
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Determining nuclear quadrupole moments of Bi and Sb from molecular data†
Jean-Pierre
Dognon
 *a and
Pekka
Pyykkö
*b
*a and
Pekka
Pyykkö
*b
aUniversité Paris-Saclay, CEA, CNRS, NIMBE, 91191, Gif-sur-Yvette, France. E-mail: jean-pierre.dognon@cea.fr
bDepartment of Chemistry, University of Helsinki, PO Box 55 (A. I. Virtasen aukio 1), FIN-00014 Helsinki, Finland. E-mail: pekka.pyykko@helsinki.fi
First published on 10th January 2023
Abstract
An independent value of −422(3) millibarn (mb) is obtained for the nuclear quadrupole moment Q(209Bi) using experimental coupling constants for diatomic BiN, BiP, BiF, BiCl, and BiI, combined with full-Dirac CCSD-T calculations of the electric field gradient q. This value lies close to two other recently published molecular results, and a full-triplet CCSDT atomic result. Based on the same approach, we obtained Q(121Sb) = −541.7(0.8) mb and Q(123Sb) = −690.6( 1.0) mb, in agreement with one recently published molecular result.
The nuclear quadrupole moment Q(209Bi) was one of the first moments determined.1 The value, in 1936, was −400 mb [1 mb = 10−31 m2]. Interest in the Q value of the neutron deficient isotopes continues, due to the nuclear physics.2 A common way to determine Q is to measure a nuclear quadrupole coupling constant (NQCC), B = eqQ/h, and to combine it with a theoretical determination of the electric field gradient (EFG), q, on the nuclear site in the system studied. This can be done for atomic, molecular, or solid-state systems and the results usually agree with each other, and with the main alternative approach of muonic or pionic systems. For a recent review see Pyykkö.3
A notable exception is Q(209Bi), where a 2001 atomic Q was −516(15) mb4 but a molecular value using the BiN and BiP diatomics by Teodoro5 was −420(8) mb. Both B experiments and both q calculations were expected to be quite accurate. Therefore the problem is still on the table (see below) and we want to have a further look at the ‘molecular’ Q(209Bi). Note that we here consider two series of molecules, the pnicogens BiN and BiP and the monohalides BiX (X = Cl, F, I) and that these series have opposite signs of B (Table 1).
| Molecule | B e (MHz) | R e (Å) | Ref. |
|---|---|---|---|
| a Experimental B0 given for BiCl and BiI. b This value of B has been calculated from the experimental results for 123Sb35Cl by using the ratio B(123Sb)/B(121Sb) = 1.27491(1).17 | |||
| BiN | 905.066(88) | 1.9349079(7) | 11 |
| BiP | 903.031 | 2.2961520(80) | 5 and 11 |
| BiF | −1150.9632 | 2.034276(1) | 12 |
| BiCl | −1027(12)a | 2.47155(7) | 13 |
| BiI | −909.5(20)a | 2.80053(8) | 14 and 15 |
| SbN | 649.669(9) | 1.8356707(2) | 16 |
| SbP | 620.35(1) | 2.2054454(5) | 16 |
| SbF | −586.802(1) | 1.917584(28) | 17 |
| SbCl | −515.124b | 2.335472(13) | 17 |
The case of antimony still remains difficult. The reference value has fluctuated widely over the years. The old, atomic standard values for 121,123Sb were −360(40) mb and −490(50) mb,6 respectively. Later, Voss et al. from an experimental and theoretical analysis of the configuration 5p2 6s supported a value of −440(30) mb for 121Sb.7 Solid-state calculations by Svane8 first suggested that the 121Sb value should be changed to −669 mb. Then molecular calculations by Demovič et al.9 and Haiduke et al.10 produced mutually consistent values of −556 mb and −543 mb, respectively. Both determinations were based on diatomic data for SbN, SbP, SbF and SbCl by Cooke and co-workers, reported in Table 1. This second series of four molecules has been studied in this work.
