
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Revisiting the origin of the bending in group 2 metallocenes AeCp2 (Ae = Be–Ba)†
Tetiana
Sergeieva
a,
T. Ilgin
Demirer
a,
Axel
Wuttke
b,
Ricardo A.
Mata
 *b,
André
Schäfer
*a,
Gerrit-Jan
Linker
*c and
Diego M.
Andrada
*a
*b,
André
Schäfer
*a,
Gerrit-Jan
Linker
*c and
Diego M.
Andrada
*a
aDepartment of Chemistry, Saarland University, Campus Saarbrücken, 66123 Saarbrücken, Germany. E-mail: diego.andrada@uni-saarland.de; andre.schaefer@uni-saarland.de
bInstitute for Physical Chemistry, Georg-August-University Göttingen, Tammannstrasse 6, D-37077 Göttingen, Germany. E-mail: ricardo.mata@chemie.uni-goettingen.de
cMESA+ Institute for Nanotechnology, University of Twente, 7522 NB Enschede, The Netherlands. E-mail: g.linker@utwente.nl
First published on 24th May 2023
Abstract
Metallocenes are well-established compounds in organometallic chemistry, and can exhibit either a coplanar structure or a bent structure according to the nature of the metal center (E) and the cyclopentadienyl ligands (Cp). Herein, we re-examine the chemical bonding to underline the origins of the geometry and stability observed experimentally. To this end, we have analysed a series of group 2 metallocenes [Ae(C5R5)2] (Ae = Be–Ba and R = H, Me, F, Cl, Br, and I) with a combination of computational methods, namely energy decomposition analysis (EDA), polarizability model (PM), and dispersion interaction densities (DIDs). Although the metal–ligand bonding nature is mainly an electrostatic interaction (65–78%), the covalent character is not negligible (33–22%). Notably, the heavier the metal center, the stronger the d-orbital interaction with a 50% contribution to the total covalent interaction. The dispersion interaction between the Cp ligands counts only for 1% of the interaction. Despite that orbital contributions become stronger for heavier metals, they never represent the energy main term. Instead, given the electrostatic nature of the metallocene bonds, we propose a model based on polarizability, which faithfully predicts the bending angle. Although dispersion interactions have a fair contribution to strengthen the bending angle, the polarizability plays a major role.
Introduction
More than seventy years ago, Kealy, Pauson, Miller, Tebboth and Tremaine described ferrocene Fe(Cp)2 for the first time, laying the foundation for research on the metallocene family.1–10 Over the years, these compounds have evolved from only a curiosity into well-recognized reagents in organometallic chemistry, with applications ranging from coordination chemistry to homogenous catalysis and even industrial processes.11 To date, many examples of sandwich- or half-sandwich-type complexes with the formula E(Cp)n (n = 1–4) have been prepared and structurally characterized, in which E is a main-group element or a transition metal.11 In particular, their structures have drawn much attention as the understanding of the bonding provides guidelines for engineering their stoichiometric and catalytic reactivity.Attempts for modelling metallocene structures were developed parallel to the structural elucidation of ferrocene, and understanding the chemical bonding between the central atom and the cyclopentadienyl ligands is challenging using the existing heuristic models.7–9 Originally, the bond between the central iron atom and the Cp rings was assumed to be an electron sharing σ-type (C–Fe–C), given the lack of X-ray analysis, although the possibility of an ionic interaction ([Fe2+] [Cp−]2) was discussed.1 However, the surprisingly high thermal stability and its remarkable chemical inertness toward acids and bases could not be explained with these proposed bonding modes. Independently, Fischer3 and a group of scientists including Wilkinson, Rosenblum, Whiting, and Woodward10 proposed the metal–ligand interaction as a π-complexation. The subsequent analysis by Pfab, Eiland and Pepinsky confirmed the η5 binding pattern and the 6π electron aromatic character of the Cp ligands.4 Shortly after, Orgel used molecular orbital (MO) theory to explain the Fe–Cp bonding in ferrocene. The binding interaction was explained as formed by two “covalent–ionic” bonds resulting from the mixing of metals with Cp− orbitals, and two “donor” bonds, when electrons located at the dx2−y2, dxy orbitals of iron donate into vacant antibonding orbitals of the Cp ligands.5
The first electronic structure description was reported about 20 years after the structure elucidation.12,13 Although its η5 coordination was reproduced at the Hartree–Fock (HF) level, the Fe–C bond lengths were poorly predicted.14 Years later, Koch, Jørgensen, and Helgaker achieved a better theoretical structure by performing CCSD and CCSD(T) calculations.15 Many theoretical calculations have been reported to provide quantitative and qualitative insights into the bonding nature of metallocenes. The extension of the Dewar–Chatt–Duncanson model for ferrocene allows for the discussion of the chemical bond in terms of donor–acceptor interactions. With a strong electrostatic nature (51%), the covalent part is predicted as π-donation from MOs of the Cp− ligands to the empty orbitals of the Fe2+ ion and a back donation from the Fe to the π* orbitals of the ligands.16
The replacement of Fe with main-group elements from the s- or p-block, in particular with alkaline earth metals of group 2 (Ae), or heavy elements of group 14, has attracted much attention in the light of their preferred oxidation state of +2, ever since Fischer and co-workers reported magnesocene,17 calcocene,18 stannocene,19 and plumbocene,20 just a few years after the discovery of ferrocene.21–24 A common observation was a more labile E–Cp bond, which was explained by the weaker π-type interaction in view of the absence of d-orbitals on the bonding. Notably, the bonding nature changes from 49% covalent in Cp2Fe to being predominantly ionic in the case of s-block metallocenes and slightly more covalent when the central atom is a p-block element.16,25
Aside from the bond nature, numerous reports attempted to explain the geometrical model of sandwich complexes. A general observation in metallocene chemistry is that structures have either a coplanar or a bent orientation of the Cp rings relative to the central atom. While the classical ferrocene Fe(Cp)2 and Fe(Cp*)2 ligands are clearly coplanar, other complexes exhibit a bent structure, i.e. Sn(Cp)2 and Pb(Cp)2. Such a distortion has been ascribed to the presence of a lone-pair at the central atom following the traditional valence-shell electron-pair-repulsion (VSEPR) model.26 However, this does not account for the subtleties of orbital interactions that influence molecular shapes and thus cannot justify bend geometries of many other complexes, the so-called non-VSEPR structures, among them are heavy alkaline earth metals (Ca, Sr, and Ba) sandwich compounds.
Numerous models aiming at a general understanding of the structure, bonding and reactivity of such molecules have been proposed. The findings from experimental and computational investigations regarding the reason for the bending of metallocenes of heavier alkaline earth metals of group 2, which does not follow the valence shell electron pair repulsion theory model's prediction, can be categorized into four groups (Fig. 1):
 | ||
| Fig. 1 Illustration of models used to explain the bending of metallocenes. (A) Molecular orbital theory model (i). (B) Polarization model (ii). (C) Weak interaction concept (iii). (D) Agostic interaction model (iv). | ||
(i) Molecular orbital (MO) theory model. In 1953, Walsh27 proposed a molecular orbital diagram and linked the angle of AB2 molecules as a function of molecular orbital energies. Later, Hayes addressed the bending of heavy alkaline-earth dihalides by evoking this diagram, although with some modifications.28 The authors suggested to take unoccupied d-orbitals of Ca, Sr and Ba into account. This was supported by the energetics of the s-, p- and d-orbitals of these elements. The d-orbitals of Be and Mg lay energetically above the level of the p-orbitals and hence do not have a strong participation in binding. Thus, the symmetry of the valence p-orbitals leads to a coplanar structure for a better orbital overlap. In contrast, the energetic arrangement of Ca, Sr and Ba orbitals is different, which results in the contribution of the d-orbitals to the Ae–Cp bonding with an optimal overlap corresponding to bent geometries.
