
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceInsight at the atomic scale of corrosion inhibition: DFT study of 8-hydroxyquinoline on oxidized aluminum surfaces
Fatah
Chiter
 *ab,
Dominique
Costa
b,
Nadine
Pébère
a,
Philippe
Marcus
*b and
Corinne
Lacaze-Dufaure
*a
*ab,
Dominique
Costa
b,
Nadine
Pébère
a,
Philippe
Marcus
*b and
Corinne
Lacaze-Dufaure
*a
aCIRIMAT, Université de Toulouse, CNRS, INPT-ENSIACET 4, allée Emile Monso, BP 44362, 31030 Toulouse Cedex 4, France. E-mail: fatah.chiter@toulouse-inp.fr; corinne.dufaure@ensiacet.fr
bPSL University, CNRS – Chimie ParisTech, Institut de Recherche de Chimie Paris/Physical Chemistry of Surfaces Group, 11 rue Pierre et Marie Curie, 75005 Paris, France. E-mail: philippe.marcus@chimieparistech.psl.eu
First published on 5th January 2023
Abstract
8-Hydroxyquinoline (8-HQ) is a promising organic molecule for the corrosion protection of aluminum and its alloys in the replacement of chromate salts. On the aluminum surface, the presence of an oxide layer naturally formed can influence the inhibition efficiency which depends on molecule–surface interactions. In the present study, we performed quantum chemical calculations on native 8-HQ, tautomer and 8-Q (deprotonated, H-abstracted or radical) molecules, adsorbed on an oxidized aluminum surface (γ-Al2O3(111)/Al(111)). All species have the ability to interact strongly with the oxidized aluminum surface and can form stable and dense organic films. The bonding strength of different species of 8-HQ on oxidized aluminum surfaces is more favorable for 8-Q and tautomer species than for the native 8-HQ molecule. On the surface, the native 8-HQ molecule is physisorbed, forming H-bonds, in contrast to the tautomer and 8-Q species that show the predominance of chemisorption modes, involving both H-bonds and covalent bonds at the molecule/substrate interface. The dispersion energy significantly contributes to the adsorption mechanism and increases with increasing molecular surface coverage, due to attractive molecule–molecule interactions. Regardless of surface coverage and considered reaction mechanisms, the 8-Q species is able to enhance the stability of all aluminum sites, and thus to slow down the anodic reaction. In contrast, the native molecule and the tautomeric form have no significant effect or even weakened the stability of aluminum surface atoms.
1 Introduction
Aluminum and its alloys show good corrosion resistance, due to the protection conferred by the native oxide film spontaneously formed on their surface in various environments. However, this natural protection depends on the composition and thickness of the oxide film. Thin oxide films can be damaged or destroyed in contact with aggressive medium containing chlorides ions or in acidic solution, which can weaken the corrosion resistance of the aluminum substrate. One of the traditional solutions to mitigate the corrosion is the use of chromate salts. However, their use in industrial applications is now strongly restricted due to the high toxicity and carcinogenic risks of these compounds.Many experimental and theoretical works have been devoted to evaluate and understand the corrosion inhibition efficiency of organic molecules as alternatives to replace chromate salts under various conditions.1–7 It is well-known that 8-hydroxyquinoline (8-HQ) molecules have a strong affinity with metals, forming chelates via their O and N atoms in solution and organic films on metals. They are also used for analytical chemistry, electronic, and photoelectronic applications and emergency technologies.8–15 Therefore, 8-HQ is also considered as a promising candidate to inhibit the corrosion of aluminum and its alloys. Previous works on aluminum alloys16,17 in acidic and alkaline solutions showed that the inhibition efficiency increases with increasing organic inhibitor concentration, time contact and pH value. The effect of the protective organic film on the surfaces and its role in the corrosion inhibition mechanism were also demonstrated.17–22 8-HQ molecules strengthen the stability of the native aluminum oxide film formed on aluminum surfaces in neutral solution.23,24 The inhibitor prevents the destruction of the oxide film and limits the adsorption of aggressive chloride ions. This result was explained assuming the adsorption of the molecule on the metal surface. The molecule has the ability to prevent pitting corrosion of the aluminum in neutral23,25 and acidic23,26 solutions. 8-HQ doped sol-gel coatings also improve the corrosion resistance of 2024 aluminum alloy.27 The molecule forms highly insoluble complexes on the surface and stops the metal dissolution.25 The adsorption of 8-HQ on the metal surface leads to the formation of a protective organic layer, which blocks the active metal sites and also prevents the adsorption of chloride ions.26 However, Balaskas et al.28 compared the efficiency of 8-HQ, benzotriazole (BTAH) and 2-mercaptobenzothiazole (2-MBT) molecules as potential corrosion inhibitors and they found that 2-MBT is the most efficient inhibitor for the 2024 aluminum alloy. The synergistic effect of 8-HQ and BTAH molecules was also studied, and combining the two molecules improves the corrosion resistance of the 2024 alloy.29
Understanding of the 8-HQ-metal interactions at the atomic scale has been the subject of several works, considering only the oxide-free metal surfaces. Alq3 complexes adsorbed on metal surfaces such as Al, Cu, Mg, Co, Ag and Au have been the subject of DFT30–33 and experimental34–37 studies. The complexes are composed of one Al atom (cation) and three 8-Q molecules (deprotonated 8-HQ). The majority of these studies mainly concern the electronic structure and the creation of a dipole at the Alq3/metal interfaces. A weak interaction was found for Alq3/Cu,35,36 Alq3/Au35 and Alq3/Co37 systems, attributed to a strong anisotropy of intermolecular interactions and physisorbed molecules. Bulteau et al.38 studied at the atomic scale (DFT calculations) the adsorption of Alq3 complexes on the Al(111) surface and sequential adsorption of the dehydrogenated 8-HQ molecules, until the formation of Alq3 complexes on the surface. The formation of Alq3 complexes from free dehydrogenated molecules on the Al(111) surface leads to more stable structures than the direct adsorption of Alq3 on the Al(111) surface. Combining density functional theory (DFT) and non-contact atomic force microscopy (nc-AFM) methods, Zhang et al.39 interpreted the intermolecular interaction of self-assembled 8-HQ molecules on the Cu(111) surface under AFM conditions. The DFT calculations were focused on low density selected ‘flat-lying’ 8-HQ adsorption configurations assembled on the substrate surface, in relation with AFM analysis.
In our previous studies,40,41 we investigated the adsorption characteristics of the 8-HQ molecule under different forms on the oxide-free Al(111) surface for different surface coverages. These features are assumed to determine the inhibitor efficiency of the molecule. Regardless of the nature of the species, the molecule could be chemisorbed on the Al(111) bare surface, with the strongest adsorption of 8-Q H-abstracted species. The molecules interact with the Al surface atoms via covalent bonds, involving O, N and C atoms. The results revealed a significant contribution of dispersion energy to the adsorption mechanism. However the value depends on the adsorption species and surface coverage. Globally, upon increasing the surface coverage, the dispersion energy decreased at the molecule/Al(111) interface and increased at the molecule/molecule in the organic film. The protective property of the organic films on the Al substrate was studied,41 considering the molecular oxygen (O2) reduction on the partially and completely covered Al surface, which is one of the elementary reactions occurring during the corrosion process of aluminum. The results indicate that O2 dissociation can occur on uncovered areas, whereas the reduction reaction does not occur on the Al(111) surface covered by a compact organic film. A study of the influence of the chemical modification (substitution of H atom by Br and SO3− group) on the reactivity of the 8-HQ molecule with Al cations for the formation of chelates and their interaction with an Al(111) surface was performed combining DFT and MD calculations.42 The bonding of the different species with Al3+ is characterized as ionic with a weak covalent character. Moreover, all species showed a strong adsorption on the Al(111) surface, driven by O and N atoms present in the native form, in addition to the O atoms of the SO3− group. Recently, You and Liu43 performed a density functional theory based molecular dynamic (DFT-MD) simulations to investigate the interaction mechanism of 8-HQ molecules on the Al2Cu alloy surface and also to address the molecular mechanism oxidation of pristine and 8-HQ coated alloy surfaces. The authors showed that under ambient conditions, O2 molecules interact strongly with Al sites on the pristine surface alloy and this interaction is strengthened by the presence of Cu. In contrast, the oxidation rate decreases on 8-HQ coated alloy surfaces and it is more pronounced at higher coverages.
