
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Substitution of Ca2+ and changes in the H-bond network near the oxygen-evolving complex of photosystem II†
Manoj
Mandal
 *a,
Keisuke
Saito
bc and
Hiroshi
Ishikita
*bc
*a,
Keisuke
Saito
bc and
Hiroshi
Ishikita
*bc
aDepartment of Chemical and Biological Sciences, S. N. Bose National Centre for Basic Sciences, Kolkata 700106, West Bengal, India. E-mail: mandalmanojcu@gmail.com
bResearch Center for Advanced Science and Technology, The University of Tokyo, 4-6-1 Komaba, Meguro-ku, Tokyo 153-8904, Japan. E-mail: hiro@appchem.t.u-tokyo.ac.jp
cDepartment of Applied Chemistry, The University of Tokyo, 7-3-1 Hongo, Bunkyo-ku, Tokyo 113-8654, Japan
First published on 6th February 2023
Abstract
Ca2+, which provides binding sites for ligand water molecules W3 and W4 in the Mn4CaO5 cluster, is a prerequisite for O2 evolution in photosystem II (PSII). We report structural changes in the H-bond network and the catalytic cluster itself upon the replacement of Ca2+ with other alkaline earth metals, using a quantum mechanical/molecular mechanical approach. The small radius of Mg2+ makes W3 donate an H-bond to D1-Glu189 in Mg2+-PSII. If an additional water molecule binds at the large surface of Ba2+, it donates H-bonds to D1-Glu189 and the ligand water molecule at the dangling Mn, altering the H-bond network. The potential energy profiles of the H-bond between D1-Tyr161 (TyrZ) and D1-His190 and the interconversion between the open- and closed-cubane S2 conformations remain substantially unaltered upon the replacement of Ca2+. Remarkably, the O5⋯Ca2+ distance is shortest among all O5⋯metal distances irrespective of the radius being larger than that of Mg2+. Furthermore, Ca2+ is the only alkaline earth metal that equalizes the O5⋯metal and O2⋯metal distances and facilitates the formation of the symmetric cubane structure.
The reaction center of the water-splitting enzyme photosystem II (PSII) is formed by two structurally similar but electrostatically different protein subunits, D1 and D2.1,2 To oxidize substrate water molecules, PSII uses the electron transfer pathway that proceeds from the catalytic Mn4CaO5 cluster via redox active D1-Tyr161 (TyrZ) to the oxidized chlorophyll pair, [PD1PD2]˙+ (≈PD1˙+3–6). [PD1PD2]˙+ forms after electronic excitation of the reaction center chlorophylls7,8 and subsequent electron transfer occurs via pheophytin and the initial quinone QA to the secondary quinone QB. As electron transfer occurs, the oxidation state of the oxygen-evolving complex, Sn, increases from S0 to S3 in the order S0 → S1 → S2 → S3 → S0 (e.g.,9,10). O2 evolves during the S3 to S0 transition after S3 absorbs a photon.
The Mn4CaO5 cluster has four water molecules as ligands, W1 and W2 at the dangling Mn (Mn4) and W3 and W4 at Ca2+, which are also candidates for substrate water molecules (e.g.,11,12). Ca2+ has seven ligand groups (O1, O2, O5, D1-Asp170, D1-Ala344, W3, and W4).1 Ca2+ and Mg2+ are the most abundant alkaline earth metals in biological systems. In PSII, Ca2+ is a prerequisite for O2 evolution.13–19 Previously, it was speculated that Ca2+ was the origin of the distorted cubane structure (e.g.,20). However, the distortion of the Mn4CaO5 cluster remains even upon the removal of Ca2+.21–23 Indeed, not Ca2+ but dangling Mn4 is most responsible for the distortion of the cluster shape.23 The S2 to S3 transition is inhibited in Ca2+-depleted PSII.13,24–26 Ca2+ depletion not only causes the alteration of the H-bond network at the Mn4O5 and TyrZ moieties23 but also decreases the redox potential (Em) of TyrZ significantly due to reorientation of the water molecules in the H-bond network, making electron transfer from the Mn4CaO5 cluster to TyrZ uphill.27
Replacement of Ca2+ with any metals except Sr2+ inhibits O2 evolution,13–17 although the inhibition mechanism may depend on the metals. The geometry of the catalytic site in Sr2+-substituted PSII (Sr2+-PSII) resembles that of native PSII (Ca2+-PSII).28,29 The Em values for the artificial clusters with Sr2+ are also similar to those with Ca2+.30–32 The Em value for the Mn4BaO5 cluster in Ba2+-substituted PSII (Ba2+-PSII) is also considered to be similar to that for the Mn4CaO5 cluster in native PSII based on the observation of the normal thermoluminescence S2QA˙− band.33 Fourier transform infrared (FTIR) studies by Kimura et al. showed that the double difference S2/S1 spectrum was not affected significantly upon the substitution of Ca2+ with Mg2+ and Sr2+, whereas the vibrational modes of the carboxylate ligand residue disappeared upon substitution with Ba2+ in the PSII membrane from spinach.33 According to FTIR studies by Suzuki et al.,34 more than three carboxylate residues, except D1-Glu189 and the carboxyl terminus of the D1 protein, D1-Ala344, were perturbed upon Sr2+ substitution. FTIR studies by Strickler et al. also suggested that D1-Ala344 was not involved in the perturbation observed upon Sr2+ substitution.35
S2 can form in Mg2+-substituted PSII (Mg2+-PSII) but not in Ba2+-PSII.36 Vrettos et al. reported that Mg2+ and Ba2+ are unlikely to bind competitively with Ca2+.14 It was proposed that Ba2+ led to the deformation of the proton-conducting H-bond network.37,38 Although the radius of Ca2+ is one of the key factors,14,32 it remains unclear what property of Ca2+ is specifically required for O2-evolving activity among alkaline earth metals. Previous theoretical studies by Vogt et al. showed the detailed geometry of the Mn4SrO5 cluster in S1, S0, S–1, and S–2 in Sr2+-PSII.29 On the other hand, not only the geometry of the Mn4SrO5 cluster but also the energetics of the H-bond network in S2, in which the significance of Ca2+ is pronounced, remains unclear. FTIR studies suggested that the S2 to S3 transition involves the migration of the proton of a ligand water molecule toward D1-Asp61,39 which is in line with mutational studies (mutated to the other 19 residues).40 Theoretical studies also showed that a low-barrier H-bond forms between the ligand water molecule W1 and D1-Asp61 specifically in S2.41,42 The replacement of Ca2+ with the other redox-inactive divalent metals is unlikely to affect the H-bond between W1 and D1-Asp61, as the Ca2+ binding site is not directly involved in the W1⋯D1-Asp61 moiety. In contrast, the redox-active TyrZ⋯D1-His190 pair is directly involved in the H-bond network of the Ca2+ binding site.23 Because TyrZ forms a low-barrier H-bond with D1-His19043 and is directly involved in the H-bond network of the ligand water molecules (W3 and W4) at Ca2+,23,27 Ca2+-substitution may affect the low-barrier H-bond formation between TyrZ and D1-His190. However, to the best of our knowledge, the influence of Ca2+ on the TyrZ⋯D1-His190 H-bond has not been specifically reported.
