
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
B–H⋯π and C–H⋯π interactions in protein–ligand complexes: carbonic anhydrase II inhibition by carborane sulfonamides†
Jindřich
Fanfrlík‡
 *a,
Jiří
Brynda‡
a,
Michael
Kugler
a,
Martin
Lepšík
a,
Klára
Pospíšilová
a,
Josef
Holub
b,
Drahomír
Hnyk
b,
Jan
Nekvinda
b,
Bohumír
Grüner
b and
Pavlína
Řezáčová
*a
*a,
Jiří
Brynda‡
a,
Michael
Kugler
a,
Martin
Lepšík
a,
Klára
Pospíšilová
a,
Josef
Holub
b,
Drahomír
Hnyk
b,
Jan
Nekvinda
b,
Bohumír
Grüner
b and
Pavlína
Řezáčová
*a
aInstitute of Organic Chemistry and Biochemistry of the Czech Academy of Sciences, Flemingovo nam. 2, 166 10, Prague 6, Czech Republic. E-mail: fanfrlik@uochb.cas.cz; pavlina.rezacova@uochb.cas.cz
bInstitute of Inorganic Chemistry of the Czech Academy of Sciences, 250 68 Husinec-Řež, Czech Republic
First published on 3rd January 2023
Abstract
Among non-covalent interactions, B–H⋯π and C–H⋯π hydrogen bonding is rather weak and less studied. Nevertheless, since both can affect the energetics of protein–ligand binding, their understanding is an important prerequisite for reliable predictions of affinities. Through a combination of high-resolution X-ray crystallography and quantum-chemical calculations on carbonic anhydrase II/carborane-based inhibitor systems, this paper provides the first example of B–H⋯π hydrogen bonding in a protein–ligand complex. It shows that the B–H⋯π interaction is stabilized by dispersion, followed by electrostatics. Furthermore, it demonstrates that the similar C–H⋯π interaction is twice as strong, with a slightly smaller contribution of dispersion and a slightly higher contribution of electrostatics. Such a detailed insight will facilitate the rational design of future protein ligands, controlling these types of non-covalent interactions.
Introduction
C–H⋯π is a weak hydrogen bond between a non-classical donor (typically aliphatic or aromatic groups) and a non-classical acceptor (e.g. electron density of phenyl rings) that can stabilize crystals of small organic molecules or proteins.1 This interaction may also involve inorganic compounds, e.g. C–H groups of carbon-substituted three-dimensional boron hydrides (carboranes).2 In contrast, the B–H groups of carboranes act as hydrogen-bond acceptors due to the low electronegativity of boron. The hydridic nature of boron-attached hydrogens thus results in dihydrogen-bond formation.3–7 Nevertheless, B–H groups can also form a weak dispersion-driven non-covalent contact with weak acceptors, referred to as B–H⋯π,8,9 which has recently been observed by means of X-ray crystallography in the boron-hydride organometallic complex.10 Comparing the C–H⋯π and B–H⋯π interactions of neutral carboranes, the latter has been calculated to be two times weaker in model systems.9 In this work, we study these interactions in protein–ligand complexes by means of X-ray crystallography and quantum-mechanical computations. The protein studied is carbonic anhydrase II (CA II), and the ligands are sulfonamide inhibitors terminated with three isomers of carborane cages (ortho – compound 1, meta – compound 2 and para – compound 3; Fig. 1(A)). | ||
| Fig. 1 (A) The structural formulae of the carborane propyl-sulfonamide compounds 1, 2 and 3 and the values of the inhibition constant Ki for the inhibition of CA II. (B) Details of the crystal structure of compound 1 bound in the active site of CA II (PDB code 6YZT). Interacting residues are marked with sticks. The inhibitor and the residue Phe131 occupy two alternative conformations: the major conformation (an occupancy of 0.8) is depicted in stick representation, whereas the minor conformation (an occupancy of 0.2) is in line representation. Polar interactions are represented by blue dashed lines, while the black dashed line marks the potential B–H⋯π interaction between the carborane cage and Phe131. | ||
Carbonic anhydrases (CAs) are metalloenzymes catalyzing the interconversion between CO2 and bicarbonate. These enzymes participate in many physiological processes and some of their isoforms are involved in human pathologies, such as cancer development (isoform CA IX).11,12 CAs have thus been targets for inhibitor development; dozens of diverse families of CA inhibitors (CAIs) have been prepared.13 Due to this wealth of gathered information, these systems are an attractive model for biophysical studies of protein–ligand binding.14 Human CAs can be efficiently and selectively inhibited by carboranes linked to a sulfamide or sulfonamide moiety.15–18 More than 30 X-ray crystal structures of carborane-containing inhibitors with CA II or CA IX have been determined at high or even atomic resolution, which has provided insight into the structural basis of the interactions of carborane with enzyme active sites.15 Here, we focus on a series of carborane propyl-sulfonamides that differ in the geometric positions of carbon atoms within the carborane cage and on their affinities toward CA II (Fig. 1(A)). Some compounds have exhibited selective inhibitory activity toward CA IX over CA II,19 and the structural basis of this selectivity has been explained by a detailed crystallographic study of compound 1 in these complexes.17 Interestingly, the crystal structure of 1 in complex with CA II (PDB code 6YZT) has revealed that compound 1 has acquired two alternative binding orientations in the CA II active site and the minor conformation (with an occupancy factor of 0.2) is accompanied by a rearrangement of the Phe131 side chain (Fig. 1(B)). The mutual position and distance between the carborane cage and the side chain of Phe131 indicate a potential B–H⋯π interaction (the dashed black line in Fig. 1(B)).