All these results are relevant to understand the reactivity of the 8-HQ molecule and its complexes toward the bare Al(111) and Al2Cu surfaces. They described the surface chemistry of the inhibitor-surface bonding and the formation of organo-metallic and organic films. However, the surface states of the metal have a significant impact on the inhibition efficiency of the molecules,44–48 and taking into account the native oxide film in the simulation is missing to clarify the 8-HQ/aluminum interactions in neutral and alkaline reacting phases. Therefore, in order to gain deeper insights into molecules/Al surface chemistry, including the role of the surface oxide, we performed quantum chemical DFT calculations on the interaction mechanism of 8-HQ molecules on an oxidized aluminum surface (γ-Al2O3(111)/Al(111)), i.e. the Al(111) surface covered by an ultrathin hydroxylated oxide film. The adsorption energy and the contribution of the dispersion forces to the adsorption mechanism and the adsorption configurations are systematically investigated from low to saturation molecular surface coverage. In addition, the electronic property analysis evidences the types of chemical bonds at the molecule–surface interface. The vacancy formation energies are calculated to estimate the corrosion inhibition power of the 8-HQ molecule on an oxidized Al surface.
2 Computational details
All calculations were performed in the framework of DFT with the periodic plane-wave basis set code VASP (Vienna Ab initio Simulation Package).49–52 All results were obtained with projector-augmented-wave potentials using a 450 eV plane-wave cut-off.53,54 The Generalized Gradient Approximation (GGA) of the Perdew–Burke–Ernzehof (PBE) functional55,56 was used for the exchange-correlation term. We used Methfessel–Paxton smearing57 with a smearing value of 0.1 eV. To describe the lateral molecule–molecule interactions, which play an important role in full surface coverage, we used the empirical dispersion correction of Grimme, known as PBE-D2.58 However, the PBE-D2 approach describes adequately the lateral intermolecular interactions, but overestimates the molecule oxide interactions. To avoid the latter term, we set the C6 coefficient of Al to zero (CAl6 = 0), because in the oxide the Al atoms are in a +3 oxidation state.48 These calculations are called PBE-D0 hereafter. The results obtained using this scheme were used to calculate the overestimation of the adsorption energies calculated by PBE-D2. Atomic positions were relaxed with the conjugate gradient (CG) algorithm until forces on each moving atom were less than 0.02 eV Å−1. All atoms were free to relax, except the two bottom Al(111) metal layers that were kept fixed at the calculated lattice parameter of Al which is 4.06 Å, in good agreement with the experimental value of 4.05 Å.59 The dimensions of the unit cell used in calculations were 9.90, 8.57 and 36.53 Å in the x, y and z directions, respectively, and it corresponds to the supercell of an Al(111) metal surface, and the vacuum region was set to more than 18 Å, to minimize the interactions in the z direction. Hence the Brillouin-zone sampling60 was set to the 5× 5 × 1 k-point grid.
supercell of an Al(111) metal surface, and the vacuum region was set to more than 18 Å, to minimize the interactions in the z direction. Hence the Brillouin-zone sampling60 was set to the 5× 5 × 1 k-point grid.
2.1 Oxidized aluminum surface model
The structure of the model is shown in Fig. 1. The completely oxidized aluminum surface model is taken from previous publications,47,48,61,62 where it is described in greater detail.61,62 The model consists of Al(111) metal substrate supporting an ultrathin γ-Al2O3(111) aluminum oxide film, with 5 Å thickness. The oxide surface exhibits a hydroxyl density of 14.4 OH per nm2, with 60% and 40% in μ2-OH and μ1-OH modes, respectively (Fig. 1). In μ2-OH mode, the OH group is in the bridge site between two Al surface ions, while in the μ1-OH mode, the OH group is coordinated to one Al surface ion as illustrated in Fig. 1b. The hydroxylated surface exhibits different distinct aluminum sites that can interact with adsorbates (organic inhibitors), and these sites could have different reactivity due to different chemical environments as shown in Fig. 1c. Each aluminum surface atom is bonded to four to six oxygen atoms. Some of them are coordinated to three OH group and one to three Odn atoms, where the subscripts dn indicates oxygen atoms located below (dn) the surface Al layer. Other aluminum atoms are coordinated to four OH groups and one Odn atom. | ||
| Fig. 1 Al(111) surface covered by ultrathin γ-Al2O3(111) hydroxylated oxide. Snapshots correspond to (a) side view, (b) top view, with encircled μ1-OH surface group and (c) chemical environment of the Al surface atoms. (a) Grey: Al oxide; grey hatched: Al metal. (b and c) Red: oxygen surface; red hatched: oxygen under surface; white: hydrogen. | ||
2.2 8-HQ molecules
The physical and chemical properties of the 8-hydroxyquinoline (8-HQ) molecule (Fig. 2) have been well described in the literature.63–66 In the bulk structure, 8-HQ crystallizes in the orthorhombic space group Fdd2.63 In dilute solution, according to the pH and the nature of the solvent, the molecule can exist under different species in monomeric form separated by the solvent molecules. The native 8-HQ molecule (Fig. 2a) is the predominant form in neutral solutions and can coexist with the tautomeric form (Fig. 2b) in a polar solvent.64–66 The native 8-HQ molecule is more stable than the tautomer species, with an energy difference of 0.50 eV40 in vacuum. Therefore, we investigated the interaction of these two forms with the hydroxylated oxidized aluminum surface. We also studied the adsorption of the deprotonated, H-abstracted and radical of the 8-HQ molecule denoted 8-Q (Fig. 2c), because it can be present in alkaline solutions, where the metal is passivated by a native oxide film. This was also motivated by the literature results that show the affinity of this species to interact strongly with metals.31–33,39–42 In calculations, the 8-Q is considered as a radical species without the H atom on the hydroxyl group of the native 8-HQ molecule. But we also considered the 8-Q H-abstracted or deprotonated species from the dissociation of the native 8-HQ molecule on the surface, which can lead to: (i) the release of H2 molecules or (ii) the formation of water molecules with the OH surface group. Other forms of the molecule could exist under certain conditions which are not considered here, such as an 8-HQ molecule without intramolecular H-bonding which is less stable by 0.37 eV in vacuum than the native molecule with an intramolecular H-bond. The protonated 8-HQ molecule present in acidic solution at pH < 3.866 is not studied in our case. The energy of the standalone molecules in vacuum was obtained in a simulation box with dimensions of 20 Å × 20 Å × 20 Å, using the same computational conditions as described above, except for the radical form, where the spin polarization was implemented to obtain the energy minimum. | ||
| Fig. 2 8-HQ molecule under different forms. (a) Native 8-HQ molecule, (b) tautomer species and (c) 8-Q species. | ||
2.3 Energetics
The adsorption energy of the molecules on the oxidized aluminum surface was calculated as:| Eads = [E(molecule/slab) − E(slab) − nE(molecule)]/n | (1) |
The adsorption energy for the 8-Q under mechanisms  (eqn (2)) or 8-HQ + * → 8-Qads + H2Oads (eqn (3)), and thus corresponding to H-abstracted or deprotonated species, respectively, was also calculated considering the 8-HQ native molecule as the reference energy:
(eqn (2)) or 8-HQ + * → 8-Qads + H2Oads (eqn (3)), and thus corresponding to H-abstracted or deprotonated species, respectively, was also calculated considering the 8-HQ native molecule as the reference energy:
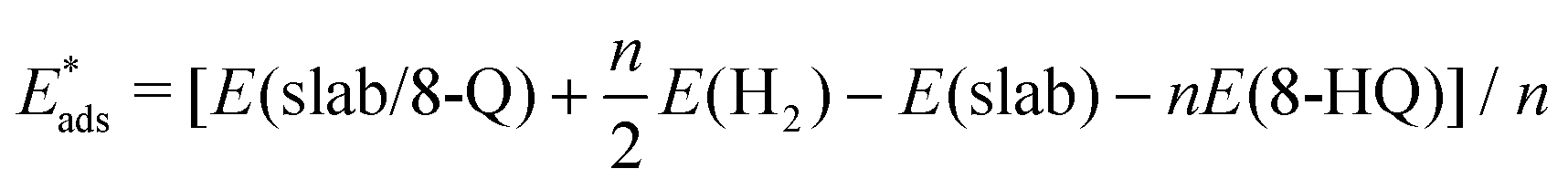 | (2) |
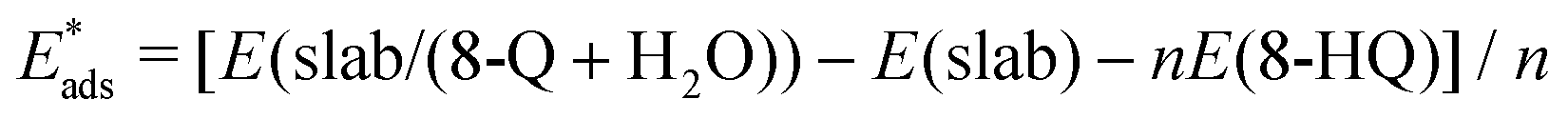 | (3) |
As the van der Waals interactions were included according to the empirical dispersion correction of Grimme (PBE-D2), we calculated its contribution to the adsorption mechanism as follows:
| Edisp = [EvdW(molecule/slab) − EvdW(slab) − nEvdW(molecule)]/n | (4) |
In order to show the ability of the 8-HQ inhibitor to slow down the corrosion of the oxidized aluminum surface, we also analyzed the defect formation energy, which corresponds to the creation of one aluminum vacancy in the hydroxylated surface without (uncoated) and with (coated) inhibitor, according to eqn (5) and (6), respectively.