To understand the specificity of Ca2+ in PSII, we investigated the local geometry of the metal-substituted Mn4MO5 cluster (M = Mg2+, Sr2+, and Ba2+) in S2 with Mn1(III)Mn2(IV)Mn3(IV)Mn4(IV) (open-cubane S2 conformation) by adopting a quantum mechanical/molecular mechanical (QM/MM) approach based on the native Ca2+-PSII crystal structure. As proton transfer occurs most effectively in the well-ordered H-bond network44,45 and the water molecules in the focusing H-bond network are less disordered in molecular dynamics simulations,46 comparisons of the H-bond networks among the metal-substituted PSIIs based on the QM/MM-optimized geometries are, therefore, the best starting point.
Methods
Coordinates and atomic partial charges
The atomic coordinates were obtained from the X-ray structure of PSII from Thermosynechococcus vulcanus (PDB code, 3ARC).1 The positions of all heavy atoms were fixed during the optimization of the positions of H atoms with CHARMM.47 All titratable groups (e.g., acidic and basic groups) were ionized. D1-His337 was considered to be protonated.48 Atomic partial charges of the amino acids and cofactors were obtained from the CHARMM2249 parameter set and previous studies,45 respectively.QM/MM calculations
The unrestricted density functional theory method was employed with the B3LYP functional (commonly used for PSII by, e.g., Amin,50 Batista,51 Guidoni,52 Ishikita,45 Pace,53 Siegbahn,54 Yamaguchi,55 and their coworkers as summarized in ref. 56) and LACVP* basis sets (LANL2DZ (double ζ quality basis set with the Los Alamos effective core potential) for Mn, Mg, Ca, Sr, and Ba atoms and 6-31G* for other atoms)57 using the QSite58 program if not otherwise specified. The M06 functional was also used to evaluate the contributions of dispersion correction. See Table S1 (ESI†) for the results obtained with other functional/basis sets. All water molecules assigned in the crystal structure were included in the present study. FTIR spectra and theoretical calculations by Nakamura and Noguchi suggested that the ligand water molecules, W1 and W2, are H2O in S1 and S2.48 pKa calculations by Saito et al. showed that W1 and W2, were H2O and pKa(W2) was only marginally (∼1 pKa unit) lower than pKa(W1) in water, whereas pKa(W1) was significantly lower than pKa(W2) in the PSII protein environment due to the presence of the proton acceptor, D1-Asp61.42 In the open-cubane S2 conformation, H2O at W2 forms a low-barrier H-bond with D1-Asp61, although the proton remains at this moiety.41 In the present study, W1 and W2 were modeled as H2O. Counter ions were added to neutralize the entire system. In the QM region, all atomic coordinates were fully relaxed (i.e., not fixed at the positions in the crystal structure). For the MM region, the atomic charges of CHARMM22 were used for amino acid groups to facilitate comparison between their influences on the active site in the QM region and their influences on Em for further purposes, as Em can be calculated using the atomic charges of CHARMM22 and solving the Poisson–Boltzmann equation (e.g.,59). In the MM region, only the H atom positions were optimized using the OPLS2005 force field, which is mandatory in QSite. In the MM region, the heavy atom positions were fixed to avoid unexpected conformational changes (e.g., caused by the absence of water molecules unassigned in the original crystal structure). In QSite, the QM and MM regions interact via electrostatic and van der Waals interactions.The Mn4CaO5 cluster was considered to be in the S2 states with antiferromagnetically coupled Mn ions; the resulting Mn oxidation states (Mn1, Mn2, Mn3, Mn4) and the total spin, S, were (III, IV, IV, IV) and S = 7/2 (↑↓↑↑) in S2, respectively. It should be noted that the difference in S (e.g., S = 1/2 in S2,60 high, low, ferromagnetic, and antiferromagnetic) did not affect the values; for example, (i) the resulting geometry61,62 and (ii) the potential-energy profile of proton transfer41 are not crucial to the spin configurations as far as the protein electrostatic environment is fully included.63
The initial-guess wavefunctions were obtained using ligand field theory64 implemented in the QSite program. For native PSII, the QM region was defined as the Mn4MO5 cluster (including the ligand side-chains of D1-Asp170, D1-Glu189, D1-His332, D1-Glu333, D1-Asp342, and CP43-Glu354; ligand carboxy-terminal group of D1-Ala344; and ligand water molecules, W1–W4), O4–water chain (W539, W538, and W393),45,65 Cl-1 binding site (Cl−, W442, W446, and the side-chains of D1-Asn181 and D2-Lys317), second-sphere ligands (side-chains of D1-Asp61 and CP43-Arg357), H-bond network of TyrZ (side-chains of D1-Tyr161, D1-His190, and D1-Asn298), including the diamond-shaped water cluster (W5, W6, and W7).43,66 The QM region defined in the present study is one of the largest among theoretical studies of PSII, which essentially covers the entire H-bond network of the ligand water molecules at the Ca2+ moiety (summarized in ref. 56).
To obtain the potential energy profiles of the O⋯H+⋯N bond for TyrZ⋯D1-His190, the QM/MM-optimized geometry was used as the initial geometry. The H atom under investigation was moved between the O and N moieties by 0.05 Å, after which the geometry was optimized by constraining the distance between O–H+ and H+–N, and the energy was calculated. This procedure was repeated until the H atom reached the O moieties. To obtain the potential energy profiles of the Mn1⋯O5 and O5⋯Mn4 bonds for the open- and closed-cubane S2 conformations, the QM/MM-optimized geometry of the open-cubane S2 conformation was used as the initial geometry. The O5 was moved toward the Mn4 moiety by 0.05 Å, after which the geometry was optimized by constraining the Mn1⋯O5 distance, and the energy was calculated.
