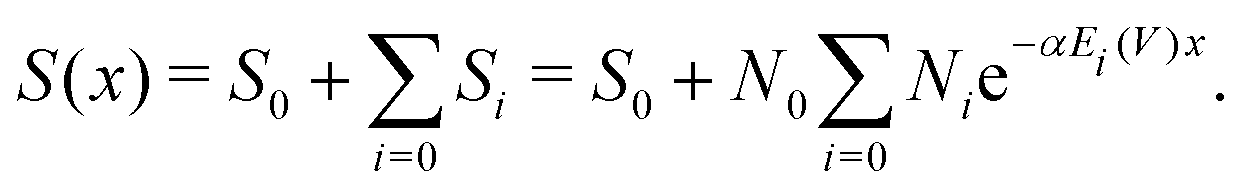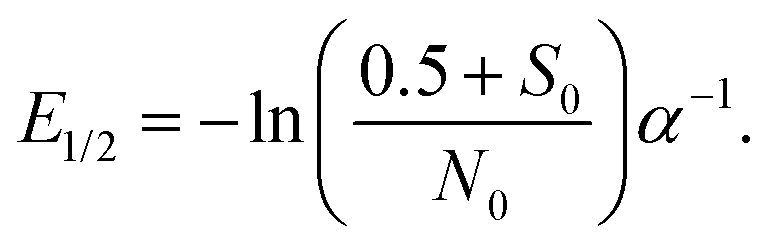Bio-SAXS of single-stranded DNA-binding proteins: radiation protection by the compatible solute ectoine†
Received
28th October 2022
, Accepted 4th January 2023
First published on 4th January 2023
Abstract
Small-angle X-ray scattering (SAXS) can be used for structural determination of biological macromolecules and polymers in their native states (e.g. liquid phase). This means that the structural changes of (bio-)polymers, such as proteins and DNA, can be monitored in situ to understand their sensitivity to changes in chemical environments. In an attempt to improve the reliability of such experiments, the reduction of radiation damage occurring from exposure to X-rays is required. One such method, is to use scavenger molecules to protect macromolecules against radicals produced during radiation exposure, such as reactive oxygen species (ROS). In this study we investigate the feasibility of applying the compatible solute, osmolyte and radiation protector Ectoine (THP(B)), as a scavenger molecule during SAXS measurements of the single-stranded DNA-binding protein Gene-V Protein (G5P/GVP). In this case, we monitor the radiation induced changes of G5P during bio-SAXS measurments and the resulting microscopic energy-damage relation was determined from microdosimetric calculations by Monte-Carlo based particle scattering simulations with TOPAS/Geant4 and a custom target-model. This resulted in a median-lethal energy deposit of pure G5P at 4 mg mL−1 of E1/2 = 7 ± 5 eV, whereas a threefold increase of energy-deposit was needed under the presence of Ectoine to reach the same level of damage. This indicates that Ectoine increases the possible exposure time before radiation-damage to G5P is observed. Furthermore, the dominant type of damage shifted from aggregation in pure solutions towards a fragmentation for solutions containing Ectoine as a cosolute. These results are interpreted in terms of indirect radiation damage by reactive secondary species, as well as post-irradiation effects, related to preferential-exclusion of the cosolute from the protein surface. Hence, Ectoine is shown to provide a non-disturbing way to improve structure-determination of proteins via bio-SAXS in future studies.
1 Introduction
Biological small-angle X-ray scattering (bio-SAXS) is an experimental technique to obtain structural information of biomolecules in solution.1,2 This means that structural changes of, for example, proteins can be observed in their native environment, or in relation to varying chemical conditions, co-solutes or temperature, which could be considered a major advantage over high-resolution techniques such as macromolecular crystallography or cryo-transmission-electron microscopy.3 However, an inherent problem for bio-SAXS, especially at the synchrotron, is radiation induced damage. Radiation damage to the macromolecules, during X-ray exposure, can lead to structural changes of the target molecules, and thus the resulting X-ray scattering signal.4,5 These changes can often start off subtle, but can quickly result in much more severe changes. Therefore, radiation damage is often the main limiting factor when it comes to maximum exposure time of such samples, meaning the ability to achieve an optimal signal-to-noise ratio (SNR) may not always be possible. The reasons for this is the indirect damage produced by reactive oxygen species (ROS), such as hydroxyl radicals, formed upon radiolysis of water.6 The presence of ROS leads to an increase in radiation damage in solution, compared with the crystalline state. To mitigate this indirect damage, radical scavengers can be introduced into solutions. These scavengers often are organic compounds with low-molecular weight which provide high reactivity towards ROS. Ideally, such scavenger molecules contribute little to the SAXS signal and should not influence the native state of the macromolecule under investigation. For instance, hydroxyl radical scavenger, which are frequently applied in radiation research studies, are alcohols such as 2-propanol.7 However, such scavengers can destabilize hydrogen bonding, influence protein hydration, and the structure of the bound water molecules. This means that the use of these scavengers can compromise the results of structural studies (e.g. those performed using SAXS), as they can disturb protein structures.8 Such destabilizing cosolutes are classified as chaotropes. Another popular scavenger molecule is dimethyl sulfoxide (DMSO) which is well known to cause precipitation and denaturation of proteins.9 Both properties are major drawbacks during bio-SAXS. More common scavengers used during bio-SAXS, despite their various limitations, are dithiothreitol (DTT), tris(2-carboxyethyl)phosphine (TCEP), ascorbic acid and glycerol, as summarized by Castellvi et al.3 In brief, ascorbic acid tends to modify the pH of the buffer which in turn might cause unwanted side effects during scattering experiments. DTT and TCEP are strong reducing agents and are therefore not applicable to proteins containing disulfide bonds. The presence of glycerol increases the viscosity of the solvent and can therefore lead to an experimentally and equipment-wise problematic pressure increase. To overcome these issues, Castelvi et al. tested the effectiveness of various nucleosides and nucleobases as scavenger.3 These bases showed promising results in terms of scavenging properties, but tend to have other drawbacks for an important range of samples and techniques: a drawback of technical nature mentioned by Castelvi et al., is their UV absorption around 280 nm which complicates the often performed concentration monitoring at this wavelength via UV-Vis spectroscopy. More importantly, the nucleoside and nucleobase derived scavengers tend to interact with oligonucleotides or other samples containing larger DNA, RNA or especially DNA-binding proteins. These DNA-binding proteins are of vital importance in maintaining DNA integrity within cells. They can be categorized into single-stranded DNA and double-stranded DNA-binding proteins. In this study we will focus on single-stranded DNA-binding proteins, which are involved in the repair of DNA double-strand breaks (DSB), one of the most lethal DNA lesions. One of the most frequent repair processes of this lesion in double-stranded DNA (dsDNA) is homologous recombination, during which large tracks of single-stranded DNA (ssDNA) are involved. Here, the role of ssDNA-binding proteins is to promote induction of ssDNA at the sites of the DSB.10 For example, it was observed, that the exposure of eukaryotic cells to radiation leads to the recruitment of repair proteins to DNA damage sites within (15–60) min after exposure.11,12 Hence, to facilitate the study of these proteins, and their interaction with ssDNA or damaged dsDNA, by bio-SAXS, the availability of a non-interfering and well understood scavenger is highly beneficial.