| ΔEpristinedefect = Edefect(Alm−1) + E(Al3+ + 3e−) − Eperfect(Alm) | (5) |
 | (6) |
Thus, the defect formation energy difference between the coated and uncoated surfaces was calculated as:
 | (7) |
2.4 Charge density analysis
The charge density difference is expressed as follows:| Δρ(r) = ρ(r)slab/molecule − (ρ(r)slab + ρ(r)molecule) | (8) |
The electron changes were determined on each atom as:
| ΔN(x) = Nads(x) − Nvac(x) | (9) |
3 Results and discussion
In the following, the analysis of the most stable adsorption configurations obtained for a native 8-HQ molecule, tautomer and 8-Q species on models of the hydroxylated oxidized aluminum surface is presented. Several envisaged initial adsorption bonding geometries were investigated for different surface coverages. For instance, the molecule plane can stand parallel, tilted or perpendicular to the surface at low surface coverage, however, for high coverage the almost perpendicular adsorption mode is more favorable. As the hydroxylated oxidized aluminum surface contains several different adsorption sites, we studied different combinations of initial geometries for molecules at high surface coverage. Thus, we focused our analysis on the perpendicular adsorption mode, where the molecule is directed toward the surface with specific chemical groups close to the surface, i.e. OH group and N atom for the native form, O atom and NH group for the tautomer species and O and N atoms for the 8-Q species. The energy criterion trends as the adsorption energy and van der Waals energy contributions at the molecule-surface interface and between the molecules, combined to the bond distances (H-bond and covalent bonds) and electronic structure tools were utilized to distinguish between chemisorbed, weakly chemisorbed and physisorbed modes68 of the different adsorbed species on the hydroxylated oxidized aluminum surface. Consistency between all of these parameters reveals the physisorption of the native 8HQ molecule and chemisorption of the tautomer and 8-Q (radical, H-abstracted or deprotonated) species on the substrate surface.3.1 Energy trends
It is known that the corrosion inhibition efficiency of the organic molecules can be evaluated by the quantification of their adsorption energy and bonding strength on the substrate surfaces.47,48,69,70 The molecules have to be adsorbed strongly on the surface at high coverage to favor the formation of a protective organic film. This organic film first plays the role of barrier avoiding the interaction of aggressive species, such as chlorides with the substrate surface. It moreover must stop the electrochemical reactions taking place in the corrosion process. All energies based on PBE-D2 and PBE-D0 methods are compiled in Table 1 for different adsorption species, surface coverages and considered reaction mechanisms on the hydroxylated oxidized aluminum surface. We notice that the energies calculated via the PBE-D2 functional are overestimated by about 0.20 eV compared to that calculated by the PBE-D0 scheme, without any change in the adsorption mechanisms and adsorption modes. The adsorption energy calculated by eqn (1) and the contribution of the dispersion forces to the bonding, quantified by the calculation of the dispersion energy by eqn (4), for the most stable configurations of native 8-HQ, tautomeric and 8-Q (radical) species adsorbed on the hydroxylated oxidized aluminum surface at molecular densities of 1.18, 2.36 and 3.54 molecule nm−2 are reported in Fig. 3.| Coverages (molecule nm−2) | Energy (eV molecule−1) | 8-HQ | Tautomer | 8-Q | ||
|---|---|---|---|---|---|---|
| Radical | H-abstracted | Deprotonated | ||||
| 1.18 | E ads | −0.98 (−0.76) | −2.06 (−1.88) | −3.75 (−3.55) | −1.90 | −1.87 |
| E disp | −0.59 (−0.36) | −0.49 (−0.29) | −0.54 (−0.30) | |||
| 2.36 | E ads | −1.09 (−0.87) | −1.73 (−1.50) | −3.20 (−2.99) | −1.34 | −1.40 |
| E disp | −0.80 (−0.55) | −0.72 (−0.48) | −0.66 (−0.43) | |||
| 3.54 | E ads | −1.12 (−0.91) | −1.69 (−1.48) | −2.91 (−2.71) | −1.05 | −1.17 |
| E disp | −1.10 (−0.86) | −1.04 (−0.83) | −1.05 (−0.81) | |||
 | ||
| Fig. 3 PBE-D2 calculated adsorption (Eads) and dispersion (Edisp) energies per molecule for the native 8-HQ, tautomeric and 8-Q (radical) species adsorbed on the γ-Al2O3(111)/Al(111) surface at different surface coverages. | ||
The calculated adsorption energies are negative regardless of the adsorbed species and coverage of the surface, meaning that the adsorption process is exothermic. This reveals the reactivity of 8-HQ molecules to interact with the hydroxylated oxidized aluminum surface and their ability to form stable organic layers on the substrate surface. However, the values of Eads and Edisp and their evolution as a function of the surface coverage depend on the adsorbed species.
For the native 8-HQ molecule (Fig. 4), the adsorption energy based on PBE-D2, is −0.97, −1.09 and −1.12 eV molecule−1 for coverages of 1.18, 2.36 and 3.45 molecule nm−2, respectively, and it is higher with the PBE-D0 approach by 0.21 ± 0.01 eV molecule−1 (Table 1), due to cancellation of the dispersion energy contribution between the molecules and the surface. H-bonds and dispersion forces drive the adsorption of 8-HQ. For the more dense organic film, the molecules binding can be attributed to dispersion forces (Fig. 3). The contribution of the dispersion energy to the adsorption energy increases as a function of the surface coverage and it is −0.59, −0.80 and −1.10 eV molecule−1 for surface coverages of 1.18, 2.36 and 3.54 molecule nm−2, respectively, representing respectively 61%, 73% and 98% of Eads. This gain of dispersion energy comes from the cohesion energy of the organic layer, which is higher for a dense organic film due to molecule–molecule interactions. The high contribution of H-bonds and dispersion interactions, together with a moderate adsorption energy, suggest a physisorption adsorption mode. Similar results were obtained for the native 8-HQ molecule on oxide-free Al(111) metal surfaces,40,41 where it was demonstrated that the molecules were adsorbed in both physisorption (−0.90 eV molecule−1) and chemisorption (−1.11 eV molecule−1) modes.40 On the oxide-free Al(111) surface, the adsorption energy was dominated by a high contribution of vdW interactions.
