
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Why does thionating a carbonyl molecule make it a better electron acceptor?†
Yi-Lin
Wu
 * and
Anna I.
Wright
* and
Anna I.
Wright
School of Chemistry, Cardiff University, Park Place, Cardiff CF10 3AT, UK. E-mail: wuyl@cardiff.ac.uk
First published on 15th December 2022
Abstract
The past decade has witnessed a surge of biomedical and materials applications of thiocarbonyl molecules (R2C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) S), such as in photodynamic therapy, organic field-effect transistors, and rechargeable batteries. The success of these applications originates from thiocarbonyl's small optical gap in the visible region and the enhanced electron affinity compared to the carbonyl analogues (R2CO). Although these observations seem to be contrary to the implication based on a simple electronegativity consideration (2.58 for sulfur and 3.44 for oxygen), a natural bond orbital (NBO) analysis gives a straightforward explanation for the LUMO-lowering effect of CO → CS substitution. In comparison to the valence (2p)C/(2p)O interactions in CO, the higher 3p orbital of sulfur and its weaker overlap with the 2p level of carbon result in a weaker antibonding interaction in
S), such as in photodynamic therapy, organic field-effect transistors, and rechargeable batteries. The success of these applications originates from thiocarbonyl's small optical gap in the visible region and the enhanced electron affinity compared to the carbonyl analogues (R2CO). Although these observations seem to be contrary to the implication based on a simple electronegativity consideration (2.58 for sulfur and 3.44 for oxygen), a natural bond orbital (NBO) analysis gives a straightforward explanation for the LUMO-lowering effect of CO → CS substitution. In comparison to the valence (2p)C/(2p)O interactions in CO, the higher 3p orbital of sulfur and its weaker overlap with the 2p level of carbon result in a weaker antibonding interaction in 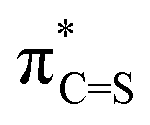 NBO, a prominent contributor to the LUMO. Such an analysis also provides a semi-quantitative understanding of the electronic effect of substituents on or in π-conjugation with a (thio)carbonyl functionality. The intuitive concepts uncovered here offer a simple rule to predict the electronic properties of π-conjugated molecules that incorporate heavy heteroelements and would facilitate materials development.
NBO, a prominent contributor to the LUMO. Such an analysis also provides a semi-quantitative understanding of the electronic effect of substituents on or in π-conjugation with a (thio)carbonyl functionality. The intuitive concepts uncovered here offer a simple rule to predict the electronic properties of π-conjugated molecules that incorporate heavy heteroelements and would facilitate materials development.
Introduction
The discovery of Lawesson's reagent1,2 for replacing oxygen with sulfur in a carbonyl functionality marks the boost of research interests in thiocarbonyl molecules (R2CS, also known as thiones) in the second half of the 20th century. Early studies focused on their reactivities;3 it was also quickly recognised that the easily accessible π–π* and n–π* excited states present thiocarbonyls with rich photophysics and intramolecular photochemistry.4–6 While these seminal reports revealed the fundamental properties of thiocarbonyl compounds, their applications in biomedical and materials chemistry were only uncovered recently.
The rapid singlet-to-triplet intersystem crossing of photoexcited thiocarbonyl compounds enables their use to sensitise oxygen into the reactive singlet form (1O2) to induce targeted cellular damage. This photosensitisation process can be achieved using site-selectively modified nucleobases or synthetic chromophores with heavy-atom-free thiocarbonyl functionalities.7–10 Given the long lifetime and high energy of the triplet excited states, thiocarbonyl chromophores also display photocatalytic activities comparable to common organometallic photocatalysts.11 For materials applications, Seferos and co-workers reported that the carrier mobility of n-type organic field-effect transistors (OFET) based on perylene diimide derivatives can be enhanced by thionation.12 Subsequently, performance enhancement has been observed for OFET using other thionated organic semiconductors, such as naphthalene diimide13–15 and iso-diketopyrrolopyrrole.16,17 Successful applications of thiocarbonyls for photovoltaic devices have also been demonstrated.17–19 Due to their good redox activity, electrical conductivity, and cycling stability, thionated organic semiconductors were also found to be promising cathode materials in lithium-ion batteries.20–22
It is conceivable that the versatile applications of thiocarbonyl compounds stem from their low optical gap (usually in the visible region) and enhanced electron affinity compared to carbonyl materials. Both properties are related to the lower energy level of the lowest unoccupied molecular orbital (LUMO), which often manifests experimentally in the more positive electrochemical reduction potentials of thiocarbonyls. Indeed, modifying the aromatic imide/amide electron acceptors into the corresponding thiocarbonyls resulted in a marked positive shift of the reduction potential by several hundred millivolts per thionation (Fig. 1; see Section 1 in ESI† for other examples).23–26 These results, together with some earlier electrochemical data of non-chromophoric thiocarbonyls, suggest that the enhanced electron affinity is a general feature for this class of molecule. However, besides a proposal based on polarity comparison,27 there is no discussion on the origin of the LUMO-lowering effect by thionation to the best of our knowledge. In fact, the enhanced electron affinity of thiocarbonyls might be seen as counterintuitive if one considers the lower electronegativity of sulfur (2.58 on the Pauling scale) than oxygen (3.44).28–31
 | ||
| Fig. 1 Comparison of electrochemical reduction potential (EREDvs. SCE) and LUMO energy (ELUMO, if reported) of selected (thio)imide and (thio)amide chromophores.23–26 | ||
Since the electron affinity, photoredox driving force, or electron transportation mobility of these thiocarbonyl materials are critically related to the LUMO level, it is important to elucidate the origin of the enhanced electron-accepting power from a theoretical point of view. An insightful understanding will facilitate the development of high-performance materials. By recognising the LUMO of all molecules in Fig. 1 features π-antibonding between the C and O/S centres of (thio)carbonyl, we took a minimalist approach and focused on the construction of the π-antibonding orbital  (X = O or S) using the natural bond orbital (NBO) analysis.32 The LUMO-lowering effect of thiocarbonyls was found to originate from the weak interaction between (2p)C and (3p)S orbitals. We also demonstrate that this simple analysis can provide instinctive rationalisation to the effect of π-conjugated substituents on the electron-accepting power of (thio)carbonyl derivatives.
