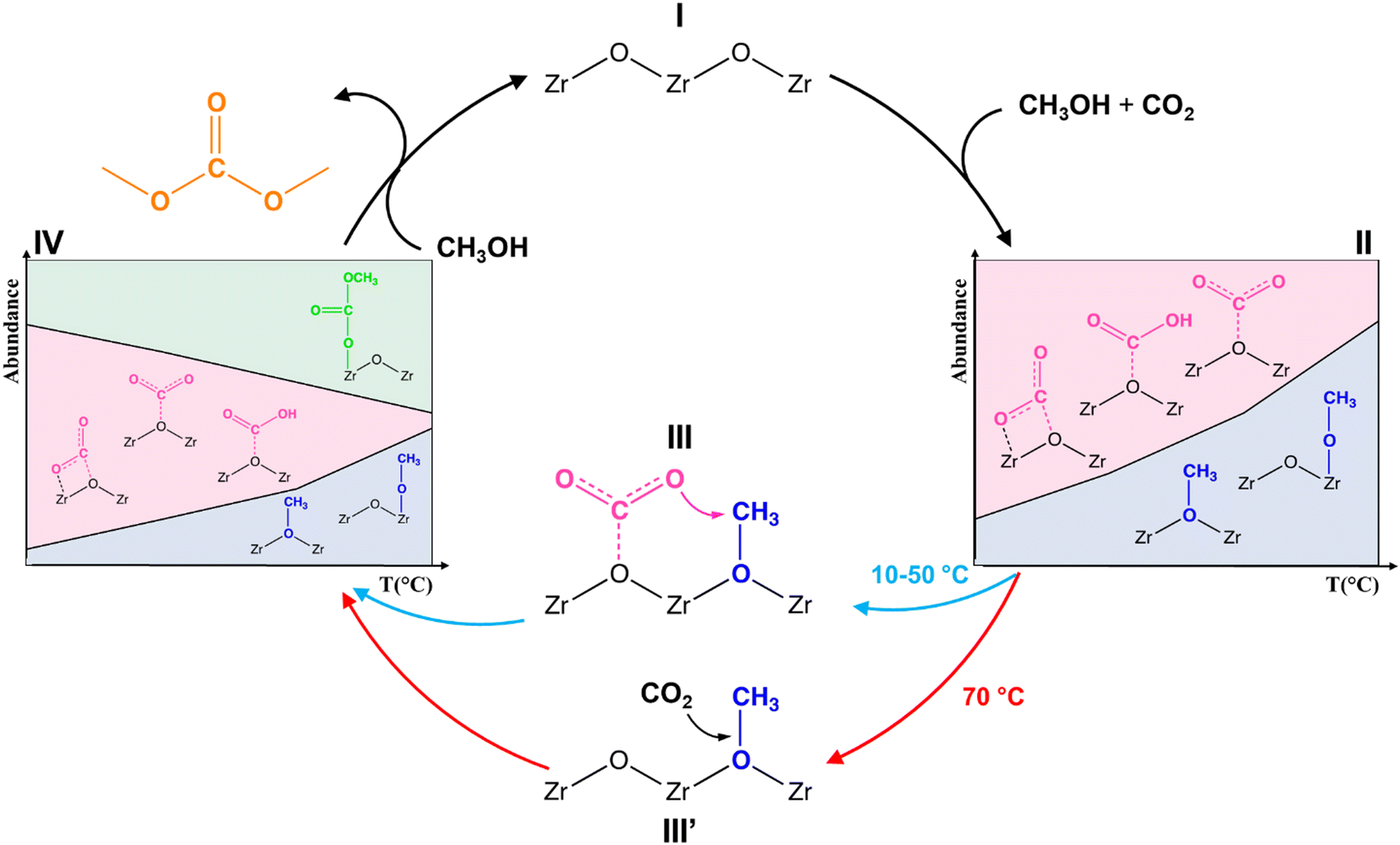 | ||
| Fig. 7 Proposed mechanism of DMC formation from CO2 and CH3OH adsorbed over ZrO2. Diagrams in steps II and IV refer to the concentrations reported in Fig. 6. | ||
Conclusions
MCR-ALS was successfully applied to liquid phase ATR-IR spectra collected during DMC synthesis over ZrO2. Five pure components, together with their respective temporal concentration evolution, were extracted during CO2/MeOH adsorption. Reactant activation was observed to strongly depend on temperature. Low temperatures (<30 °C) favoured formation of stable carbonates limiting methanol activation to its molecular physisorption, hence reducing MMC formation. In contrast, higher temperatures (≥50 °C) improved methanol dissociation, thus forming reactive methoxide species at the expense of the stability of carbonates. Moreover, whilst the coexistence of carbonates and methoxide species at 10–50 °C makes us hypothesize the already known Langmuir–Hinshelwood reaction mechanism involving an interaction between the two surface species, the abundance of methoxides at higher temperature together with the low levels of carbonates and the pronounced MMC formation suggested that the interaction between methoxide and CO2 could occur prior to carbonate formation. This observation may imply that MMC could be produced through an alternative reaction pathway, involving the Eley–Rideal mechanism with the direct interaction between CO2 from the solution and methoxide species, preventing the formation of stable carbonates. This assumption, however, requires to be verified through detailed kinetic studies as carbonates may still form, but they can also be consumed at a high rate to allow their detection. It is noteworthy that for closing the catalytic cycle, DMC decomposition over ZrO2 must be prevented prior to its desorption. This seems to be promoted by the liquid phase. Indeed, the liquid phase ATR-IR experiments demonstrated an increased stability of DMC compared to analogous gas phase experiments, where the decomposition of DMC over ZrO2 occurs. Despite the ambient pressure conditions and the presence of a solvent for these experiments, the detected intermediate species were observed in the whole temperature regime explored, thus lowering the temperature can be considered beneficial in terms of “trapping” species otherwise promptly reacting.Conflicts of interest
There are no conflicts to declare.Acknowledgements