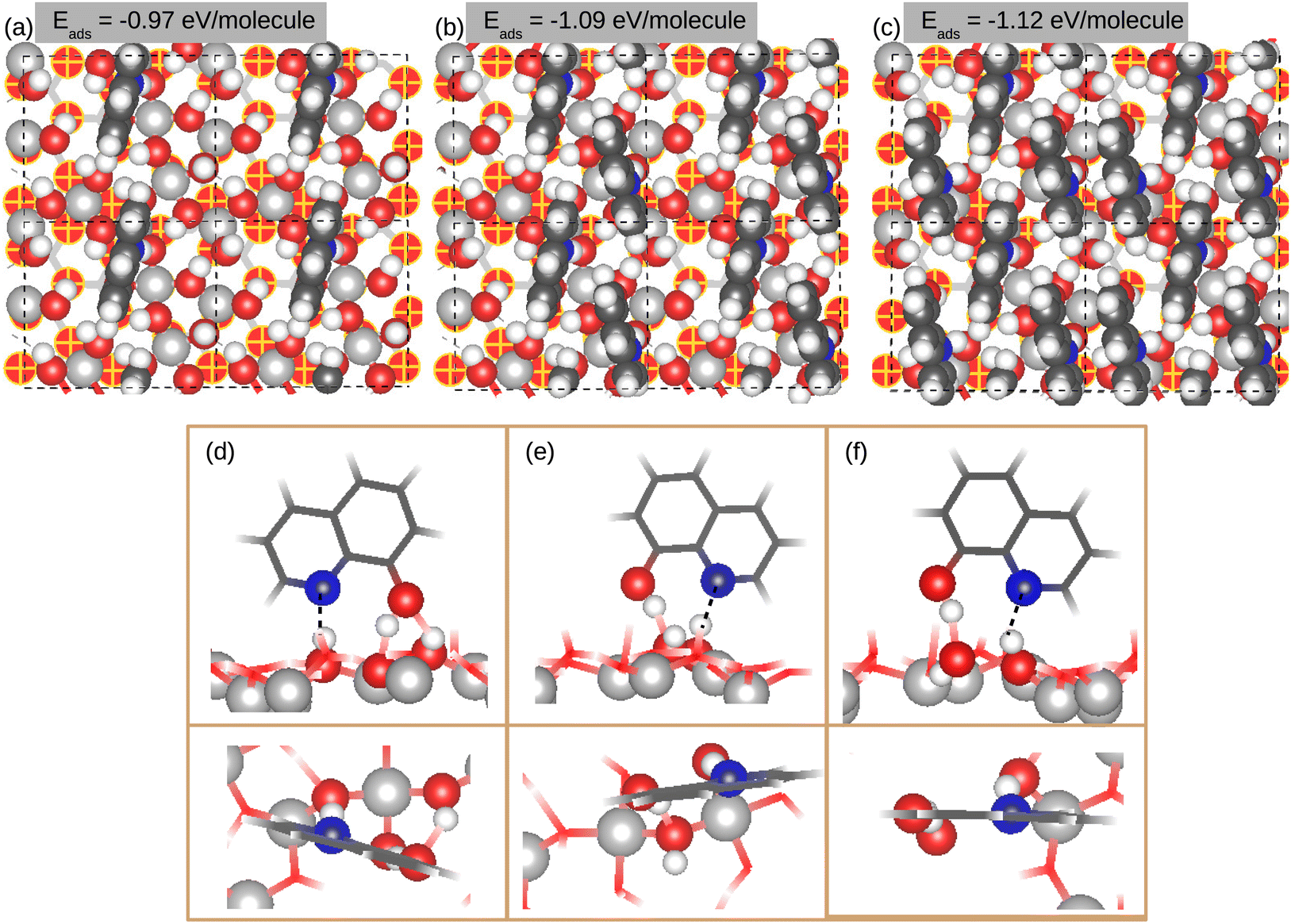 | ||
| Fig. 4 Top views of the most stable adsorption configurations of the native 8-HQ molecules on the γ-Al2O3(111)/Al(111) surface, with their adsorption energies (PBE-D2 approach). Snapshots (a–c) correspond to surface coverages of 1.18, 2.36 and 3.54 molecule nm−2, respectively. Snapshots (d–f) illustrate side and top views for the different adsorption features of the species. | ||
The adsorption energy of tautomer species is lower than that of native 8-HQ molecules whatever the surface coverage. This means that the binding of the tautomer species on the hydroxylated oxidized aluminum surface is stronger than in the case of the native form which leads to a more stable organic film on the surface. The competitive reactivity of both species is similar to what was found on oxide-free Al(111) metal surfaces.40 The reactivity of the tautomer species comes from the O atom and NH group instead of the N atom and OH group in the native 8-HQ. We calculated adsorption energies with a PBE-D2 (PBE-D0) approach of −2.06 (−1.88), −1.73 (−1.50) and −1.69 (−1.48) eV molecule−1 for surface coverages of 1.18, 2.36 and 3.54 molecule nm−2, respectively. These values of the adsorption energies are therefore a first indication of the predominance of the chemisorption of the tautomer species. At a coverage of 3.54 molecule nm−2, shown in Fig. 5(d)–(f), the molecules are less strongly bonded with a decrease of adsorption energy (in absolute value). However, the bonding of the molecule remains strong to form stable and dense organic films. The values of the dispersion energy calculated for the adsorption of tautomer species are of the same order of magnitude as those calculated for the adsorption of the native form at each coverage. However, in terms of contribution to the adsorption energies, they correspond to 23, 42 and 62% for tautomer species, due to the strong chemical bonding (61, 73, and 98% for the native form) for surface coverages of 1.18, 2.36 and 3.54 molecule nm−2, respectively.
 | ||
| Fig. 5 Top views of the most stable adsorption configurations of tautomer species on the γ-Al2O3(111)/Al(111) surface, with their adsorption energies (PBE-D2 approach). Snapshots (a)–(c) correspond to surface coverages of 1.18, 2.36 and 3.54 molecule nm−2, respectively. Snapshots (d)–(f) illustrate side and top views for the different adsorption features of the species. | ||
The 8-Q species only chemisorbs, with adsorption energies of −3.75, −3.20 and −2.91 eV molecule−1 for surface coverages of 1.18, 2.36 and 3.54 molecule nm−2, respectively, considering the direct adsorption of the species (radical 8-Q) from the reacting phase to the surface substrate (eqn (1)). These results indicate that among 8-HQ molecule species, the 8-Q species has the strongest bonding with the hydroxylated oxidized aluminum surface, and are suitable to mitigate the corrosion processes. We calculated values for the dispersion energies that contribute to the adsorption energy to about 14, 20 and 36% for surface coverages of 1.18, 2.36 and 3.54 molecule nm−2, respectively. Considering the native 8-HQ molecule as the reference energy (eqn (2) and (3)), we obtained adsorption energies values of −1.90, −1.34 and −1.05 eV molecule−1 for the 8-Q H-abstracted mechanism, and −1.87, −1.40 and −1.17 eV per molecule for 8-Q and water co-adsorption mechanism for a surface coverage of 1.18, 2.36 and 3.54 molecule nm−2, respectively. These results also indicate that whatever the reaction mechanisms, the deprotonation of the native form is favorable to obtain 8-Q species adsorbed on the surface. As for the tautomer species, we note a decrease of the bonding strength (Eads decrease in absolute value) when increasing surface coverage, due to the different reactivity of the aluminum sites, which leads to an inhomogeneity in the adsorption configurations (Fig. 6). However, the formation of a dense organic film remains an exothermic process.