Results
H-bond network
QM/MM calculations show that the difference in the geometry is predominantly observed at the M binding moiety of the Mn4MO5 cluster (Fig. 1). Among the three metal-substituted PSIIs, Sr2+-PSII is closest to Ca2+-PSII, as the H-bond patterns are conserved between the two PSIIs (Fig. 1b and c). The only remarkable difference is observed at the slightly longer distances between M and the ligand water molecules in Sr2+-PSII than in Ca2+-PSII, 2.42–2.44 Å for Ca2+ and 2.55–2.56 Å for Sr2+, predominantly due to the radii (Table 1).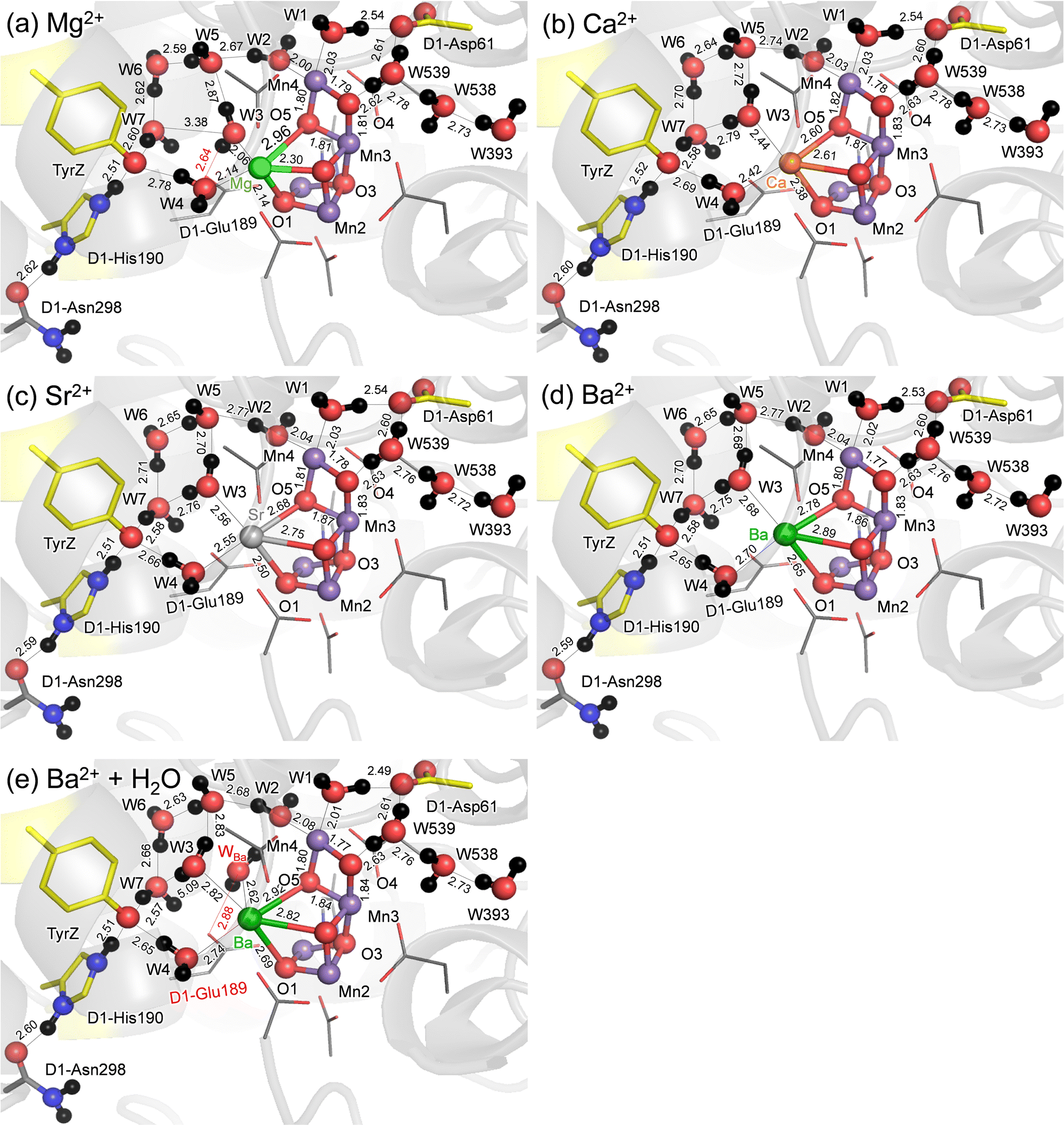 | ||
| Fig. 1 QM/MM-optimized geometries in the open-cubane S2 conformations. (a) Mg2+-PSII. (b) Ca2+-PSII. (c) Sr2+-PSII. (d) Ba2+-PSII. (e) Ba2+-PSII with an additional water molecule at Ba2+ (WBa). Dotted lines indicate H-bonds. Bond distances are in Å. | ||
| Mg2+ | Ca2+ | Sr2+ | Ba2+ | Ba2+ + water | |
|---|---|---|---|---|---|
| a See ref. 67. b The surface area of Ca2+ is normalized to 1. | |||||
| Ionic radiusa | 0.66 | 0.99 | 1.12 | 1.34 | 1.34 |
| (Surface area ratiob) | (0.44) | (1) | (1.28) | (1.83) | (1.83) |
| W3⋯M | 2.06 | 2.44 | 2.56 | 2.68 | 2.82 |
| W4⋯M | 2.14 | 2.42 | 2.55 | 2.70 | 2.74 |
| O1⋯M | 2.14 | 2.38 | 2.50 | 2.65 | 2.69 |
| O2⋯M | 2.30 | 2.61 | 2.75 | 2.89 | 2.82 |
| O5⋯M | 2.96 | 2.60 | 2.68 | 2.78 | 2.92 |
| O5⋯Mn1 | 2.93 | 3.08 | 3.08 | 3.12 | 3.15 |
| O5⋯Mn4 | 1.80 | 1.82 | 1.81 | 1.80 | 1.80 |
The small radius of Mg2+ shortens the Mg2+⋯W3 and Mg2+⋯W4 distances (to 2.06 and 2.14 Å, respectively) (Table 1). The small radius of Mg2+ also breaks the H-bond between W3⋯W7 (3.26 Å), which is energetically compensated for by the H-bond formation between W3 and D1-Glu189 (2.66 Å). Eventually, the alteration in the H-bond network occurs with respect to the Ca2+- and Sr2+-PSIIs (Fig. 1a), which may be associated with the inhibited O2-evolving activity in Mg2+-PSII. In contrast, the large radius of Ba2+ increases the Ba2+⋯W3 and Ba2+⋯W4 distances significantly (to 2.68–2.70 Å, Table 1). However, the H-bond pattern of the H-bond network essentially remains unchanged with respect to the Ca2+- and Sr2+-PSIIs (Fig. 1d). The surface area of Ba2+, which is 1.8 times larger than that of Ca2+, may allow the third water molecule (WBa) to bind at Ba2+ in addition to W3 and W4 (Table 1). To evaluate the existence of the extra water molecule, WBa is placed at the resulting cavity near Ba2+. QM/MM calculations show that WBa bridges the gap between D1-Glu189 and W2 via H-bonds (Fig. 1e). Thus, the alteration of the H-bond network is pronounced if the third water molecule is incorporated at the Ba2+ site. Note that no remarkable difference in the QM/MM-optimized geometry is observed when considering dispersion correction (Table S2, ESI†).