BiN, SbN, BiP and SbP are triple-bonded, closed-shell, short-bond systems with a substantial gap between the highest occupied and lowest empty molecular orbital. They are consequently easiest to describe with a single-configuration starting point. The monohalide BiX (X = F, Cl, I) and SbX (X = F, Cl) are, in a non-relativistic picture, open-shell 3∑− states and were found to be a good example of Hund's case (b). In the relativistic picture, due to strong spin–orbit coupling, they are very near the Hund's case (c) limit, with the consequence of a 1∑+ ground state, that could be considered as a relativistic closed-shell.
In a diatomic molecule, the total electric-field gradient q at the nucleus X can be decomposed into nuclear and electronic contributions.
| q(X) = qnucl(X) + qel(X) |
All calculations were carried out applying CV3Z triple-zeta Dyall basis sets with core correlating functions (see ref. 21 and 22 and the appropriate references) in their fully uncontracted form. We used a Gaussian charge distribution model to describe the charge of nuclei. Furthermore, all the present calculations have been carried out at the experimental value of the equilibrium bond distances, Re (Table 1). Note that the use of available experimental (Be, Re) takes care of the vibrational effects. Within the DFT framework, the electronic contribution to the q value is obtained from an analytic gradient formulation as implemented in the DIRAC program. For the CCSD-T approach, the electronic contribution to the q value is obtained by adding the q analytic HF value to the contribution of the electronic correlation to q (qel(X) = qel.,HF(X) + qel.,corr.(X)). In order to get the correlation correction to q we used an approach proposed by Pernpointner et al.23 fitting the correlation energy obtained at different field strengths (between ±1.0 × 10−6 a.u. and ±1.0 × 10−10 a.u.) to an nth order polynomial. The correlation correction to q is obtained from the first order response energy23 (see ESI† for details). In a final step, Q was calculated from the indirect method introduced by Belpassi et al.24 which corresponds to the calculation of slope from a least squares regression line through all (q,B) points of the Bi and Sb molecular series BIN, BiP, BiF, BiCl, BiI and SbN, SbP, SbF, SbCl according to: (B/MHz) = 0.2349647 (q/a.u.) (Q/mb) with B the experimental nuclear quadrupole coupling constant (see ESI† for details). This original approach combining on the one hand the polynomial determination of the correlation correction to q and on the other hand the indirect method to determine Q makes it possible to minimize the impact of errors in each stage of the evaluation of the Q value (see ESI† for details).
The graphs are reported in Fig. 1 and 2 for the Bi and Sb molecules, respectively, in a closed-shell ground-state picture.
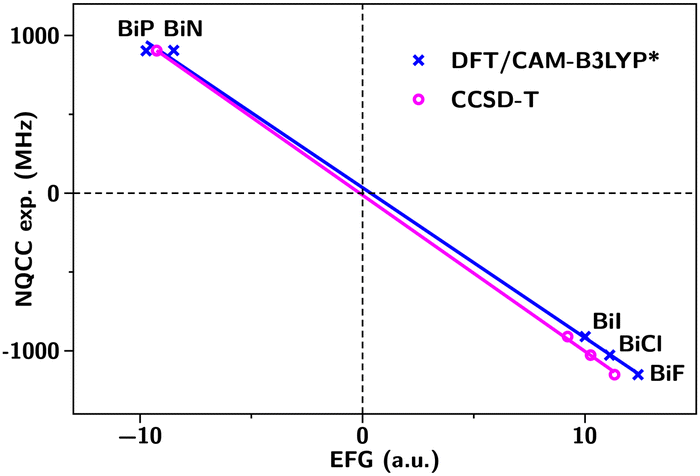 | ||
| Fig. 1 The experimental NQCC in MHz as a function of the calculated EFG in atomic units for bismuth-based molecules. | ||
 | ||
| Fig. 2 The experimental NQCC in MHz as a function of the calculated EFG in atomic units for antimony-based molecules. | ||
For the Bi-containing molecules, we obtained a good agreement between CAM-B3LYP* functional and the CCSD-T value. The determined Q(209Bi) value from the linear fit is −405(9) mb from the DFT/CAM-B3LYP* calculations and −422(3) mb from the CCSD-T calculation. Here the error limits are statistical standard errors. These values are in agreement with the recent determinations of Teodoro et al.5 at −420(8) mb based on the direct approach for each molecule BiN and BiP separately. Shee et al.25 obtain from the same two molecules −415.1 mb. Skripnikov et al.26 published a new ‘atomic’, FS-CCSDT Q(209Bi) of −418(6) mb. Our results are also consistent with recent work by Liu and Cheng27 who obtained values in the range of −411 to −422 mb for the same five molecules using an atomic mean-field spin–orbit approach within the exact two-component theory (SOX2CAMF).