 | ||
| Fig. 6 Top views of the most stable adsorption configurations of 8-Q under radical, H-abstracted and deprotonated forms on the γ-Al2O3(111)/Al(111) surface, with their adsorption energies (PBE-D2 approach for radical species). Snapshots (a–c) correspond to surface coverages of 1.18, 2.36 and 3.54 molecule nm−2, respectively. Snapshots (d–f) illustrate side and top views for the different adsorption features of the species. | ||
To summarize, the 8-HQ molecules interact strongly with the hydroxylated oxidized aluminum surface and show the ability to form dense organic layers. However, their bonding strength and adsorption mode depend on the molecule species present in the reacting phase and the reaction mechanism. The adsorption strength increases in the order native 8-HQ < tautomer species < 8-Q species. The contribution of the dispersion forces to the adsorption energy is higher for the dense organic film and increases as 8-Q species < tautomer species < native 8-HQ. The differences in molecule-surface bonds formation (covalent bonds and H-bonds) are developed in the following, according to the adsorbed species, surface atoms of the adsorption sites and surface coverage.
3.2 Adsorption configurations
The native 8-HQ molecule interacts with the hydroxylated oxidized aluminum surface by the formation of two or three H-bonds, according to the adsorption sites on the surface and surface coverage. The adsorption mechanism involves both an N atom and OH group of the molecule, and μ1-OH and μ2-OH surface groups. The OH molecule group can act as an H-bond donor and acceptor (Fig. 4(d)) or only H-bond donor (Fig. 4(e) and (f)), whereas the N atom always acts as an H-bond acceptor (Fig. 4(d)–(f)). The results reveal no hierarchy in the reactivity of OH surface groups toward the 8-HQ molecule, which is important to the formation of a dense organic layer. Indeed, both the OH group and N atom form H-bonding with μ1-OH and μ2-OH groups in Fig. 4(d), with the μ2-OH group in Fig. 4(e) and with μ1-OH in Fig. 4(f). In Fig. 4(d), three H-bonds are formed between the molecule and the OH surface groups One H-bond is between the OH group (H-donor) of the molecule and oxygen of μ1-OH surface group, with an OH–OH bond length of 1.51 Å. The second H-bond is between the oxygen of the molecule (H-acceptor) and μ2-OH surface group, with an OH–OH bond length of 2.06 Å. The third H-bond is between the N atom (H-acceptor) of the molecule and μ2-OH surface group, with an OH–N bond length of 1.78 Å. Overall, the OH–OH bond lengths for the H-bond donor is shorter than the H-bond acceptor and these bond lengths increase slightly with increasing surface coverage. In contrast, the OH–N bond length remains similar at different coverages. This adsorption configuration is common to the three surface coverages studied. In Fig. 4(e) and (f), the molecules form two H-bonds and involve μ1-OH and μ2-OH surface sites. The adsorption configuration in Fig. 4(e) is common for both 2.36 and 3.45 molecule nm−2 surface coverages, involving μ2-OH surface groups, while the configuration in Fig. 4(f) is identified for 3.45 molecule nm−2 surface coverage, involving μ1-OH surface groups. The OH–OH and OH–N bond lengths range from 1.58 to 1.88 Å, and from 1.68 to 2.05 Å, respectively.
According to the surface coverage, the tautomer species can form several bonds. Some molecules can adsorb with the formation of Al–O and Al–N bonds and H-bonding. The results reveal a mix of both chemisorption and physisorption modes of tautomer species on hydroxylated oxidized aluminum surface at higher coverage, while the chemisorption mode is preferred for low (1.18 molecule nm−2) and intermediate (2.36 molecule nm−2) coverages. Fig. 5(a) shows the most stable configuration obtained at low coverage (1.18 molecule nm−2), and the adsorption mode is displayed in Fig. 5(d). The tautomer species forms bond via an O atom with an Al site (almost top site), with an O–Al bond length of 1.75 Å. This is an indication of O-Al covalent bonding. The chemisorption of the molecule is also enhanced by the formation of H-bonding between the NH group of the molecule and OH surface group, with an NH–OH bond length of 1.94 Å.
Fig. 5(b) shows the most stable configuration of the tautomer obtained for a surface coverage of 2.36 molecule nm−2. Some molecules are adsorbed in a similar geometry (NH–OH bond length of 2.00 Å and O–Al bond length of 1.75 Å) as the one found at low coverage (Fig. 5(d)). Other molecules displayed in Fig. 5(e) also show similar adsorption behaviour on adjacent surface sites as for the first configuration. Chemisorption on the surface via O–Al chemical bond and NH–OH H-bond, with bond lengths of 1.95 and 1.89 Å, respectively, is observed.
For a higher surface coverage of 3.54 molecule nm−2, the configuration is shown in Fig. 5(c). In the organic film, some molecules are chemisorbed in similar configurations as previously described for low and intermediate surface coverages (Fig. 5(d) and (e)). Other molecules are shown in Fig. 5(f). They interact only by the formation of H-bonding (three H-bonds). The tautomer species is physisorbed (or weakly chemisorbed) via the formation of OH–O–HO H-bonds, with bond lengths of 1.62 and 1.82 Å. The NH group also forms a H-bond with a NH–OH bond length of 1.78 Å.
3.3 Charge density analysis
In order to bring out more information about the bonding strength and to confirm the type of molecule-surface bonds, we investigated the electronic properties of the molecule/surface system for different surface coverages. Analysis of the charge density difference (Δρ(r)), was performed using eqn (8) for different adsorption configurations. Fig. 7 shows the Δρ(r) plots for different adsorption snapshots of native 8-HQ, tautomeric and 8-Q species on the hydroxylated oxidized aluminum surface. For better visualization and interpretation of the results, we kept only one molecule on the surface and plotted a density isosurface of ±0.0009 e Å−3. In Fig. 7, snapshots a–c, d–f and g–i correspond to the different adsorption configurations obtained for the native 8-HQ molecule, tautomer and 8-Q species, respectively, on the oxidized aluminum surfaces. The Bader charge distribution (number of electrons in each atom) in the systems was evaluated by eqn (9) and the results are compiled in Table 2. | ||
| Fig. 7 Charge density variation for different adsorption snapshots of the most stable configurations obtained for 8-HQ molecules (native, tautomer and 8-Q species) on the γ-Al2O3(111)/Al(111) surface at different coverages. Blue and green colors correspond to the accumulation and depletion of charge density, respectively, with an isosurface of ±0.0009 e Å−2. | ||
| Adsorption species | Part of systems | Surface coverage | ||
|---|---|---|---|---|
| 1.18 | 2.36 | 3.54 | ||
| Native 8-HQ | ΔN (molecule) | within ±0.06 | ||
| ΔN (metal) | −0.34 | −0.34 | −0.04 | |
| ΔN (oxide) | +0.31 | +0.33 | +0.04 | |
| Tautomer species | ΔN (molecule) | +0.78 | +0.54 | +0.40 |
| ΔN (metal) | −0.28 | −0.05 | −0.05 | |
| ΔN (oxide) | −0.50 | −0.49 | −0.35 | |
| 8-Q species | ΔN (molecule) | +0.80 | +0.79 | +0.79 |
| ΔN (metal) | −0.07 | +1.05 | +0.63 | |
| ΔN (oxide) | −0.73 | −1.84 | −1.42 | |
For the native molecule in Fig. 7(a)–(c), the molecule-surface H-bond formation is confirmed by the charge density analysis. For all configurations, one H-bond is evidenced by charge density accumulation (blue color) on the N atom and charge density depletion (green color) on the hydrogen atom of the OH surface group. Another H-bond is evidenced by charge density depletion on the H atom of the OH of the 8-HQ and charge density accumulation on the O atom of the OH surface group.