(X = O or S) using the natural bond orbital (NBO) analysis.32 The LUMO-lowering effect of thiocarbonyls was found to originate from the weak interaction between (2p)C and (3p)S orbitals. We also demonstrate that this simple analysis can provide instinctive rationalisation to the effect of π-conjugated substituents on the electron-accepting power of (thio)carbonyl derivatives.
Method
Since the electrochemical reduction potential (ERED), as a measure of the electron-accepting power of a molecule, is empirically found to correlate with the energy of lowest unoccupied molecular orbitals (ELUMO) computed by density functional theory (DFT),33,34 we analyse the title question by examining the effect of CO → CS substitution on LUMO. This canonical molecular orbital can be expressed in terms of a complete orthonormal set of localised NBOs. For (thio)carbonyl compounds, such a combination can be explicitly written as: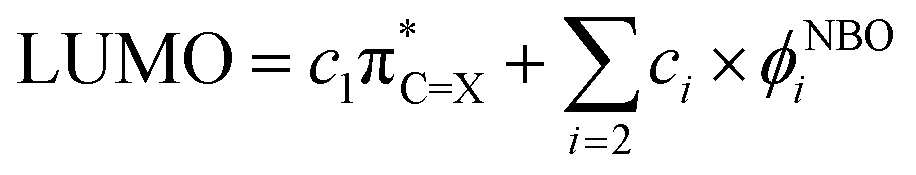 | (1) |
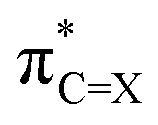 (X = O or S) is the antibonding π* NBO of the (thio)carbonyl functional group, and ϕNBOi are other NBOs of the molecule, often also of the π* antibonding character or lone-pair orbitals of the π symmetry. The coefficient ci represents the percentage (100 × ci2) of each NBO to LUMO. Furthermore, the
(X = O or S) is the antibonding π* NBO of the (thio)carbonyl functional group, and ϕNBOi are other NBOs of the molecule, often also of the π* antibonding character or lone-pair orbitals of the π symmetry. The coefficient ci represents the percentage (100 × ci2) of each NBO to LUMO. Furthermore, the 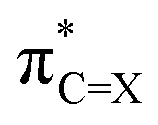 NBO is composed of the natural atomic hybrid orbitals (NHOs) of carbon and oxygen/sulfur (hC and hX, respectively) of essentially pure (>99%) p character (pπ, which would be pz if the molecule is set at the xy plane):
NBO is composed of the natural atomic hybrid orbitals (NHOs) of carbon and oxygen/sulfur (hC and hX, respectively) of essentially pure (>99%) p character (pπ, which would be pz if the molecule is set at the xy plane): | (2a) |
 | (2b) |
By defining the O/S atom and the rest of the molecule as two fragments, the composition of molecular orbitals (MOs) can alternatively be expressed in terms of fragment orbitals (FOs) in a linear combination of fragment orbitals (LCFO-MO) approach. Complementary to the NBO analysis, the effect of O/S on the LUMO was examined using this orbital composition analysis considering the entire molecule as opposed to the local (thio)carbonyl functionality. The analysis was performed using Multiwfn version 3.8(dev)38 based on the same DFT-optimised molecules. The coordinates of the fragments were extracted from the optimised whole molecules without re-optimisation, and the wavefunctions of these three systems were then computed considering their respective spin configuration. FOs contribution to each MO was evaluated using the charge decomposition analysis (CDA)39,40 module with the Mulliken method implemented in Multiwfn.
The electrochemical reduction potentials for discussion are the half-wave potentials quoted from the literature and referenced against the saturated calomel electrode (SCE), using known conversion factors,41 if necessary, for values reported against other references, such as the ferrocenium/ferrocene couple.