(ii) Polarization model. Klemperer and co-workers observed a permanent electric dipole moment for monomeric dihalides of heavy alkaline earth elements, suggesting a bent arrangement.29 This finding was linked to the polarized-ion model, where large cations may be significantly polarized by anions due to charge–dipole and dipole–dipole interactions. In fact, these simple classical arguments were drawn by Debye to give a qualitative description for the angle in H2O.30 As such, molecular bending is related to the polarizability of the central atom and the polarizing power of the ligands. Gigli applied the polarizable Rittner type ion model to develop a quantitative prediction of dihalide dimer geometries.31 By splitting charge–dipole and dipole–dipole moment interactions into individual contributions, it was proposed that the major role in stabilization of linear or bent geometries is dependent on the magnitude of the induced dipole moment of the central cation. Among a large range of dipole moment values applied to the system, barium halides always showed a bent configuration, while strontium analogues adopt a linear form when the induced dipole is significantly lower.
(iii) Weak interaction concept. An alternative explanation for the bending of alkaline-earth metal complexes with Cp* ligands was discussed by Andersen et al.32 The bending energy was found to be relatively small (0.5 kcal mol−1 for [Ca(Cp*)2]) and the tilting was described in terms of maximizing the van der Waals (VdW) attractions between the methyl groups of two Cp* rings. Bosnich and co-workers re-evaluated this “weak interaction” concept and expanded the list of compounds to complexes such as SrCp*2, BaCp*2, SmCp*2, and EuCp*2.33 The conclusion regarding the importance of the VdW interactions was made based on the calculations of ΔEVdW, which referred to the VdW energy difference between the linear and bent geometries. The increase of ΔEVdW values with an increase of the metal radius was ascribed to the weakening of VdW attractive forces in a linear geometry. However, Huffman and co-workers postulated that the length of intramolecular methyl-methyl contacts is in the range of 3.55–3.59 Å, regardless the radii of the central atoms (Ca, Ba, Yb, Sm, and Eu) in sandwich complex bearing Cp* ligands.34 One could interpret this finding in the way that metallocenes with large central cations and longer M–Cp distances should be more bent to enhance VdW interactions. It should be noted, however, that many studies do not rule out the polarizability model and consider these strengths to reinforce each other.32,35
(iv) Agostic interaction model. This hypothesis has been formulated relatively recently, after recognizing the importance of three-center–two-electron (3c2e) C–H⋯[E] bonds,36 thus an interaction between a C–H bond and a metal center with relatively high Lewis acidic character. Evidence of intermolecular agostic interactions in metallocenes of alkaline earth metals (CaCp* and BaCp*) has been invoked by Huffman as an explanation for the molecular structure and lattice pattern observed by X-ray diffraction.34 However, the absence of a clear trend in the packing arrangement cannot provide a clear picture of bending behaviour in the crystals. A similar conclusion was drawn by Pal et al., based on the performed calculation of noncovalent interactions (NCIs) and topological analysis within the quantum theory of atoms in molecules (QTAIM) for MgCp*2 and CaCp*2 complexes.37 Although the geometrical, topological and NBO analysis interpretations of the C–H⋯Mg/Ca interactions to be pregostic, other forces such as VdW attraction between two Cp* rings were proposed to be the driving force of bending.
In view of the fact that the questions about the bonding situation in alkaline earth metal metallocenes were often controversially discussed because of vaguely defined concepts, we aim to address this in terms of well-established quantum chemical expressions, in the current work. The set of compounds in this study consists of unsubstituted metallocenes Ae(Cp)2, their methylated derivatives Ae(Cp*)2, and their penta-halogenated analogues Ae(C5R5), where R refers to F, Cl, Br, and I. We performed a series of analyses using energy decomposition analysis (EDA), the calculation of dispersion interaction density (DID) and polarizability to evaluate the existing concepts on the structure of metallocenes.
Computational details
General
Geometry optimizations were performed using the Gaussian 16 C01 software suite.38 The geometry optimizations for unsubstituted complexes with the general structure Ae(Cp)2 were carried out using density functional theory (DFT) BP86,39–41 B3LYP,42,43 M06-2X44 functionals with Grimme dispersion corrections D345 and the Becke-Jonson damping function46 in combination of def2-TZVPP47 basis sets without any symmetry restrictions. Substituted metallocenes [Ae(C5R5)2] (Ae = Be-Ba and R = Me, F, Cl, Br, and I) were optimized at the B3LYP-D3(BJ)/def2-TZVPP level of theory. The stationary points were located with the Berny algorithm48 using redundant internal coordinates. Analytical Hessians were computed to determine the nature of stationary points (one and zero imaginary frequencies for transition states and minima, respectively)49 and to calculate unscaled zero-point energies (ZPEs) as well as thermal corrections and entropy effects using the standard statistical-mechanics relationships for an ideal gas.To further evaluate bond dissociation energies (De), single-point energy calculations using the LCCSD(T)50–58 methods were performed on B3LYP-D3(BJ)/def2-TZVPP optimized geometries using the program package Molpro2019.1.59 The cc-pVTZ basis set was used for carbon hydrogen, fluorine, chlorine and bromine, the cc-pCVTZ basis set was used for beryllium, magnesium and calcium, and the cc-pVTZ-PP60 basis set was used for strontium, barium and iodine.61,62 The LCCSD(T) calculations were carried out using Pipek-Mezey localized orbitals.63 The domains were determined with the use of natural population analysis criteria, with NPA = 0.03.
The natural bond orbital (NBO)64,65 partial charges were computed at B3LYP-D3(BJ)/def2-TZVPP using NBO 7.0.66
We calculate the atomic polarizability of the metal atom in the metallocenes in support of the polarizability model for the chemical angle. Single point calculations were performed, at the aforementioned optimized structures, using the local properties module LoProp67 of the Molcas software68 at the B3LYP/ANO-RCC-VTZP level of theory.42,43
Energy decomposition analysis
The nature of the chemical bonds was investigated by means of the energy decomposition analysis (EDA) method, which was developed by Morokuma69 and Ziegler and Rauk.70,71 The bonding analysis focuses on the instantaneous interaction energy ΔEint of a bond A–B between two fragments A and B in the particular electronic reference state and in the frozen geometry AB. This energy is divided into four main components (eqn (1)):| ΔEint = ΔEelst + ΔEPauli + ΔEorb + ΔEdisp | (1) |
| ΔE(−De) = ΔEint + ΔEprep | (2) |
![[V with combining circumflex]](https://www.rsc.org/images/entities/i_char_0056_0302.gif) , given by equation (eqn (3)):
, given by equation (eqn (3)):| VΨi = viΨi | (3) |
 | (4) |
Dispersion interaction density
Dispersion interaction densities (DIDs) were computed as proposed at the PAO-LMP2/cc-pCVDZ&cc-pVDZ level of theory.77 The Voxel DIDs78 are plotted using the ParaView Software.79Results and discussion
This section is divided as follows: first, the results and discussion on the geometry and bond dissociation of different metallocenes are presented (Section A). We then discuss the chemical bonding by dispersion interactions, and energy decomposition analysis (Section B). We finish with a model to predict the bending angle in metallocenes based on the polarizability model.A. Geometry and bond energies
 | ||
| Fig. 2 Optimized geometries of group 2 metallocenes [Ae(Cp)2] (Ae = Be–Ba) at the B3LYP-D3(BJ)/def2-TZVPP level of theory along with β angles in [°]. | ||
Table 1 presents the selected geometrical parameters, bond dissociation energies and natural atomic partial charges of the central element (Ae). As expected, the distances between Cp rings (geometrical centre) and central atoms are elongated with the increase of the radii of the central element in the series Mg–Ba and range from 2.00 to 2.72 Å. The values of β angles (Table 1) predicted magnesocene being coplanar, while strontocene and barocene are bent, regardless of the functional being used. This is in agreement with previously reported findings.84–87 Notably, the calcocene geometry significantly depends on the choice of the DFT level of theory. An optimization of Ca(Cp)2 with B3LYP leads to a coplanar structure, while BP86 and M06-2X furnished a bent structure (see the ESI,† Table S1). To examine the reliability of considered DFT methods for the prediction of calcocene bending situation, we carried out the rigid scan of PES along the Cp–Ca–Cp angle (140–180°) at the LCCSD/cc-pCVTZ&cc-pVTZ level of theory. The flat pattern of the PES (ESI,† Fig. S2) makes the estimation of functional performance to be difficult. Furthermore, the absence of experimental evidence of bending for monomeric calcocene88 and significant debates on this topic among theoretical reports85,89,90 also cannot provide an unambiguous answer regarding the bending of CaCp2.