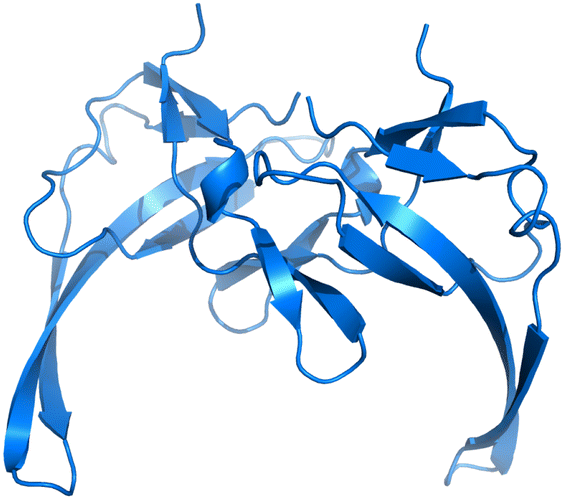
Therefore, we have chosen to study the well understood Gene-V protein (G5P), which serves as a model system for sequence-nonspecific interaction of single-stranded DNA-binding proteins with ssDNA (Fig. 1).13–15 G5Ps binding to ssDNA is formed by two G5P monomers which are tightly interlocked.17 A G5P monomer has a molecular weight of 9.7 kDa and consists of 87 amino acids in a single polypeptide chain which are mostly in the β-conformation, organized as a three-stranded sheet and a two-stranded ribbon with a broad connecting loop.18,19 The DNA-binding fold motif is shared with the ssDNA-binding domain of human replication protein A.19
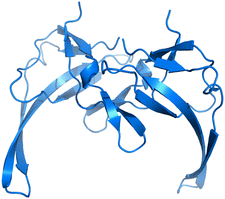 |
| | Fig. 1 G5P dimer created from the PDB structure 1GVP.16 | |
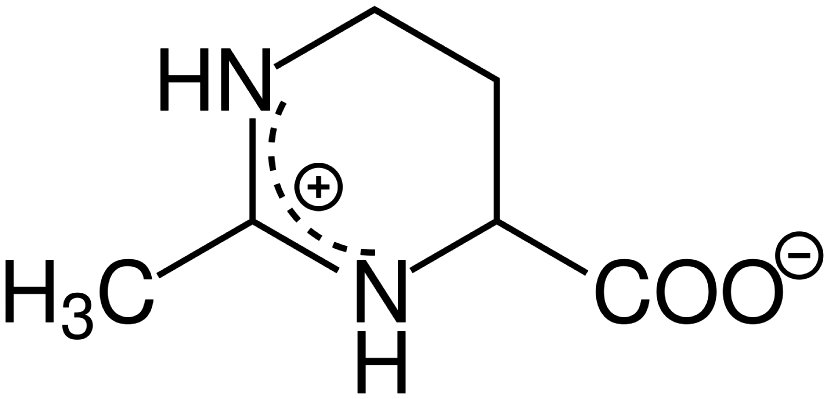
As a candidate for a ssDNA-binding protein compatible scavenger we have chosen the osmolyte and compatible solute Ectoine ((S)-2-methyl-1,4,5,6-tetrahydropyrimidine-4-carboxylic acid, THP(B), Fig. 2). Hereby, the description as compatible solute refers to the fact that Ectoine can be accumulated in molar concentrations within cells without disturbing their metabolism.20,21 These high concentrations are found in the cytosol of some halophilic microorganisms, where Ectoine is used to counter the osmotic pressure to survive in environments with high salinity, e.g. salt lakes. Furthermore, in contrast to the chaotropes mentioned above, Ectoine is a so called kosmotropic or water-structure forming substance, which strengthens intermolecular hydrogen bonding.22,23 Thus, the combination of Ectoine being a compatible solute, a kosmotrop22–24 and a hydroxyl radical scavenger21,25,26 at the same time, makes it likely to be suitable for the efficient protection of proteins during bio-SAXS based structure determination. Furthermore, Ectoines influence on G5P binding to ssDNA was characterized previously,15 and is well understood in terms of it's protective effects for DNA against ionizing radiation,25 UV radiation,21 as well as repeated freeze-thaw cycles.27 Therefore we have identified Ectoine as an ideal candidate to be evaluated as protective cosolute during bio-SAXS measurements in combination with the single-stranded DNA-binding protein G5P. The synchrotron based bio-SAXS measurements are accompanied by Monte-Carlo based particle scattering simulations with the Geant4/TOPAS framework to obtain a complete picture of the damage processes, including the relation between protein damage and the microscopic energy deposit.28
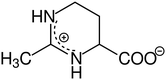 |
| | Fig. 2 Chemical structure of the compatible solute, radical scavenger and zwitterion Ectoine in aqueous solution. | |
2 Materials and methods
2.1 G5P production and purification
Expression and purification of Gene-V Protein G5P/GVP. (Swissprot: P69544, 87 AA, Mw 9688 Da) The G5P plasmid PET-30b transformed into Xl-1 blue was obtained from previous workers29 and grown on 2YT agar with 50 μg mL−1 Kanamycin. 16 mL overnight culture was inoculated into 1.6 L 2YT medium. Protein expression was induced with 1 mM IPTG at an OD600 = 0.6 for 4 h. Cells were collected, resuspended in 24 mL lysis buffer (PBS with 75 mM NaCl) and sonicated. Cell debris were removed and the supernatant was separated with an ÄKTA FPLC system using a Resource Q anionexchange column (both GE Healthcare, USA). Protein was found in the flowthrough which was purified with 1 mL NiNTA suspension (Qiagen, Netherlands). Resin was washed with 15 mL buffer and protein eluted with 5 mL buffer with 250 mM Imidazole. Protein concentration was determined by BCA assay. A purity of at least 85% was achieved as determined by SDS-PAGE (compare ESI†). The protein was stored at −20 °C.
2.2 Sample preparation and handling
All samples were prepared in 1 × PBS with three G5P concentrations of 4 mg mL−1, 2 mg mL−1 and 1 mg mL−1. Additonal to pure G5P samples, the samples with a 20 mM concentration of the scavenger Ectoine (Sigma, 99.5% purity) were prepared for all three G5P concentrations. After preparation the samples were shipped on dry ice to the Diamond light source were they were kept frozen upon arrival until the bio-SAXS measurements were performed at the B21 beamline.30
2.3 Small angle X-ray scattering
The small angle X-ray scattering (SAXS) experiments were performed at Diamond Light Source beamline B21.30 Samples were injceted into a capillary (1.5 mm diameter), and exposed to X-rays of an energy of 13.0 keV (0.9524 Å), with a beam size 0.05 × 0.05 mm (FWHM) and a flux of 2 × 1012 photons s−1. Scattered radiation was recorded on an EigerX 4M (Dectris) positioned at a sample to detector distance of 3698.8 mm. For each sample, data was collected every second to observe changes occurring to the samples with sufficient time resolution. Data processing was performed using the DAWN software package, where the data was scaled to absolute intensity using water, and subsequently background subtracted for contributions coming from the capillary and PBS.31
2.4 Data processing and analysis
Analysis of the X-ray scattering data was performed using McSAS3, a Monte Carlo method to extract form-free size distributions. Correction and background subtraction of the SAXS data was performed with DAWN.31 SAXS data analysis was performed with BioXTAS RAW 2.1.2.32 The determination of the damage induction threshold was performed by similary testing. Similarity testing was performed in RAW with the correlation map (CorMap) test method and Bonferroni multiple testing activated.32 Results with p < 0.01 for testing of similarity were classified as being substantially different (Table 1).
Table 1 Results of the similarity analysis (CorMap test) of the SAXS curves to determine the onset of radiation damage to G5P under the absence (Pure) and presence (Ect.) of Ectoine and their ratio for different protein concentrations
| Protein conc. |
Damage after time |
Ratio |
| Pure |
Ect. |
Ect./Pure |
| 4 mg mL−1 |
9 s |
24 s |
2.7 |
| 2 mg mL−1 |
12 s |
39 s |
3.3 |
| 1 mg mL−1 |
19 s |
109 s |
5.7 |
Curve fitting with McSAS3 and size distribution histograms.
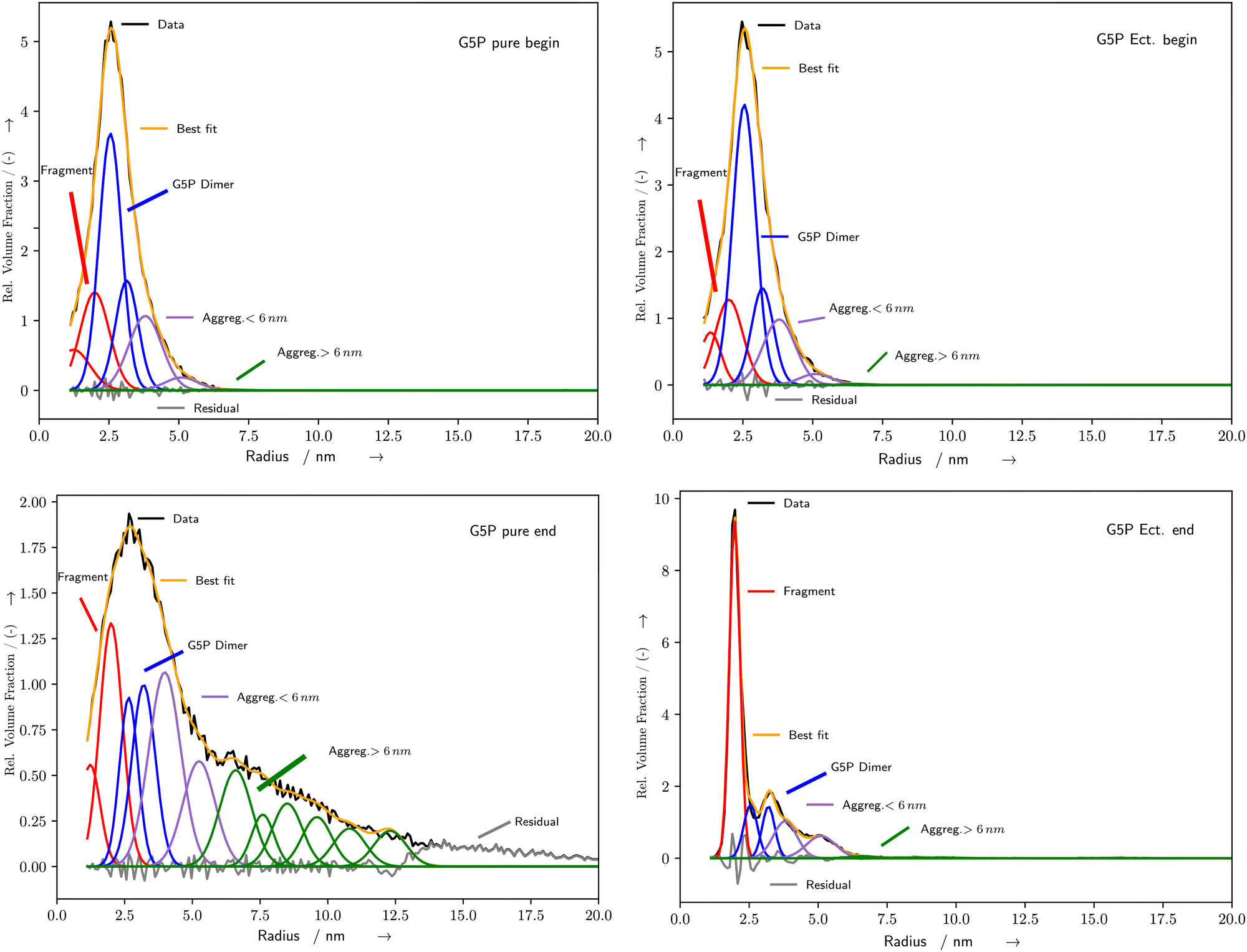
SAXS scattering patterns were fitted with McSAS333 to obtain a size distributions without specific assumptions about the microscopic form. Hereby the following parameters were used to fit all presented curves: nbins = 1000, data_range = (0.0312–2.93) nm−1, model = mcsas_sphere, contributions = 1000, modelDtype = default, radius = auto, sld = 10.946 × 10−6 Å−2, sld_solvent = 9.946 × 10−6 Å−2, maxIter = 200![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000, convCrit = 1, nRep = 50. From the output, histograms were generated with 1000 bins and volume bin weighting (compare Fig. 4). To distinguish the different species in the histograms of the different volumes obtained with McSAS3, twelve position-constrained Gaussian peaks were fitted to all curves (Fig. 4). The initial positions were set with respect to the maximum of the distribution, which is assumed to correspond to the undamaged G5P dimer (Fig. 1). The Gaussians were assigned relative to this undamaged G5P dimer around 2.5–3.1 nm (blue): the fragments below the dimer (red), the small aggregates above the dimer (purple), and the aggregates at r > 6 nm (green). The positions of the exact peaks are not of relevance here, since we are interested in the relative evolution of the different species over time, respective dose. From the position of the maximum of the G5P sample at a concentration of 4 mg mL−1 without additional scavenger (pure sample), the fitted radius (r = 2.5 nm) for the calculation of the diffusion constant, as described below, was determined. This radius (r) obtained from fitting the McSAS sphere model are related to the radius of gyration (rG) as
000, convCrit = 1, nRep = 50. From the output, histograms were generated with 1000 bins and volume bin weighting (compare Fig. 4). To distinguish the different species in the histograms of the different volumes obtained with McSAS3, twelve position-constrained Gaussian peaks were fitted to all curves (Fig. 4). The initial positions were set with respect to the maximum of the distribution, which is assumed to correspond to the undamaged G5P dimer (Fig. 1). The Gaussians were assigned relative to this undamaged G5P dimer around 2.5–3.1 nm (blue): the fragments below the dimer (red), the small aggregates above the dimer (purple), and the aggregates at r > 6 nm (green). The positions of the exact peaks are not of relevance here, since we are interested in the relative evolution of the different species over time, respective dose. From the position of the maximum of the G5P sample at a concentration of 4 mg mL−1 without additional scavenger (pure sample), the fitted radius (r = 2.5 nm) for the calculation of the diffusion constant, as described below, was determined. This radius (r) obtained from fitting the McSAS sphere model are related to the radius of gyration (rG) as| | 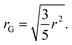 | (1) |
Eqn 1 allows us to compare the fitted r values with the calculations of rG by the Pepsi-SAXS software34 (compare ESI†) from G5P structures which can be found in the protein database (structure: 1GVP16Fig. 1), as a monomer and dimer. The rG of the monomer resulted in 1.46 nm without water of hydration, and in 2.16 nm including the water of hydration. The rG of the dimer resulted in 1.84 nm and 2.65 nm respectively. Whereby the dimer value is in good agreement with the results from the fitting of the initial scattering curve. The time and dose dependent relative contributions of these species are shown in Fig. 4.