Methods
Crystallization, structure determination and refinement
Complexes of CA II with compounds were prepared by the addition of a one- to twofold molar excess of the compounds (dissolved in 100% DMSO) to a 20–25![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) mg ml−1 protein solution in 50mM Tris–H2SO4, pH 7.8. The final concentration of DMSO in crystallization drops did not exceed 10%. Crystals were prepared by the hanging-drop vapor-diffusion method at 18 °C using EasyXtal® 15-well plates (Qiagen). Drops containing 2 μl of the complex solution were mixed with 1 μl of precipitant solution. Subsequently, these mixtures were equilibrated over a reservoir containing 1ml of the precipitant solution. The precipitation solution consisted of 1.6M sodium citrate and 50mM tris–H2SO4, pH 7.8. Crystals formed within one to three weeks. Prior to data collection, the crystals were soaked for 10s in the reservoir solution supplemented with 20% (v/v) sucrose and stored in liquid nitrogen. X-ray diffraction data at 100K were collected on BL14.1 and BL14.2, operated by the Helmholtz-Zentrum Berlin (HZB) at the BESSY II electron storage ring (Berlin-Adlershof, Germany).20 Diffraction data were processed using the XDS suite of programs.21,22 Crystal parameters and data-collection statistics are summarized in the ESI† (Table S1).
mg ml−1 protein solution in 50mM Tris–H2SO4, pH 7.8. The final concentration of DMSO in crystallization drops did not exceed 10%. Crystals were prepared by the hanging-drop vapor-diffusion method at 18 °C using EasyXtal® 15-well plates (Qiagen). Drops containing 2 μl of the complex solution were mixed with 1 μl of precipitant solution. Subsequently, these mixtures were equilibrated over a reservoir containing 1ml of the precipitant solution. The precipitation solution consisted of 1.6M sodium citrate and 50mM tris–H2SO4, pH 7.8. Crystals formed within one to three weeks. Prior to data collection, the crystals were soaked for 10s in the reservoir solution supplemented with 20% (v/v) sucrose and stored in liquid nitrogen. X-ray diffraction data at 100K were collected on BL14.1 and BL14.2, operated by the Helmholtz-Zentrum Berlin (HZB) at the BESSY II electron storage ring (Berlin-Adlershof, Germany).20 Diffraction data were processed using the XDS suite of programs.21,22 Crystal parameters and data-collection statistics are summarized in the ESI† (Table S1).
The crystal structures of the CA II complexes with compounds 2 and 3 were determined by the difference Fourier technique. Coordinates from the 4Z1N entry in the PDB23 were used as models for the CA II complexes. Atomic coordinates of inhibitor molecules were generated by quantum-mechanical (QM) optimizations in the Turbomole package24 by means of the density functional theory (DFT) method augmented with empirical dispersion correction25 using the B-LYP functional and the SVP basis set. The geometric library for the compounds was generated using the Libcheck program.26 The Coot program27 was used for inhibitor fitting, model rebuilding, and the addition of water molecules. Refinement was carried out with the Refmac5 program28 with 5% of the reflections reserved for cross-validation. The structures were first refined with isotropic atomic-displacement parameters (ADPs). After the addition of solvent atoms and zinc ions, building inhibitor molecules in the active site, and several alternate conformations for a number of residues, anisotropic ADPs were refined for nearly all atoms. The refinement of ADPs was not carried out for spatially overlapping atoms in segments with alternate conformations or for oxygen atoms of water molecules with an unrealistic ratio of ellipsoid axes. The quality of the crystallographic model was assessed with MolProbity.29 The final refinement statistics are summarized in the ESI† (Table S1). All the figures representing structures were created using PyMOL.30 Contacts were analyzed using the Contact program included in the CCP4 suite.31 Structure factors and coordinates were deposited in the PDB under the accession codes 8AA6 and 8AAE for the CAII complex with 2 and 3, respectively.