| Ae | [Ae(Cp)2] | [Ae(Cp*)2] | [Ae(C5F5)2] | [Ae(C5Cl5)2] | [Ae(C5Br5)2] | [Ae(C5I5)2] |
|---|---|---|---|---|---|---|
| a The dissociation energies (De) considering the [Ae(C5R5)2] → Ae2+ + 2 C5R5− dissociation. | ||||||
| C5R5–Ae bond lengths, Å | ||||||
| Be | — | 1.649 | 1.635 | 1.639 | 1.656 | 1.676 |
| Mg | 2.000 | 1.959 | 2.028 | 1.987 | 1.981 | 1.983 |
| Ca | 2.351 | 2.315 | 2.367 | 2.337 | 2.336 | 2.328 |
| Sr | 2.542 | 2.500 | 2.559 | 2.520 | 2.524 | 2.524 |
| Ba | 2.722 | 2.687 | 2.731 | 2.702 | 2.713 | 2.718 |
| C5R5–Ae–C5R5 angles, deg | ||||||
| Be | — | 179.9 | 179.9 | 179.9 | 179.9 | 179.9 |
| Mg | 179.9 | 179.8 | 180.0 | 179.9 | 179.9 | 179.8 |
| Ca | 178.1 | 158.1 | 150.2 | 151.1 | 154.9 | 163.9 |
| Sr | 165.1 | 149.3 | 155.6 | 145.4 | 148.0 | 153.6 |
| Ba | 138.2 | 139.4 | 142.2 | 138.5 | 142.4 | 148.5 |
| D e, kcal mol−1a | ||||||
| Be | 713.3{724.2} | 725.0{725.7} | 648.1{654.8} | 629.7{633.6} | 625.3{626.9} | 628.3{627.8} |
| Mg | 576.8{574.1} | 583.6{573.6} | 506.7{502.5} | 493.1{484.7} | 492.7{482.0} | 501.7{484.0} |
| Ca | 501.8{500.6} | 503.6{495.0} | 439.3{435.3} | 429.6{420.7} | 428.8{418.3} | 436.2{420.8} |
| Sr | 464.7{467.2} | 465.5{460.4} | 406.5{406.6} | 398.2{393.9} | 397.3{392.0} | 403.5{393.9} |
| Ba | 437.0{441.3} | 439.4{436.5} | 385.0{386.0} | 378.1{376.0} | 376.4{374.3} | 381.6{376.1} |
| NPA charges | ||||||
| Be | +1.61 | +1.72 | +1.55 | +1.68 | +1.71 | +1.74 |
| Mg | +1.80 | +1.88 | +1.72 | +1.81 | +1.81 | +1.80 |
| Ca | +1.78 | +1.79 | +1.72 | +1.74 | +1.71 | +1.67 |
| Sr | +1.81 | +1.81 | +1.75 | +1.77 | +1.76 | +1.72 |
| Ba | +1.78 | +1.77 | +1.72 | +1.76 | +1.76 | +1.74 |
The calculated bond dissociation energies (De) suggest that the strength of Ae–Cp bonds is decreasing when going down the group from Mg to Ba. This finding is in agreement with the previously published data that Cp complexes of the heavier alkaline earth metals have a tendency to dissociate.91 Theoretically predicted partial charges at the central atoms Ae do not change within the series Mg–Ba and are determined to be approximately +1.8 au.
 | ||
| Fig. 3 Optimized geometries of metallocenes for [Ae(Cp*)2] (Ae = Be–Ba) at the B3LYP-D3(BJ)/def2-TZVPP level of theory along with angles in degrees. Ae = Be–Ba and Cp* = a methylated cyclopentadienyl anion. Hydrogens are omitted for clarity. | ||
 | ||
| Fig. 4 Optimized geometries of group 2 metallocenes [Ae(C5F5)2] (Ae = Be–Ba) at the B3LYP-D3(BJ)/def2-TZVPP level of theory along with angles in [°]. | ||
Natural partial charges are comparable with those obtained for Cp and Cp*. Extreme cases such as [Be(C5F5)2] show a less positively charged central atoms than the rest of the molecules in the series. In terms of the bond dissociation energies De, the halogenated systems bear between 100 and 50 kcal mol−1 less than Cp congeners. This suggests that the electrostatic interaction should not be strongly affected, while other physical factors might be playing a role for the weaker interaction.
B. Chemical bonding and bending
In the absence of the centre metal, we have computed the energy between the C5R5 groups in two different geometries, bent (140°) and coplanar (180°), at different distances (Fig. 5A and B). While the Cp system shows a lower energy for the coplanar structure at short distances, at longer distances the bent structure becomes more favourable. Notably, in the case of Cp*, the coplanar structure is more stable at short distances, while at longer distances the bent structure sets in (at 4.7 Å) since the energy penalty becomes negligible. These results indicate that the cation is necessary for a bent structure where the dispersion interaction might not be the main determinant of the distortion. To expand aforementioned ideas, we performed a rigid scan of PES for planar (180°) vs. bent (140°) (C5R5)22− structures at different distances CpX(centroid)–CpX(centroid) ranging from 4.0 to 6.0 Å (Fig. 5C and D), which covers the range of atomic radii considered. Firstly, the preference of (C5F5)22− to be coplanar at any point of the curve (Fig. 5D), which is in sharp contrast to the behaviour of [Ae(C5F5)2] (Table 1), indicating that the presence of the central atom has an important role. The same conclusion can be outlined analysing the trends for (C5I5)22−. In contrast, (Cp)22− and (Cp*)22− indeed behave very similarly to analogous [Ae(Cp)2] and [Ae(Cp*)2] complexes, now suggesting that the central atom has a lower impact on the bending. Furthermore, an inspection of plots (Fig. 5B) reveals that the bending of (Cp*)22− strongly depends on the distance between aromatic rings. (Cp*)22− is bent when the distance is 4.8–6.0 Å, while planar at 4.0–4.7 Å. This finding contradicts a Kaltsoyannis argument about the necessity of the cation for bending, since the model they utilized was limited to specific CpX–CpX lengths. At this point, one cannot exclude the dispersion to be a driving force of the bending.