H-bond between TyrZ and D1-His190
The distance between Ca2+ and D1-Tyr161 (TyrZ) is 4.8 Å.1 Ca2+ substitution induces deformations in the shape of a cluster of water molecules near TyrZ (i.e., W3, W5, W6, and W7, Fig. 1), which are essential for forming the low-barrier H-bond between TyrZ and D1-His190.43 Nevertheless, TyrZ⋯D1-His190 remains short (∼2.5 Å, Fig. 1). The potential-energy profile for the H-bond indicates that TyrZ and D1-His190 form low-barrier H-bonds in all metal-substituted PSIIs (Fig. 2). It seems likely that the difference in the radius does not affect the formation of the low-barrier H-bond between TyrZ and D1-His190 if the H-bond network is maintained.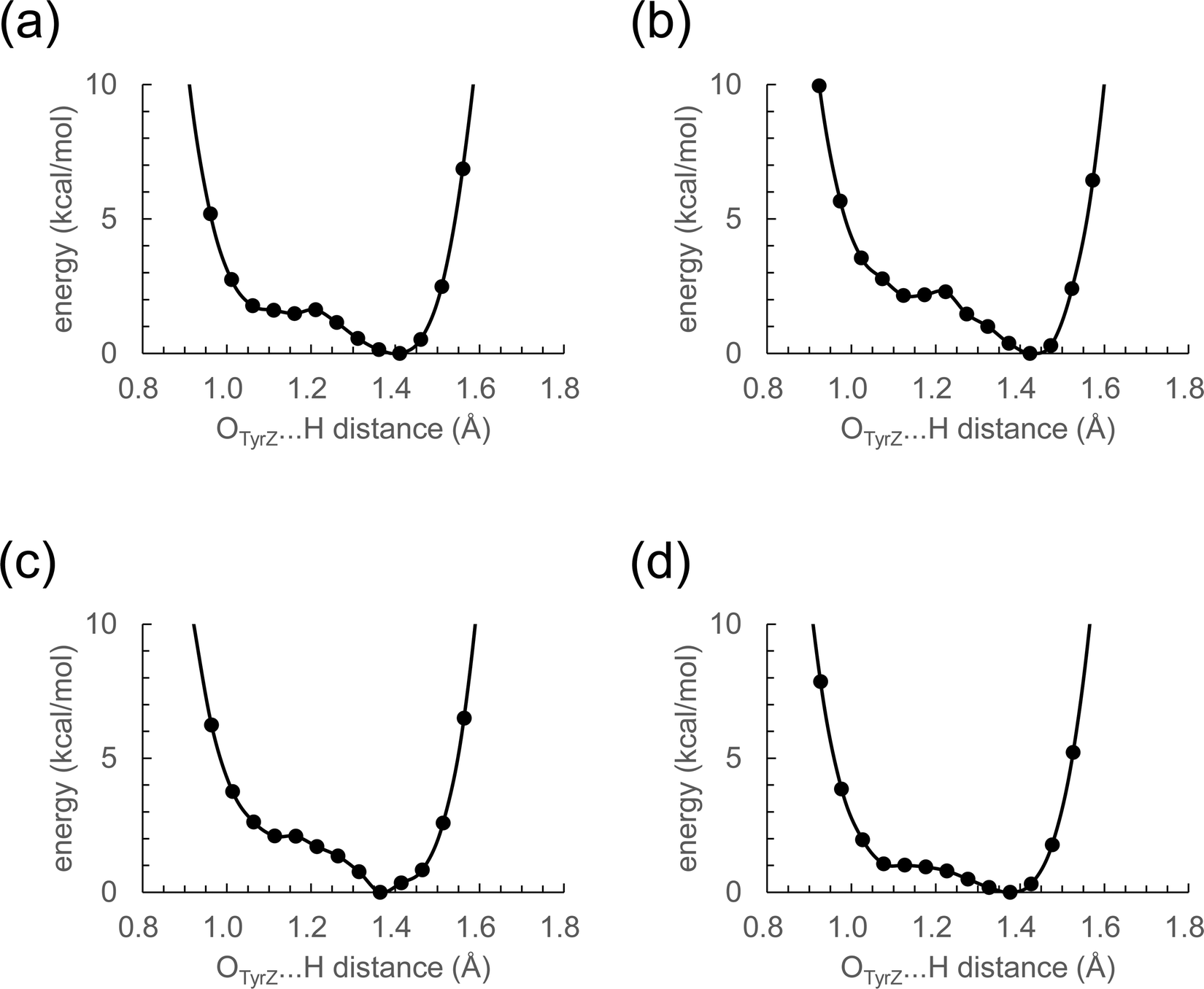 | ||
| Fig. 2 Potential-energy profile of the H-bond between TyrZ and D1-His190 in S2. (a) Mg2+-PSII. (b) Ca2+-PSII. (c) Sr2+-PSII. (d) Ba2+-PSII. | ||
Energetics of the open- and closed-S2 cubane conformations
In S2, the Mn1(III)Mn2(IV)Mn3(IV)Mn4(IV) state adopts the open-cubane S2 conformation, where the Mn1(III)⋯O5 distance is larger than the O5⋯Mn4(IV) distance. In contrast, the Mn1(IV)Mn2(IV)Mn3(IV)Mn4(III) state adopts the closed-cubane S2 conformation, where the Mn1(IV)⋯O5 distance is shorter than the O5⋯Mn4(III) distance.68 In electron paramagnetic resonance (EPR) spectroscopy for the Mn4CaO5 cluster, the g = 2 multiline and g > 4.1 signals are observed (e.g.,69). The g > 4.1 signals are classified into two cases: the g = 4.1 and g = 4.8 signals. Recent QM/MM calculations showed that the g = 4.1 signal corresponds to the closed-cubane conformation.70 However, only the open-cubane S2 conformation was identified in the XFEL structures, but not the closed-cubane S2 conformation,71–73 probably because the open-cubane S2 conformation is energetically more stable than the closed-cubane S2 conformation.23,50,62,74 This also holds true for Mg2+-PSII, Sr2+-PSII, and Ba2+-PSII: the differences in the alkaline-earth-metal and the H-bond network do not affect the stability of the open-cubane S2 conformation with respect to the closed-cubane S2 conformation (Fig. 3).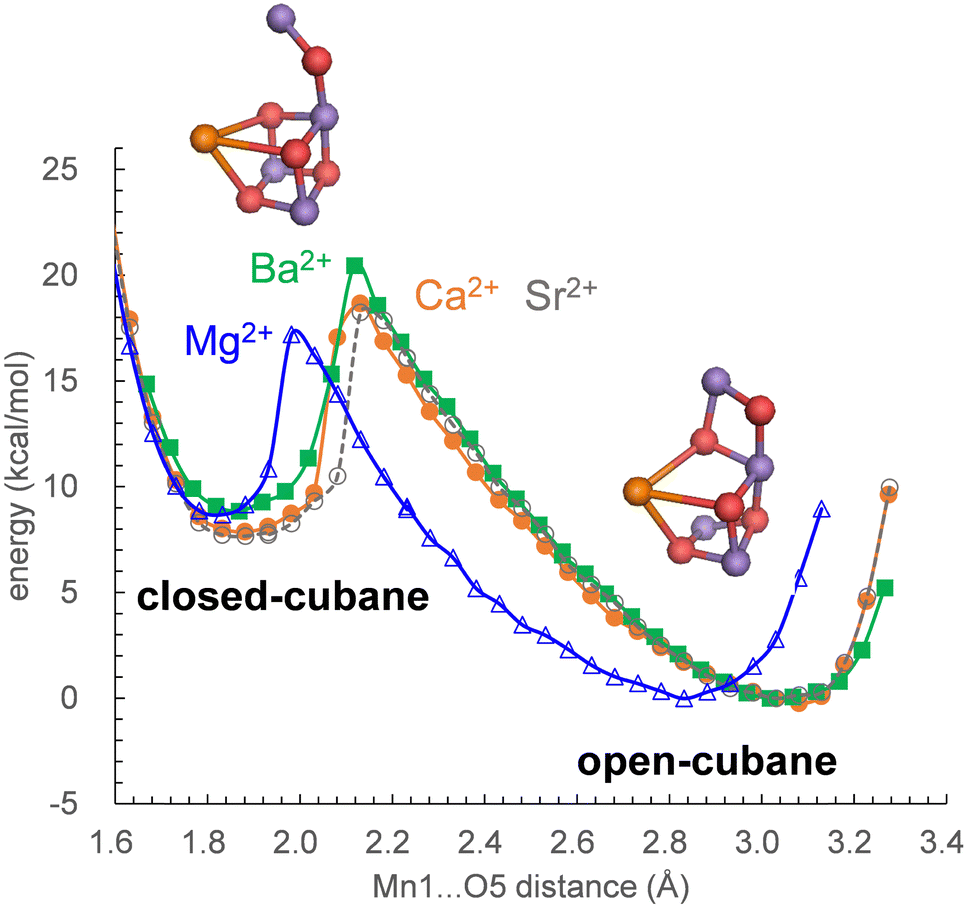 | ||
| Fig. 3 Potential-energy profile for the O5 position along the Mn1⋯O5 and O5⋯Mn4 axes. The local energy minimum at the short Mn1⋯O5 distance (∼1.8 Å) corresponds to the closed-cubane S2 conformation, whereas the local energy minimum at the long Mn1⋯O5 distance (∼3.0 Å) corresponds to the open-cubane S2 conformation. The geometry was fully QM/MM-optimized at each point. Blue solid curve with open triangles: Mg2+-PSII; orange solid curve with closed circles: Ca2+-PSII; gray dotted curve with open circles: Sr2+-PSII; green solid curve with closed squares: Ba2+-PSII. | ||
Discussion
Mn K-edge X-ray absorption spectroscopy21 and ENDOR22 studies suggested that the (electronic) structure of the Mn4CaO5 cluster remains essentially unaltered upon Ca2+ depletion. Consistently, the present QM/MM calculations show that Ca2+-substitution/-depletion does not substantially affect the electronic structure of the Mn4MO5 cluster75 (see also Table S3, ESI†).The potential-energy profiles for the interconversion between the open- and closed-cubane S2 conformations, namely, the energy difference between the open- and closed-cubane S2 conformations, are similar in native Ca2+-PSII and metal-substituted PSIIs (Fig. 3), which suggests that the inhibition of the interconversion of the two S2 conformations is not responsible for the inhibition of O2 evolution upon replacement of Ca2+. Thus, the difference in the pKa value for ligand-water deprotonation between alkaline earth metals may still be one of the plausible hypotheses for the inhibition mechanism.14,76,77
