
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Microhydrated clusters of a pharmaceutical drug: infrared spectra and structures of amantadineH+(H2O)n†
Martin Andreas Robert
George
 and
Otto
Dopfer
*
and
Otto
Dopfer
*
Institut für Optik und Atomare Physik, Technische Universität Berlin, Hardenbergstr. 36, 10623 Berlin, Germany. E-mail: dopfer@physik.tu-berlin.de
First published on 23rd January 2023
Abstract
Solvation of pharmaceutical drugs has an important effect on their structure and function. Analysis of infrared photodissociation spectra of amantadineH+(H2O)n=1–4 clusters in the sensitive OH, NH, and CH stretch range by quantum chemical calculations (B3LYP-D3/cc-pVTZ) provides a first impression of the interaction of this pharmaceutically active cation with water at the molecular level. The size-dependent frequency shifts reveal detailed information about the acidity of the protons of the NH3+ group of N-protonated amantadineH+ (AmaH+) and the strength of the NH⋯O and OH⋯O hydrogen bonds (H-bonds) of the hydration network. The preferred cluster growth begins with sequential hydration of the NH3+ group by NH⋯O ionic H-bonds (n = 1–3), followed by the extension of the solvent network through OH⋯O H-bonds. However, smaller populations of cluster isomers with an H-bonded solvent network and free N–H bonds are already observed for n ≥ 2, indicating the subtle competition between noncooperative ion hydration and cooperative H-bonding. Interestingly, cyclic water ring structures are identified for n ≥ 3, each with two NH⋯O and two OH⋯O H-bonds. Despite the increasing destabilization of the N–H proton donor bonds upon gradual hydration, no proton transfer to the (H2O)n solvent cluster is observed up to n = 4. In addition to ammonium cluster ions, a small population of microhydrated iminium isomers is also detected, which is substantially lower for the hydrophilic H2O than for the hydrophobic Ar environment.
1. Introduction
1-Amantadine (1-C10H15NH2, 1-amino adamantane, Ama, Fig. 1) is the amino derivative of adamantane (C10H16, Ada) and thus belongs to the diamondoid family, a fundamental class of rigid and strain-free cycloalkanes.1,2 These nm-sized H-passivated nanodiamonds are rather stable compounds3–5 and offer a wide range of applications, e.g., in molecular electronics, astrochemistry, materials and polymer sciences, chemical synthesis, and medical sciences.6–19 Since several amines and protonated molecules have been detected in the interstellar medium and the presence of diamondoids has been suggested due to their high stability,20–26 it is likely that Ama and its protonated ion are also present.7,18,19 In terms of pharmaceutical applications, Ama is one of the best known commercially available diamondoids, being successfully marketed as antiviral and antiparkinsonian agents.27–30 Indeed, Ama is effective in prevention and treatment of influenza A infections,31–35 Parkinson,36–38 multiple sclerosis, depression, and cocaine addiction.39–43 In all postulated mechanisms, the biochemically active form is N-protonated Ama, AmaH+, or its dimethylated derivative memantine.44,45 The latter has been among the 100 best-selling drugs worldwide (>109 $ in 2013).46 Beside its pharmaceutical applications, Ama is an interesting diamondoid derivative because it is a suitable target for studying ionization- and protonation-induced rearrangement processes of the adamantyl cage.47,48 To this end, it has been shown that the replacement of H by the electron-donating NH2 group at the apical 1-position largely preserves the chemical properties of diamond-like structures, but greatly reduces the barrier to opening the C10H15 cage.47,48 Similarly, protonation of Ama can also lead to the formation of open-cage bicyclic iminium ions.49 | ||
| Fig. 1 Calculated equilibrium structures (in Å and degrees) of H2O, Ama, AmaH+(I,II), and AmaH+(I,II)(H2O)(I,II) in their ground electronic state (B3LYP-D3/cc-pVTZ). All bond lengths are shown in ESI† (Fig. S22). | ||