Results and discussion
Since all the examples in Fig. 1 are aromatic (thio)imides and (thio)amides, we would like to first verify if structurally simpler thiocarbonyls also possess stronger electron-accepting power. Our analysis begins with propan-2-one (1a, acetone) and propan-2-thione (1b). Electrochemical reduction potentials of these two molecules have not been reported but can be estimated to be at ERED < −2.1 V for 1a42 and ERED = −1.99 V for 1b43 (Table 1). Consistent with these values, DFT calculations at the level of ωB97X-D/def2-TZVP reveal a higher LUMO energy for 1a (1.58 eV) than 1b (−0.05 eV).|
|
|||||
|---|---|---|---|---|---|
| |〈hC|F|hO〉| (eV) | |〈hC|F|hS〉| (eV) | E LUMO (eV) | E RED (V vs. SCE) | Ref. | |
| a Value of thiofenchone. b Values of N-(3-pentyl) molecules, E(Fc+/Fc) = +0.46 V41 used for conversion. c Two |〈hC|F|hO〉| are 6.77 and 6.78 eV. d Values of 2-piperdino-N-phenyl molecules. | |||||
| 1a | 7.21 | 1.58 | <−2.1 | 42 | |
| 1b | 5.44 | −0.05 | −1.99a | 43 | |
| 2a | 7.13 | −0.71 | −1.14 | 44 | |
| 2b | 5.31 | −1.44 | −0.82 | 44 | |
| 3a | 6.83 | −0.93 | −1.36b | 45 | |
| 3b | 6.86 | 4.84 | −1.31 | −0.98b | 45 |
| 4a | 6.78c | −0.49 | −1.52d | 11 | |
| 4b | 6.83 | 4.79 | −0.84 | −1.20d | 11 |

Comparison between the LUMO and 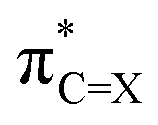 (X = O or S) clearly shows the predominant contribution of this antibonding NBO to LUMO (Fig. 2, 79% for 1a and 83% for 1b; see also Fig. S1, ESI† for the LUMO of other molecules). Analysis of the composing atomic hybrids to this NBO should reveal the origin of the lower
(X = O or S) clearly shows the predominant contribution of this antibonding NBO to LUMO (Fig. 2, 79% for 1a and 83% for 1b; see also Fig. S1, ESI† for the LUMO of other molecules). Analysis of the composing atomic hybrids to this NBO should reveal the origin of the lower  energy (0.69 eV) compared to
energy (0.69 eV) compared to  (2.62 eV). As illustrated in Fig. 3a, the energy of the atomic hybrids follows the sequence of hC > hS > hO (or basically (pπ)C > (pπ)S > (pπ)O), in line with the common anticipation based on the electronegativity of these elements. The extent of interaction between the composing NHOs, represented by the off-diagonal Fock (Kohn–Sham) matrix element, is smaller between hC and hS (|〈hC|F|hS〉| = 5.44 eV for 1b, Table 1) than between hC and hO (|〈hC|F|hO〉| = 7.21 eV for 1a). The weaker interaction between hC and hS can be understood considering (i) the longer CS bond length and (ii) mismatch between the (2pπ)C and (3pπ)S orbitals, resulting in the weaker (2pπ)C/(3pπ)S overlap, compared to the stronger (2pπ)C/(2pπ)O overlap. Consequently, the
(2.62 eV). As illustrated in Fig. 3a, the energy of the atomic hybrids follows the sequence of hC > hS > hO (or basically (pπ)C > (pπ)S > (pπ)O), in line with the common anticipation based on the electronegativity of these elements. The extent of interaction between the composing NHOs, represented by the off-diagonal Fock (Kohn–Sham) matrix element, is smaller between hC and hS (|〈hC|F|hS〉| = 5.44 eV for 1b, Table 1) than between hC and hO (|〈hC|F|hO〉| = 7.21 eV for 1a). The weaker interaction between hC and hS can be understood considering (i) the longer CS bond length and (ii) mismatch between the (2pπ)C and (3pπ)S orbitals, resulting in the weaker (2pπ)C/(3pπ)S overlap, compared to the stronger (2pπ)C/(2pπ)O overlap. Consequently, the  energy is lower than
energy is lower than  , resulting in a lower LUMO of 1b.
, resulting in a lower LUMO of 1b.
 | ||
Fig. 2 LUMO (a),  NBO (b), and hC and hS NHOs of NBO (b), and hC and hS NHOs of  (c) of propan-2-thione (1b). Colour code for molecular structure: C = grey, H = white, S = yellow. Orbitals are plotted at isosurface = 0.04 a.u. (c) of propan-2-thione (1b). Colour code for molecular structure: C = grey, H = white, S = yellow. Orbitals are plotted at isosurface = 0.04 a.u. | ||
 | ||
Fig. 3 (a) Development of bonding  and antibonding and antibonding  NBOs of a (thio)carbonyl functional group from their atomic hybrids hC and hX NHOs. Left: X = O, right: X = S. (b) Orbital interaction diagrams for the formation of frontier MOs of 1 considering the FOs of O/S atom and propan-2-ylidene. Dashed lines connect MOs to FOs that show contributions greater than >15%. NBOs of a (thio)carbonyl functional group from their atomic hybrids hC and hX NHOs. Left: X = O, right: X = S. (b) Orbital interaction diagrams for the formation of frontier MOs of 1 considering the FOs of O/S atom and propan-2-ylidene. Dashed lines connect MOs to FOs that show contributions greater than >15%. | ||
An alternative way to track the effect of O/S substitution on LUMO energy can be done by evaluating the contribution of the fragment orbitals (FOs) of O/S and the rest of the molecule to the overall molecular orbitals (MOs).39,40 We consider the O/S and propan-2-ylidene fragments both in the triplet configuration, without losing generality, to form the CX double bond in 1a and 1b (Fig. 3b). The LUMO, composed of the FOs of the π symmetry, is raised above the highest singly occupied FO of propan-2-ylidene. The extent of energy increase (ΔE in Fig. 3b, signifying the antibonding character of LUMO) is smaller for 1b than 1a. This reflects a weaker overlap between (2pπ)C/(3pπ)S orbitals than (2pπ)C/(2pπ)O and is consistent with the NBO/NHO analysis discussed above. It is of interest to note that the HOMO of 1b is primarily contributed by the lone pair on sulfur. This unique characteristic should be a consequence of the higher 3p level of S than 2p of O, contributing to the rich photochemical processes of thiocarbonyl's 1(n–π) excited state.5,49 Despite the wealth of information available in orbital interaction diagrams, we will, however, focus on NBO analysis in the rest of this study for its simple and straightforward chemical picture.