The PSII crystal structure shows that Ca2+ has seven ligand groups (O1, O2, O5, D1-Asp170, D1-Ala344, W3, and W4).1 In particular, O1, O2, and O5 form the Ca2+ binding site of the Mn4CaO5 cluster. Most distances with M increase as the radius of the alkaline earth metal increases (Fig. 4a). However, the O5⋯M distance is exceptional. Intriguingly, the O5⋯M distance in Ca2+-PSII is the shortest among all metal-substituted PSIIs. Thus, Ca2+ is the alkaline earth metal that interacts most strongly with the Mn4O5 host region. The short O5⋯Ca2+ distance, even shorter than the O5⋯Mg2+ distance, suggests that Ca2+ may function most cooperatively with the Mn sites of the Mn4O5 cubane among all alkaline earth metals.
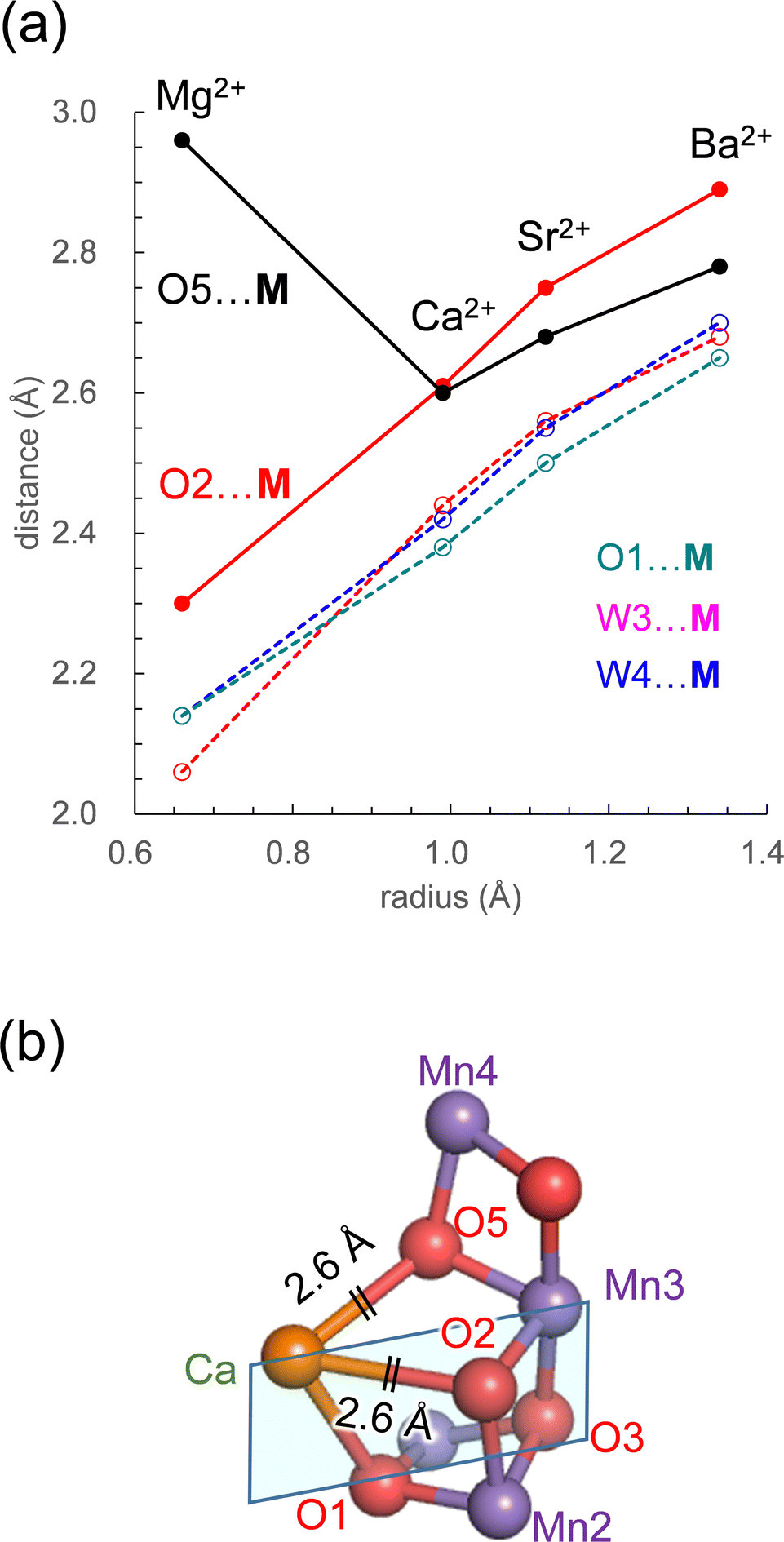 | ||
| Fig. 4 (a) Dependence of the distances with M on the radius in the open-cubane S2 conformation: O5⋯M (black solid line); W3⋯M (red dotted line); W4⋯M (blue dotted line); O1⋯M (green dotted line). (b) The open-cubane Mn4CaO5 structure and the [M (Ca2+)–O1–O3–Mn3] plane (sky blue square). See Table 1 for ionic radii. | ||
The O5⋯M and O2⋯M distances are identical (2.6 Å) only in Ca2+-PSII, which suggests that the shape of the open-cubane Mn3CaO4 region is most symmetric with respect to the [M (Ca2+)–O1–O3–Mn3] plane among all metal-substituted PSIIs (Fig. 4b). The result presented here indicates that the binding of Ca2+ is not the origin of the distorted cubane structure (e.g.,20), but it minimizes the distortion of the cluster with respect to other alkaline earth metals, leading to the symmetric shape of the open-cubane Mn3CaO4 region. The minimized distortion with Ca2+ may indicate that the Mn4CaO5 cluster is most stable among all alkaline earth metal clusters. Thus, Ca2+ may contribute to the remarkably large turnover number of 105 for the Mn4CaO5 cluster in native PSII.78
In summary, no significant difference is observed in the core structure or the characteristics of the H-bond between TyrZ and D1-His190 among the metal-substituted PSII (Fig. 2). This is consistent with the recent observations of synthetic Mn4Ca clusters, in which Ca2+ can be structurally and energetically replaced by other metal ions (e.g., Y and Gd).79
The characteristics of Sr2+-PSII are closest to those of Ca2+-PSII among Mg2+, Sr2+, and Ba2+-PSIIs (Fig. 3). The H-bond pattern of the H-bond network of only Sr2+-PSII is also consistent with that of Ca2+-PSII (Fig. 1). The calculated pKa value of a ligand water molecule at the Mn4MO5 cluster at the same level for Ca2+ and Sr2+ (∼13) in the absence of the PSII protein environment (i.e., gas phase).77 In contrast, the pKa value for the ligand water molecule differs by 2 units between Mg2+ and Ba2+,76 which largely originates from the difference in the radius. The difference in pKa alters the H-bond distances with the ligand water molecules, whereas the difference in the radius alters the distance between the metal center and the ligand water molecule. These differences are ultimately pronounced in the difference in the H-bond pattern of W3 in Mg2+-PSII. In Mg2+-PSII, W3 donates an H-bond to D1-Glu189, but the H-bond between W3 and W7 disappears, altering the H-bond network with respect to Ca2+- and Sr2+-PSIIs (Fig. 1a). In contrast, an increase in the radius compared to that of Ba2+ does not induce an alteration in the H-bond pattern of the H-bond network (Fig. 1d). Indeed, the difference in the pKa value for the ligand water molecule among Ca2+ (12.8), Sr2+ (13.2), and Ba2+ (13.4) is very small,76 which cannot explain the