Neutral Ama has been well characterized by infrared (IR), Raman and electron momentum spectroscopy as well as computational techniques.50–54 It consists of a primary pyramidal NH2 group attached to the adamantyl backbone, which gives the molecule the special chemical properties of a diamond-like structure.39,55 In addition, the Ama⋯DNA interaction has been investigated by Raman spectroscopy.56 Ionization of Ama into the electronic ground state of the radical cation occurs by electron ejection from the nonbonding N lone pair of the NH2 group (HOMO) with a vertical ionization energy roughly estimated as 8.6 eV by its low-resolution electron momentum spectrum.51 Recently, we have studied Ama+Ln radical cation clusters (with L = Ar, N2, H2O) generated in a supersonic electron-ionization plasma expansion with IR photodissociation (IRPD) spectroscopy and quantum chemical calculations.47,48,57 These studies reveal that the adamantyl cage can open upon ionization, thus producing two distonic bicyclic iminium ions, Ama+(II,III), in addition to the canonical nascent Ama+(I) ion with an intact C10H15 cage. Due to the strong p/π-electron donating character of the NH2 group, the barriers for cage-opening are much lower than for Ada+. Due to its higher reaction barriers, no open-cage isomers of Ada+ have yet been detected in both IR and optical spectra.19,25,58 For Ama+, the canonical cage isomer Ama+(I) (E0 = 46 kJ mol−1) is generated by vertical ionization of Ama, and the resulting activation of the C–C bond adjacent to the NH2 group opens the cage and forms the primary radical Ama+(II) (E0 = 87 kJ mol−1) via a conversion of one of the cyclohexane rings from a boat to a chair configuration. A subsequent 1,2 H-shift with reconversion from chair to boat forms the much more stable tertiary radical Ama+(III) (E0 = 0 kJ mol−1).
In addition to ionization, we have recently investigated protonated amantadine (AmaH+) by IRPD spectroscopy (Ar-tagging) and quantum chemical calculations and identified, in addition to the canonical N-protonated AmaH+(I) ammonium ion (E0 = 0 kJ mol−1), an only slightly less stable open-cage bicyclic iminium ion (II) (E0 = 3 kJ mol−1).49 This open-cage isomer II is probably not generated by direct protonation of Ama but by ionization of Ama followed by cage opening (by the mechanism described above) and subsequent addition of H. However, the dominant population corresponds clearly to the ammonium ion (I), which is the relevant structure for pharmaceutical applications in solution. Apart from our previous letter,49 which provides the first spectroscopic characterization of the structure of AmaH+, there have been no spectroscopic studies of this fundamental bio-active molecular ion, despite its pharmaceutical importance. However, AmaH+ has been analysed by mass spectrometry to obtain information on its fragmentation and proton affinity.59 Furthermore, there are several spectrophotometric, NMR and IR studies on amantadine hydrochloride salt (Ama·HCl), but these are mainly concerned with analytical applications of this molecular drug.52,60–63
Herein, we analyse IRPD spectra of size-selected AmaH+(H2O)n=1–4 clusters by dispersion-corrected density functional theory (DFT) calculations at the B3LYP-D3/cc-pVTZ level to characterize the initial microhydration steps of this important bio-active molecular ion. This dual approach provides direct access to the interaction potential between the ion and the solvent molecules, as demonstrated in previous studies of microhydrated cations in our laboratory.57,58,64–87 Thus, the AmaH+(H2O)n=1–4 data provide a first impression of the interaction of this pharmaceutically relevant ion with water solvent molecules at the molecular level. Specifically, the IRPD spectra recorded in the informative OH, NH, and CH stretch range yield information about the potential of the ion–ligand interaction in terms of the binding site and the strength of the interaction, as well as the structure of the hydrogen-bonded (H-bonded) solvent network and the acidity of the N–H bonds of the protonated NH2 group of AmaH+ (and also of the iminium isomer). In previous work,57 we compared microhydration of the Ama+ radical cation with that of aniline+ (An+),88,89 aminobenzonitrile+ (ABN+),75 and CH3NH2+ (computational data)90 to reveal the differences of