Calculation of the diffusion constant of G5P.
The diffusion constant (D in m2 s−1) of a protein can be estimated from the empirical formula given by Tyn and Gusek,35| | 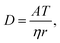 | (2) |
with the constant A = 5.78 × 10−25 kg m2 s−2 K−1, the temperature T = 288 K, the dynamic viscosity η = 0.001 N s m−2 at T = 293 K, and r = 2.5 nm as estimated from the SAXS measurements (see above). This results in a diffusion constant of G5P at D288K = 66.6 μm2 s−1 The diffusion constant at room temperature (298 K) is only slightly higher DRT = 67.7 μm2 s−1, so eventual variation due to an temperature increase during the measurement are neglectable. The average displacement due to diffusion in one dimension (λ) is given by28| | 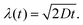 | (3) |
with D288K and t = 500 s, the duration over which the samples were analysed and measured, the average displacement results in λ(t = 500 s) = 258.0 μm.
2.5 Particle scattering simulations
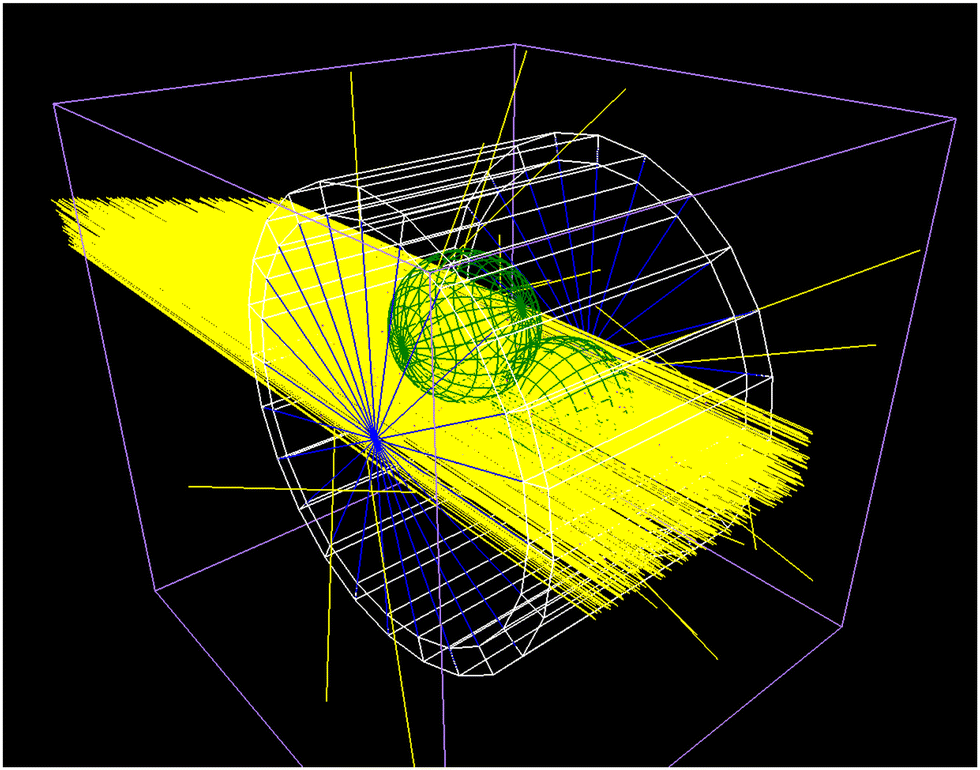
To obtain the inelastic scattering events and energy deposits in the irradiated capillary containing the protein solution, Monte Carlo simulations (MCS) of the scattering processes were performed. Thereby the Geant4 10-06-p3 MCS framework36 and the TOPAS 3.7 interface37 was used to simulate the X-rays passing through the sample holder, the subsequent production of secondary particles, their interaction with the proteins and the surrounding water. The scattering events were simulated by applying the G4emLivermore models in the capillary. Within water the G4emDNA models were applied.38 The cut length was set to 1 μm and all atomic deexcitation processes were activated. A complete list of activated scattering processes can be found in our previous work,39 as well as general details concerning dose calculation and the complete simulation methodology.28,40 The capillary used during the experiment at Diamond B21 beamline was modelled from quartz glass over a length of 1.0 mm, an outer diameter of 1.7 mm and an inner diameter of 1.5 mm. The inner part was filled with water (Fig. 6).
The quartz glass was simulated as G4_Silicon_Dioxide with a density of 2.32 g cm−3 and the water within as G4_Water with a density of 1.0 g cm−3. 109 X-ray photons with an initial energy of 13 keV were emitted perpendicular to a plane (1.0 mm × 0.25 mm), corresponding to the beam profile during the SAXS experiments at B21. Hereby the longer axis of the emitting plane was parallel to the z-length of the capillary as given above. The simulations were repeated ten times to obtain standard deviation and standard error of the resulting energy deposit histograms (Fig. 7). During the simulation the energy deposit was recorded in the capillary wall, the medium, and thousand target spheres which were randomly distributed within the liquid. For the scoring within the target spheres the parallel world feature of TOPAS was used, to allow them to overlap. The rational behind this approach is described in the following subsection.
Determination of the energy-deposit distribution at different locations.
The average displacement of a protein during irradiation is related to the accessible volume within the capillary.28 This accessible volume can be modeled in a first approximation by a sphere with radius λ. Hereby λ depends on the diffusion behaviour of the protein (eqn (3)). During a bio-SAXS experiment, the energy deposit caused by inelastic scattering events within such a sphere, is the average energy deposit per volume at a protein, which resides within the center of this sphere at the beginning of the irradiation. Therefore, these spheres are called in the following target spheres. Due to the inhomogeneous nature of the energy deposit within the capillary, the targets spheres receive different amounts of energy deposited by the inelastic scattering processes, which depends on their position within the capillary. By randomly distributing target spheres within the irradiated liquid and simulating the irradiation, inelastic scattering events and energy deposit, the distribution of energy deposit per protein per volume can be determined (compare Fig. 7). This method was described in detail for electron irradiation of plasmid DNA pUC19 in our previous works.28,40
The microscopic target volume of the protein.
The target volume of the protein (VG5P) is defined as the volume of and around the protein, where an inelastic scattering event and resulting energy deposit can lead to direct or indirect damage at the protein. According to our previous work,28 this can be calculated as| | 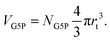 | (4) |
Hereby NG5P = 1381 corresponds to the number of atoms of the protein, whereby each of them can be ionized independently, leading to direct damage, or become the target of an radical attack, causing indirect damage. Therefore each atom of the protein corresponds to an independent sensitive volume and has to be accounted for individually. Inelastic scattering events and the resulting energy deposit within this volume associated to the individual atoms relates directly to a certain probability of induction of radiation damage at one of its corresponding molecular bonds. The contribution to the total sensitive volume of the whole protein by each atom can be best modeled by a sphere with rt = 7.5 nm, as rationalized in detail previously.28 From this, the energy deposit within the different target volumes, at different locations in the capillary can be calculated with a dependence on the time, together with their individual energy deposit per primary incoming particle (Esim, Fig. 7) as obtained by the particle-scattering simulations as| | 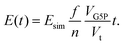 | (5) |
With the number of simulated photons (n = 109), the fluence of the beamline (f = 2 × 1012 photons s−1), the target sphere 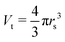 with rs = 258 μm, and the sensitive volume of G5P (VG5P = 2.44 × 106 nm3, eqn (4)). These results are used to calculate the microscopic energy-damage relation in the following section.
with rs = 258 μm, and the sensitive volume of G5P (VG5P = 2.44 × 106 nm3, eqn (4)). These results are used to calculate the microscopic energy-damage relation in the following section.