Molecular modeling
The alignment of the three crystal structures of 1, 2 and 3 bound to CA II showed high similarity between the structures. The only larger difference concerned the Phe131 residue. To minimize the effects of the minor differences in the structures, we selected a single protein structure (the 1/CA II complex, PDB code 6YZT17), into which the other two inhibitors and Phe113 were seeded based on the alignment. All the rotamers of the carborane cages of 1 and 2 (including the two orientations of 1) were modeled because B–H and C–H groups are difficult to distinguish by X-ray crystallography.32 The most stable conformations obtained from QM/MM optimization (see below) were further used for scoring. Both alternative orientations were considered for compounds 1 and 3. The hydrogen atoms of the protein were added by the Reduce and Leap programs in AMBER 14,33 with individual protonation of all the histidines assigned on the basis of the visual inspection of their surroundings. The H atoms of the inhibitors were added manually. The sulfonamide moiety binds to the Zn2+ of CA II and is thus considered in a deprotonated NH− form.34 The ff14SB force field35 was used for the protein. A general AMBER force field (GAFF)36 for the organic parts of the inhibitor was combined with the tested boron parameters from the universal force field (UFF) for the ligand.6 Hydrogen atoms were relaxed by annealing from 900 K to 0 K at the molecular mechanics (MM) level in AMBER 14. The cooling runs using a Berendsen thermostat were 4 ps long with a 1 fs step. The complexes were optimized employing the hybrid QM/MM methodology and mechanical embedding. The QM part comprised His94, His96, His119, Phe131, Thr198, Zn2+, and the inhibitor. The QM part was treated at the DFT-D3 level using B3LYP/TZVPP.25 The rest of the system was treated at the MM level by AMBER. The environment was described by the generalized Born (IGB7) implicit solvent model.33 The coupling between QM and MM was done by Cuby437 which uses Turbomole 7.024 for QM and AMBER 1433 for MM. Residues farther than 5 Å from the inhibitor were frozen during the optimization.The score was computed as the sum of the gas-phase interaction energy (ΔEint), the interaction solvation free energy (ΔΔGsolv), and the change of the conformational “free” energy of the ligand and protein  .38 ΔEint was computed using the QM/MM methodology described above. For solvation free-energy calculations, the implicit solvent IGB7 model was utilized.
.38 ΔEint was computed using the QM/MM methodology described above. For solvation free-energy calculations, the implicit solvent IGB7 model was utilized.  was computed as the “free”-energy change between the monomer taken from the optimized protein–ligand complex and the monomer structure optimized in solution by the same method. The “free” energy (G′) was defined as the sum of the gas-phase energy (E) and the solvation free energy (G).
was computed as the “free”-energy change between the monomer taken from the optimized protein–ligand complex and the monomer structure optimized in solution by the same method. The “free” energy (G′) was defined as the sum of the gas-phase energy (E) and the solvation free energy (G).