an aromatic, aliphatic, and cycloaliphatic substituent on the acidity and intermolecular interaction of the NH protons of primary amine cations. For Ama+, the same preferential cluster growth was found as for An+ and ABN+ but with stronger NH⋯O H-bonds due to the higher acidity of the N–H bonds of Ama+. In contrast, the NH⋯O H-bonds of CH3NH2+ to H2O are much stronger than those of Ama+ due to the higher positive charge on the NH2+ group. Therefore, a particular question to be addressed is the change in microhydration due to the additional proton attached to the NH2 group of AmaH+ and the resulting change in the NH⋯O H-bonds. Moreover, in another study on Ama+Ln clusters (with L = Ar, N2, H2O), we have shown that the population of the bicyclic iminium ion Ama+(III) is significantly smaller in a H2O environment than in a nonpolar environment such as Ar or N2, due to higher rearrangement barriers between Ama+(II) and Ama+(III) (1,2 H-shift) for H2O. Since the open-cage isomer AmaH+(II) is assumed to be formed by H addition to the distonic Ama+(III) ion, the population of AmaH+(II) in a H2O environment should also be much lower, which shall be verified herein. Another important aspect is the competition between the formation of the H-bonded solvent network (which is strongly favoured by nonadditive cooperative three-body interactions) and the internal cation hydration with individual ligands bound by charge–dipole forces (which suffer from noncooperative three-body forces).64 A further topic to be addressed is potential hydration-induced proton transfer from the NH3+ group to the solvent cluster, which becomes progressively more favourable for larger n due to the increasing proton affinity of (H2O)n clusters (e.g., PA = 691, 808, 862, 900, 904, and 908 kJ mol−1 for n = 1–6)91–95 approaching that of Ama (measured and computed as 949 and 957 kJ mol−1, respectively).49,91
2. Experimental and computational techniques
The IRPD spectra of mass-selected AmaH+(H2O)n clusters (n = 1–4) shown in Fig. 2 are recorded in a tandem quadrupole mass spectrometer coupled to an electron ionization (EI) source.64,96 The clusters are produced in a pulsed supersonic plasma expansion by electron and/or chemical ionization of Ama followed by three-body hydration reactions. The expansion gas is generated by passing a carrier gas mixture (9 bar) composed of Ar and 5%H2/He in the ratio 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 through a reservoir containing solid Ama (Sigma-Aldrich, >97%) heated to 80 °C. The filaments of the EI source are powered by a 220 V offset voltage, which sets an upper limit on the kinetic energy of the electrons hitting the molecules in the expansion. To produce hydrated AmaH+ clusters, distilled water is added to the gas line just in front of the sample reservoir. One possible production route for AmaH+(H2O)n involves ionization of H2 (eqn (1)) followed by exothermic proton transfer reactions (eqn (2) and (3)) to generate AmaH+ and three-body collisions to form AmaH+(H2O)n clusters (eqn (4)):97,98
:1 through a reservoir containing solid Ama (Sigma-Aldrich, >97%) heated to 80 °C. The filaments of the EI source are powered by a 220 V offset voltage, which sets an upper limit on the kinetic energy of the electrons hitting the molecules in the expansion. To produce hydrated AmaH+ clusters, distilled water is added to the gas line just in front of the sample reservoir. One possible production route for AmaH+(H2O)n involves ionization of H2 (eqn (1)) followed by exothermic proton transfer reactions (eqn (2) and (3)) to generate AmaH+ and three-body collisions to form AmaH+(H2O)n clusters (eqn (4)):97,98| H2 + e → H2+ + 2e | (1) |
| H2+ + H2 → H3+ + H | (2) |
| Ama + H3+ → AmaH+ + H2 | (3) |
| AmaH+(H2O)n−1 + H2O + M → AmaH+(H2O)n + M | (4) |
| (H2O)n+1 + e → (H2O)nH+ + OH + 2e | (5) |
| Ama + (H2O)nH+ → AmaH+ + (H2O)n | (6) |
| Ama + e → Ama+ + 2e | (7) |
| Ama+ + H2 → AmaH+ + H | (8) |
 | ||
| Fig. 2 IRPD spectra of AmaH+(H2O)n=1–4 in the 2600–4000 cm−1 range recorded in the H2O loss channel are compared to the IRPD spectrum of AmaH+Ar.49 The position, widths, and assignments of the transitions observed (A–Z) are listed in Table 1. The bands that can only be assigned to the iminium isomers are highlighted in red. | ||