Calculation of the microscopic energy-damage relation.
In standard irradiation experiments all irradiated molecules are assumed to receive a similar macroscopic dose. However, in reality the proteins receive a broad asymmetric distribution of energy deposits, as determined by the simulation (compare Fig. 7). As shown in our previous work,28 the decrease of the amount of undamaged, irradiated species (S – Survival rate) with a dependence on external dose or energy (Fig. 4) can be expressed as the sum of the survival rate Si weighted by the number of proteins Ni and their respective energy deposit Ei received per photon. By exclusion of non-linear effects, implying that all Si obey a single-hit model, the damage-coefficient α is independent of Ei.28 Thus, the measured survival rate can be written as the sum of the survival rates (Si) obeying all the same energy-damage relation with the same damage-coefficient (α):| | 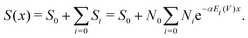 | (6) |
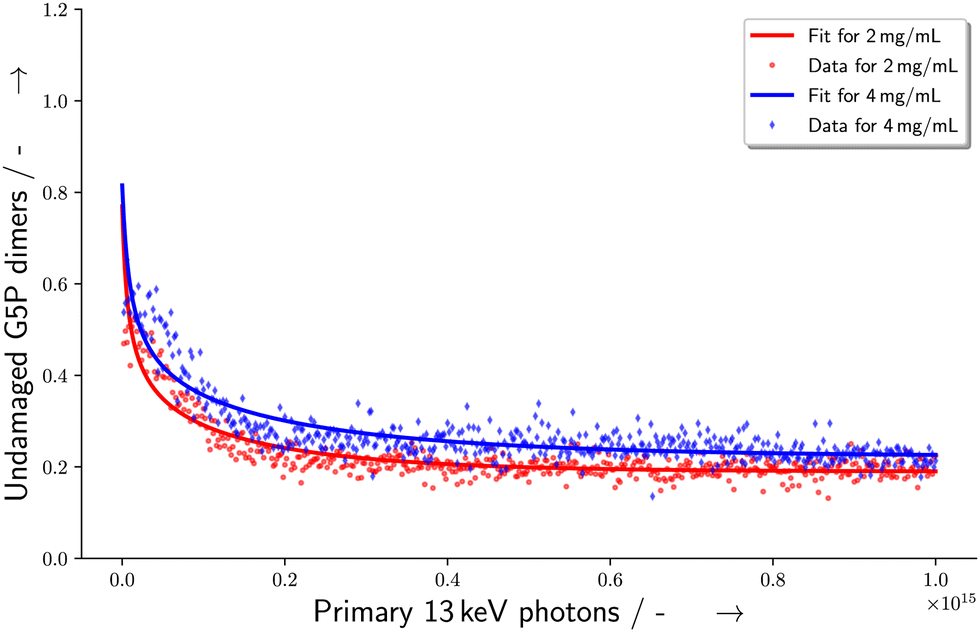
This makes it possible to perform a single parameter fit for α even for the situation of varying energy deposits Ei over the sample geometry. Here, S equals the experimentally determined amount of undamaged species with a dependence on the amount (x) of primary X-ray photons. Ni represents the relative number of proteins receiving the corresponding energy deposit Ei as determined from the particle scattering simulation and shown in the histogram in Fig. 7. Furthermore, N0 describes the scattering signal at the beginning of the measurement, corresponding to the species which are assigned to the G5P dimer, being suspected to change. On the other hand, S0 represents the contributions to the scattering curve located at the fitted radius of the G5P dimer which are not stemming from the undamaged dimer, and are assumed to be constant. The fitting results are shown in Fig. 8.
Since eqn (6) allows to perform a single parameter fit even in the case of varying energy deposit,28 the median lethal energy deposit (E1/2, 50% undamaged proteins) with S(x) = 0.5 can be calculated as:
| | 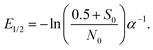 | (7) |
The fit leads to
α = 0.11 ± 0.02 eV
−1, and a median lethal energy deposit of
E1/2 = 7.0 ± 0.8 eV at a protein concentration of 4 mg mL
−1. For G5P at at a concentration of 2 mg mL
−1. the results are
α = 0.16 ± 0.02 eV
−1, and
E1/2 = 4.0 ± 0.3 eV, respectively. The uncertainty is calculated by the fitting algorithm (python-scipy). A detailed discussion of this methodology can be found in our previous work.
28
3 Results
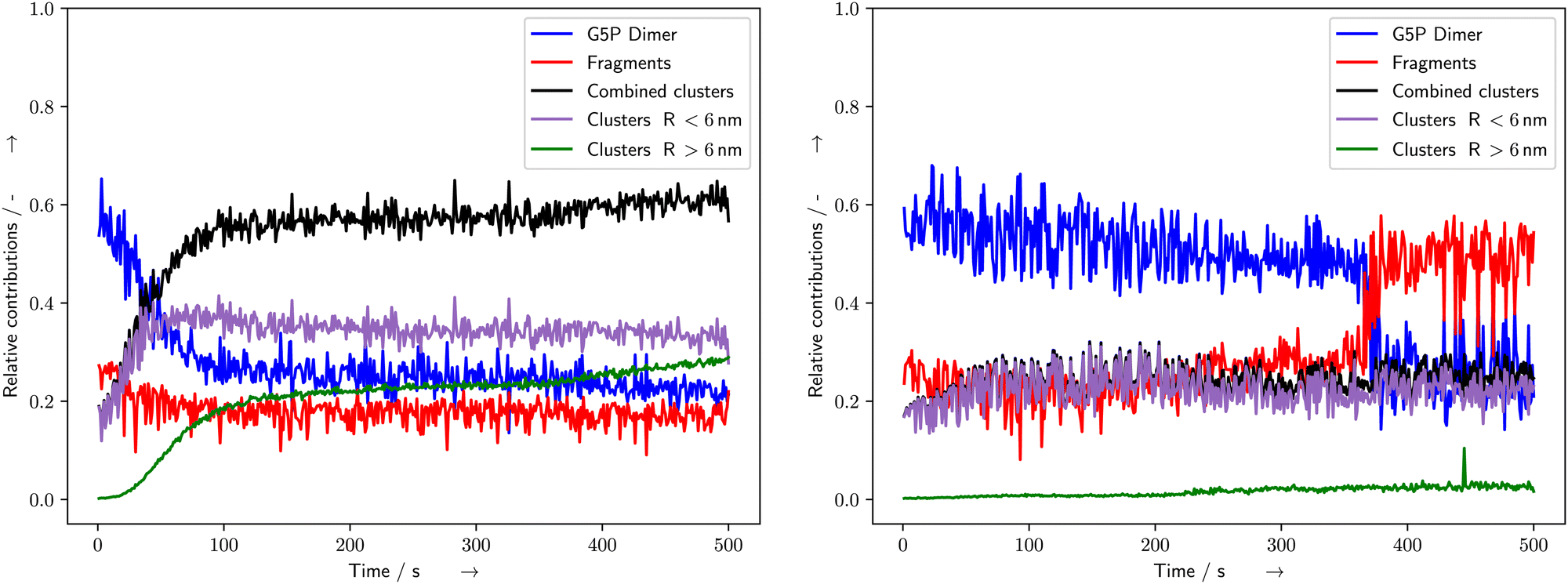
During bio-SAXS measurements of G5P the resulting scattering curves (Fig. 3) and related histograms (Fig. 4) of the fitted radius (r) showed different energy-deposit/dose responses in dependence of protein concentration and the presence or absence of the compatible solute Ectoine (Fig. 5). For pure G5P solutions in 1 × PBS the scattering curves showed strongest variation in the region at and above r ≥ 2.5 nm (Fig. 4 left, top and bottom), which is mostly associated with unfolding and aggregation of the proteins. Under the presence of Ectoine, the overall change in the SAXS curves was decreased as revealed by the CorMap test (Table 1). Furthermore, the region where most of the damage occurred shifted towards (Fig. 4 right, top and bottom) r < 2.5 nm, when Ectoine was present.