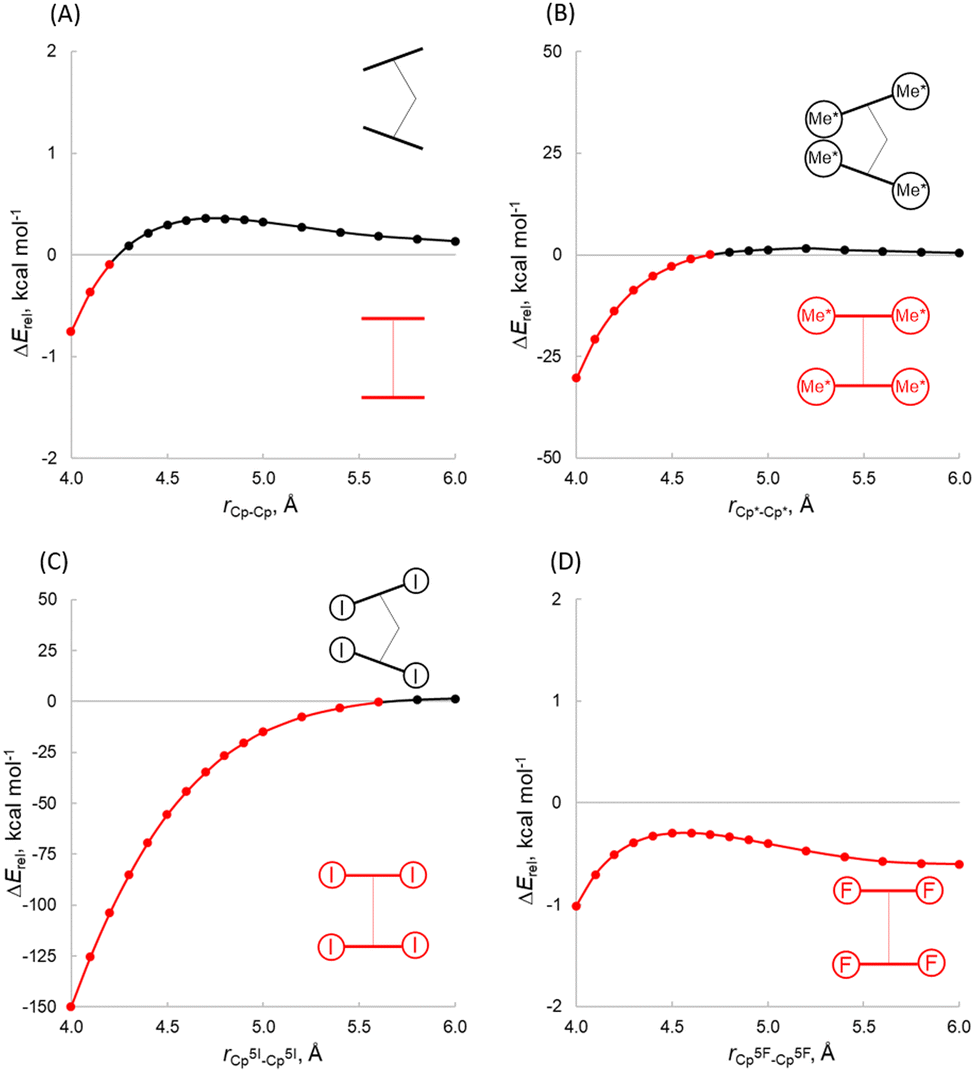 | ||
| Fig. 5 Rigid potential energy surface scans of planar vs. bent metalloceneanions (Cp)22− as a function of the CpX–CpX distance performed at the B3LYP-D3(BJ)/def2-TZVPP level of theory (ΔErel = Ebent − Elinear). | ||
From the optimised structures, we calculated at the PAO-LMP2/cc-pCVTZ&cc-pVTZ level the dispersion interactions between the C5R5 rings (R = H and Me). Given the local character of occupied and virtual orbitals in the local correlation treatments, the intermolecular effects due to double excitations from occupied orbitals of one unit into virtual orbitals of the same unit and intermolecular effects due to excitations involving orbitals from both units can be divided. Additionally, the interactions (Cp⋯Cp) can be dissected into dispersion effects, exchange dispersion, and ionic contributions.99–101Table 2 provides the results of the energy decomposition of the local correlation approach, and the exemplary dispersion interaction density (DID)77 profiles of Be(Cp)2, Be(Cp*)2, Ca(Cp)2 and Ca(Cp*)2 are shown in Fig. 6. As expected, the dispersion interaction between the rings is stronger for the beryllium complexes and drastically diminishes with the heavier analogues. The Cp* system doubles the amount of the dissection interaction in Cp systems, in good agreement with the higher dissociation energy values. The DID profile shows that the π–π interaction between the rings dominates the dispersion profile in most cases, but for Cp* starting from Ca the close C–H contacts rule the interaction.
| ΔEintra | ΔEdisp ex | ΔEdisp | ΔEionic | |
|---|---|---|---|---|
| Be(Cp)2 | −18.1 | −0.1 | −10.8 | −7.2 |
| Mg(Cp)2 | −6.3 | 0.0 | −4.5 | −1.8 |
| Ca(Cp)2 | −3.8 | 0.0 | −2.8 | −1.0 |
| Sr(Cp)2 | −2.6 | 0.0 | −2.0 | −0.6 |
| Ba(Cp)2 | −9.2 | 0.0 | −2.2 | −7.0 |
| Be(Cp*)2 | −30.6 | −0.1 | −20.5 | −9.9 |
| Mg(Cp*)2 | −12.4 | 0.0 | −9.4 | −2.9 |
| Ca(Cp*)2 | −11.2 | 0.0 | −6.7 | −4.5 |
| Sr(Cp*)2 | −7.9 | 0.0 | −4.9 | −3.0 |
| Ba(Cp*)2 | −39.0 | 0.0 | −4.9 | −34.1 |
 | ||
| Fig. 6 Dispersion interaction density (DID) plots calculated at the PAO-LMP2/cc-pCVTZ&cc-pVTZ level of theory. The brown zones indicate regions of electron density in a molecule which interacts strongly by dispersion interactions with the other molecule. Blue stands for weaker/diffuse contributions. | ||
To gain a quantitative insight into the role of the main orbital interaction, we performed the energy decomposition analysis (EDA) of the complexes maintaining the same C5R5–Ae distance given by the full optimization but changing the CpX–Ae–CpX angle from 140 to 180°. The EDA is a useful tool to assess the nature of the chemical bond and to identify the driving forces behind the binding interaction.102 The nature of the energy components has been a matter of debate, given the path-dependent nature.103–105 However, the fragmentation schemes used in a consistent manner generate reliable trends.106,107 We have used three fragment schemes to take into account the interaction between the Cp– moieties. The result of splitting ΔEint is shown in Fig. 7. Inspection of the EDA terms revealed that the trend in stabilizing bent structures by ΔEorb, ΔEdisp and ΔEelst terms counteract by the ΔEPauli forcing metallocenes to adopt the coplanar configuration and the interplay between these attractive and repulsive components determines the extent of deformation. Notably, the electrostatic interaction favours a coplanar structure for most of the metallocenes, with the exception of Cp. This finding shows that the previously controversial orbital interaction and dispersion in fact accompany each other promoting a bent structure. Note, however, that the dispersion interaction represents a minor factor (1%) in comparison to the orbital interaction.