The ΔE values of the selected motif were decomposed using the symmetry-adapted perturbation-theory (SAPT) methodology.39 The simplest truncation of SAPT (SAPT0) decomposition was performed with the recommended jun-cc-pVDZ basis set using the PSI4 program.40
Synthesis
For a synthesis using two different methods and detailed characterization of compounds 1, 2 and 3 see previous articles.18,19Results and discussion
X-ray crystal structures
To obtain structural information on the interaction of the other studied compounds with the CA II active site, the crystal structures of CA II in complex with 2 and 3 were determined in this work at a high resolution of 1.09 Å and 1.15 Å, respectively (Table S1, ESI†). Unlike compound 1 (Fig. 2(A)), compound 2 was modeled as a single conformation with full occupancy (Fig. 2(B)). Compound 3 was modeled in two alternative orientations within the CA II active site: conformations A and B with the occupancy of 0.7 and 0.3, respectively (Fig. 2(C)).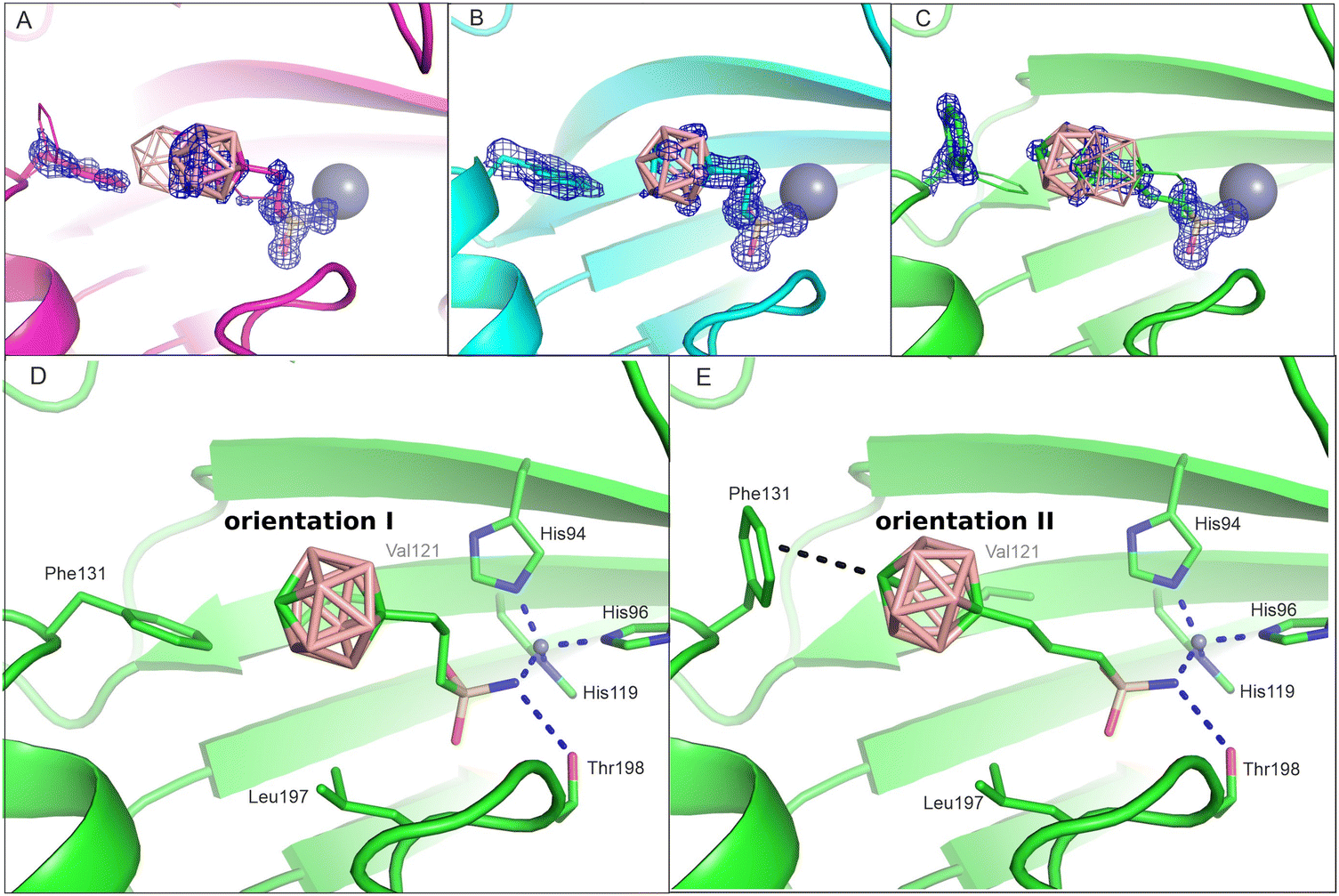 | ||
| Fig. 2 The active site of CA II with the bound compounds 1 (panel A), 2 (B) and 3 (C). The inhibitor residue Phe131 occupies two alternative conformations. Major populations are depicted in stick representation, whereas minor conformations are shown in line representation. The 2Fo-Fc map is contoured on 1.2 sigma in blue (for OMIT difference maps see Fig. S1, ESI†). On this level, electron density is shown for the major conformation of the ligand and the side chain of Phe131. Panels D and E depict two inhibitor orientations in CA II active sites: orientations I and II exemplified by compound 3. Interacting residues are highlighted as sticks and labeled. The dashed lines have the same meaning as in Fig. 1. | ||
A comparison of the crystal structures of all the three isomers (Fig. 2(A)–(C)) shows that the interaction of the sulfonamide head moiety with the catalytic zinc ion and residues at the bottom of the active-site cavity is conserved, with differences being located in the position of the carborane cage. For ligands 1 and 3, we can identify two possible ligand orientations: orientations I and II (Fig. 2(D) and (E)). Orientation I is characterized by the staggered position of the S–C and S=O bonds in the –CH2–CH2–SO2–NH− moiety, which enables the binding of the carborane cage to a preformed hydrophobic pocket formed by residues Val121 and Phe131. This orientation is more prevalent in our crystal structures and can be found in all three structures as: (a) the major conformation A of compound 1, (b) a single conformation of 2, and (c) the minor conformation B of compound 3. Orientation II has an extended propyl linker (eclipsed position of the S–C and S![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bonds in the –CH2–CH2–SO2–NH− moiety) and is only possible when the Phe131 side chain changes its rotamer position such that the phenyl ring is more exposed to the solvent (Fig. 2). This orientation occurs as: (a) the minor conformation B of compound 1 and (b) the major conformation A of compound 3.