The charge density difference for the different adsorption configurations of tautomer species adsorbed on the hydroxylated oxidized aluminum surface is shown in Fig. 7(d)–(f). The strong bonding of the molecule to the surface is confirmed by the formation of covalent and H-bonds that indicate chemisorption and physisorption (or weakly chemisorbed) modes of the species. Fig. 7(d) and (e) correspond to the chemisorption of the tautomer on the surface with O–Al covalent bonds and H-bonding between NH and OH groups. This is confirmed by the charge density accumulation (blue color) between the O atom of the molecule and an Al surface atom. H-bond is evidenced by the charge density depletion (green color) on the hydrogen atom of the NH molecule group and the charge density accumulation on the O atom of the OH surface group. Fig. 7(f) confirms the H-bond formation for the physisorption mode of the species on the surface by charge density accumulation on the O atom and charge density depletion on the hydrogen atoms of the OH surface groups. In addition, a second H-bond is evidenced by the charge depletion and accumulation in a H atom of NH and O atom of OH groups, respectively. However, even without showing the covalent bonds, the configuration (Fig. 7(f)) shows strong redistribution of electrons in the molecule, therefore can be classified as weakly chemisorbed.
For 8-Q species shown in Fig. 7(g)–(i), the charge density difference confirms the chemisorption of the species on the oxidized aluminum surface by the formation of covalent and H-bonds. In Fig. 7(g), the O–Al–N bridge bonds are evidenced by the charge density accumulation between O, N and Al atoms involved in the chemical interaction. In addition an H-bond is formed by the O atom of the molecule and H atom of the hydroxyl surface group, evidenced by the charge density accumulation and depletion in the O and H atoms, respectively. The charge density in Fig. 7(h) and (i) with similar adsorption behavior on adjacent surface reactive sites confirm the formation of O–Al covalent bonds by the charge accumulation between them and H-bond by the charge density accumulation and depletion in the N and H atoms, respectively, of the molecule atom and OH surface group.
The Bader charge analysis in Table 2 reveals that the electron transfer and its distribution depends on the adsorption species and surface coverage. The electron distribution shows no significant electron transfer between the molecule and the substrate for the native 8-HQ molecule, again accounting for a physisorption mode of the species on the hydroxylated oxidized aluminum surface. Independently of the surface coverage, the electron changes in the molecules are within ±0.06 e molecule−1, with a slight redistribution of the electrons in the molecule after adsorption. However, the physisorption of the molecule induces a charge reorganisation in the substrate for surface coverages of 1.18 and 2.36 molecule nm−2. We thus find an electron transfer of about 0.33 electrons from the metallic part to the oxide.
The charge analysis for the tautomer species shows an electron transfer from the substrate to the molecules, whatever the surface coverage. However, the electron transfer decreases for high surface coverage, due to the difference in the adsorption configurations of the molecules on the surface, increasing surface coverage. We calculated values of 0.78, 0.54 and 0.40 e molecule−1 for surface coverages of 1.18, 2.36 and 3.54 molecule nm−2, respectively. The electrons transfer comes mainly from the oxide part, with slight contribution of the Al of the metallic part, except at low coverage. Indeed for surface coverage of 1.18 molecule nm−2, 0.28 and 0.50 electron are transferred from the metal and the oxide, respectively, to each molecule, whereas only about 0.05 electron comes from the metal part for surface coverages of 2.36 and 3.54 molecule nm−2.
The Bader charge analysis shows that the largest charge changes are for the adsorption of the 8-Q species. This reveals that the organic film and the metal part of the substrate are electron acceptors, while the oxide part is an electron donor. The oxide gives globally 0.73, 1.84 and 1.42 e molecule−1 for coverages of 1.18, 2.36 and 3.54 molecule nm−2, while the electron transfer from the oxide to the adsorption species has an average value of 0.79 ± 0.01 e molecule−1. The electron gain in the metal part is only significant when increasing coverage, and the values are 1.05 and 0.63 e molecule−1 for surface coverages of 2.36 and 3.54 molecule nm−2, whereas it remains unchanged for surface coverages of 1.18 molecule nm−2, with a value of 0.07 e molecule−1.
Through this analysis, we can also see that for tautomer and 8-Q species at high coverage, the electron transfer is shared between the molecules proportionally to their bonding strength on the surface. We recall that the organic films at coverages of 2.36 and 3.54 molecule nm−2 were composed by a combination of different adsorption configurations, with an inhomogeneity in the adsorption features. Thus, the molecule that gains more electrons coincides with the molecule with the highest bonding strength. For example, the charge gains are 0.73, 0.28 and 0.18 e for each molecule in the organic film formed at a coverage of 3.54 molecule nm−2 by the tautomer species, which corresponds to three molecules adsorbed on the surface, and the bonding energies for the same molecules in the organic film are about 4.44, 2.62, and 2.39 eV, respectively. A similar trend is found for the 8-Q species, where the charge gains are about 0.90, 0.88 and 0.57 e for the three molecules, correlating to bonding energies of 7.04, 5.94 and 5.88 eV, respectively. We can also emphasize a charge redistribution in the molecules after their chemisorption on the surface. All atoms of the molecules (N and O atoms) involved directly in the formation of covalent bonds with the Al surface atoms underwent strong electron changes, which confirms the covalent nature of the bonds. For the 8-Q species, the O and N atoms gain about 0.26 ± 0.02 and 0.12 ± 0.01 e, respectively, according to the surface coverage. The chemisorption of the tautomer species via O–Al covalent bond leads to an electron gain of about 0.55 e for the O atom.
3.4 Vacancy formation energy
In order to investigate further the corrosion inhibition efficiency of native 8-HQ, tautomer and 8-Q molecules, related to a slow down of the dissolution of aluminum atoms present in the hydroxylated oxide, we calculated the vacancy formation energy difference (ΔΔEdefect) between the coated and uncoated surfaces, using eqn (7). This shows whether the molecules can mitigate the anodic reaction and their ability to decrease or inhibit the surface atoms dissolution. It corresponds to the creation of one aluminum vacancy on the surface with and without the presence of the molecules. We considered all surface atoms for each adsorption species and increase the surface coverage. However for better interpretation of the data, we calculated an average value for each surface coverage. The results are shown in Fig. 8. A negative value of ΔΔEdefect, means that the extraction of an Al atom of the surface could be easier in the presence of the organic layer. This can accelerate the dissolution of the atom surface. However, the dissolution process depends also on the kinetics of the reactions in relation with the activation energy for Al extraction. In contrast, ΔΔEdefect > 0 means that the hydroxylated oxidized surface is stabilized by the presence of the organic layer and the dissolution of the surface atoms can be slowed down from a thermodynamical point of view and independently of the kinetics of the reactions. | ||
| Fig. 8 Average vacancy formation energy (eV per atom) difference between coated and pristine oxidized aluminum surface at different molecular surface coverages. (a) Native 8-HQ species, (b) tautomer species and (c) 8-Q species. | ||
Regardless of the surface coverage, the adsorption of the native 8-HQ molecule on oxidized aluminum surfaces leads to no significant change in the average ΔΔEdefect. We calculated average values of 0.07, −0.10 and 0.01 eV per Al atom for surface coverages of 1.18, 2.36 and 3.54 molecule nm−2, respectively. These results indicate that the native species do not have the ability to improve the stability of the surface aluminum atoms. Locally, the sensitivity of the Al atoms to the anodic reaction remains similar to without the inhibitor and for certain atoms is almost weakened. The adsorption of the tautomer form leads to more sensitivity of the aluminum surface atoms toward the dissolution, even if the tautomer species shows a stronger bonding than the native molecules on the hydroxylated oxidized aluminum surface. The average defect formation energies are negative, and could mean the acceleration of the anodic reaction with a decrease of this effect for full surface coverage. We calculated values of −0.33, −0.37 and −0.27 eV per Al atom for a surface coverage of 1.18, 2.36 and 3.54 molecule nm−2. Globally, we can conclude that both the native 8-HQ molecule and its tautomer form do not slow down the dissolution of aluminum, despite their strong interaction with the hydroxylated oxide surface. However, the presence of dense organic films formed for the highest surface coverage could act as a physical barrier and induce an activation barrier for the Al extraction.