Fluoren-9-one (2a) and fluoren-9-thione (2b) were selected as the model system to analyse the effect of thionation on the electron-accepting power of aromatic molecules (Table 1). Due to conjugation with the aromatic units, the reduction potential of 2 is easier to measure under common electrochemical conditions; ERED = −0.82 V was reported for 2b, which is less negative than 2a (ERED = −1.14 V).44 The easier reduction of the thiocarbonyl is, again, consistent with its lower LUMO energy (−0.71 eV for 2a and −1.44 eV for 2b).
Although the LUMO of 2 delocalises to the benzene units, since the 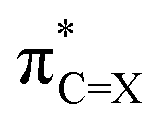 NBO has a significant contribution (35% for 2a and 52% for 2b) and is the only differing NBO component contributing to LUMO, it is instructive to analyse the atomic hybrids of
NBO has a significant contribution (35% for 2a and 52% for 2b) and is the only differing NBO component contributing to LUMO, it is instructive to analyse the atomic hybrids of  . Similar to the aliphatic (thio)carbonyl, a smaller |〈hC|F|hS〉| was found for 2b, resulting in the lower
. Similar to the aliphatic (thio)carbonyl, a smaller |〈hC|F|hS〉| was found for 2b, resulting in the lower  energy, in turn a lower LUMO compared to 2a.
energy, in turn a lower LUMO compared to 2a.
Analyses of aromatic imides 3 and 4 also reveal a smaller |〈hC|F|hS〉| ≅ 4.8 eV than |〈hC|F|hO〉| ≅ 6.8 eV of the (thio)carbonyl units. This result is consistent with that found for 1 and 2 and explains the lower LUMO and easier reduction of thioimides (Table 1 and Fig. 1). Furthermore, since 3b and 4b contain both CO and CS units in the same molecule, their NHO Fock matrix elements were evaluated on the same ground, adding to the reliability of the above comparison. It is worth noting that  NBO often shows a high contribution to LUMO (∼24% for
NBO often shows a high contribution to LUMO (∼24% for  , relative to ∼12% for
, relative to ∼12% for  in the case of 3 and 4; see also above for 2). The significant weight of
in the case of 3 and 4; see also above for 2). The significant weight of  reflects the pronounced LUMO-lowering effect by CO → CS substitution, and is consistent with the positive shift in g values of the radical anion of these molecules, resultant from a large spin density on the heavy sulfur atom.45,46,50
reflects the pronounced LUMO-lowering effect by CO → CS substitution, and is consistent with the positive shift in g values of the radical anion of these molecules, resultant from a large spin density on the heavy sulfur atom.45,46,50
Taken together, the NBO analysis of (thio)carbonyl 1–4 indicates that the LUMO-lowering of these molecules originates from weaker (2pπ)C/(3pπ)S interactions, despite sulfur in CS being less electronegative than oxygen in CO (see also Fig. S2–S4 in ESI† for the effect of electronegativity on molecular electrostatic potential surface and Mulliken charges). This analysis also confirms that thiocarbonyl's stronger electron-accepting power (or easier reduction) is a general phenomenon but not a result of particular substituent patterns. We should point out that we have chosen molecules with nearly planar structures. Such a selection is only for the ease of visualisation of NBOs of the σ and π characters; it would not affect the discussion. Inspired by the effect of LUMO-lowering from π-type atomic hybrids, we wondered what the electronic effect would be if the O → S exchange takes place on a site π-conjugated to carbonyl but not on carbonyl itself.
Using methyl ester [RC(O)OCH3] and methyl thioester [RC(O)SCH3] as the example, our above query can be reformulated as comparing the electron withdrawing/donating power of –OCH3 and –SCH3 substituents. In this regard, it is conceivable that Hammett substituent constants σp could be used for estimation, since these substituents are π-conjugated with the carbonyl functionality. It was reported that –OCH3 (σp = −0.27) displays a moderate electron-donating character, whereas –SCH3 (σp = 0.00) in comparison is somewhat electron withdrawing. This observation is, again, contrary to the simple consideration based on electronegativity.
To investigate the electronic effect of conjugated O/S, we used isochromen-1-one (5a) and isothiochromen-1-one (6a), whose reduction potentials have been reported, as the model system for comparison (Table 2).46 The “thioester” version of the isochromenone derivative (6a) displays a noticeably less negative ERED = −1.27 V compared to ERED = −1.41 V for “ester” 5a. The enhancement in the electron-accepting power by –C(O)O– → –C(O)S– is clear and consistent with the implication of the Hammett substituent constants σp of –OCH3 and –SCH3 and the relative LUMO energy.