inactivity of Ba2+-PSII. Only if a water molecule additionally binds at Ba2+ as the third ligand water molecule does it donate H-bonds to D1-Glu189 and W2, altering the H-bond network (Fig. 1e). According to recent X-ray free electron laser (XFEL) structures, a water molecule (O6) was inserted between W2 and D1-Glu189 during the S2 to S3 transition.72,73 However, in Mg2+-PSII, W3 is closer to O5 and already donates an H-bond to D1-Glu189, which may inhibit O6 insertion (i.e., the S2 to S3 transition). In Ba2+-PSII, the binding site of the third ligand water molecule overlaps with the O6 binding site in the XFEL Ca2+-PSII structure. Thus, Ba2+ with a third ligand water molecule may restrict the insertion of an extra water molecule (e.g.,71,72) in the S2 to S3 transition. The low-barrier H-bond between TyrZ and D1-His190 remains unaffected even in the Mg2+- and Ba2+-PSIIs irrespective of the Ca2+/metal binding site being relatively close to TyrZ (Fig. 2). Although Mg2+ and Ba2+ may not bind competitively with Ca2+,14 the observed alteration in the “external” environment of the catalytic center (e.g., ligand structure and H-bond network) may be associated with the inhibition mechanism for O2 evolution if the metal-substituted PSIIs are properly assembled.
More importantly, Ca2+ exclusively minimizes the “internal” structure of the catalytic center. Ca2+ is the unique alkaline earth metal that (i) has the shortest O5⋯M distance (irrespective of the radius being larger than Mg2+) and interacts most strongly with the Mn4O5 host region and (ii) equalizes the O5⋯M and O2⋯M distances (2.6 Å) and facilitates the formation of the symmetric cubane structure (Fig. 4a). The resulting distortion-free Mn4CaO5 cluster is energetically advantageous with respect to the other metal-substituted clusters, which may contribute to the remarkably large turnover number of 105![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) in native PSII.78 This may be one of the reasons why Ca2+ is the preferred redox-inactive site in nature as the catalytic O2-evolving center.
in native PSII.78 This may be one of the reasons why Ca2+ is the preferred redox-inactive site in nature as the catalytic O2-evolving center.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank Keisuke Kawashima and Dr Toyokazu Ishida for useful discussion. This research was supported by JSPS KAKENHI (JP18H05155, JP18H01937, JP20H03217, and JP20H05090 to H. I. and 16H06560 and 18H01186 to K. S.), JST CREST (JPMJCR1656 to H. I.), and the Interdisciplinary Computational Science Program in CCS, University of Tsukuba. M. M. thanks Department of Biotechnology, Government of India for Ramalingaswami Re-entry Fellowship scheme (Ref. No. BT/RLF/Re-entry/41/2020, Dated: 15/12/2022) and S. N. Bose National Centre for Basic Sciences, Kolkata for computational facilities.References
- Y. Umena, K. Kawakami, J.-R. Shen and N. Kamiya, Nature, 2011, 473, 55–60 CrossRef CAS PubMed.
- M. Suga, F. Akita, K. Hirata, G. Ueno, H. Murakami, Y. Nakajima, T. Shimizu, K. Yamashita, M. Yamamoto, H. Ago and J. R. Shen, Nature, 2015, 517, 99–103 CrossRef CAS PubMed.
- S. E. J. Rigby, J. H. A. Nugent and P. J. O'Malley, Biochemistry, 1994, 33, 10043–10050 CrossRef CAS PubMed.
- B. A. Diner, E. Schlodder, P. J. Nixon, W. J. Coleman, F. Rappaport, J. Lavergne, W. F. J. Vermaas and D. A. Chisholm, Biochemistry, 2001, 40, 9265–9281 CrossRef CAS PubMed.
- T. Okubo, T. Tomo, M. Sugiura and T. Noguchi, Biochemistry, 2007, 46, 4390–4397 CrossRef CAS PubMed.
- M. Mandal, K. Kawashima, K. Saito and H. Ishikita, J. Phys. Chem. Lett., 2020, 11, 249–255 CrossRef CAS PubMed.