 | ||
| Fig. 7 Energy decomposition analysis at the BP86-D3(BJ)/TZ2P level of theory for the [Ae(C5R5)2] complexes. Ae = Be–Ba and R = H, Me, and F–I. Energy values are given in kcal mol−1. (A) Orbital interaction, (B) electrostatic interaction, (C) dispersion interaction, and (D) Pauli repulsion. | ||
Deeper insights into the nature of the orbital interaction are available from the combination of EDA with natural orbitals for chemical valence calculations (EDA-NOCV).108,109 This method deconstructs the orbital term (ΔEorb) into components (ΔEorbρ(i)) that provide an energetic estimation of a given deformation density (ρ(i)), which is related to a particular electron flow channel, and consequently the amount of charge transferred, Δq(i) = |ν(i)|, for the bonding between the interacting fragments. The most interesting results are obtained by breaking down the orbital term ΔEorb. Fig. 8 shows the orbital diagram for the most important orbital interaction in a coplanar situation (D5d). In this situation, s and dz2 can interact with the a1g combination of the C5R5 rings. The p orbitals interact with the e1u and a2u orbitals, while the remaining d orbitals can interact with the e1g C5R5 orbitals. Fig. 9 represents the shape of the deformation densities ν1–ν6, showing the charge flow and the most important fragment orbitals which are involved in the pairwise donor–acceptor bonding for the MgCp2 metallocene complexes. The scheme in Fig. 8 can be associated with the interactions displayed in Fig. 9. The color coding red to blue illustrates the direction of the charge flow. EDA-NOCV reveals that the major interaction originates from the participation of s and p orbitals, which is because the symmetry keeps the complexes co-planar. When going down in the group from Mg to Ba, the contribution of d-orbitals is increasing triggering the sandwiches to bent their geometries (ESI,† Table S2 and Fig. S3–S7). For instance, the acceptor contributions of the formally empty d orbital to the total orbital interaction are Mg(Cp)2 (19.2%), Ca(Cp)2 (49.5%), Sr(Cp)2 (46.9%), and Ba(Cp)2 (47.2%). With the contribution of the d orbitals in addition to the small contribution of the dispersion interactions, we benchmarked the model based on the polarizability of the central atom.
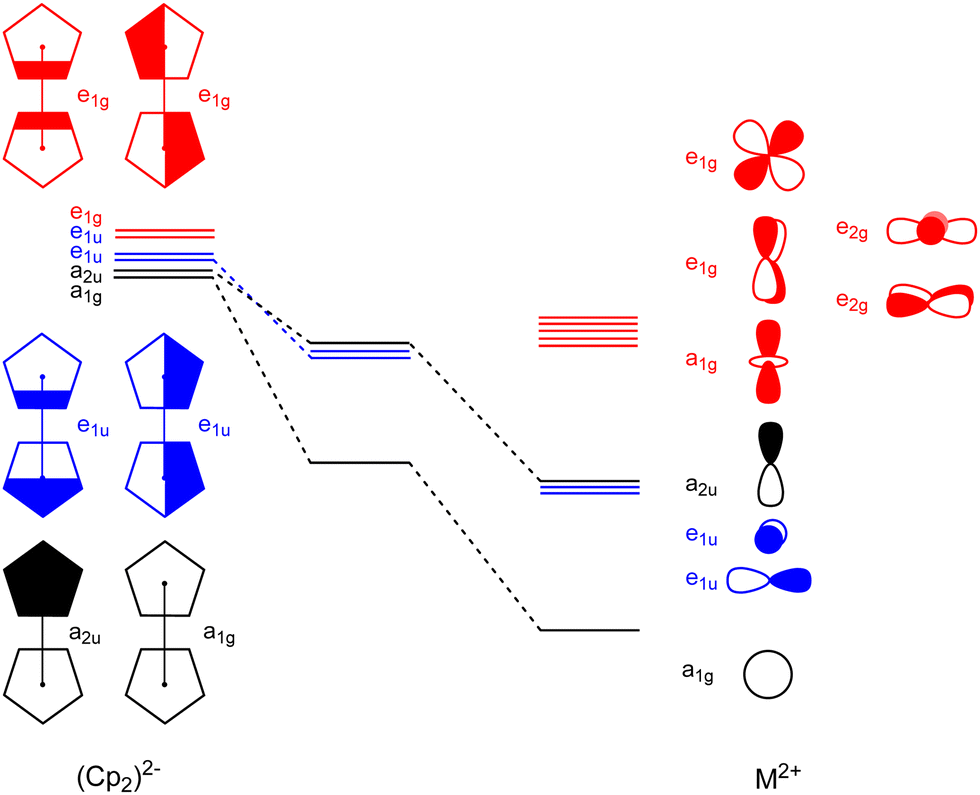 | ||
| Fig. 8 Schematic orbital correlation diagram for the coplanar complex Mg(Cp)2. | ||
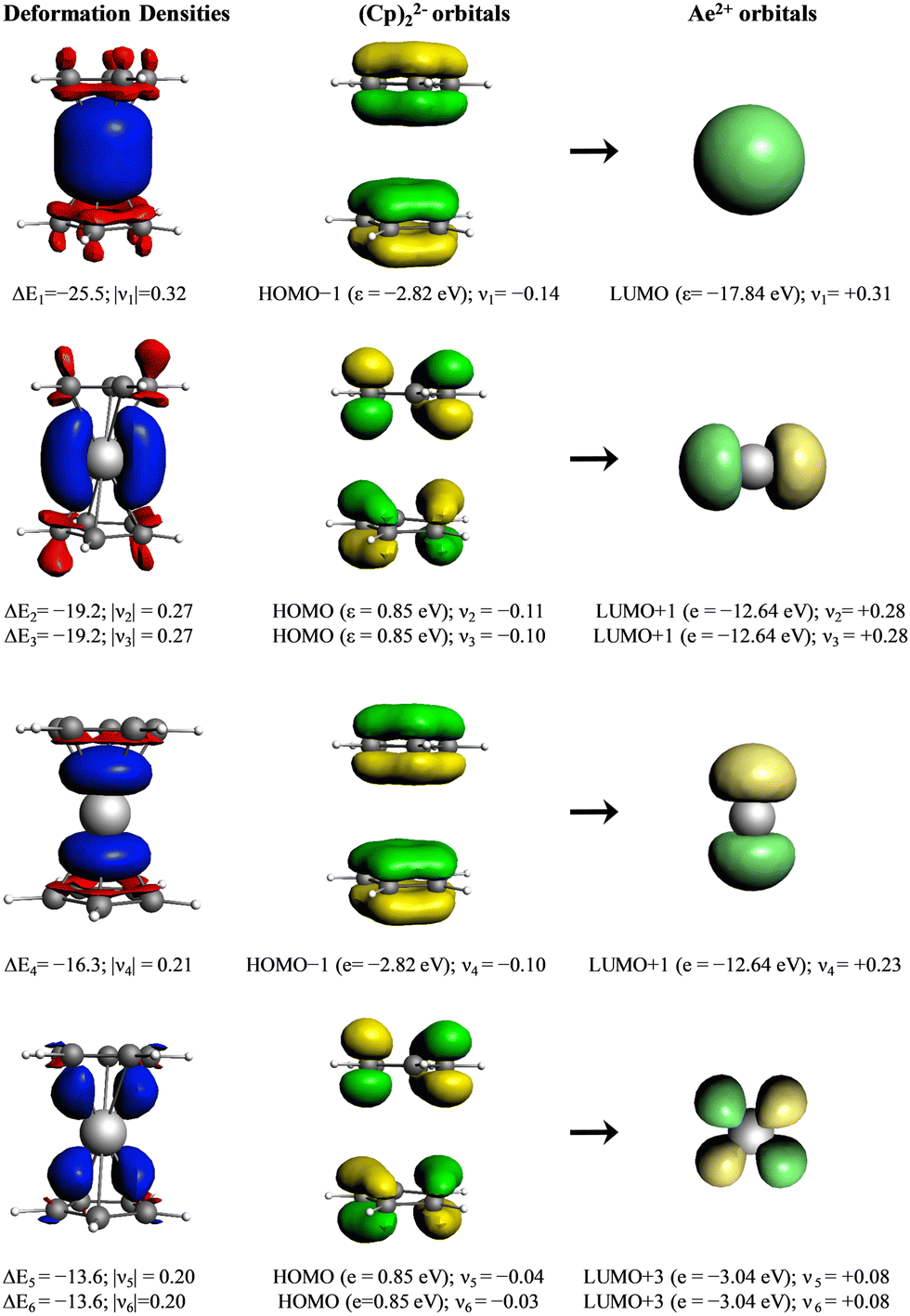 | ||
| Fig. 9 Plot of the deformation densities Δρ of the pairwise orbital interactions between Mg2+ in its A10 electronic state and (Cp2)2−, associated energies ΔE (in kcal mol−1) and eigenvalues ν (in a.u.). The red color shows the charge outflow, whereas the blue color shows the charge density accumulation. The shape of the most important interacting occupied and vacant orbitals of the fragments. | ||
 | ||
| Fig. 10 CpX–Ae–CpX angle depending on the polarizability of the constituent metal centre atom, where Ae = Ca, Sr, and Ba and CpX is the indication of the H substitution in the Cp molecules (see the text). | ||
Conclusions
In this work, we have addressed the long-standing dichotomy of the experimentally observed metallocene coplanar and bent structures. Under (quasi) gas phase conditions, the lightest group 2 metallocenes such as Be and Mg exhibit a coplanar arrangement of the Cp ligands, while the geometry becomes bent for heavier atoms such as Ca, Sr and Ba. Our calculation of the interaction of the C5R5 rings in the absence of the central atom suggests a coplanar structure as the most favourable at short distances, while at longer distances the coplanar and bent structures are energetically similar. This is a consequence of a strong interaction between the π systems of the rings. Nonetheless, the energy decomposition analysis suggests a strong ionic character (ca. 70%) of the bond between the metal and Cp and only a minor role of dispersion