|
|
|||||||
|---|---|---|---|---|---|---|---|
| |〈hC1|F|hO1〉| (eV) | |〈hC1|F|hS1〉| (eV) | |〈hC|F|hO2〉| (eV) | |〈hC|F|hS2〉| (eV) | E LUMO (eV) | E RED (V vs. SCE) | Ref. | |
| a Superscripted 1 and 2 in the chemical structure are for indexing purpose. b Natural hybrid orbital hC of the (thio)carbonyl C1 atom. c h C of the aromatic C2 atom. | |||||||
| 5a | 6.99 | 4.11b | −0.11 | −1.41 | 46 | ||
| 5b | 4.93 | 4.19b | −0.49 | −1.07 | 46 | ||
| 6a | 6.86 | 3.16b | −0.16 | −1.27 | 46 | ||
| 6b | 4.76 | 3.46b | −0.79 | −0.72 | 46 | ||
| 7a | 6.91 | 4.11c | −0.33 | −1.65 | 47 | ||
| 7b | 4.98 | 4.14c | −1.03 | −1.12 | 47 | ||
| 8a | 6.91 | 3.16c | −0.35 | −1.62 | 48 | ||

While the  NBO contributes significantly to the LUMO of 5a and 6a (20%, along with other antibonding CC NBOs), the lone-pair NBO of the conjugated O/S atom also has 5–9% importance. This lone-pair orbital orients perpendicular to the molecular plane, adopting the π symmetry, and can mix with the (2pπ)C of carbonyl (Fig. 4).51 The off-diagonal Fock matrix elements between these π-symmetric atomic hybrid hC of carbonyl and hO2/hS2 of σ-bonded O/S is smaller for 6a (|〈hC1|F|hS2〉| = 3.16 eV) than for 5a (|〈hC1|F|hO2〉| = 4.11 eV), reflecting again the weaker (2pπ)C/(3pπ)S overlap (Table 2). This smaller |〈hC1|F|hS2〉| results in a lower LUMO energy, an interpretation derived similarly to the above NHO Fock matrix analysis for CO/CS molecules (1–4). Therefore, even the S lone-pair orbital of “thioester” is not formally part of
NBO contributes significantly to the LUMO of 5a and 6a (20%, along with other antibonding CC NBOs), the lone-pair NBO of the conjugated O/S atom also has 5–9% importance. This lone-pair orbital orients perpendicular to the molecular plane, adopting the π symmetry, and can mix with the (2pπ)C of carbonyl (Fig. 4).51 The off-diagonal Fock matrix elements between these π-symmetric atomic hybrid hC of carbonyl and hO2/hS2 of σ-bonded O/S is smaller for 6a (|〈hC1|F|hS2〉| = 3.16 eV) than for 5a (|〈hC1|F|hO2〉| = 4.11 eV), reflecting again the weaker (2pπ)C/(3pπ)S overlap (Table 2). This smaller |〈hC1|F|hS2〉| results in a lower LUMO energy, an interpretation derived similarly to the above NHO Fock matrix analysis for CO/CS molecules (1–4). Therefore, even the S lone-pair orbital of “thioester” is not formally part of  as the Lewis structure represents, its weaker coupling to the carbonyl still makes the LUMO lower compared to the “ester” analogue (Section 5 in ESI†). While dπ–pπ interactions have been proposed for sulfur-containing molecules,52 such an effect is not obvious in all molecules under investigation, likely due to the high energy of sulfur's 3d level. The possibility of such an interaction will be probed in our future study. We also note that using the planar and cyclic model system simplifies the visualisation and avoids conformation complexity; we can generally expect thioester-type molecules to have a stronger electron-accepting power based on this analysis.
as the Lewis structure represents, its weaker coupling to the carbonyl still makes the LUMO lower compared to the “ester” analogue (Section 5 in ESI†). While dπ–pπ interactions have been proposed for sulfur-containing molecules,52 such an effect is not obvious in all molecules under investigation, likely due to the high energy of sulfur's 3d level. The possibility of such an interaction will be probed in our future study. We also note that using the planar and cyclic model system simplifies the visualisation and avoids conformation complexity; we can generally expect thioester-type molecules to have a stronger electron-accepting power based on this analysis.
 | ||
Fig. 4 Top row: LUMO (a),  NBO (b), lone-pair NBO of sulfur (c), and hC & hO NHOs of NBO (b), lone-pair NBO of sulfur (c), and hC & hO NHOs of  and lone-pair hS NHO of sulfur (d) of isothiochromen-1-one (6a). Bottom row: LUMO (e), and lone-pair hS NHO of sulfur (d) of isothiochromen-1-one (6a). Bottom row: LUMO (e),  NBO (f), lone-pair NBO of sulfur (g), and hC & hO NHOs of NBO (f), lone-pair NBO of sulfur (g), and hC & hO NHOs of  and lone-pair hS NHO of sulfur (h) of thioxanthen-9-one (8a). The π-symmetric NHO of the carbon next to S (C2 in Table 2) is also shown in panel h. Colour code for molecular structure: C = grey, H = white, O = red, S = yellow. Orbitals are plotted at isosurface = 0.04 a.u. and lone-pair hS NHO of sulfur (h) of thioxanthen-9-one (8a). The π-symmetric NHO of the carbon next to S (C2 in Table 2) is also shown in panel h. Colour code for molecular structure: C = grey, H = white, O = red, S = yellow. Orbitals are plotted at isosurface = 0.04 a.u. | ||
In comparison to CO → CS replacement, the effect on ERED/LUMO is weaker for –C(O)O– → –C(O)S–, as the O/S atom is not directly modifying the  orbital. Therefore, among the four possible S-replaced isochromenone derivatives, ERED was found to be 6b > 5b > 6a > 5a, with doubly thionated 6b displaying the greatest electron-accepting power. The effect on the LUMO of these formally σ-bonded O/S decreases, as one would expect, when it is further away from the carbonyl. Using xanthen-9-one (7a) and thioxanthen-9-one (8a) for illustration, the |〈hC2|F|hS2〉| between the π hybrids of the heteroatom and the directly attached carbon is smaller for 8a, consistent with the above analysis (Table 2); however, the ERED and ELUMO are found to be comparable for these two molecules.47,48 Such a similarity indicates marginal enhancement of the electron-accepting power by thionation at this remote position. In fact, the lone-pair NBO of the conjugated O/S atom shows a minimal contribution to LUMO (2–3% for 7 and 8, see also the small MO lobes on sulfur in Fig. 4e). This comparison highlights the limitation of thionation for LUMO-lowering and the explanation power of simple NBO analysis.