O bonds in the –CH2–CH2–SO2–NH− moiety) and is only possible when the Phe131 side chain changes its rotamer position such that the phenyl ring is more exposed to the solvent (Fig. 2). This orientation occurs as: (a) the minor conformation B of compound 1 and (b) the major conformation A of compound 3.
Molecular modeling
The binding of the studied inhibitors to CA II has been examined by quantum mechanics-based scoring function,38,41,42 which was shown previously to describe reliably the CA II metalloenzyme-carborane system.34 Ranking was performed using the most stable (see Tables S2 and S3, ESI†) of the five possible rotamers for each orientation I or II of inhibitors 1 and 2 bound to CA II. Considering the more favorable scores of the two orientations of 1 and 3 (Table 1 in bold), the obtained ranking of the compounds (the scores of −10.6 > −12.0 > −13.3 kcal mol−1 for compounds 1, 2 and 3, respectively) matched the order of experimental binding free energies (the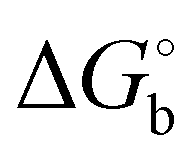 of −8.5, −9.0 and −9.2 kcal mol−1, respectively, Table 1). It should be noted here that entropic effects, coming for example from two possible conformations of the propyl linker of bound 1 and 3, which are not treated in the present version of the scoring function, could add a higher penalty to the score of 2 with respect to those of 1 and 3. Compound 1, the weakest-binding inhibitor, had the least favorable score because of the least favorable interactions with the CA II active site as reflected in the least negative ΔEint value of −194.5 kcal mol−1 (Table 1). The score of 3 was the most favorable due to the strongest interactions with the protein (the most negative ΔEint of −203.6 kcal mol−1). Considering the conserved positions of the sulfonamide head moiety, the catalytic zinc ion and the residues of the active-site cavity in the modeled structures, the only difference was in the position of the C–H vertex of the carborane cage. The C–H vertex of 3 was oriented toward Phe131, thus replacing the B–H···π interaction of the 1:CA II complex with the C–H⋯π interaction, which implies that the latter is stronger. The stronger interaction (ΔEint) is only partly offset (163.3 vs. 169.8 kcal mol−1) by solvation (ΔΔGsolv; Table 1).
of −8.5, −9.0 and −9.2 kcal mol−1, respectively, Table 1). It should be noted here that entropic effects, coming for example from two possible conformations of the propyl linker of bound 1 and 3, which are not treated in the present version of the scoring function, could add a higher penalty to the score of 2 with respect to those of 1 and 3. Compound 1, the weakest-binding inhibitor, had the least favorable score because of the least favorable interactions with the CA II active site as reflected in the least negative ΔEint value of −194.5 kcal mol−1 (Table 1). The score of 3 was the most favorable due to the strongest interactions with the protein (the most negative ΔEint of −203.6 kcal mol−1). Considering the conserved positions of the sulfonamide head moiety, the catalytic zinc ion and the residues of the active-site cavity in the modeled structures, the only difference was in the position of the C–H vertex of the carborane cage. The C–H vertex of 3 was oriented toward Phe131, thus replacing the B–H···π interaction of the 1:CA II complex with the C–H⋯π interaction, which implies that the latter is stronger. The stronger interaction (ΔEint) is only partly offset (163.3 vs. 169.8 kcal mol−1) by solvation (ΔΔGsolv; Table 1).
 ) and scores. The total score was computed as the sum of the following terms: the interaction energy (ΔEint), the interaction solvation free energy (ΔΔGsolv), and the change of the conformational ‘free’ energy
) and scores. The total score was computed as the sum of the following terms: the interaction energy (ΔEint), the interaction solvation free energy (ΔΔGsolv), and the change of the conformational ‘free’ energy 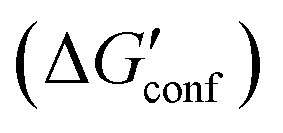 of the ligand (L) and protein (P). The more favorable scores of the two orientations of 1 and 3 are in bold. All energies are in kcal mol−1 and Ki in nM
of the ligand (L) and protein (P). The more favorable scores of the two orientations of 1 and 3 are in bold. All energies are in kcal mol−1 and Ki in nM
| Complex | Orientation | Occupancy in crystal | K i |
|
Score | ΔEint | ΔΔGsolv |
|
|
|---|---|---|---|---|---|---|---|---|---|
a As determined in ref. 15.
b
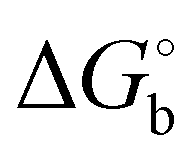 is derived from Ki values using is derived from Ki values using  . .