Commenting on the Q(209Bi) of −516 mb by Bieroń and Pyykkö in 2001,4 it was obtained by about 20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 Configuration State Functions (CSF), thought at the time to be adequate. Single, some double and no triple substitutions were included. Currently over 4 million CSFs are possible and still don’t provide a complete convergence for the 5-valence-electron system Bi.28
000 Configuration State Functions (CSF), thought at the time to be adequate. Single, some double and no triple substitutions were included. Currently over 4 million CSFs are possible and still don’t provide a complete convergence for the 5-valence-electron system Bi.28
We compare our Q(Bi) determination with the (previous) Group-15 case of Q(Sb). The Be and Re are listed in Table 1. Concerning the Q(121Sb) we observed a significant deviation between the CAM-B3LYP* value at −474(3) mb and the CCSD-T value at −541.7(0.8) mb. In 2006, from a CCSD-T calculation, Haiduke et al.10 proposed a recommended value of −543(11) mb for 121Sb. Their values were calculated using only the SbN and SbP closed-shell molecule results due to too large errors in the SbF and SbCl open-shell calculations. In the same time, Demovič et al.9 recommended the value of (−556 ± 24) mb from direct method on the SbN, SbP, SbF, SbCl series with IOTC CCSD(T) calculations. In closed-shell picture, the Haiduke et al.10 value is in good agreement with our new value. We can also notice the quality of calculated EFGs confirmed by an R2 of 1 and the intercept at origin that does not deviate substantially from zero. Any deviation from zero intercept would indicate a systematic error in the method applied. This consistency of values can also be seen as an argument in favor of relativistic closed-shell monohalides.
To go further and interpret the origin of calculated values, we performed an analysis of the electronic contributions to the q value of the individual orbitals. These results are illustrated in Fig. 3 and 4. These analyses are only possible from analytical calculation of the EFG. Hence they were done with DFT calculations with CAM-B3LYP* functional.
 | ||
| Fig. 3 Electronic contribution from individual orbitals to qel (CAM-B3LYP*/CV3Z) for bismuth-based molecules. | ||
 | ||
| Fig. 4 Electronic contribution from individual orbitals to qel (CAM-B3LYP*/CV3Z) for antimony-based molecules. | ||
A first general comment is that the main contribution to EFG is due to valence electrons. This has already been outlined, e.g., by Neese et al.29 for the HI and HAt compounds for which most of the observable field gradient comes from the valence region. The contribution from the 6s(Bi) is quite similar in BiN (6s(Bi)/2s(N) mixing) and BiP (6s(Bi)/3s(P) mixing). The negative overall value of q is due to the large negative contribution given by the mixing of 6p(Bi) and 2p(N) (σ bonding orbital) in BiN and by the 6p(Bi) orbital in BiP. In contrast, in BiF and BiCl, the 6p(Bi) π orbital provides a significant positive contribution to EFG that is partially counterbalanced by a negative EFG contribution from σ bonding orbitals (2p(F)/3p(Cl)/5p(I)) mixing with the 6p(Bi) orbitals resulting in a significant positive total EFG value. In SbN and in SbP the negative EFG value originates from the 5p(Sb)/2p(N) and 5p(Sb)/3p(P) mixing respectively (σ bonding orbitals). In the SbX series of molecules, the large positive value is due to the high contribution of the 5p(Sb) π orbital that is partly compensated, by negative contribution from the mixing of the 5p(Sb) with the 2p(F)/3p(Cl) (σ bonding orbitals).