- S. Vasil'ev, P. Orth, A. Zouni, T. G. Owens and D. Bruce, Proc. Natl. Acad. Sci. U. S. A., 2001, 98, 8602–8607 CrossRef PubMed.
- S. Vasil’ev and D. Bruce, Biophys. J., 2006, 90, 3062–3073 CrossRef PubMed.
- E. Schlodder and H. T. Witt, J. Biol. Chem., 1999, 274, 30387–30392 CrossRef CAS PubMed.
- H. Suzuki, M. Sugiura and T. Noguchi, Biochemistry, 2005, 44, 1708–1718 CrossRef CAS PubMed.
- J. R. Shen, Annu. Rev. Plant Biol., 2015, 66, 23–48 CrossRef CAS PubMed.
- D. J. Vinyard and G. W. Brudvig, Annu. Rev. Phys. Chem., 2017, 68, 101–116 CrossRef CAS PubMed.
- A. Boussac and A. W. Rutherford, Biochemistry, 1988, 27, 3476–3483 CrossRef CAS.
- J. S. Vrettos, D. A. Stone and G. W. Brudvig, Biochemistry, 2001, 40, 7937–7945 CrossRef CAS PubMed.
- A. Boussac, F. Rappaport, P. Carrier, J. M. Verbavatz, R. Gobin, D. Kirilovsky, A. W. Rutherford and M. Sugiura, J. Biol. Chem., 2004, 279, 22809–22819 CrossRef CAS PubMed.
- C. F. Yocum, Coord. Chem. Rev., 2008, 252, 296–305 CrossRef CAS.
- V. K. Yachandra and J. Yano, J. Photochem. Photobiol., B, 2011, 104, 51–59 CrossRef CAS PubMed.
- T. Ono, A. Rompel, H. Mino and N. Chiba, Biophys. J., 2001, 81, 1831–1840 CrossRef CAS PubMed.
- C.-I. Lee, K. V. Lakshmi and G. W. Brudvig, Biochemistry, 2007, 46, 3211–3223 CrossRef CAS PubMed.
- K. Kawakami, Y. Umena, N. Kamiya and J.-R. Shen, J. Photochem. Photobiol., B, 2011, 104, 9–18 CrossRef CAS PubMed.
- M. J. Latimer, V. J. DeRose, V. K. Yachandra, K. Sauer and M. P. Klein, J. Phys. Chem. B, 1998, 102, 8257–8265 CrossRef CAS PubMed.
- T. Lohmiller, N. Cox, J. H. Su, J. Messinger and W. Lubitz, J. Biol. Chem., 2012, 287, 24721–24733 CrossRef CAS PubMed.
- K. Saito and H. Ishikita, Biochim. Biophys. Acta, 2014, 1837, 159–166 CrossRef CAS PubMed.
- T.-A. Ono and Y. Inoue, FEBS Lett., 1988, 227, 147–152 CrossRef CAS.
- M. Sivaraja, J. Tso and G. C. Dismukes, Biochemistry, 1989, 28, 9459–9464 CrossRef CAS PubMed.
- A. Boussac, J.-L. Zimmermann and A. W. Rutherford, Biochemistry, 1989, 28, 8984–8989 CrossRef CAS PubMed.
- K. Saito, M. Mandal and H. Ishikita, Biochemistry, 2020, 59, 3216–3224 CrossRef CAS PubMed.
- F. H. Koua, Y. Umena, K. Kawakami and J. R. Shen, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 3889–3894 CrossRef CAS PubMed.
- L. Vogt, M. Z. Ertem, R. Pal, G. W. Brudvig and V. S. Batista, Biochemistry, 2015, 54, 820–825 CrossRef CAS PubMed.
- E. Y. Tsui, R. Tran, J. Yano and T. Agapie, Nat. Chem., 2013, 5, 293–299 CrossRef CAS PubMed.
- P.-H. Lin, M. K. Takase and T. Agapie, Inorg. Chem., 2015, 54, 59–64 CrossRef CAS PubMed.
- K. Saito, M. Nakagawa, M. Mandal and H. Ishikita, Photosynth. Res., 2021, 148, 153–159 CrossRef CAS PubMed.
- Y. Kimura, K. Hasegawa and T.-A. Ono, Biochemistry, 2002, 41, 5844–5853 CrossRef CAS PubMed.
- H. Suzuki, Y. Taguchi, M. Sugiura, A. Boussac and T. Noguchi, Biochemistry, 2006, 45, 13454–13464 CrossRef CAS PubMed.
- M. A. Strickler, L. M. Walker, W. Hillier and R. J. Debus, Biochemistry, 2005, 44, 8571–8577 CrossRef CAS PubMed.
- T.-a Ono and Y. Inoue, Biochim. Biophys. Acta, 1990, 1020, 269–277 CrossRef CAS.
- Z. Guo and B. A. Barry, J. Phys. Chem. B, 2017, 121, 3987–3996 CrossRef CAS PubMed.
- Z. Guo, J. He and B. A. Barry, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, 5658–5663 CrossRef CAS PubMed.
- R. J. Debus, Biochemistry, 2014, 53, 2941–2955 CrossRef CAS PubMed.
- H. Kuroda, K. Kawashima, K. Ueda, T. Ikeda, K. Saito, R. Ninomiya, C. Hida, Y. Takahashi and H. Ishikita, Biochim. Biophys. Acta, 2021, 1862, 148329 CrossRef CAS PubMed.
- K. Kawashima, T. Takaoka, H. Kimura, K. Saito and H. Ishikita, Nat. Commun., 2018, 9, 1247 CrossRef PubMed.
- K. Saito, M. Nakagawa and H. Ishikita, Commun. Chem., 2020, 3, 89 CrossRef CAS PubMed.
- K. Saito, J.-R. Shen, T. Ishida and H. Ishikita, Biochemistry, 2011, 50, 9836–9844 CrossRef CAS PubMed.
- A. A. Stuchebrukhov, Phys. Rev. E, 2009, 79, 031927 CrossRef PubMed.
- K. Saito, A. W. Rutherford and H. Ishikita, Nat. Commun., 2015, 6, 8488 CrossRef CAS PubMed.
- N. Sakashita, H. C. Watanabe, T. Ikeda, K. Saito and H. Ishikita, Biochemistry, 2017, 56, 3049–3057 CrossRef CAS PubMed.
- B. R. Brooks, R. E. Bruccoleri, B. D. Olafson, D. J. States, S. Swaminathan and M. Karplus, J. Comput. Chem., 1983, 4, 187–217 CrossRef CAS.
- S. Nakamura and T. Noguchi, J. Am. Chem. Soc., 2017, 139, 9364–9375 CrossRef CAS PubMed.
- A. D. MacKerell, Jr., D. Bashford, R. L. Bellott, R. L. Dunbrack, Jr., J. D. Evanseck, M. J. Field, S. Fischer, J. Gao, H. Guo, S. Ha, D. Joseph-McCarthy, L. Kuchnir, K. Kuczera, F. T. K. Lau, C. Mattos, S. Michnick, T. Ngo, D. T. Nguyen, B. Prodhom, W. E. Reiher, III, B. Roux, M. Schlenkrich, J. C. Smith, R. Stote, J. Straub, M. Watanabe, J. Wiorkiewicz-Kuczera, D. Yin and M. Karplus, J. Phys. Chem. B, 1998, 102, 3586–3616 CrossRef PubMed.