|
|||||||||
| 1:CA II | I | 0.8 | 622 ± 87 | −8.47 | −8.7 | −190.1 | 168.0 | 5.6 | 7.8 |
| II | 0.2 | −10.6 | −194.5 | 163.3 | 7.4 | 13.4 | |||
| 2:CA II | I | 1.0 | 254 ± 23 | −9.01 | −12.0 | −197.7 | 171.5 | 6.7 | 7.5 |
| 3:CA II | I | 0.3 | 171 ± 17 | −9.24 | −10.1 | −196.6 | 171.8 | 6.9 | 7.8 |
| II | 0.7 | −13.3 | −203.6 | 169.8 | 7.2 | 13.3 | |||



Compound 2 only had a single conformation in orientation I in the crystal structure. In such an arrangement, the C–H vertex of 2 still interacted with the π ring of Phe131, albeit in a less favourable tilted arrangement (as opposed to an ideal perpendicular arrangement). In contrast to other inhibitors, orientation I of compound 2 enabled the formation of C–Hδ+⋯δ−H–B dihydrogen bonds (Fig. 3), a typical non-covalent interaction of heteroboranes with proteins.3
 | ||
| Fig. 3 The models of the active site of CA II with the bound compounds 1 (A), 2 (B) and 3 (C) in orientation I (crystal structures of orientations II are shown in Fig. 2). B–H⋯π and C–H···π are shown as dot-dashed lines. Hydrogen contacts are shown as dashed lines. Hydrogen separations are in Å. | ||
The applied scoring function did not rank correctly the orientations of 1. The orientation II with a minor occupancy of 0.2 had erroneously a more favorable score by 1.9 kcal mol−1, which was due to more favorable interaction (ΔEint) and desolvation (ΔΔGsolv) terms. This may be caused by inherent limitations of the methodology used, such as the lack of explicit solvation or dynamical treatment and entropy effects.
To gain an even deeper insight into the nature of the B–H⋯π and C–H⋯π interactions evidenced in CA II complexes with 1 and 3, we further dissected the gas-phase interaction energy ΔE into individual terms using the SAPT methodology on small models derived from the crystal structures.39 The results showed favorable interactions of the carborane cages of both inhibitors with the side chain of Phe131 but more so for C–H⋯π (−3 kcal mol−1) than for B–H⋯π (−1.6 kcal mol−1) (Table 2). The leading term was dispersion (Edisp) as expected for weak hydrogen bonding.43 It was more pronounced for B–H⋯π than for C–H⋯π (74 and 66% of the sum of all attractive terms, respectively). The second largest attractive term was electrostatics (Eelec), which was more important for C–H⋯π than for B–H⋯π (26 and 17% of the sum of all attractive terms, respectively). It is worth mentioning that the Eelec for B–H⋯π is attractive, which has confirmed the amphiphilic noncovalent bonding character of the B–H group.9,44 In both cases, the induction term (Eind) was of little importance (below 10% of the sum of all attractive terms). The exchange-repulsive term (Eexch) was more repulsive for B–H⋯π (3.1 kcal mol−1) than for C–H⋯π (1.7 kcal mol−1). The larger repulsion of B–H⋯π might be due to the smaller spatial requirement of the acidic Hδ+ atom of the C–H group in comparison with the hydridic Hδ− atom of the B–H group. Moreover, the C atom is also smaller than the B atom (van der Waals radius of C being smaller by 0.22 Å45 than that of B), which overall reduces the spatial requirement of the entire C–H vertex.
| Model |
|
|
|---|---|---|
| Interaction | C–H⋯π | B–H⋯π |
| E disp | −3.1 (66%) | −3.5 (74%) |
| E elec | −1.2 (26%) | −0.8 (17%) |
| E ind | −0.4 (8%) | −0.4 (9%) |
| E exch | 1.7 | 3.1 |
| Total | −3.0 | −1.6 |

Conclusions
B–H⋯π and C–H⋯π non-covalent interactions have been examined in a series of carborane propyl-sulfonamide inhibitors binding to CA II using a combination of X-ray crystallography and quantum-chemical calculations. Based on the X-ray structures, the interactions of the sulfonamide moiety were conserved, while the carborane cages had different orientations and interactions. In the case of the ortho-carborane moiety (compound 1), the B–H⋯π interaction with Phe131 was minor and the affinity was the lowest in the series. In contrast, for para-carborane (compound 3), the C–H⋯π interaction with Phe131 was the major conformation, with the highest affinity attained. The difference in the affinities was a result of the difference in the strength of these two interactions as determined by quantum-mechanical calculations. A detailed understanding of B–H⋯π and C–H⋯π non-covalent interactions will be used for the future rational control over protein–ligand binding.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
This work was supported by the project ChemBioDrug CZ.02.1.01/0.0/0.0/16_019/0000729 from the European Regional Development Fund (OP RDE) and the project 2114409S from the Czech Science Foundation under a grant awarded to B. G and by project National Institute for Cancer Research (Programme EXCELES, ID Project No. LX22NPO5102) – Funded by the European Union – Next Generation EU.References
- G. R. Desiraju and T. Steiner, The weak hydrogen bond in structural chemistry and biology, Oxford University Press, Oxford, 2006, ISBN 9780198509707 Search PubMed.