MS acknowledges Compagnia di San Paolo for the financial support to this project, through the “Bando Internazionalizzazione 2018” competitive call, Dr T. Fovanna for indispensable support with the ATR-IR setup construction and data collection, and Dr A. Martini for the unvaluable help provided for the MCR-ALS analysis. DF is thankful to PSI for funding (CROSS project).References
- J. Rockström, W. Steffen, K. Noone, A. Persson, F. S. Chapin, E. F. Lambin, T. M. Lenton, M. Scheffer, C. Folke, H. Joachim, B. Schnellhuber, B. Nykvist, C. A. de Wit, T. Hughes, S. van der Leeuw, H. Rodhe, S. Sörlin, P. K. Snyder, R. Costanza, U. Svedin, M. Falkenmark, L. Karlberg, R. W. Corell, V. J. Fabry, J. Hansen, B. Walker, D. Liverman, K. Richardson, P. Crutzen and J. K. Foley, A safe operating space for humanity, Futur. Nat. Doc. Global Change, 2013, 461, 491–501 Search PubMed.
- R. Cassia, M. Nocioni, N. Correa-Aragunde and L. Lamattina, Climate change and the impact of greenhouse gasses: CO2 and NO, friends and foes of plant oxidative stress, Front. Plant Sci., 2018, 9, 1–11 CrossRef PubMed.
- IPCC, IPCC Third Assessment Report (TAR), Ipcc, 2001, 995.
- X. Wang, F. Zhang, L. Li, H. Zhang and S. Deng, Carbon dioxide capture, 2021, vol. 58 Search PubMed.
- J. Li, Y. Hou, P. Wang and B. Yang, A Review of carbon capture and storage project investment and operational decision-making based on bibliometrics, Energies, 2019, 12, 23 CrossRef CAS.
- P. Styring, D. Jansen, H. de Coninck, H. Reith and K. Armstrong, Carbon Capture and Utilisation in the green economy, 2011 Search PubMed.
- M. Aresta, A. Dibenedetto and A. Angelini, From CO2 to Chemicals, Materials, and Fuels: The Role of Catalysis, Encycl. Inorg. Bioinorg. Chem., 2014, 1–18 CAS.
- The Royal Society, The potential and limitations of using carbon dioxide, R. Soc., 2017, 1–18.
- M. Aresta, Carbon Dioxide as Chemical Feedstock, Wiley-VCH Verlag GmbH & Co. KGaA, 2010 Search PubMed.
- International Energy Agency, Putting CO2 to Use, Energy Rep., 2019, 86 Search PubMed.
- M. Aresta, A. Dibenedetto and A. Angelini, The changing paradigm in CO2 utilization, J. CO2 Util., 2013, 3–4, 65–73 CrossRef CAS.
- T. Tabanelli, D. Bonincontro, S. Albonetti and F. Cavani, Conversion of CO2 to Valuable Chemicals: Organic Carbonate as Green Candidates for the Replacement of Noxious Reactants, Elsevier B.V., 1st edn, 2019, vol. 178 Search PubMed.
- W. Zhou, K. Cheng, J. Kang, C. Zhou, V. Subramanian, Q. Zhang and Y. Wang, New horizon in C1 chemistry: Breaking the selectivity limitation in transformation of syngas and hydrogenation of CO2 into hydrocarbon chemicals and fuels, Chem. Soc. Rev., 2019, 48, 3193–3228 RSC.