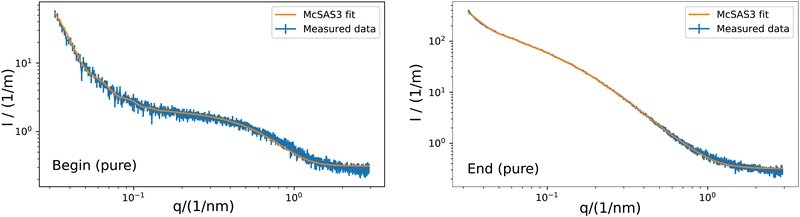 |
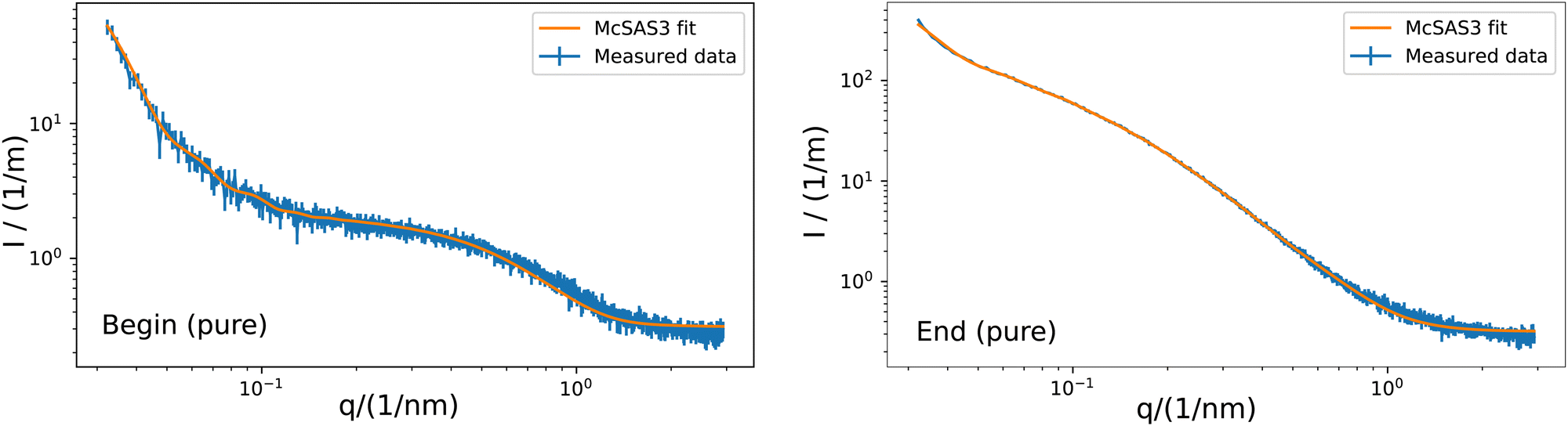
| | Fig. 3 Example of the fitting of a sphere model with McSAS3 to the obtained scattering curve of the G5P sample without scavenger at a concentration of 4 mg mL−1. McSAS at the beginning (left) and and end (right) of the measurement. Hereby q is the scattering vector and I the intensity. Additional curves and fits are shown in the ESI.† The related histograms are shown in Fig. 4. | |
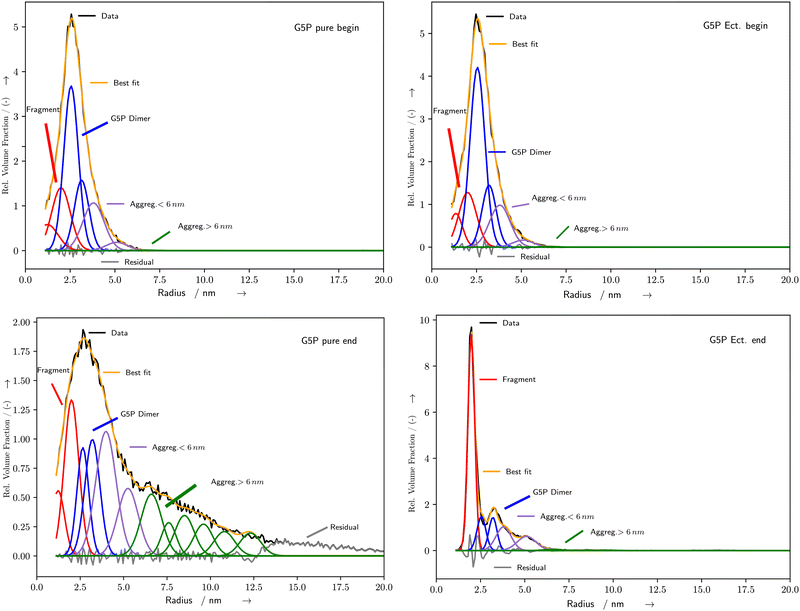 |
| | Fig. 4 Exemplary histograms of the fitted radii for pure (left) and Ectoine (right) containing solutions at the beginning (top) and end (bottom) of the irradiation. Black curves represent the output of McSAS calculated from the measured data. The blue Gaussians are representing the undamaged G5P dimer, while red are fragments, purple small aggregations and green aggregates with r > 6 nm. The orange curve is the sum of the fits (best fit) while the grey curve is the residuum. For detail see the text. Additional fits and corresponding SAXS curves are shown in the ESI.† | |
 |
| | Fig. 5 Time evolution of the fits of the different Gaussians corresponding to the various G5P species at concentrations of 4 mg mL−1 resulting in the distribution of the different fitted radii for pure (left) and Ectoine containing solutions (right). The curves for 2 mg mL−1 are shown in the ESI.† | |
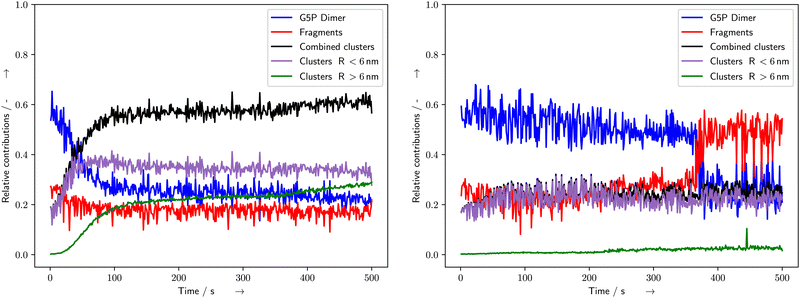
The fitting of the SAXS scattering curves by McSAS3 provided the distribution of radius values for further analysis (Fig. 4). The subsequent fitting of Gaussian curves provided a quantitative measure of the time and energy-deposit dependent evolution of the radius values. For the pure solutions with 2 mg mL−1 and 4 mg mL−1 G5P, the species associated with the G5P dimer showed a steady decrease during the measurement, while the clusters increased (Fig. 5 blue and black curves, respectively). For the corresponding samples that contained Ectoine this damage associated decrease was much lower until a strong change was observed around 360–390 s for G5P concentrations of 4 mg mL−1 (Fig. 5 blue and red curve) and at around t = 470 s for 2 mg mL−1 G5P (compare ESI†). The pure solutions showed the strongest increase for radius values above 3.1 nm (Fig. 5 left, green curve). In contrast, under the presence of Ectoine this behavior was nearly absent (Fig. 5 right, green curve). Here, mostly an increase of the fragments (r < 2.5 nm) was observed (Fig. 5 right, red curve). For the solutions with 1 mg mL−1 G5P concentrations no stable fits of the histogram for the time evolution of the different components could be achieved, therefore a detailed analysis had to be omitted for theses curves.
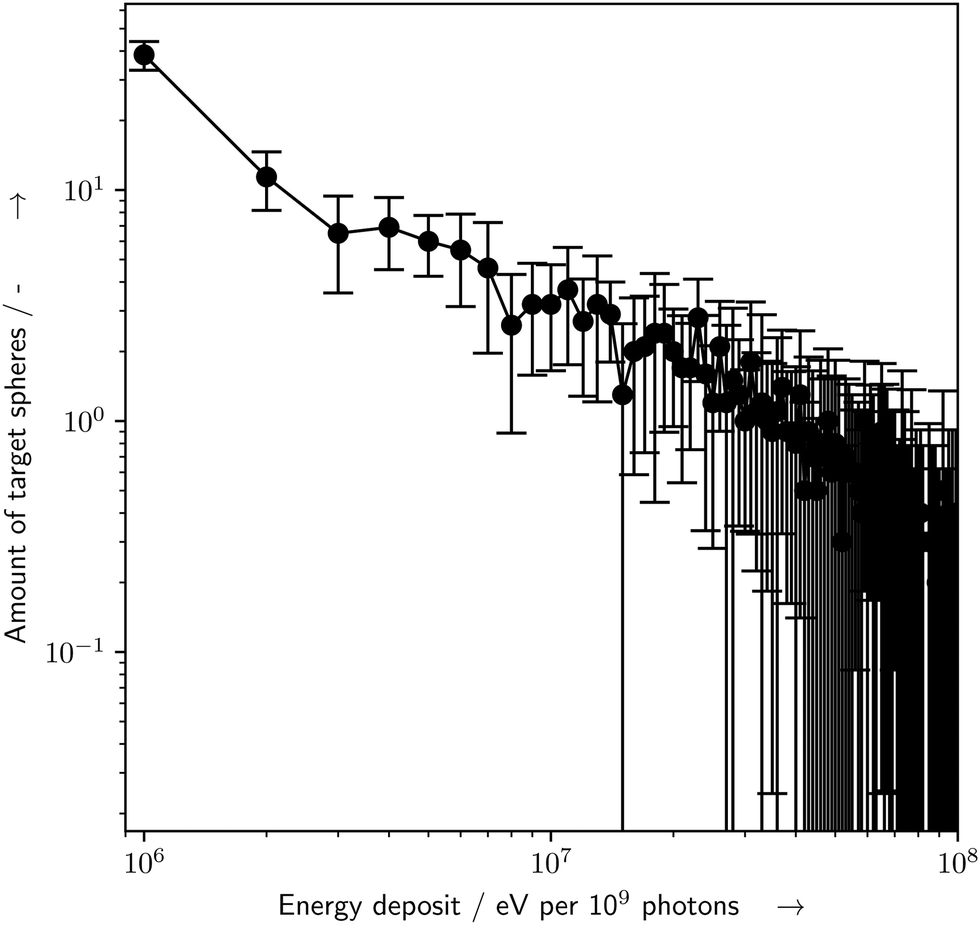
The particle-scattering simulations were used to determine the energy deposit distribution within the capillary (Fig. 6). The energy deposit distribution throughout the target spheres (Fig. 7) was determined by the custom target model, whereby about 17% of all simulated spheres experienced an inelastic scattering event and an energy deposit event.