orbital. Therefore, among the four possible S-replaced isochromenone derivatives, ERED was found to be 6b > 5b > 6a > 5a, with doubly thionated 6b displaying the greatest electron-accepting power. The effect on the LUMO of these formally σ-bonded O/S decreases, as one would expect, when it is further away from the carbonyl. Using xanthen-9-one (7a) and thioxanthen-9-one (8a) for illustration, the |〈hC2|F|hS2〉| between the π hybrids of the heteroatom and the directly attached carbon is smaller for 8a, consistent with the above analysis (Table 2); however, the ERED and ELUMO are found to be comparable for these two molecules.47,48 Such a similarity indicates marginal enhancement of the electron-accepting power by thionation at this remote position. In fact, the lone-pair NBO of the conjugated O/S atom shows a minimal contribution to LUMO (2–3% for 7 and 8, see also the small MO lobes on sulfur in Fig. 4e). This comparison highlights the limitation of thionation for LUMO-lowering and the explanation power of simple NBO analysis.
Finally, it is worth pointing out that the use of π-symmetric NHO hC,O,S in this study is reminiscent of the Hückel molecular orbital (HMO) theory. In the Hamiltonian matrix of HMO calculations, the Coulomb integral αC represents the energy of individual (2pπ)c atomic orbital of carbon, and the resonance integral βCC characterises the coupling between the neighbouring (2pπ)c orbitals. By modifying the integral values of carbon through αX = αC + kX × βCC and βXY = kXY × βCC, the property of the frontier orbitals of heteroatom-containing π systems can be estimated in the Hückel framework (kX and kXY are proportional constants for heteroatom X). The resonance integral βCX of (thio)carbonyl can be conceptually related to 〈hC|F|hX〉 in the present study. Values of βCO = 1.06βCC and βCS = 0.81βCC have been suggested by Van-Catledge.53 The smaller βCS is in agreement with the smaller |〈hC|F|hS〉| in Tables 1 and 2, and the ratio of |〈hC|F|hO〉|/|〈hC|F|hS〉| similar to βCO/βCS. Therefore, it may be suggested that the HMO analysis of the title problem could provide a qualitative understanding with sufficient accuracy.
Despite the simplicity and elegance of the HMO method, its treatment of heteroatom relies heavily on the parameters it uses and the underlying assumptions. For instance, in the same report by Van-Catledge, βC–O = 0.66βCC and βC–S = 0.69βCC were suggested between pπ orbitals of singly-bonded C and O/S atoms;53 the larger βC–S is contrary to the Fock matrix elements in Table 2 and the anticipation for the weaker 2p/3p orbital interactions. Furthermore, a wide range of kCO = 1–2 can be found in the collection of HMO parameters curated by Purcell and Singer.54 The ambiguity in parameter selection obscures the use of HMO method in the present study. We therefore recommend using the DFT-based NBO/NHO analysis to obtain a more reliable understanding of the electronic effect of heteroatom substitution on π-conjugated molecules. This computationally economic analysis provides intuitive and clear chemical insights similar to what one might obtain from HMO without the aforementioned problems.
Conclusions
The origin of the superior electron affinity of thionated carbonyls was elucidated by NBO analyses. The use of localised orbitals (NBOs and NHOs) for analysing global molecular properties (electron-accepting power) is semi-quantitatively satisfactory as the NBO contributes predominantly to the LUMO of (thio)carbonyl molecules. Despite the lower electronegativity of sulfur (hence higher atomic orbital levels), the smaller (2pπ)C/(3pπ)S interaction results in the weaker antibonding character in
NBO contributes predominantly to the LUMO of (thio)carbonyl molecules. Despite the lower electronegativity of sulfur (hence higher atomic orbital levels), the smaller (2pπ)C/(3pπ)S interaction results in the weaker antibonding character in  , lower LUMO level, and less negative reduction potential of thiocarbonyl molecules compared with the corresponding carbonyls. This chemically straightforward interpretation/analysis can be extended to understand the enhanced electron-accepting power of carbonyl derivatives with π-conjugated sulfur atoms. The focus on π-symmetric atomic hybrids in this NBO analysis shares the same philosophy as the Hückel theory but avoids relying on the empirical parameters. Therefore, the present strategy can be applied to a broader range of heteroatom-containing molecules or materials.55,56 Once the effect of heteroatom is evaluated and understood at the atomic level using a model system, this knowledge will provide a useful basis for rapid estimation of the molecular reactivity and electronic property induced by uncommon functional groups, such as acyl metalloids and phosphine sulfides,57,58 and facilitate materials discovery.