- S. Saeidi, N. A. S. Amin and M. R. Rahimpour, Hydrogenation of CO2 to value-added products – A review and potential future developments, J. CO2 Util., 2014, 5, 66–81 CrossRef CAS.
- H. Yang, C. Zhang, P. Gao, H. Wang, X. Li, L. Zhong, W. Wei and Y. Sun, A review of the catalytic hydrogenation of carbon dioxide into value-added hydrocarbons, Catal. Sci. Technol., 2017, 7, 4580–4598 RSC.
- W. Wang, S. Wang, X. Ma and J. Gong, Recent advances in catalytic hydrogenation of carbon dioxide, Chem. Soc. Rev., 2011, 40, 3703–3727 RSC.
- G. A. Olah, G. K. S. Prakash and A. Goeppert, Anthropogenic chemical carbon cycle for a sustainable future, J. Am. Chem. Soc., 2011, 133, 12881–12898 CrossRef CAS PubMed.
- T. Sakakura, J.-C. Choi and H. Yasuda, Transformation of Carbon Dioxide, Chem. Rev., 2007, 107, 2365–2387 CrossRef CAS PubMed.
- M. Aresta, A. Dibenedetto and E. Quaranta, Reaction mechanisms in carbon dioxide conversion, 2015 Search PubMed.
- D. Delledonne, F. Rivetti and U. Romano, Developments in the production and application of dimethylcarbonate, Appl. Catal., A, 2001, 221, 241–251 CrossRef CAS.
- F. Aricò and P. Tundo, Dimethyl carbonate as a modern green reagent and solvent, Russ. Chem. Rev., 2010, 79, 479–489 CrossRef.
- N. Keller, G. Rebmann and V. Keller, Catalysts, mechanisms and industrial processes for the dimethylcarbonate synthesis, J. Mol. Catal. A: Chem., 2010, 317, 1–18 CrossRef CAS.
- A. H. Tamboli, A. A. Chaugule and H. Kim, Catalytic developments in the direct dimethyl carbonate synthesis from carbon dioxide and methanol, Chem. Eng. J., 2017, 323, 530–544 CrossRef CAS.
- P. Kongpanna, V. Pavarajarn, R. Gani and S. Assabumrungrat, Techno-economic evaluation of different CO2-based processes for dimethyl carbonate production, Chem. Eng. Res. Des., 2015, 93, 496–510 CrossRef CAS.
- S. H. Pyo, J. H. Park, T. S. Chang and R. Hatti-Kaul, Dimethyl carbonate as a green chemical, Curr. Opin. Green Sustainable Chem., 2017, 5, 61–66 CrossRef.
- A. H. Tamboli, A. A. Chaugule and H. Kim, Catalytic developments in the direct dimethyl carbonate synthesis from carbon dioxide and methanol, Chem. Eng. J., 2017, 323, 530–544 CrossRef CAS.
- M. Zhang, Y. Xu, B. L. Williams, M. Xiao, S. Wang, D. Han, L. Sun and Y. Meng, Catalytic materials for direct synthesis of dimethyl carbonate (DMC) from CO2, J. Cleaner Prod., 2021, 279, 123344 CrossRef CAS.
- S. Y. Zhao, S. P. Wang, Y. J. Zhao and X. Bin Ma, An in situ infrared study of dimethyl carbonate synthesis from carbon dioxide and methanol over well-shaped CeO2, Chinese Chem. Lett., 2017, 28, 65–69 CrossRef CAS.
- A. H. Tamboli, N. Suzuki, C. Terashima, S. Gosavi, H. Kim and A. Fujishima, Direct dimethyl carbonates synthesis over CeO2 and evaluation of catalyst morphology role in catalytic performance, Catalysts, 2021, 11, 1–16 CrossRef.
- B. A. V. Santos, C. S. M. Pereira, V. M. T. M. Silva, J. M. Loureiro and A. E. Rodrigues, Kinetic study for the direct synthesis of dimethyl carbonate from methanol and CO2 over CeO2 at high pressure conditions, Appl. Catal., A, 2013, 455, 219–226 CrossRef CAS.
- T. Akune, Y. Morita, S. Shirakawa, K. Katagiri and K. Inumaru, ZrO2 Nanocrystals As Catalyst for Synthesis of Dimethylcarbonate from Methanol and Carbon Dioxide: Catalytic Activity and Elucidation of Active Sites, Langmuir, 2018, 34, 23–29 CrossRef CAS PubMed.
- Citation Report graphic, source: https://www.webofscience.com/, (accessed 28 April 2022).
- K. Taek Jung and A. T. Bell, An in situ infrared study of dimethyl carbonate synthesis from carbon dioxide and methanol over zirconia, J. Catal., 2001, 204, 339–347 CrossRef.