- M. Amin, R. Pokhrel, G. W. Brudvig, A. Badawi and S. S. Obayya, J. Phys. Chem. B, 2016, 120, 4243–4248 CrossRef CAS PubMed.
- M. Askerka, J. Wang, D. J. Vinyard, G. W. Brudvig and V. S. Batista, Biochemistry, 2016, 55, 981–984 CrossRef CAS PubMed.
- M. Capone, D. Narzi and L. Guidoni, Biochemistry, 2021, 60, 2341–2348 CrossRef CAS PubMed.
- S. Petrie, R. Terrett, R. Stranger and R. J. Pace, Chem. Phys. Chem., 2020, 21, 785–801 CrossRef CAS PubMed.
- P. E. Siegbahn, Biochim. Biophys. Acta, 2013, 1827, 1003–1019 CrossRef CAS PubMed.
- M. Shoji, H. Isobe, J.-R. Shen, M. Suga, F. Akita, K. Miyagawa, Y. Shigeta and K. Yamaguchi, Chem. Phys. Lett., 2019, 730, 416–425 CrossRef CAS.
- M. Mandal, K. Saito and H. Ishikita, J. Phys. Soc. Jpn., 2022, 91, 091012 CrossRef.
- P. J. Hay and W. R. Wadt, J. Chem. Phys., 1985, 82, 299–310 CrossRef CAS.
- QSite, 2012, version 5.8, Schrödinger, LLC, New York, NY.
- K. Saito, T. Ishida, M. Sugiura, K. Kawakami, Y. Umena, N. Kamiya, J.-R. Shen and H. Ishikita, J. Am. Chem. Soc., 2011, 133, 14379–14388 CrossRef CAS PubMed.
- J. L. Zimmermann and A. W. Rutherford, Biochemistry, 1986, 25, 4609–4615 CrossRef CAS.
- W. Ames, D. A. Pantazis, V. Krewald, N. Cox, J. Messinger, W. Lubitz and F. Neese, J. Am. Chem. Soc., 2011, 133, 19743–19757 CrossRef CAS PubMed.
- H. Isobe, M. Shoji, S. Yamanaka, Y. Umena, K. Kawakami, N. Kamiya, J. R. Shen and K. Yamaguchi, Dalton Trans., 2012, 41, 13727–13740 RSC.
- K. Saito and H. Ishikita, Biochim. Biophys. Acta, 2019, 1860, 148059 CrossRef CAS PubMed.
- G. Vacek, J. K. Perry and J. M. Langlois, Chem. Phys. Lett., 1999, 310, 189–194 CrossRef CAS.
- T. Takaoka, N. Sakashita, K. Saito and H. Ishikita, J. Phys. Chem. Lett., 2016, 7, 1925–1932 CrossRef CAS PubMed.
- K. Kawashima, K. Saito and H. Ishikita, Biochemistry, 2018, 57, 4997–5004 CrossRef CAS PubMed.
- R. C. Weast, CRC Handbook of Chemistry and Physics, CRC Press, West Palm Beach, FL, 1978 Search PubMed.
- D. A. Pantazis, W. Ames, N. Cox, W. Lubitz and F. Neese, Angew. Chem., Int. Ed., 2012, 51, 9935–9940 CrossRef CAS PubMed.
- A. W. Rutherford, Biochim. Biophys. Acta, 1985, 807, 189–201 CrossRef CAS.
- K. Saito, H. Mino, S. Nishio and H. Ishikita, PNAS Nexus, 2022, 1, pgac221 CrossRef PubMed.
- M. Suga, F. Akita, M. Sugahara, M. Kubo, Y. Nakajima, T. Nakane, K. Yamashita, Y. Umena, M. Nakabayashi, T. Yamane, T. Nakano, M. Suzuki, T. Masuda, S. Inoue, T. Kimura, T. Nomura, S. Yonekura, L. J. Yu, T. Sakamoto, T. Motomura, J. H. Chen, Y. Kato, T. Noguchi, K. Tono, Y. Joti, T. Kameshima, T. Hatsui, E. Nango, R. Tanaka, H. Naitow, Y. Matsuura, A. Yamashita, M. Yamamoto, O. Nureki, M. Yabashi, T. Ishikawa, S. Iwata and J. R. Shen, Nature, 2017, 543, 131–135 CrossRef CAS PubMed.
- J. Kern, R. Chatterjee, I. D. Young, F. D. Fuller, L. Lassalle, M. Ibrahim, S. Gul, T. Fransson, A. S. Brewster, R. Alonso-Mori, R. Hussein, M. Zhang, L. Douthit, C. de Lichtenberg, M. H. Cheah, D. Shevela, J. Wersig, I. Seuffert, D. Sokaras, E. Pastor, C. Weninger, T. Kroll, R. G. Sierra, P. Aller, A. Butryn, A. M. Orville, M. Liang, A. Batyuk, J. E. Koglin, S. Carbajo, S. Boutet, N. W. Moriarty, J. M. Holton, H. Dobbek, P. D. Adams, U. Bergmann, N. K. Sauter, A. Zouni, J. Messinger, J. Yano and V. K. Yachandra, Nature, 2018, 563, 421–425 CrossRef CAS PubMed.
- M. Suga, F. Akita, K. Yamashita, Y. Nakajima, G. Ueno, H. Li, T. Yamane, K. Hirata, Y. Umena, S. Yonekura, L. J. Yu, H. Murakami, T. Nomura, T. Kimura, M. Kubo, S. Baba, T. Kumasaka, K. Tono, M. Yabashi, H. Isobe, K. Yamaguchi, M. Yamamoto, H. Ago and J. R. Shen, Science, 2019, 366, 334–338 CrossRef CAS PubMed.
- J. Yang, M. Hatakeyama, K. Ogata, S. Nakamura and C. Li, J. Phys. Chem. B, 2014, 118, 14215–14222 CrossRef CAS PubMed.
- K. Saito, M. Mandal and H. Ishikita, Biochemistry, 2020, 59, 3216–3224 CrossRef CAS PubMed.
- G. W. Brudvig, Phil. Trans. R. Soc. Lond. B, 2008, 363, 1211–1219 CrossRef CAS PubMed.
- F. Pitari, D. Bovi, D. Narzi and L. Guidoni, Biochemistry, 2015, 54, 5959–5968 CrossRef CAS PubMed.
- M. M. Najafpour, G. Renger, M. Hołyńska, A. N. Moghaddam, E.-M. Aro, R. Carpentier, H. Nishihara, J. J. Eaton-Rye, J.-R. Shen and S. I. Allakhverdiev, Chem. Rev., 2016, 116, 2886–2936 CrossRef CAS PubMed.
- R. Yao, Y. Li, Y. Chen, B. Xu, C. Chen and C. Zhang, J. Am. Chem. Soc., 2021, 143, 17360–17365 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2cp05036f |
| This journal is © the Owner Societies 2023 |