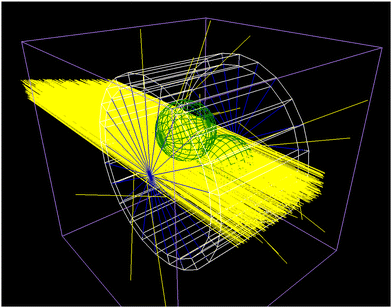 |
| | Fig. 6 Example of the Geant4/TOPAS particle scattering simulations with the newly developed bio-SAXS extension. Shown is the capillary (white), the water (blue), two target spheres (green) and 1000 incoming X-rays (yellow). For details see the text. | |
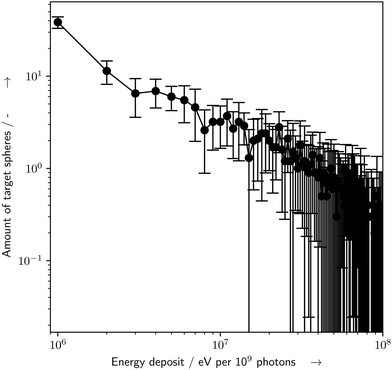 |
| | Fig. 7 Histogram of amount of target spheres which received a certain amount of energy deposit (E) per 109 primary photons, as determined by the particle scattering simulations with Geant4/TOPAS and the presented bio-SAXS extension. Of the 1000 simulated target spheres, only spheres which received an E > 0 eV are shown. Errorbars represent the standard deviation for n = 10 repeated simulations, binsize = 106 eV. For details see the text. | |
4 Discussion
In general, the observed changes in the X-ray scattering profile can be attributed to aggregation, fragmentation, conformational changes and unfolding of the proteins.1 Aggregation is induced by the formation of intermolecular crosslinks and leads to an increase in scattering at low values of the scattering vector (q) and to a decrease at high q values.41 Fragmentation is caused by the breakage of covalent bonds in the protein-backbones. It is expected to show contrary evolution of the scattering curves, namely a decrease of scattering for low q and an increase at high q values. Whereby a breakage of the main-chain, and therefore fragmentation, can be made undetectable by simultaneously occurring formation of intramolecular crosslinks.42 Here, formation of crosslinks stabilizes these otherwise “fragmented” parts, and prevents them to diffuse away from each other. Furthermore, protein unfolding can lead to an increase of the radius as well. In the literature radiation-induced unfolding is mostly associated with modifications of the side-chains, and depends strongly on the specific amino acid sequence involved.42 Here, ˙OH-radicals can modify groups related to the secondary structure of a protein, trigger changes in the tertiary structure, and thus, induce unfolding processes.43 However, the partial or total unfolding of a single G5P protein can be masked in solutions when aggregation occurs at the same time, as both changes can result in an increase of the scattered intensity at low-q. Therefore it must be taken care to consider the possibility of intermixing of these effects during analysis. A criteria to distinguish both effects is, that an unfolding can only lead to a limited increase of the radius, due to the finite nature of the peptide-chain within one dimer, while in theory, aggregation has not such a well defined upper bound.
Therefore, it can be concluded directly from the pure solution data, that aggregation plays a dominant part. Especially since it can be observed, that with increasing time, the aggregation evolves from smaller aggregates (r < 6 nm) towards structures with r > 6 nm (Fig. 4). This behavior is expected for solutions with high protein concentrations due to the elevated likelihood to encounter other damaged proteins by diffusion, leading to aggregation. According to Hopkins and Thorne,1 aggregation can occur due to site-specific damage, for example reduction of amino acids at the protein surface, leading to partially charged groups. In general, the binding of G5P to ssDNA is a result of electrostatic ion pairing of the DNA backbone with the positive charged lysine and arginine G5P side-chains and stacking of the aromatic side-chains on the nucleobases.44 Since these parts are already prone to interact with partially charged subgroups of DNA, it is possible that they play an important role during aggregation with other partially charged groups in other proteins, where radicals have already formed upon reactions with ROS. For pure G5P with a concentration of 1 mg mL−1 the situation is less clear due to a generally lower SNR, here subcomponents between 4–8 nm appear upon radiation exposure (compare ESI†). This subcomponents are much narrower in width, and might be attributed to the aforementioned unfolded G5P dimer, which might be only clearly visible here, due to a decreased likelihood of forming bigger aggregates. However this interpretation, concerning the data originating from G5P concentrations of 1 mg mL−1, should be taken to be more uncertain due to the lower SNR and resulting unstable fitting. Still, fragmentation can be observed for all G5P concentrations (Fig. 4 left, red curves), but with clearly less contribution to the total decrease of the amount of undamaged G5P dimer, in comparison to the formation of aggregates (Fig. 4 left, purple and green curves). This might be due to the intrinsic stabilization of proteins by radiation induced intra-molecular and inter-molecular crosslinks, which can counteract fragments from separating them-self, even when a strand-break was produced in the protein-backbone.
To interpret these observation in terms of a microscopic picture of the involved physico-chemical processes on the molecular scale it is necessary to understand how much energy is deposited within a single protein. To determine this microscopic energy-damage relation, additional microdosimetric calculations were performed. Therefore a new TOPAS extension was developed, which is presented in this study for the first time. This extension is freely available online,45 and a continuously updated version can be found in our Github repository.46 The extension for Geant4/TOPAS allows for direct calculation of the energy deposit distribution within the capillaries used in bio-SAXS measurements. Here, the inclusion of target spheres allows to determine the energy deposit within the target volume of the protein. Therefore it provides a flexible alternative approach to the methodology of Hopkins and Thorne1 to estimate the effects of diffusion and beamline parameters on radiation damage during bio-SAXS experiments. To perform the calculations only the beam parameter, the capillary properties, and the diffusion constants of the proteins are needed. Details on the method can be found in the methods sections. Before considering the concrete values of the calculated median-lethal energy deposits, their uncertainties have to discussed. The values were calculated by applying a microscopic target-model, which depends on the radius of the target sphere of 7.5 nm as an input parameter. It originates from a study of α and dose mean-lineal energy ratio of different radiation types performed by Lindborg et al.47 For the simulated and measured values included in their study uncertainties up to 20% were reported. Thus, the radius of the target spheres has a similar uncertainty, which propagates to the uncertainty of the volume of the target sphere as 60%, being clearly the dominant contribution towards the total uncertainty budget of the median lethal energy values.28 Hence, the median lethal energy deposits values have to be reported as E4 mg/mL1/2 = 7 ± 5 eV at a G5P concentration of 4 mg mL−1 and E2 mg/mL1/2 = 4 ± 3 eV at 2 mg mL−1, respectively. These values are very similar to the values for electron irradiated plasmid DNA pUC19 with EpUC191/2 = 6 ± 4 eV.28 Whereby in case of the plasmid DNA this median lethal energy deposit refers to the induction of a so called single-strand break (SSB) at the sugar–phosphate backbone. On the other hand, for G5P the damage refers to all type of processes which lead to a decrease of the undamaged G5P dimer, may it be either aggregation, unfolding or fragmentation. The question arises which physico-chemical mechanisms lead to these damages? To give an answer one has to estimate which energy is needed to trigger which degradation pathway. The presented results showed (Fig. 8), that only an energy deposit of on average less than 10 eV within the target volume of the protein is required to cause a damage event with 50% probability. Here it is worth noting, that on the nano-scale the energy deposit over the target volume is not homogeneously distributed due to the stochastic nature of the involved scattering processes.48 By considering that the energy thresholds for ionization of water or biomolecules are roughly around 10 eV, and the electronic excitations of water or energies of resonances for dissociative electron attachment (DEA) or dissociative electron transfer processes (DET) are even lower, in some cases even as low as 0 eV,49 so that even these reported low energies could lead to one, or in rare cases, even to multiple damaging events. However, most of the species involved in the damage at the target, such as low energy electrons (LEE) and ROS, have a rather short reaction range of a couple of nanometers only.28 Both species can be produced from processes described by the net-ionization reaction6,50
| | | γ + 2H2O → H2O+ + e− + H2O → H3O+ + ˙OH + e−. | (8) |
Thus, as a starting point of this damage process an ionization event within the aforementioned 7.5 nm around the damage endpoint can be assumed. Therefore, it is most likely that most of the observed damage stems from single-hit events, highlighting their high efficiency in causing structural changes to the G5P dimer as reflected in the change of the observed SAXS scattering patterns. To get a more detailed insight into the chemical modifications involved, complementary techniques such as near-ambient pressure X-ray photoelectron spectroscopy (NAP-XPS) are needed, which allow for
in situ monitoring of chemical changes during irradiation of biomolecules.
6 Nevertheless, some conclusions can be drawn from the change in amount and type of radiation damage to G5P under the presence of the cosolute Ectoine, as will be discussed in the following.
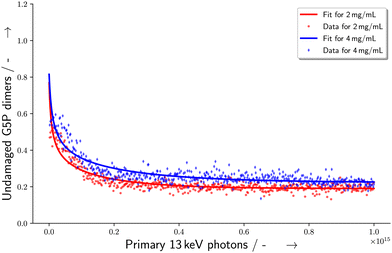 |
| | Fig. 8 Change in the number of undamaged G5P dimers with dependence on the amount of primary X-ray photons with 13 keV incident energy. The curve is a fit according to eqn (6). | |
In the presence of Ectoine the degradation behaviour changes completely. Here, aggregation is almost absent (Fig. 4 right, green curve). This absence of aggregation might be explained by two types of mechanisms: either it can be prevented due to reduction of the type of damage which is the starting point of aggregation behavior. Namely creation of reactive sites on the surface of the proteins by either direct damage via ionization events, or by indirect damage from radical species. Due to Ectoine's property as a radical scavenger as well as its influence on the scattering of low-energy electrons (LEE) at water, it is quite likely that a large part of the damage reduction is due to prevention of the occurrence of radiation damage directly.25 The second mode of action might prevent aggregation after a reactive species was already produced at the surface. To form an aggregate, the protein needs to encounter a reaction partner. Thus, a decrease in the probability from influencing diffusion behavior or shielding of the reactive sides might lead to such an outcome. In general, both types of mechanisms could take place simultaneously, leading to a synergetic protective effect of Ectoine. However to test this hypothesis and clarify whether the latter contributes to the observed effects, additional studies are needed.