, lower LUMO level, and less negative reduction potential of thiocarbonyl molecules compared with the corresponding carbonyls. This chemically straightforward interpretation/analysis can be extended to understand the enhanced electron-accepting power of carbonyl derivatives with π-conjugated sulfur atoms. The focus on π-symmetric atomic hybrids in this NBO analysis shares the same philosophy as the Hückel theory but avoids relying on the empirical parameters. Therefore, the present strategy can be applied to a broader range of heteroatom-containing molecules or materials.55,56 Once the effect of heteroatom is evaluated and understood at the atomic level using a model system, this knowledge will provide a useful basis for rapid estimation of the molecular reactivity and electronic property induced by uncommon functional groups, such as acyl metalloids and phosphine sulfides,57,58 and facilitate materials discovery.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was undertaken using the supercomputing facilities at Cardiff University operated by Advanced Research Computing at Cardiff (ARCCA) on behalf of the Cardiff Supercomputing Facility and the HPC Wales and Supercomputing Wales (SCW) projects. We acknowledge the support of the latter, which is part-funded by the European Regional Development Fund (ERDF) via the Welsh Government. We thank the financial support from the School of Chemistry at Cardiff University and the UK Engineering and Physical Sciences Research Council (EPSRC) through grants EP/W03431X/1. A. I. W. is supported by EPSRC through Doctoral Training Partnerships (DTP). Y.-L. W. thanks Professors K. D. M. Harris and J. A. Platts (Cardiff) for fruitful discussion.Notes and references
- I. Thomsen, K. Clausen, S. Scheibye and S. O. Lawesson, Org. Synth., 1984, 62, 158–164 CrossRef.
- T. Ozturk, E. Ertas and O. Mert, Chem. Rev., 2007, 107, 5210–5278 CrossRef.
- P. Metzner, in Organosulfur Chemistry I, ed. P. C. B. Page, Springer, Berlin, Heidelberg, 1999, pp. 127–181 Search PubMed.
- R. P. Steer and V. Ramamurthy, Acc. Chem. Res., 1988, 21, 380–386 CrossRef.
- J. D. Coyle, Tetrahedron, 1985, 41, 5393–5425 CrossRef.
- A. Ohno, Y. Ohnishi and G. Tsuchihashi, J. Am. Chem. Soc., 1969, 91, 5038–5045 CrossRef.
- V.-N. Nguyen, Y. Yan, J. Zhao and J. Yoon, Acc. Chem. Res., 2021, 54, 207–220 CrossRef CAS.
- L. A. Ortiz-Rodríguez and C. E. Crespo-Hernández, Chem. Sci., 2020, 11, 11113–11123 RSC.
- T.-G. Chen, X.-Q. Zhang, J.-F. Ge, Y.-J. Xu and R. Sun, Spectrochim. Acta, Part A, 2022, 270, 120783 CrossRef CAS PubMed.
- Y. L. Lee, Y. T. Chou, B. K. Su, C. C. Wu, C. H. Wang, K. H. Chang, J. A. A. Ho and P. T. Chou, J. Am. Chem. Soc., 2022, 144, 17249–17260 CrossRef CAS PubMed.
- A. I. Wright, B. M. Kariuki and Y.-L. Wu, Eur. J. Org. Chem., 2021, 4647–4652 CrossRef CAS.
- A. J. Tilley, C. Guo, M. B. Miltenburg, T. B. Schon, H. Yan, Y. Li and D. S. Seferos, Adv. Funct. Mater., 2015, 25, 3321–3329 CrossRef CAS.
- A. Welford, S. Maniam, E. Gann, L. Thomsen, S. J. Langford and C. R. McNeill, Org. Electron., 2018, 53, 287–295 CrossRef CAS.
- D. Zheng, M. Zhang and G. Zhao, Phys. Chem. Chem. Phys., 2017, 19, 28175–28181 RSC.
- D. Nava, Y. Shin, M. Massetti, X. Jiao, T. Biskup, M. S. Jagadeesh, A. Calloni, L. Duò, G. Lanzani, C. R. McNeill, M. Sommer and M. Caironi, ACS Appl. Energy Mater., 2018, 1, 4626–4634 CrossRef PubMed.
- H. Zhang, K. Yang, K. Zhang, Z. Zhang, Q. Sun and W. Yang, Polym. Chem., 2018, 9, 1807–1814 RSC.
- D. Gendron, F. Maasoumi, A. Armin, K. Pattison, P. L. Burn, P. Meredith, E. B. Namdas and B. J. Powell, RSC Adv., 2017, 7, 10316–10322 RSC.
- H. Zhang, M. Liu, W. Yang, L. Judin, T. I. Hukka, A. Priimagi, Z. Deng and P. Vivo, Adv. Mater. Interfaces, 2019, 6, 1901036 CrossRef.
- K. Rundel, Y.-h Shin, A. S. R. Chesman, A. C. Y. Liu, A. Welford, L. Thomsen, M. Sommer and C. R. McNeill, J. Phys. Chem. C, 2019, 123, 12062–12072 CrossRef CAS.
- B. Zhang, X. Yang, B. He, Q. Wang, Z. Liu, D. Yu and G. He, J. Mater. Chem. A, 2021, 9, 14444–14450 RSC.
- B. Zhang, Y. Zhang, X. Yang, G. Li, S. Zhang, Y. Zhang, D. Yu, Z. Liu and G. He, Chem. Mater., 2020, 32, 10575–10583 CrossRef CAS.
- A. Iordache, V. Maurel, J.-M. Mouesca, J. Pécaut, L. Dubois and T. Gutel, J. Power Sources, 2014, 267, 553–559 CrossRef CAS.
- X. Chen, A. A. Sukhanov, Y. Yan, D. Bese, C. Bese, J. Zhao, V. K. Voronkova, A. Barbon and H. G. Yaglioglu, Angew. Chem., Int. Ed., 2022, 61, e202203758 CAS.