interactions, representing only about 1% of the total stabilizing interactions. The further dissection of the orbital term reveals six main orbital contributions which consist of donations of the different π-orbitals into the s, p and d orbitals on the metal centers. Notably, the contribution of the d orbitals becomes dominant for Ca, Sr and Ba. Thus, the strong d-orbital dependence and associated charge deformations give prevalence to the notion, put forward originally by Debye, that the ability to form induced dipoles at the centre atom is connected to the stabilization of the bent molecule. In this manner, the bending angle can be accurately assessed by the polarizability of the central Ae2+ atom. Exactly how the angle changes with the polarizability will depend on the ligand, as it influences the ratio of the different forces at play (e.g., Pauli repulsion and electrostatic interactions). The clearest example within the complexes studied is itself the cyclopentadienyl ring derivatives [Ae(Cp)2], which exhibit less Pauli repulsion and favorable electrostatic interactions.Author contributions
T. S., T. I. D., A. W., G.-J. L., and D. M. A. performed the calculations. G.-J. L., R. A. M., A. S., and D. M. A. acquired funding and contributed methodologies. T. S., T. I. D., A. S., G.-J. L., and D. M. A. prepared the manuscript and the ESI.† All authors contributed to the interpretation of the computed data and the writing and editing of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
D. M. A. and A. S. thank Prof. Dr David Scheschkewitz, Prof. Dr Guido Kickelbick and Saarland University for support. D. M. A., T. S. and T. I. D. thank the European Research Council, ERC, (ERC Starting grants, EU805113) for funding. A. S. thanks the Deutsche Forschungsgemeinschaft, DFG, (Emmy Noether-program, SCHA1915/3-1/2) for funding. G.-J. L. thanks Prof. Dr Piet Th. van Duijnen for support.References
- T. J. Kealy and P. L. Pauson, Nature, 1951, 168, 1039–1040 CrossRef CAS.
- S. A. Miller, J. A. Tebboth and J. F. Tremaine, J. Chem. Soc., 1952, 632–635 RSC.
- E. O. Fischer and W. Pfab, Z. Naturforsch. B, 1952, 7, 377–379 CrossRef.
- P. F. Eiland and R. Pepinsky, J. Am. Chem. Soc., 1952, 74, 4971 CrossRef CAS.
- J. D. Dunitz and L. E. Orgel, Nature, 1953, 171, 121–122 CrossRef CAS.
- J. D. Dunitz, L. E. Orgel and A. Rich, Acta Crystallogr., 1956, 9, 373–375 CrossRef CAS.
- P. L. Pauson, J. Organomet. Chem., 2001, 637–639, 3–6 CrossRef CAS.
- G. B. Kauffman, J. Chem. Educ., 1983, 60, 185 CrossRef CAS.
- H. Werner, Angew. Chem., Int. Ed., 2012, 51, 6052–6058 CrossRef CAS PubMed.
- G. Wilkinson, M. Rosenblum, M. C. Whiting and R. B. Woodward, J. Am. Chem. Soc., 1952, 74, 2125–2126 CrossRef CAS.
- L. Wirtz and A. Schäfer, Chem. – Eur. J., 2021, 27, 1219–1230 CrossRef CAS PubMed.
- H. P. Lüthi, J. Ammeter, J. Almlöf and K. Korsell, Chem. Phys. Lett., 1980, 69, 540–542 CrossRef.
- P. S. Bagus, U. I. Walgren and J. Almlof, J. Chem. Phys., 1976, 64, 2324 CrossRef CAS.
- H. P. Lüthi, J. H. Ammeter, J. Almlöf and K. Faegri, Jr., J. Chem. Phys., 1982, 77, 2002 CrossRef.
- H. Koch, P. Jørgensen and T. Helgaker, J. Chem. Phys., 1996, 104, 9528–9530 CrossRef CAS.
- J. Frunzke, M. Lein and G. Frenking, Organometallics, 2002, 21, 3351–3359 CrossRef CAS.
- E. O. Fischer and W. Hafner, Z. Naturforsch. B, 1954, 9, 503–504 CrossRef.
- K. Ziegler, H. Froitzheimkuhlhorn and K. Hafner, Chem. Ber., 1956, 89, 434–443 CrossRef CAS.
- E. O. Fischer and H. Grubert, Z. Naturforsch. B, 1956, 11b, 423–424 CAS.
- E. O. Fischer and H. Grubert, Z. Anorg. Allg. Chem., 1956, 286, 237–242 CrossRef CAS.
- P. Jutzi and N. Burford, Chem. Rev., 1999, 99, 969–990 CrossRef CAS PubMed.
- P. H. M. Budzelaar, J. J. Engelberts and J. H. van Lenthe, Organometallics, 2003, 22, 1562–1576 CrossRef CAS.
- P. Jutzi and G. Reumann, J. Chem. Soc., Dalton Trans., 2000, 2237–2244 RSC.
- T. P. Hanusa, Organometallics, 2002, 21, 2559–2571 CrossRef CAS.