For the 8-Q species, the average value of ΔΔEdefect is positive for all studied surface coverages and involved Al sites. These results reveal clearly that the presence of the organic layer slows down the extraction of Al atoms from the surface of the substrate. ΔΔEdefect increases with the surface coverage, showing the beneficial effect of a compact organic layers on the substrate surface. The average defect formation energy differences (between coated and uncoated surfaces) are about 0.73, 1.34 and 1.46 eV per Al atom for surface coverages of 1.18, 2.36 and 3.54 molecule nm−2, respectively. However, a difference in the local effect of the inhibitor to slow down the Al extraction was evidenced. ΔΔEdefect changes with the aluminum surface sites. ΔΔEdefect is in the range of 0.13 to 1.91 eV per Al atom for a coverage of 1.18 molecule nm−2, of 0.56 to 2.38 eV per Al atom for a coverage of 2.36 molecule nm−2 and 0.57 to 2.71 eV per Al atom for a coverage of 3.54 molecule nm−2. We can conclude that among the native 8-HQ, tautomer and 8-Q molecules, the 8-Q species shows suitable features to improve the corrosion resistance of the aluminum surface.
4 Conclusions
Dispersion corrected Density Functional Theory (DFT-D) was used to investigate the molecule–surface interactions and to study the ability of the molecules to form a protective organic film to prevent metal dissolution. The adsorption of the 8-HQ molecule and its tautomeric and 8-Q species on the hydroxylated oxidized aluminum (γ-Al2O3(111)/Al(111)) surface was studied from low to high surface coverage. The structural and energetic trends were analysed to determine the most stable adsorption configurations. The electronic structure analysis was used to provide more details about the molecule–surface interactions. The reactive sites, adsorption configurations and modes are highlighted for different species, i.e. native 8-HQ, tautomer and 8-Q species at surface coverages from 1.18 to 3.54 molecule nm−2. The defect formation energy calculated with and without inhibitors on the surface is a good indicator to show if the molecules have the ability to slow down the dissolution of surface atoms, thereby mitigating the anodic reaction on the metal.The adsorption strength of different species increases from the native 8-HQ to the deprotonated species. The dispersion energy increases as a function of surface coverage. This molecule–molecule interaction becomes dominant for the highest coverage for the native form. However, in terms of contribution to the adsorption energy, it increases as 8-Q species < tautomer species < native 8-HQ.
The reactive sites are the N atom and OH group for the native form, the O atom and NH group for the tautomer species, and the O and N atoms for the 8-Q species. The adsorption strength is enhanced by the synergy of different bond formations. Al sites on the hydroxylated oxidized aluminum surface show different reactivities, but all are able to interact with the molecules, forming covalent bonds. In addition, the OH oxide surface groups play a significant role in the determination of the adsorption modes and configurations, forming H-bonding according to the adsorption species. The native 8-HQ molecule is physisorbed on a hydroxylated oxide surface, whereas the tautomer and 8-Q species are chemisorbed.
Despite the ability of all species to interact strongly with the oxide surface and their ability to form stable organic films, only the 8-Q species prevents the extraction of Al atoms from the oxide surface. In contrast, the native form does not significantly change the average defect formation energy difference between the coated and uncoated surfaces and the tautomer species can favor the extraction of the surface metal atoms. However, the two last species could form a physical barrier, which may induce kinetic activation energy for Al extraction. We can conclude that among the native 8-HQ, tautomer and 8-Q molecules, the 8-Q species provide the best protection against aluminum corrosion.
Author contributions
F. Chiter: methodology; validation; investigation; formal analysis; visualization; writing original draft; writing review & editing. D. Costa: supervision; writing – review & editing. N. Pébère: supervision; writing – review & editing. P. Marcus: supervision; writing – review & editing. C. Lacaze-Dufaure: resources; supervision, writing – review; editing & funding acquisition & project management.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was performed using HPC resources from CALMIP (Grant p12174) and from CINES (Grant c2013097076). It was supported by the National Research Agency (ANR support number ANR-2011 JS08 015 01).References
- R. L. Twite and G. P. Bierwagen, Prog. Org. Coat., 1998, 33, 91–1000 CrossRef CAS.
- P. B. Raja and M. G. Sethuraman, Mater. Lett., 2008, 62, 113–116 CrossRef CAS.
- Y. Yin, T. Liu, S. Chen, T. Liu and S. Cheng, Appl. Surf. Sci., 2008, 255, 2978–2984 CrossRef CAS.
- G. Gece, Corros. Sci., 2011, 53, 3873–3898 CrossRef CAS.
- S. A. Kulinich and A. S. Akhtar, Russ. J. Non-Ferrous Metals, 2012, 53, 176–203 CrossRef.
- S. Gangopadhyay and P. A. Mahanwar, J. Coat. Technol. Res., 2018, 15, 789–807 CrossRef CAS.
- I. Milošev, T. Bakarič, S. Zanna, A. Seyeux, P. Rodič, M. Poberžnik, F. Chiter, P. Cornette, D. Costa, A. Kokalj and P. Marcus, J. Electrochem. Soc., 2019, 166, C3131–C3146 CrossRef.
- A. Rajagopal and A. Kahn, J. Appl. Phys., 1998, 84, 355 CrossRef CAS.
- N. Johansson, T. Osada, S. Stafström and W. R. Salaneck, J. Chem. Phys., 1999, 111, 2157 CrossRef CAS.
- M. Stößel, J. Staudigel, F. Steuber, J. Blässing and J. Simmerer, Appl. Phys. Lett., 2000, 76, 115 CrossRef.
- M. D. Halls and H. B. Schlegel, Chem. Mater., 2001, 13, 2632–2640 CrossRef CAS.
- E. R. de Sousa, E. P. Marques, E. N. Fernandes, J. Zhangb and A. L. B. Marques, J. Braz. Chem. Soc., 2006, 17, 177–183 CrossRef CAS.
- Y. Q. Zhan, M. P. de Jong, F. H. Li, V. Dediu, M. Fahlman and W. R. Salaneck, Phys. Rev. B: Condens. Matter Mater. Phys., 2008, 78, 045208 CrossRef.
- C. Barraud, P. Seneor, R. Mattana, S. Fusil, K. Bouzehouane, C. Deranlot, P. Graziosi, L. Hueso, I. Bergenti, V. Dediu, F. Petroff and A. Fert, Nat. Phys., 2010, 6, 615–620 Search PubMed.
- Z. Yuanyuan, C. Hongrui, S. Qingrong, C. Hongli, Y. Weiqing and M. Menglin, J. Chem. Res., 2021, 10, 623–634 Search PubMed.
- H. N. Soliman, Corros. Sci., 2011, 53, 2994–3006 CrossRef CAS.
- A. S. Yaro and H. A. Dahyool, Iraqi J. Chem. Pet. Eng., 2009, 10, 19–25 Search PubMed.
- C. N. Njoku, W. Bai, M. Meng, I. O. Arukalam, D. I. Njoku and Y. Li, Mater. Res. Express, 2019, 6, 115706 CrossRef CAS.
- A. C. Balaskas, I. A. Kartsonakis, L.-A. Tziveleka and G. C. Kordas, Prog. Org. Coat., 2012, 74, 418–426 CrossRef CAS.