- L. M. Kozycz, C. Guo, J. G. Manion, A. J. Tilley, A. J. Lough, Y. Li and D. S. Seferos, J. Mater. Chem. C, 2015, 3, 11505–11515 RSC.
- N. Pearce, E. S. Davies, W. Lewis and N. R. Champness, ACS Omega, 2018, 3, 14236–14244 CrossRef CAS PubMed.
- E. Ripaud, D. Demeter, T. Rousseau, E. Boucard-Cétol, M. Allain, R. Po, P. Leriche and J. Roncali, Dyes Pigm., 2012, 95, 126–133 CrossRef CAS.
- R. H. Abeles, R. F. Hutton and F. H. Westheimer, J. Am. Chem. Soc., 1957, 79, 712–716 CrossRef CAS.
- K. M. Pelzer, L. Cheng and L. A. Curtiss, J. Phys. Chem. C, 2017, 121, 237–245 CrossRef CAS.
- I. V. Martynov, Russ. J. Inorg. Chem., 2008, 53, 579–582 CrossRef.
- A. Campero and J. A. D. Ponce, ACS Omega, 2020, 5, 12046–12056 CrossRef CAS PubMed.
- Considering Mulliken's definition of electronegativity: χM = (IP + EA)/2, where IP and EA are the ionisation potential and electron affinity, respectively, the importance of IP (or EHOMO by Koopmans’ theorem) in electronegativity is apparent. Likely due to such a HOMO-dependence, electronegativity of the constituent elements is a poor indicator for the electrochemical reduction potential (related to ELUMO).
- E. D. Glendening, C. R. Landis and F. Weinhold, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2011, 2, 1–42 Search PubMed.
- D. D. Méndez-Hernández, P. Tarakeshwar, D. Gust, T. A. Moore, A. L. Moore and V. Mujica, J. Mol. Model., 2012, 19, 2845–2848 CrossRef PubMed.
- J. Conradie, J. Phys.: Conf. Ser., 2015, 633, 012045 CrossRef.
- E. D. Glendening, J. K. Badenhoop, A. E. Reed, J. E. Carpenter, J. A. Bohmann, C. M. Morales, P. Karafiloglou, C. R. Landis and F. Weinhold, NBO 7.0, 2018 Search PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery, Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, T. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revision D.01, Gaussian, Inc., Wallingford CT, 2013 Search PubMed.
- W. Humphrey, A. Dalke and K. Schulten, J. Mol. Graphics, 1996, 14, 33–38 CrossRef CAS PubMed.
- T. Lu and F. Chen, J. Comput. Chem., 2011, 33, 580–592 CrossRef PubMed.
- M. Xiao and T. Lu, J. Adv. Phys. Chem., 2015, 4, 111–124 CrossRef CAS.
- S. Dapprich and G. Frenking, J. Phys. Chem., 1995, 99, 9352–9362 CrossRef.
- N. G. Connelly and W. E. Geiger, Chem. Rev., 1996, 96, 877–910 CrossRef.
- H. Adkins and F. W. Cox, J. Am. Chem. Soc., 1938, 60, 1151–1159 CrossRef.
- C. P. Klages and J. Voß, Chem. Ber., 1980, 113, 2255–2277 CrossRef.
- L. Lunazzi, G. Maccagnani, G. Mazzanti and G. Placucci, J. Chem. Soc. B, 1971, 162–166 RSC.
- N. Pearce, E. S. Davies, R. Horvath, C. R. Pfeiffer, X.-Z. Sun, W. Lewis, J. McMaster, M. W. George and N. R. Champness, Phys. Chem. Chem. Phys., 2018, 20, 752–764 RSC.
- J. Voss, G. Kupczik and H. Stahncke, J. Chem. Res., 2009, 283–286 CrossRef.
- H. J. Timpe and K. P. Kronfeld, J. Photochem. Photobiol., A, 1989, 46, 253–267 CrossRef.
- P.-A. Muller and E. Vauthey, J. Phys. Chem. A, 2001, 105, 5994–6000 CrossRef.
- A. Maciejewski and R. P. Steer, Chem. Rev., 1993, 93, 67–98 CrossRef.
- J. Voss and R. Edler, J. Chem. Res., 2007, 226–228 CrossRef.
- Although there is no intrinsic bias for the two lone-pair orbitals to have symmetrical (sp3-like “rabbit ears”) or unsymmetrical (σ and π) characters in the NBO theory, the latter is typically found as the consequence of the search for the “maximum occupancy” orbitals. The σ/π orientation is also convenient for the present study since the O/S atoms are conjugated with the molecular π-system.
- M. Paramasivam, R. K. Chitumalla, J. Jang and J. H. Youk, Phys. Chem. Chem. Phys., 2018, 20, 22660–22673 RSC.
- F. A. Van-Catledge, J. Org. Chem., 1980, 45, 4801–4802 CrossRef CAS.
- W. P. Purcell and J. A. Singer, J. Chem. Eng. Data, 1967, 12, 235–246 CrossRef CAS.
- W. Zhang, Y. Liu and G. Yu, Adv. Mater., 2014, 26, 6898–6904 CrossRef CAS PubMed.
- X. Feng, Y. Bai, M. Liu, Y. Li, H. Yang, X. Wang and C. Wu, Energy Environ. Sci., 2021, 14, 2036–2089 RSC.
- A. Holownia, C. N. Apte and A. K. Yudin, Chem. Sci., 2021, 12, 5346–5360 RSC.
- M. Hayashi, Chem. Lett., 2021, 50, 1–6 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2cp05186a |
| This journal is © the Owner Societies 2023 |