The relative sudden decrease in undamaged protein around 380 s, (4 mg mL−1 G5P, Fig. 4 right) and 470 s (2 mg mL−1 G5P, data shown in the ESI†) in the presence of Ectoine, indicates a mechanism that may need some time to occur, or the accumulation of a certain amount of damage within the protein (more than a “single-hit”), to lead to fragmentation under a given condition. This could involve a meta-stable state of the protein, which might be somewhat stabilized temporarily by an increase of the stability of the intra-molecular hydrogen bonds within the protein, as resulting from the presence of the kosmotrop Ectoine. These stabilization effects are generally explained in terms of “preferential exclusion” of Ectoine from the protein surface, which leads to a preferential hydration of the protein and therefore stabilization of their structure.15,51,52 On the other hand, the questions arises why only the aggregation is effectively prevented during the whole course of the measurement, while fragmentation is only prevented until about 360 s. A possible explanation might be related to the different parts of a protein being involved either in fragmentation or aggregation. Fragmentation can happen when a covalent bond of the protein-backbone breaks. From a relative point of view, the protein-backbone is located to a high percentage within the protein volume itself. In contrast, aggregation needs formation of reactive species at the surface, and a subsequent encounter with another protein with which it then can undergo a inter-molecular interaction. Under the assumption, that Ectoine is mot likely excluded from the protein volume itself, it is reasonable, that it provides higher protection against damage occurring on the protein surface. Again, here especially the scavenging of hydroxyl radicals or other reactive species by Ectoine, is an effective way of preventing indirect damage at the protein surface. With respect to scavenging effects, the protective influence that buffers can have during bio-SAXS experiments should be evaluated with respect to their different scavenging capacities. For example, common Tris (tris(hydroxymethyl)aminomethane) based buffers provide a comparatively higher scavenging capacity than the phosphate-based buffers applied here. This is due to the scavenging properties of Tris.7 On one hand, the combination of Ectoine and Tris might lead to beneficial synergetic effects, though this would also add an additional layer of complexity to studies trying to probe radiation degradation directly. However, we can conclude, that Ectoine has shown effective protection of G5P against early radiation damage during synchrotron based bio-SAXS experiments, allowing for longer, undisturbed data collection. In comparison with other protective cosolutes, especially nucleosides and nucleobases,3 Ectoine showed a similar effectiveness according to the CorMap test (Table 1). At a concentration of 20 mM Ectoine provided better protection (ratio of 2.7–5.7) than glycerol (ratio 1.6 calculated from Table one in ref. 3) at 100 mM concentrations for lysozyme solutions. Ectoine's protection was similar to uridine (3.8) and only slightly worse than cytidine and 5-methyluridine (8.8–16.1) despite their five times higher concentrations (100 mM, all data from ref. 3). Furthermore it provides the additional beneficial property, that it can be used in combination with DNA-binding proteins, which naturally interact with nucleotides or nucleobases, as proposed by Castellvi et al.3 In contrast, Ectoine is most likely excluded from the protein surface due to the so called preferential exclusion mechanisms.15 Here it is noteworthy, that this is not the case for another biomolecule of importance, DNA. There, Ectoine was shown to accumulate at the DNA phosphate backbone, which was assumed due to its zwitterionic character.21 However, for proteins without strongly charged negative subgroups, such a behaviour is thought to be unlikely. For example Zaccai et al. provided structural evidence for the preferential exclusion of Ectoine from the surface of maltose binding protein (MBP) using small-angle neutron scattering.52 To test the hypothesis that the assumed radiation protection effects, originating from the kosmotropic properties of Ectoine, future studies should systematically investigate other chaotropes with similar scavenging capacities, so that the structure-influencing and radical scavenging properties can be separated from one another. However, for practical applications as “hypothetical protective cosolutes” in bio-SAXS, chaotropes are not so suitable, as they alter the structure of the macromolecules under investigation, due to their nature of being a chaotrope. In conclusion, Ectoine can serve as a radiation protectant and scavenger during bio-SAXS studies of a broad range of proteins, including delicate ssDNA and dsDNA-binding proteins. Furthermore, the protective effects of Ectoine, described in the present study, were observed under in vitro conditions. However, they may be of relevance in vivo as well. Especially, as Ectoine is a compatible solute, which can be accumulated in crowded cellular environment in molar concentrations, without disturbing the cells metabolism.21,24 In such a case, it may not only stabilize hydrogen-bonding,23,24 and protect DNA,21,25 but serve as multifunctional protectant of proteins against radiation induced stress, as it occurs during medical imaging, radiation therapy or long-distance space-flights.
5 Summary
Small-angle X-ray scattering measurements were performed on the ssDNA-binding protein G5P, both in the presence and absence of the compatible solute, osmolyte and radioprotectant Ectoine. The time and energy-deposit dependent evolution of the scattering curves were monitored and analysed in terms of multi-component fits. The data revealed a tendency of aggregation of G5P in pure solutions with some fragmentation, whereby the addition of Ectoine prevented aggregation of G5P effectively, while having less influence of the occurrence of fragmentation for a period of time. The resulting microscopic energy-damage relation was determined from a microdosimetric model based on Monte-Carlo particle scattering simulations and a custom target-model developed specifically for bio-SAXS experiments. These presented simulation models for Geant4/TOPAS provide an accessible way to calculate the energy deposit distribution within flexible sized capillaries as used for bio-SAXS experiments and are freely available on Github.46 The inclusion of target spheres allows to score the energy deposit within the surrounding of proteins or the volume within the capillary accessible to the proteins during the measurements. The combination of experimental data with this newly developed target model allowed for the determination of the microscopic median-lethal energy deposit for pure G5P with a concentration of 4 mg mL−1 as E1/2 = 7 ± 5 eV. In the presence of Ectoine the onset of damage according to the CorMap test was delayed by a factor of 2.7–5.7 with dependence to the protein concentration, showing the effective radiation protection of the protein during bio-SAXS measurements under the presence of Ectoine. The results indicate a protective effect from Ectoine, which acts as a scavenger of reactive oxygen species, e.g. hydroxyl radicals, and possible effects via post-irradiation mechanisms, related to preferential-exclusion of the cosolute from the protein surface. This reveals that the presence of Ectoine increases the possible exposure time during bio-SAXS experiments until radiation damage sets in. These findings may also be applicable for future bio-SAXS measurements, involving other DNA-binding proteins such as human mtSSB (hmtSSB), Replication protein A (RPA) or E. coli SBP. Thus, the addition of Ectoine provides an non-disturbing way to improve structure-determination of proteins via bio-SAXS.
Conflicts of interest
There are no conflicts of interest to declare.
Acknowledgements
We acknowledge helpful discussions with Dr Tihomir Solomun, Dr Brian Pauw, and Paul Mrkwitschka, and we would like to thank staff at Diamond Light Source beamline B21 for their help in data collection. This work was funded by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) under grant number 442240902 (HA 8528/2-1 and SE 2999/2-1). We acknowledge Diamond Light Source for time on Beamline B21 under Proposal SM29806. This work has been supported by iNEXT-Discovery, grant number 871037, funded by the Horizon 2020 program of the European Commission.
References
- J. B. Hopkins and R. E. Thorne, J. Appl. Crystallogr., 2016, 49, 880–890 CrossRef CAS PubMed.
- A. L. Brooks, D. G. Hoel and R. J. Preston, Int. J. Radiat. Biol., 2016, 92, 405–426 CrossRef CAS PubMed.
- A. Castellví, C. Pascual-Izarra, E. Crosas, M. Malfois and J. Juanhuix, Acta Crystallogr., Sect. D: Struct. Biol., 2020, 76, 971–981 CrossRef PubMed.
- C. M. Jeffries, M. A. Graewert, D. I. Svergun and C. E. Blanchet, J. Synchrotron Radiat., 2015, 22, 273–279 CrossRef CAS PubMed.
- T. R. Stachowski, M. E. Snell and E. H. Snell, J. Synchrotron Radiat., 2021, 28, 1309–1320 CrossRef CAS PubMed.
- M. B. Hahn, P. M. Dietrich and J. Radnik, Commun. Chem., 2021, 4, 1–8 CrossRef PubMed.
-
C. von Sonntag, Free-Radical-Induced DNA Damage and Its Repair, Springer, Berlin, Heidelberg, 2006 Search PubMed.
- A. Deshpande, S. Nimsadkar and S. C. Mande, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2005, 61, 1005–1008 CrossRef PubMed.
- T. Arakawa, Y. Kita and S. N. Timasheff, Biophys. Chem., 2007, 131, 62–70 CrossRef CAS PubMed.
- B. P. Schlegel, F. M. Jodelka and R. Nunez, Cancer Res., 2006, 66, 5181–5189 CrossRef CAS PubMed.
- E. Raderschall, E. I. Golub and T. Haaf, Proc. Natl. Acad. Sci. U. S. A., 1999, 96, 1921–1926 CrossRef CAS PubMed.
- P. Perucca, O. Cazzalini, O. Mortusewicz, D. Necchi, M. Savio, T. Nardo, L. A. Stivala, H. Leonhardt, M. C. Cardoso and E. Prosperi, J. Cell Sci., 2006, 119, 1517–1527 CrossRef CAS PubMed.