In this work, from extensive ab initio and DFT calculations in the relativistic four-component framework, on a series of closed-shell states of molecules containing bismuth and antimony, we have estimated the nuclear quadrupole moment of 209Bi and 121Sb at the CCSD-T level, equal to −422 mb and −542 mb, respectively. Though there has been much debate on the subject, this is a further step towards a converged value of the nuclear quadrupole moment of 209Bi and 121Sb. The proposed methodology significantly improves the consistency of the results, minimizes the impact of systematic errors, and allows to discard unreliable results.
Additionally, an analysis of the electronic contributions to the q value of individual orbitals reveals a general layout along the Bi and Sb series of molecules. 6p(Bi) or 5p(Sb) σ orbitals have a significant negative contribution to EFG while 6p(Bi) or 5p(Sb) π orbitals have a significant positive contribution to EFG. The overall value can be modulated by the mixing of these orbitals with those of N, P, F, Cl or I.
In conclusion, our five new full-Dirac CCSD-T determinations using the coupling constants for diatomic BiN, BiP, BiF, BiCl and BiI yield a Q(209 Bi) of −422(3) mb, in close agreement with the molecular values of Teodoro et al.5 or Shee et al.,25 and the new CCSDT atomic value by Skripnikov et al.26 Our more approximate molecular DFT/CAM-B3LYP* Q(Bi) of −405(9) mb also lies close. Thus we fully support the ‘World average’ of −420(17) mb in Barzakh et al.2 For the antimony isotopes we obtain Q(121Sb) = −541.7(0.8) mb and Q(123Sb) = −690.6(0.8) mb using the ratio B(123Sb)/B(121Sb) = 1.27491(1).17
We thank the referees for their careful work. J.-P. Dognon thanks the Finnish-French Maupertuis program for supporting his stays at Helsinki by travel grants.
Conflicts of interest
There are no conflicts to declare.Notes and references
- H. Schüler and T. Schmidt, Z. Phys., 1936, 99, 717–728 CrossRef.
- A. Barzakh, A. N. Andreyev, C. Raison, J. G. Cubiss, P. Van Duppen, S. Péru, S. Hilaire, S. Goriely, B. Andel, S. Antalic, M. Al Monthery, J. C. Berengut, J. Bieroń, M. L. Bissell, A. Borschevsky, K. Chrysalidis, T. E. Cocolios, T. Day Goodacre, J.-P. Dognon, M. Elantkowska, E. Eliav, G. J. Farooq-Smith, D. V. Fedorov, V. N. Fedosseev, L. P. Gaffney, R. F. Garcia Ruiz, M. Godefroid, C. Granados, R. D. Harding, R. Heinke, M. Huyse, J. Karls, P. Larmonier, J. G. Li, K. M. Lynch, D. E. Maison, B. A. Marsh, P. Molkanov, P. Mosat, A. V. Oleynichenko, V. Panteleev, P. Pyykkö, M. L. Reitsma, K. Rezynkina, R. E. Rossel, S. Rothe, J. Ruczkowski, S. Schiffmann, C. Seiffert, M. D. Seliverstov, S. Sels, L. V. Skripnikov, M. Stryjczyk, D. Studer, M. Verlinde, S. Wilman and A. V. Zaitsevskii, Phys. Rev. Lett., 2021, 127, 192501 CrossRef CAS PubMed.
- P. Pyykkö, Mol. Phys., 2018, 116, 1328–1338 CrossRef.
- J. Bieroń and P. Pyykkö, Phys. Rev. Lett., 2001, 87, 133003 CrossRef PubMed.
- T. Q. Teodoro and R. L. A. Haiduke, Phys. Rev. A: At., Mol., Opt. Phys., 2013, 88, 052504 CrossRef.
- B. Buchholz, H. D. Kronfeldt, G. Müller, M. Voss and R. Winkler, Z. Phys. A: Hadrons Nucl., 1978, 288, 247–256 CrossRef CAS.
- M. Voss, W. Weiss, B. Buchholz and R. Winkler, Z. Phys. D: At., Mol. Clusters, 1986, 1, 151–156 CrossRef CAS.
- A. Svane, Phys. Rev. B: Condens. Matter Mater. Phys., 2003, 68, 064422 CrossRef.
- L. Demovič, V. Kellö, A. J. Sadlej and S. A. Cooke, J. Chem. Phys., 2006, 124, 184308 CrossRef PubMed.