- M. Lein, J. Frunzke, A. Timoshkin and G. Frenking, Chem. – Eur. J., 2001, 7, 4155–4163 CrossRef CAS PubMed.
- M. A. Beswick, J. S. Palmer and D. S. Wright, Chem. Soc. Rev., 1998, 27, 225–232 RSC.
- A. D. Walsh, J. Chem. Soc., 1953, 2260–2266 RSC.
- E. F. Hayes, J. Phys. Chem., 1966, 70, 3740–3742 CrossRef CAS.
- L. Wharton, R. A. Berg and W. Klemperer, J. Chem. Phys., 1963, 39, 2023–2031 CrossRef CAS.
- P. Debye, Polar Molecules, The Chemical Catalogue Company, New York, 1929 Search PubMed.
- G. Gigli, J. Chem. Phys., 1990, 93, 5224–5233 CrossRef CAS.
- R. A. Andersen, R. Blom, J. M. Boncella, C. J. Burns and H. V. Volden, Acta Chem. Scand., 1987, 41, 24–35 CrossRef.
- T. K. Hollis, J. K. Burdett and B. Bosnich, Organometallics, 1993, 12, 3385–3386 CrossRef CAS.
- R. A. Williams, T. P. Hanusa and J. C. Huffman, Organometallics, 1990, 9, 1128–1134 CrossRef CAS.
- W. J. Evans, M. A. Johnston, M. A. Greci and J. W. Ziller, Organometallics, 1999, 18, 1460–1464 CrossRef CAS.
- M. Brookhart, M. L. H. Green and G. Parkin, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 6908–6914 CrossRef CAS PubMed.
- R. Pal, S. Mebs, M. W. Shi, D. Jayatilaka, J. M. Krzeszczakowska, L. A. Malaspina, M. Wiecko, P. Luger, M. Hesse, Y.-S. Chen, J. Beckmann and S. Grabowsky, Inorg. Chem., 2018, 57, 4906–4920 CrossRef CAS PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. J. A. Montgomery, J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 16, Revision C.01., Gaussian, Inc., Wallingford CT, 2019 Search PubMed.
- A. D. Becke, Phys. Rev. A, 1988, 38, 3098–3100 CrossRef CAS PubMed.
- J. P. Perdew, Phys. Rev. B: Condens. Matter Mater. Phys., 1986, 33, 8822–8824 CrossRef PubMed.
- J. P. Perdew, Phys. Rev. B: Condens. Matter Mater. Phys., 1986, 34, 7406 CrossRef PubMed.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS.
- C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785–789 CrossRef CAS PubMed.
- Y. Zhao and D. G. Truhlar, Theor. Chem. Acc., 2008, 120, 215–241 Search PubMed.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed.
- S. Grimme, S. Ehrlich and L. Goerigk, J. Comput. Chem., 2011, 32, 1456–1465 CrossRef CAS PubMed.
- F. Weigend and R. Ahlrichs, Phys. Chem. Chem. Phys., 2005, 7, 3297–3305 RSC.
- C. Y. Peng, P. Y. Ayala, H. B. Schlegel and M. J. Frisch, J. Comput. Chem., 1996, 17, 49–56 CrossRef CAS.
- J. W. McIver and A. Komornic, JACS, 1972, 94, 2625–2633 CrossRef CAS.
- G. Hetzer, P. Pulay and H. J. Werner, Chem. Phys. Lett., 1998, 290, 143–149 CrossRef CAS.
- P. Pulay, Chem. Phys. Lett., 1983, 100, 151–154 CrossRef CAS.
- S. Sæbø and P. Pulay, Chem. Phys. Lett., 1985, 113, 13–18 CrossRef.
- G. Hetzer, M. Schütz, H. Stoll and H. J. Werner, J. Chem. Phys., 2000, 113, 9443–9455 CrossRef CAS.
- S. Saebø and P. Pulay, J. Chem. Phys., 1987, 86, 914–922 CrossRef.
- S. Saebø and P. Pulay, J. Chem. Phys., 1988, 88, 1884–1890 CrossRef.
- M. Schütz, G. Hetzer and H. J. Werner, J. Chem. Phys., 1999, 111, 5691–5705 CrossRef.
- G. Rauhut, P. Pulay and H. J. Werner, J. Comput. Chem., 1998, 19, 1241–1254 CrossRef CAS.
- P. Pulay and S. Saebø, Theor. Chim. Acta, 1986, 69, 357–368 CrossRef CAS.
- H.-J. Werner, P. J. Knowles, G. Knizia, F. R. Manby, M. Schütz, P. Celani, W. Györffy, D. Kats, T. Korona, R. Lindh, A. Mitrushenkov, G. Rauhut, K. R. Shamasundar, T. B. Adler, R. D. Amos, S. J. Bennie, A. Bernhardsson, A. Berning, D. L. Cooper, M. J. O. Deegan, A. J. Dobbyn, F. Eckert, E. Goll, C. Hampel, A. Hesselmann, G. Hetzer, T. Hrenar, G. Jansen, C. Köppl, S. J. R. Lee, Y. Liu, A. W. Lloyd, R. A. Mata, A. J. May, S. J. McNicholas, W. Meyer, T. F. Miller III, M. E. Mura, A. Nicklass, D. P. O’Neill, P. Palmieri, D. Peng, K. Pflüger, R. Pitzer, M. Reiher, T. Shiozaki, H. Stoll, A. J. Stone, R. Tarroni, T. Thorsteinsson, M. Wang and M. Welborn, MOLPRO, 2019, https://www.molpro.net Search PubMed.
- J. G. Hill and K. A. Peterson, J. Chem. Phys., 2017, 147, 244106 CrossRef PubMed.
- T. H. Dunning Jr, J. Chem. Phys., 1989, 90, 1007–1023 CrossRef.
- D. E. Woon and T. H. Dunning Jr, J. Chem. Phys., 1993, 98, 1358–1371 CrossRef CAS.
- M. Schütz, H. J. Werner, R. Lindh and F. R. Manby, J. Chem. Phys., 2004, 121, 737–750 CrossRef PubMed.
- E. D. Glendening, C. R. Landis and F. Weinhold, J. Comput. Chem., 2019, 40, 2234–2241 CrossRef CAS PubMed.
- A. E. Reed, R. B. Weinstock and F. Weinhold, J. Chem. Phys., 1985, 83, 735–746 CrossRef CAS.
- E. D. Glendening, J. K. Badenhoop, A. E. Reed, J. E. Carpenter, J. A. Bohmann, C. M. Morales, P. Karafiloglou, C. R. Landis and F. Weinhold, NBO 7.0, Theoretical Chemistry Institute, University of Wisconsin, Madison, 2018 Search PubMed.
- L. Gagliardi, R. Lindh and G. Karlström, J. Chem. Phys., 2004, 121, 4494–4500 CrossRef CAS PubMed.
- F. Aquilante, J. Autschbach, A. Baiardi, S. Battaglia, V. A. Borin, L. F. Chibotaru, I. Conti, L. D. Vico, M. Delcey, I. F. Galván, N. Ferré, L. Freitag, M. Garavelli, X. Gong, S. Knecht, E. D. Larsson, R. Lindh, M. Lundberg, P. Å. Malmqvist, A. Nenov, J. Norell, M. Odelius, M. Olivucci, T. B. Pedersen, L. Pedraza-González, Q. M. Phung, K. Pierloot, M. Reiher, I. Schapiro, J. Segarra-Martí, F. Segatta, L. Seijo, S. Sen, D.-C. Sergentu, C. J. Stein, L. Ungur, M. Vacher, A. Valentini and V. Veryazov, J. Chem. Phys., 2020, 152, 214117 CrossRef CAS PubMed.