- K. T. Jung and A. T. Bell, Effects of catalyst phase structure on the elementary processes involved in the synthesis of dimethyl carbonate from methanol and carbon dioxide over zirconia, Top. Catal., 2002, 20, 97–105 CrossRef CAS.
- C. Gionco, M. C. Paganini, E. Giamello, O. Sacco, V. Vaiano and D. Sannino, Rare earth oxides in zirconium dioxide: How to turn a wide band gap metal oxide into a visible light active photocatalyst, J. Energy Chem., 2017, 26, 270–276 CrossRef.
- J. Rodríguez-Carvajal, Recent developments of the program FULLPROF, Int. Union Crystallogr., Newsl., 2001, 26, 12–19 Search PubMed.
- C. Ruckebusch and L. Blanchet, Multivariate curve resolution: A review of advanced and tailored applications and challenges, Anal. Chim. Acta, 2013, 765, 28–36 CrossRef CAS PubMed.
- A. A. Tsyganenko, L. A. Denisenko, S. M. Zverev and V. N. Filimonov, Infrared study of lateral interactions between carbon monoxide molecules adsorbed on oxide catalysts, J. Catal., 1985, 94, 10–15 CrossRef CAS.
- J. Jaumot, R. Gargallo, A. De Juan and R. Tauler, A graphical user-friendly interface for MCR-ALS: A new tool for multivariate curve resolution in MATLAB, Chemom. Intell. Lab. Syst., 2005, 76, 101–110 CrossRef CAS.
- A. L. Smith, The Coblentz Society Desk Book of Infrared Spectra, The Coblentz Society, Kirkwood, MO, 1982 Search PubMed.
- M. Bensitel, O. Saur and J. C. Lavalley, Use of methanol as a probe to study the adsorption sites of different MgO samples, Mater. Chem. Phys., 1991, 28, 309–320 CrossRef CAS.
- A. J. Tench, D. Giles and J. F. J. Kibblewhite, Adsorption and desorption of methyl alcohol on magnesium oxide, Trans. Faraday Soc., 1971, 67, 854–863 RSC.
- M. Daturi, C. Binet, J. C. Lavalley, A. Galtayries and R. Sporken, Surface investigation on CexZr1−xO2 compounds, Phys. Chem. Chem. Phys., 1999, 1, 5717–5724 RSC.
- C. Binet and M. Daturi, Methanol as an IR probe to study the reduction process in ceria-zirconia mixed compounds, Catal. Today, 2001, 70, 155–167 CrossRef CAS.
- K. Taek Jung and A. T. Bell, An in situ infrared study of dimethyl carbonate synthesis from carbon dioxide and methanol over zirconia, J. Catal., 2001, 204, 339–347 CrossRef.
- C. Morterra and L. Orio, Surface characterization of zirconium oxide. II. The interaction with carbon dioxide at ambient temperature, Mater. Chem. Phys., 1990, 24, 247–268 CrossRef CAS.
- W. Hertl, Surface Chemistry of Zirconia Polymorphs, Langmuir, 1989, 5, 96–100 CrossRef CAS.
- V. Crocellà, G. Cerrato and C. Morterra, On the adsorption/reaction of acetone on pure and sulfate-modified zirconias, Phys. Chem. Chem. Phys., 2013, 15, 13446 RSC.
- H. H. Li, Refractive Index of ZnS, ZnSe, and ZnTe and Its Wavelength and Temperature Derivatives, J. Phys. Chem. Ref. Data, 1984, 13, 103–150 CrossRef CAS.
- D. Shi, S. Heyte, M. Capron and S. Paul, Catalytic processes for the direct synthesis of dimethyl carbonate from CO2 and methanol: A review, Green Chem., 2022, 24, 1067–1089 RSC.
- V. Bolis, G. Cerrato, G. Magnacca and C. Morterra, Surface acidity of metal oxides. Combined microcalorimetric and IR-spectroscopic studies of variously dehydrated systems, Thermochim. Acta, 1998, 312, 63–77 CrossRef CAS.
- C. Morterra, G. Cerrato, V. Bolis, C. Lamberti, L. Ferroni and L. Montanaro, Surface characterization of yttria-stabilized tetragonal ZrO2. Part 2. – Adsorption of CO, J. Chem. Soc., Faraday Trans., 1995, 91, 113–123 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Literature trend on DMC synthesis over ZrO2 and CeO2; Rietveld refinement of PXRD data; volumetric adsorption of N2 at 77K; reagent feeding setup schematic; transmission IR; and MCR-ALS including DMC as the sixth component. See DOI: https://doi.org/10.1039/d2cp05800f |
| This journal is © the Owner Societies 2023 |