- L. R. Raston and W. V. Cave, Chem. – Eur. J., 2004, 10, 279–282 CrossRef PubMed.
- J. Fanfrlík, M. Lepšík, D. Hořínek, Z. Havlas and P. Hobza, ChemPhysChem, 2006, 7, 1100–1105 CrossRef PubMed.
- N. V. Belkova, E. S. Shubina and L. M. Epstein, Acc. Chem. Res., 2005, 38, 624–631 CrossRef CAS PubMed.
- R. Custelcean and J. E. Jackson, Chem. Rev., 2001, 101, 1963–1980 CrossRef CAS PubMed.
- J. Fanfrlík, A. Pecina, J. Řezáč, M. Lepšík, M. B. Sárosi, D. Hnyk and P. Hobza, ChemPhysChem, 2020, 21, 2599–2604 CrossRef PubMed.
- J. G. Planas, C. Viñas, F. Teixidor, A. Comas-Vives, G. Ujaque, A. Lledós, M. E. Light and M. B. Hursthouse, J. Am. Chem. Soc., 2005, 127, 15976–15982 CrossRef CAS PubMed.
- J. Grunenberg, Chem. – Eur. J., 2016, 22, 18678 CrossRef CAS PubMed.
- J. Fanfrlík, A. Pecina, J. Řezáč, R. Sedlak, D. Hnyk, M. Lepšík and P. Hobza, Phys. Chem. Chem. Phys., 2017, 19, 18194–18200 RSC.
- X. Zhang, H. Dai, H. Yan, W. Zou and D. Cremer, J. Am. Chem. Soc., 2016, 138, 4334–4337 CrossRef CAS PubMed.
- S. Pastorekova and R. J. Gillies, Cancer Metastasis Rev., 2019, 38, 65–77 CrossRef CAS PubMed.
- S. Zamanova, A. M. Shabana, U. K. Mondal and M. A. Ilies, Expert Opin. Ther. Pat., 2008, 29, 509–533 CrossRef PubMed.
- C. B. Mishra, M. Tiwari and C. T. Supuran, Med. Res. Rev., 2020, 40, 2485–2565 CrossRef CAS PubMed.
- V. M. Krishnamurthy, G. K. Kaufman, A. R. Urbach, I. Gitlin, K. L. Gudiksen, D. B. Weibel and G. M. Whitesides, Chem. Rev., 2008, 108, 946–1051 CrossRef CAS PubMed.
- M. Kugler, J. Nekvinda, J. Holub, S. E. Anwar, V. Das, V. Šícha, K. Pospíšilová, M. Fábry, V. Král and J. Brynda, et al. , ChemBioChem, 2021, 22, 2741–2761 CrossRef CAS PubMed.
- J. Brynda, P. Máder, V. Šícha, M. Fábry, K. Poncová, M. Bakardiev, B. Grüner, P. Cígler and P. Řezáčová, Angew. Chem., Int. Ed., 2013, 52, 13760–13763 CrossRef CAS PubMed.
- M. Kugler, J. Holub, J. Brynda, K. Pospisilova, S. E. Anwar, D. Bavol, M. Havranek, V. Kral, M. Fabry, B. Gruner and P. Rezacova, J. Enzyme Inhib. Med. Chem., 2020, 35, 1800–1810 CrossRef CAS PubMed.
- J. Dvořanová, M. Kugler, J. Holub, V. Šícha, V. Das, J. Nekvinda, S. E. Anwar, M. Havránek, K. Pospíšilová and M. Fábry, et al. , Eur. J. Med. Chem., 2020, 200, 112460 CrossRef PubMed.
- J. Nekvinda, M. Kugler, J. Holub, S. E. Anwar, J. Brynda, K. Pospíšilová, Z. Růžičková, P. Řezáčová and B. Grüner, Chem. – Eur. J., 2020, 26, 16541–16553 CrossRef CAS PubMed.
- U. Mueller, R. Forster, M. Hellmig, F. U. Huschmann, A. Kastner, P. Malecki, S. Puhringer, M. Rower, K. Sparta and M. Steffien, et al. , Eur. Phys. J. Plus, 2015, 130, 141 CrossRef.
- W. Kabsch, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2010, 66, 125–132 CrossRef CAS PubMed.
- W. Kabsch, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2010, 66, 133–144 CrossRef CAS PubMed.