- K. Morokuma, J. Chem. Phys., 1971, 55, 1236–1244 CrossRef CAS.
- T. Ziegler and A. Rauk, Inorg. Chem., 1979, 18, 1558–1565 CrossRef CAS.
- T. Ziegler and A. Rauk, Inorg. Chem., 1979, 18, 1755–1759 CrossRef CAS.
- F. M. Bickelhaupt, N. M. M. Nibbering, E. M. Van Wezenbeek and E. J. Baerends, J. Phys. Chem., 1992, 96, 4864–4873 CrossRef CAS.
- F. M. Bickelhaupt and E. J. Baerends, in Rev. Comput. Chem., ed. K. B. Lipkowitz and D. B. Boyd, 2000, vol. 15, pp. 1–86 Search PubMed.
- G. te Velde, F. M. Bickelhaupt, E. J. Baerends, C. Fonseca Guerra, S. J. A. van Gisbergen, J. G. Snijders and T. Ziegler, J. Comput. Chem., 2001, 22, 931–967 CrossRef CAS.
- J. Krijn and E. J. Baerends, Fit Functions in the HFS-Method, 1984.
- E. Van Lenthe, E. J. Baerends and J. G. Snijders, J. Chem. Phys., 1993, 99, 4597–4610 CrossRef CAS.
- A. Wuttke and R. A. Mata, J. Comp. Chem., 2017, 38, 15–23 CrossRef CAS PubMed.
- S. Löffler, A. Wuttke, B. Zhang, J. J. Holstein, R. A. Mata and G. H. Clever, Chem. Comm., 2017, 53, 11933–11936 RSC.
- J. Ahrens, B. Geveci and C. Law, in Visualization Handbook, ed. C. D. Hansen and C. R. Johnson, Butterworth-Heinemann, Burlington, 2005, pp. 717–731 Search PubMed.
- P. H. M. Budzelaar, J. J. Engelberts and J. H. van Lenthe, Organometallics, 2003, 22, 1562–1576 CrossRef CAS.
- K. Nugent, J. Beattie, T. Hambley and M. Snow, Aust. J. Chem., 1984, 37, 1601–1606 CrossRef CAS.
- P. Margl, K. Schwarz and P. E. Bloechl, J. Am. Chem. Soc., 1994, 116, 11177–11178 CrossRef CAS.
- I. Hung, C. L. B. Macdonald and R. W. Schurko, Chem. – Eur. J., 2004, 10, 5923–5935 CrossRef CAS PubMed.
- M. D. Walter, G. Wolmershäuser and H. Sitzmann, J. Am. Chem. Soc., 2005, 127, 17494–17503 CrossRef CAS PubMed.
- A. J. Bridgeman, J. Chem. Soc., Dalton Trans., 1997, 2887–2894 RSC.
- P. Jutzi and N. Burford, Chem. Rev., 1999, 99, 969–990 CrossRef CAS PubMed.
- M. Kaupp, P. V. R. Schleyer, M. Dolg and H. Stoll, J. Am. Chem. Soc., 1992, 114, 8202–8208 CrossRef CAS.
- R. Zerger and G. Stucky, J. Organomet. Chem., 1974, 80, 7–17 CrossRef CAS.
- R. Pal, S. Mebs, M. W. Shi, D. Jayatilaka, J. M. Krzeszczakowska, L. A. Malaspina, M. Wiecko, P. Luger, M. Hesse, Y.-S. Chen, J. Beckmann and S. Grabowsky, Inorg. Chem., 2018, 57, 4906–4920 CrossRef CAS PubMed.
- I. Bytheway, P. L. A. Popelier and R. J. Gillespie, Can. J. Chem., 1996, 74, 1059–1071 CrossRef CAS.
- P. Jutzi and N. Burford, Chem. Rev., 1999, 99, 969–990 CrossRef CAS PubMed.
- S. Harder, Coor. Chem. Rev., 1998, 176, 17–66 CrossRef CAS.
- R. P. Hughes, X. Zheng, C. A. Morse, O. J. Curnow, J. R. Lomprey, A. L. Rheingold and G. P. A. Yap, Organometallics, 1998, 17, 457–465 CrossRef CAS.
- K. Sünkel, Chem. Ber., 1997, 130, 1721–1730 CrossRef.
- C. Hansch, A. Leo and R. W. Taft, Chem. Rev., 1991, 91, 165–195 CrossRef CAS.
- V. M. Rayón and G. Frenking, Chem. – Eur. J., 2002, 8, 4693–4707 CrossRef.
- N. M. Edelstein, P. G. Allen, J. J. Bucher, D. K. Shuh, C. D. Sofield, N. Kaltsoyannis, G. H. Maunder, M. R. Russo and A. Sella, J. Am. Chem. Soc., 1996, 118, 13115–13116 CrossRef CAS.
- M. R. Russo, N. Kaltsoyannis and A. Sella, Chem. Commun., 2002, 2458–2459 RSC.
- M. Schütz, G. Rauhut and H. J. Werner, J. Chem. Phys. A, 1998, 102, 5997–6003 CrossRef.
- W. B. Schneider, G. Bistoni, M. Sparta, M. Saitow, C. Riplinger, A. A. Auer and F. Neese, J. Chem. Theory Comput., 2016, 12, 4778–4792 CrossRef CAS PubMed.
- G. Bistoni, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2019, e1442 Search PubMed.
- L. L. Zhao, M. von Hopffgarten, D. M. Andrada and G. Frenking, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2018, 8, e13450 Search PubMed.
- D. M. Andrada and C. Foroutan-Nejad, Phys. Chem. Chem. Phys., 2020, 22, 22459–22464 RSC.
- J. Poater, D. M. Andrada, M. Solà and C. Foroutan-Nejad, Phys. Chem. Chem. Phys., 2022, 24, 2344–2348 RSC.
- M. Sola, M. Duran and J. Poater, Theo. Chem. Acc., 2021, 140, 33 CrossRef CAS.
- M. Gimferrer, S. Danés, E. Vos, C. B. Yildiz, I. Corral, A. Jana, P. Salvador and D. M. Andrada, Chem. Sci., 2022, 13, 6583–6591 RSC.
- M. Gimferrer, S. Danés, E. Vos, C. B. Yildiz, I. Corral, A. Jana, P. Salvador and D. M. Andrada, Chem. Sci., 2023, 14, 384–392 RSC.
- M. Mitoraj and A. Michalak, J. Mol. Model., 2008, 14, 681–687 CrossRef CAS PubMed.
- M. P. Mitoraj, A. Michalak and T. Ziegler, J. Chem. Theory Comput., 2009, 5, 962–975 CrossRef CAS PubMed.
- G.-J. Linker, P. T. van Duijnen, P. H. M. van Loosdrecht and R. Broer, J. Phys. Chem. A, 2012, 116, 7219–7227 CrossRef CAS PubMed.
- G.-J. Linker, P. Th van Duijnen, P. H. M. van Loosdrecht and R. Broer, J. Phys. Chem. A, 2022, 126, 5231 CrossRef CAS PubMed.
- G.-J. Linker, P. T. van Duijnen and R. Broer, J. Phys. Chem. A, 2020, 124, 1306–1311 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2cp05020j |
| This journal is © the Owner Societies 2023 |