- A. Balaskas, I. Kartsonakis, D. Snihirova, M. Montemor and G. Kordas, Prog. Org. Coat., 2012, 72, 653–662 CrossRef.
- M. Sowa, M. Wala, A. Blacha-Grzechnik, A. Maciej, A. Kazek-Kesik, A. Stolarczyk and W. Simka, Materials, 2021, 14, 619 CrossRef CAS PubMed.
- C. Zhu, H. Yang, A. Wu, D. Zhang, L. Gao and T. Lin, J. Power Sources, 2019, 432, 55–64 CrossRef CAS.
- L. Garrigues, N. Pébère and F. Dabosi, Electrochim. Acta, 1996, 41, 1209–1215 CrossRef CAS.
- C. Casenave, N. Pébère and F. Dabosi, Mater. Sci. Forum, 1995, 599–610 CAS.
- S. V. Lamaka, M. L. Zheludkevich, K. A. Yasakau, M. F. Montemor and M. G. S. Ferreira, Electrochim. Acta, 2007, 52, 7231–7247 CrossRef CAS.
- L. Song-mei, Z. Hong-rui and L. Jian-hua, Trans. Nonferrous Met. Soc. China, 2007, 17, 318–325 CrossRef.
- K. A. Yasakau, M. L. Zheludkevich, O. V. Karavai and M. G. S. Ferreira, Prog. Org. Coat., 2008, 63, 352–361 CrossRef CAS.
- A. C. Balaskas, M. Curioni and G. E. Thompson, Surf. Interface Anal., 2015, 47, 1029–1039 CrossRef CAS.
- S. Marcelin and N. Pébère, Corros. Sci., 2015, 101, 66–74 CrossRef CAS.
- Y.-P. Wang, X.-F. Han, Y.-N. Wu and H.-P. Cheng, Phys. Rev. B: Condens. Matter Mater. Phys., 2012, 85, 144430 CrossRef.
- S. Yanagisawa and Y. Morikawa, Chem. Phys. Lett., 2006, 420, 523–528 CrossRef CAS.
- K. L. S. Yanagisawa and Y. Morikawa, J. Chem. Phys., 2008, 128, 244704 CrossRef PubMed.
- S. Yanagisawa and Y. Morikawa, J. Phys.: Condens. Matter, 2009, 21, 064247 CrossRef PubMed.
- I. G. Hill, A. J. Mäkinen and Z. H. Kafafi, Appl. Phys. Lett., 2000, 77, 1825 CrossRef CAS.
- D. Ino, K. Watanabe, N. Takagi and Y. Matsumoto, Phys. Rev. B: Condens. Matter Mater. Phys., 2005, 71, 115427 CrossRef.
- Z. Wang, A. Pronschinske and D. B. Dougherty, Org. Electron., 2011, 12, 1920–1926 CrossRef CAS.
- A. Droghetti, P. Thielen, I. Rungger, N. Haag, N. Großmann, J. Stockl, B. Stadtmuller, M. Aeschlimann, S. Sanvito and M. Cinchetti, Nat. Commun., 2016, 7, 12668–12677 CrossRef CAS PubMed.
- Y. Bulteau, N. Tarrat, N. Pébère and C. Lacaze-Dufaure, New J. Chem., 2020, 44, 15209–15222 RSC.
- J. Zhang, P. Chen, B. Yuan, W. Ji, Z. Cheng and X. Qiu, Science, 2013, 342, 1242603 Search PubMed.
- F. Chiter, C. Lacaze-Dufaure, H. Tang and N. Pébère, Phys. Chem. Chem. Phys., 2015, 17, 22243 RSC.
- F. Chiter, M.-L. Bonnet, C. Lacaze-Dufaure, H. Tang and N. Pébère, Phys. Chem. Chem. Phys., 2018, 20, 21474 RSC.
- Y. Bulteau, C. Lepetit and C. Lacaze-Dufaure, Inorg. Chem., 2020, 59, 17916–17928 CrossRef CAS PubMed.
- J. You and Z. Liu, Appl. Surf. Sci., 2021, 540, 148315 CrossRef CAS.
- F. Chiter, D. Costa, V. Maurice and P. Marcus, J. Phys. Chem. C, 2020, 124, 17048–17057 CrossRef CAS.
- F. Chiter, D. Costa, V. Maurice and P. Marcus, Appl. Surf. Sci., 2021, 537, 147802 CrossRef CAS.
- F. Chiter, D. Costa, V. Maurice and P. Marcus, J. Electrochem. Soc., 2020, 167, 161506 CrossRef CAS.
- M. Poberžnik, D. Costa, A. Hemeryck and A. Kokalj, J. Phys. Chem. C, 2018, 122, 9417–9431 CrossRef.
- M. Poberžnik, F. Chiter, I. Milošev, P. Marcus, D. Costa and A. Kokalj, Appl. Surf. Sci., 2020, 525, 146156 CrossRef.
- G. Sun, J. Kürti, P. Rajczy, M. Kertesz, J. Hafner and G. Kresse, THEOCHEM, 2003, 624, 37–45 CrossRef CAS.
- G. Kresse and J. Hafner, Phys. Rev. B: Condens. Matter Mater. Phys., 1993, 47, 558 CrossRef CAS PubMed.
- G. Kresse and J. Furthmüller, Comput. Mater. Sci., 1996, 6, 15–50 CrossRef CAS.
- G. Kresse and J. Furthmüller, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 11169–11186 CrossRef CAS PubMed.
- G. Kresse and D. Joubert, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 59, 1758–1775 CrossRef CAS.
- P. E. Blöchl, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 50, 17953 CrossRef PubMed.
- J. P. Perdew, J. A. Chevary, S. H. Vosko, K. A. Jackson, M. R. Pederson, D. J. Singh and C. Fiolhais, Phys. Rev. B: Condens. Matter Mater. Phys., 1992, 46, 6671–6687 CrossRef CAS PubMed.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865 CrossRef CAS PubMed.
- M. Methfessel and A. T. Paxton, Phys. Rev. B: Condens. Matter Mater. Phys., 1989, 40, 3616 CrossRef CAS PubMed.
- S. Grimme, J. Comput. Chem., 2006, 27, 1787–1799 CrossRef CAS PubMed.
- C. Kittel, Introduction to Solid State Physics, Wiley Sons, New York, 7th edn, 1996 Search PubMed.
- H. J. Monkhorst and J. D. Pack, Phys. Rev. B: Solid State, 1976, 13, 5188 CrossRef.
- D. Costa, T. Ribeiro, F. Mercuri, G. Pacchioni and P. Marcus, Adv. Mater. Interfaces, 2014, 1, 1300072 CrossRef.
- D. Costa, T. Ribeiro, P. Cornette and P. Marcus, J. Phys. Chem. C, 2016, 120, 28607–28616 CrossRef CAS.
- P. Roychowdhury, Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem., 1978, 34, 1047–1048 CrossRef.
- Q. S. Li and W. H. Fang, Chem. Phys. Lett., 2003, 367, 637–644 CrossRef CAS.
- A. E. Shchavlev, A. N. Pankratov and A. V. Shalabay, Int. J. Quantum Chem., 2006, 106, 876–886 CrossRef CAS.
- E. Bardez, I. Devol, B. Larrey and B. Valeur, J. Phys. Chem. B, 1997, 101, 7786–7793 CrossRef CAS.
- W. Tang, E. Sanville and G. Henkelman, J. Phys.: Condens. Matter, 2009, 21, 084204 CrossRef CAS PubMed.
- A. Kokalj, Corros. Sci., 2022, 196, 109939 CrossRef CAS.
- F. Chiter, D. Costa, V. Maurice and P. Marcus, npj Mater. Degrad., 2021, 5, 52 CrossRef CAS.
- F. Chiter, D. Costa, V. Maurice and P. Marcus, J. Electrochem. Soc., 2021, 168, 121507 CrossRef CAS.
| This journal is © the Owner Societies 2023 |