- J. E. Coleman and J. L. Oakley, CRC Crit. Rev. Biochem., 1980, 7, 247–289 CrossRef CAS PubMed.
- T. Solomun, H. Sturm, R. Wellhausen and H. Seitz, Chem. Phys. Lett., 2012, 533, 92–94 CrossRef CAS.
- M. B. Hahn, T. Solomun, R. Wellhausen, S. Hermann, H. Seitz, S. Meyer, H.-J. Kunte, J. Zeman, F. Uhlig, J. Smiatek and H. Sturm, J. Phys. Chem. B, 2015, 119, 15212–15220 CrossRef CAS PubMed.
- S. Su, Y.-G. Gao, H. Zhang, T. C. Terwilliger and A. H.-J. Wang, Protein Sci., 1997, 6, 771–780 CrossRef CAS PubMed.
- H. T. Pretorius, M. Klein and L. A. Day, J. Biol. Chem., 1975, 250, 9262–9269 CrossRef CAS.
- M. M. Skinner, H. Zhang, D. H. Leschnitzer, Y. Guan, H. Bellamy, R. M. Sweet, C. W. Gray, R. N. Konings, A. H. Wang and T. C. Terwilliger, Proc. Natl. Acad. Sci. U. S. A., 1994, 91, 2071–2075 CrossRef CAS PubMed.
- A. G. Murzin, EMBO J., 1993, 12, 861–867 CrossRef CAS PubMed.
- E. A. Galinski, H.-P. Pfeiffer and H. G. Trueper, Eur. J. Biochem., 1985, 149, 135–139 CrossRef CAS PubMed.
- M. B. Hahn, G. J. Smales, H. Seitz, T. Solomun and H. Sturm, Phys. Chem. Chem. Phys., 2020, 22, 6984–6992 RSC.
- J. Smiatek, R. K. Harishchandra, O. Rubner, H.-J. Galla and A. Heuer, Biophys. Chem., 2012, 160, 62–68 CrossRef CAS PubMed.
- M. B. Hahn, F. Uhlig, T. Solomun, J. Smiatek and H. Sturm, Phys. Chem. Chem. Phys., 2016, 18, 28398–28402 RSC.
- T. Solomun, M. B. Hahn and J. Smiatek, ChemPhysChem, 2020, 21, 1945–1950 CrossRef CAS PubMed.
- M. B. Hahn, S. Meyer, M.-A. Schröter, H.-J. Kunte, T. Solomun and H. Sturm, Phys. Chem. Chem. Phys., 2017, 19, 25717–25722 RSC.
- S. Brands, P. Schein, K. F. Castro-Ochoa and E. A. Galinski, Arch. Biochem. Biophys., 2019, 108097 CrossRef CAS PubMed.
- L. Cordsmeier and M. B. Hahn, ChemBioChem, 2022, 23, e202200391 CrossRef CAS PubMed.
- M. B. Hahn, S. Meyer, H.-J. Kunte, T. Solomun and H. Sturm, Phys. Rev. E, 2017, 95, 052419 CrossRef PubMed.
- S.-K. Lee, M. M. Maye, Y.-B. Zhang, O. Gang and D. van der Lelie, Langmuir, 2009, 25, 657–660 CrossRef CAS PubMed.
- N. P. Cowieson, C. J. C. Edwards-Gayle, K. Inoue, N. S. Khunti, J. Doutch, E. Williams, S. Daniels, G. Preece, N. A. Krumpa, J. P. Sutter, M. D. Tully, N. J. Terrill and R. P. Rambo, J. Synchrotron Radiat., 2020, 27, 1438–1446 CrossRef CAS PubMed.
- M. Basham, J. Filik, M. T. Wharmby, P. C. Y. Chang, B. El Kassaby, M. Gerring, J. Aishima, K. Levik, B. C. A. Pulford, I. Sikharulidze, D. Sneddon, M. Webber, S. S. Dhesi, F. Maccherozzi, O. Svensson, S. Brockhauser, G. Náray and A. W. Ashton, J. Synchrotron Radiat., 2015, 22, 853–858 CrossRef PubMed.
- J. B. Hopkins, R. E. Gillilan and S. Skou, J. Appl. Crystallogr., 2017, 50, 1545–1553 CrossRef CAS PubMed.
- B. R. Pauw, J. S. Pedersen, S. Tardif, M. Takata and B. B. Iversen, J. Appl. Crystallogr., 2013, 46, 365–371 CrossRef CAS PubMed.
- S. Grudinin, M. Garkavenko and A. Kazennov, Acta Crystallogr., Sect. D: Struct. Biol., 2017, 73, 449–464 CrossRef CAS PubMed.
- M. T. Tyn and T. W. Gusek, Biotechnol. Bioeng., 1990, 35, 327–338 CrossRef CAS PubMed.
- S. Agostinelli and others, Nucl. Instrum. Methods Phys. Res., Sect. A, 2003, 506, 250–303 CrossRef CAS.
- J. Perl, J. Shin, J. Schumann, B. Faddegon and H. Paganetti, Med. Phys., 2012, 39, 6818–6837 CrossRef CAS PubMed.
- S. Incerti and others, Med. Phys., 2010, 37, 4692–4708 CrossRef CAS PubMed.
- J. M. Zutta Villate and M. B. Hahn, Eur. Phys. J. D, 2019, 73, 95 CrossRef.
- M. B. Hahn, S. Meyer, M.-A. Schröter, H. Seitz, H.-J. Kunte, T. Solomun and H. Sturm, Phys. Chem. Chem. Phys., 2017, 19, 1798–1805 RSC.
- S. Kuwamoto, S. Akiyama and T. Fujisawa, J. Synchrotron Radiat., 2004, 11, 462–468 CrossRef CAS PubMed.
- W. M. Garrison, Chem. Rev., 1987, 87, 381–398 CrossRef CAS.
- S. Mazier, S. Villette, S. Goffinont, S. Renouard, J. C. Maurizot, D. Genest and M. Spotheim-Maurizot, Radiat. Res., 2008, 170, 604–612 CrossRef CAS PubMed.
- I. Rasched and E. Oberer, Microbiol. Rev., 1986, 50, 401–427 CrossRef CAS PubMed.
- M. B. Hahn, BioSAXS models for TOPAS/Geant4, 2022 DOI:10.26272/opus4-55751 , BAM Eigenverlag.
-
M. B. Hahn, TOPAS Bio-SAXS Dosimetry Extension, 2022, https://github.com/MarcBHahn/TOPAS-bioSAXS-dosimetry.
- L. Lindborg, M. Hultqvist, Å. C. Tedgren and H. Nikjoo, Phys. Med. Biol., 2013, 58, 3089 CrossRef CAS PubMed.
- D. T. Goodhead, Radiat. Prot. Dosim., 2006, 122, 3–15 CrossRef PubMed.
- E. Alizadeh and L. Sanche, Chem. Rev., 2012, 112, 5578–5602 CrossRef CAS PubMed.
- M. S. Kreipl, W. Friedland and H. G. Paretzke, Radiat. Environ. Biophys., 2008, 48, 11–20 CrossRef PubMed.
- S. N. Timasheff, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 9721–9726 CrossRef CAS PubMed.
- G. Zaccai, I. Bagyan, J. Combet, G. J. Cuello, B. Demé, Y. Fichou, F.-X. Gallat, V. M. G. Josa, S. von Gronau, M. Haertlein, A. Martel, M. Moulin, M. Neumann, M. Weik and D. Oesterhelt, Sci. Rep., 2016, 6, 31434 CrossRef CAS PubMed.
|
| This journal is © the Owner Societies 2023 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  abc,
Glen J.
Smales
abc,
Glen J.
Smales
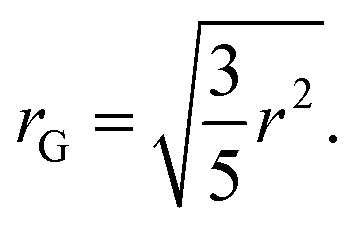

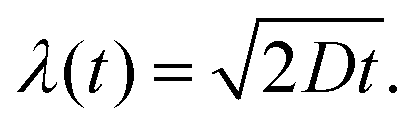
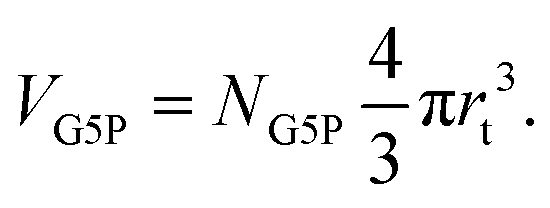
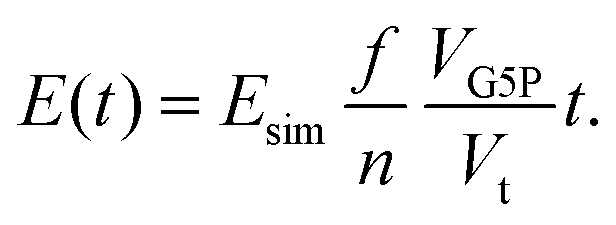
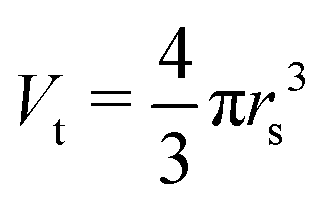 with rs = 258 μm, and the sensitive volume of G5P (VG5P = 2.44 × 106 nm3, eqn (4)). These results are used to calculate the microscopic energy-damage relation in the following section.
with rs = 258 μm, and the sensitive volume of G5P (VG5P = 2.44 × 106 nm3, eqn (4)). These results are used to calculate the microscopic energy-damage relation in the following section.