- R. L. A. Haiduke, A. B. F. da Silva and L. Visscher, J. Chem. Phys., 2006, 125, 064301 CrossRef PubMed.
- S. A. Cooke, J. M. Michaud and M. C. L. Gerry, J. Mol. Struct., 2004, 695–696, 13–22 CrossRef CAS.
- E. H. Fink, K. D. Setzer, D. A. Ramsay, J. P. Towle and J. M. Brown, J. Mol. Spectrosc., 1996, 178, 143–156 CrossRef CAS.
- P. Kuijpers, T. Törring and A. Dymanus, Chem. Phys., 1976, 18, 401–416 CrossRef CAS.
- P. Kuijpers, T. Torring and A. Dymanus, Chem. Phys., 1976, 12, 309–313 CrossRef CAS.
- R. Tischer, K. Möller and T. Törring, Chem. Phys., 1981, 62, 115–121 CrossRef CAS.
- S. A. Cooke and M. C. L. Gerry, Phys. Chem. Chem. Phys., 2004, 6, 4579–4585 RSC.
- S. A. Cooke and M. C. Gerry, J. Mol. Spectrosc., 2005, 234, 195–203 CrossRef CAS.
- DIRAC, a relativistic ab initio electronic structure program, Release DIRAC19 (2019), written by A. S. P. Gomes, T. Saue, L. Visscher, H. J. Aa. Jensen, and R. Bast, with contributions from I. A. Aucar, V. Bakken, K. G. Dyall, S. Dubillard, U. Ekström, E. Eliav, T. Enevoldsen, E. Faßhauer, T. Fleig, O. Fossgaard, L. Halbert, E. D. Hedegård, B. Heimlich-Paris, T. Helgaker, J. Henriksson, M. Iliaš, Ch. R. Jacob, S. Knecht, S. Komorovský, O. Kullie, J. K. Lærdahl, C. V. Larsen, Y. S. Lee, H. S. Nataraj, M. K. Nayak, P. Norman, G. Olejniczak, J. Olsen, J. M. H. Olsen, Y. C. Park, J. K. Pedersen, M. Pernpointner, R. di Remigio, K. Ruud, P. Sałek, B. Schimmelpfennig, B. Senjean, A. Shee, J. Sikkema, A. J. Thorvaldsen, J. Thyssen, J. van Stralen, M. L. Vidal, S. Villaume, O. Visser, T. Winther, and S. Yamamoto (available at DOI:10.5281/zenodo.3572669, see also http://www.diracprogram.org).
- C. Thierfelder, P. Schwerdtfeger and T. Saue, Phys. Rev. A: At., Mol., Opt. Phys., 2007, 76, 034502 CrossRef.
- M. J. Deegan and P. J. Knowles, Chem. Phys. Lett., 1994, 227, 321–326 CrossRef CAS.
- K. G. Dyall, Theor. Chem. Acc., 2012, 131, 1217 Search PubMed.
- K. G. Dyall, Theor. Chem. Acc., 2016, 135, 128 Search PubMed.
- M. Pernpointner and L. Visscher, J. Chem. Phys., 2001, 114, 10389–10395 CrossRef CAS.
- L. Belpassi, F. Tarantelli, A. Sgamellotti, H. M. Quiney, J. N. P. van Stralen and L. Visscher, J. Chem. Phys., 2007, 126, 064314 CrossRef PubMed.
- A. Shee, L. Visscher and T. Saue, J. Chem. Phys., 2016, 145, 184107 CrossRef PubMed.
- L. V. Skripnikov, A. V. Oleynichenko, A. V. Zaitsevskii, D. E. Maison and A. E. Barzakh, Phys. Rev. C, 2021, 104, 034316 CrossRef CAS.
- J. Liu and L. Cheng, J. Chem. Phys., 2018, 148, 144108 CrossRef PubMed.
- J. Bieroń, 2022, private communication.
- F. Neese, A. Wolf, T. Fleig, M. Reiher and B. A. Hess, J. Chem. Phys., 2005, 122, 204107 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2cp04747k |
| This journal is © the Owner Societies 2023 |