- M. R. Buemi, L. De Luca, S. Ferro, E. Bruno, M. Ceruso, C. T. Supuran, K. Pospisilova, J. Brynda, P. Rezacova and R. Gitto, Eur. J. Med. Chem., 2015, 102, 223–232 CrossRef CAS PubMed.
- R. Ahlrichs, M. Bar, M. Haser, H. Horn and C. Kolmel, Chem. Phys. Lett., 1989, 162, 165–169 CrossRef CAS.
- J. Hostas and J. Rezac, J. Chem. Theory Comput., 2017, 13, 3575 CrossRef CAS PubMed.
- A. A. Vagin, G. N. Murshudov and B. V. Strokopytov, J. Appl. Crystallogr., 1998, 31, 98–102 CrossRef CAS.
- P. Emsley and K. Cowtan, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2004, 60, 2126–2132 CrossRef PubMed.
- G. N. Murshudov, A. A. Vagin and E. J. Dodson, Acta Crystallogr., Sect. D: Biol. Crystallogr., 1997, 53, 240–255 CrossRef CAS PubMed.
- V. B. Chen, W. B. Arendall, J. J. Headd 3rd, A. D. Keedy, R. M. Immormino, G. J. Kapral, L. W. Murray, J. S. Richardson and D. C. Richardson, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2010, 66, 12–21 CrossRef CAS PubMed.
- W. DeLano, 2020 PyMOL. Retrieved from https://www.pymol.org/pymol or The PyMOL Molecular Graphics System, Version 2.0 Schrödinger, LLC.
- M. D. Winn, C. C. Ballard, K. D. Cowtan, E. J. Dodson, P. Emsley, P. R. Evans, R. M. Keegan, E. B. Krissinel, A. G. Leslie and A. McCoy, et al. , Acta Crystallogr.; Sect. D: Biol. Crystallogr, 2011, 67, 235–242 CrossRef CAS PubMed.
- A. Welch, Crystals, 2017, 7, 234 CrossRef.
- D. A. Case, V. Babin, J. T. Berryman, R. M. Betz, Q. Cai, D. S. Cerutti, T. E. Cheatham, T. A. Darden, R. E. Duke and H. Gohlke, et al., AMBER 14, University of California, San Francisco, San Francisco (USA), 2014 Search PubMed.
- A. Pecina, M. Lepšík, J. Řezáč, J. Brynda, P. Mader, P. Řezáčová, P. Hobza and J. Fanfrlík, J. Phys. Chem. B, 2013, 117, 16096–16104 CrossRef CAS PubMed.
- J. A. Maier, C. Martinez, K. Kasavajhala, L. Wickstrom, K. E. Hauser and C. Simmerling, J. Chem. Theory Comput., 2015, 11, 3696–3713 CrossRef CAS PubMed.
- J. Wang, R. M. Wolf, J. W. Caldwell, P. A. Kollman and D. A. Case, J. Comput. Chem., 2004, 25, 1157–1174 CrossRef CAS PubMed.
- J. Řezáč, J. Comput. Chem., 2016, 37(13), 1230–1237 CrossRef PubMed.
- A. Pecina, S. M. Eyrilmez, C. Köprülüoğlu, V. M. Miriyala, M. Lepšík, J. Fanfrlík, J. Řezáč and P. Hobza, ChemPlusChem, 2020, 85, 2362–2371 CrossRef CAS PubMed.
- B. Jeziorski, R. Moszynski and K. Szalewicz, Chem. Rev., 1994, 94, 1887–1930 CrossRef CAS.
- J. M. Turney, A. C. Simmonett, R. M. Parrish, E. G. Hohenstein, F. Evangelista, J. T. Fermann, B. J. Mintz, L. A. Burns, J. J. Wilke and M. L. Abrams, et al. , Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2012, 2, 556–565 CAS.
- M. Lepšík, J. Řezáč, M. Kolář, P. Pecina, P. Hobza and J. Fanfrlík, ChemPlusChem, 2013, 78, 921–931 CrossRef PubMed.
- J. Fanfrlík, A. K. Bronowska, J. Řezáč, O. Přenosil, J. Konvalinka and P. Hobza, J. Phys. Chem. B, 2010, 114, 12666–12678 CrossRef PubMed.
- M. Nishio, Phys. Chem. Chem. Phys., 2011, 13, 13873–13900 RSC.
- C. Esterhuysen, A. Hesselmann and T. Clark, ChemPhysChem, 2017, 18, 772–784 CrossRef CAS PubMed.
- M. Mantina, A. C. Chamberlin, R. Valero, Ch. J. Cramer and D. G. Truhlar, J. Phys. Chem. A, 2009, 113, 5806–5812 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2cp04673c |
| ‡ These authors contributed equally to this work. The manuscript was written through contributions of all authors. |
| This journal is © the Owner Societies 2023 |