Quantum chemical calculations are performed at the B3LYP-D3/cc-pVTZ level of theory for Ama, Ama+, AmaH+, and AmaH+(H2O)n hydrates to determine their structural, energetic, and spectroscopic properties.100 As shown for the related Ama+Ln and AmaH+Ar clusters, this computational approach reproduces the experimental IR spectra and binding energies with satisfactory accuracy and is an efficient compromise between accuracy and computing time.47–49,57 Relative energies and equilibrium binding energies (Ee, De) are corrected for harmonic zero-point vibrational energies to yield E0 and D0. Gibbs free energies (G) are evaluated at 298.15 K. Harmonic vibrational frequencies are scaled by factors of 0.9618, 0.9491, and 0.9630 for CH, NH, and OH stretch frequencies, respectively.47,48,57 The frequencies in the fingerprint range required for the determination of the overtones and combination bands are scaled with a factor of 0.9732.47 Computed IR stick spectra are convoluted with Gaussian line profiles (fwhm = 10 cm−1) to facilitate convenient comparison with the measured IRPD spectra. Natural bond orbital (NBO) analysis is employed to evaluate the charge distribution and charge transfer in AmaH+(H2O)n as well as the second-order perturbation energies (E(2)) of donor–acceptor orbital interactions involved in the H-bonds.101,102 Cartesian coordinates and energies of all relevant structures are available in ESI.†
3. Experimental results
3.1 Mass spectra
Typical mass spectra of the ion source show dehydrogenated, ionized, and protonated Ama (m/z 150–152) as well as their clusters with Ar and H2O (Fig. S1, ESI†). In addition, the typical fragment ions of Ama+ (F+ at m/z 57, 94, 108, 135) are detected, consistent with the standard EI mass spectrum of Ama and previous mass spectra, confirming the identity of m/z 151 and 152 as Ama+ and AmaH+.47–49,52 To confirm the chemical composition of the generated AmaH+(H2O)n clusters, collision-induced dissociation (CID) experiments are performed, revealing mainly dissociation of H2O molecules and ruling out significant isobaric mass contamination (Fig. S2 and S3, ESI†).3.2 Overview of IRPD spectra
The IRPD spectra of AmaH+(H2O)n=1–4 are compared in Fig. 2 to the previously recorded AmaH+Ar spectrum,49 and the observed bands and their vibrational assignments are listed in Table 1. The investigated spectral range (2600–4000 cm−1) covers aliphatic CH stretch modes (B–G, νCHn, 2800–3000 cm−1), and H-bonded (A, H–K, , 2600–3200 cm−1) and free NH stretch modes (N–P,
, 2600–3200 cm−1) and free NH stretch modes (N–P,  , 3200–3400 cm−1) of the NH3+ ammonium group of AmaH+, NH stretch modes of the NH2+ amino group of AmaH+ (Q, R, V, νNH2, 3300–3500 cm−1), bound (S, T, W,
, 3200–3400 cm−1) of the NH3+ ammonium group of AmaH+, NH stretch modes of the NH2+ amino group of AmaH+ (Q, R, V, νNH2, 3300–3500 cm−1), bound (S, T, W, 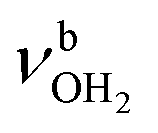 , 3300–3600 cm−1) and free (X–Z,
, 3300–3600 cm−1) and free (X–Z,  , 3600–3900 cm−1) OH stretch modes of the H2O ligands, and combination bands and overtones of the OH, NH, and CH bending fundamentals (L, M, U, 3100–3500 cm−1). The IRPD spectra show a clear and distinct dependence on the cluster size n and therefore provide detailed information about the hydration motifs, both in terms of the H2O binding sites and the structures of the hydration network.
, 3600–3900 cm−1) OH stretch modes of the H2O ligands, and combination bands and overtones of the OH, NH, and CH bending fundamentals (L, M, U, 3100–3500 cm−1). The IRPD spectra show a clear and distinct dependence on the cluster size n and therefore provide detailed information about the hydration motifs, both in terms of the H2O binding sites and the structures of the hydration network.
| Peak | Modeb | AmaH+Ara | AmaH+H2O | AmaH+(H2O)2 | AmaH+(H2O)3 | AmaH+(H2O)4 |
|---|---|---|---|---|---|---|
a Ref. 49.
b Stretching (ν), bending (β).
c FR = Fermi resonance with  . .
|
||||||
| A |

|
2754 (24) | 2712 (13) | 2680 (11) | ||
| B | ν CHn | 2821 (8) | 2823 (11) | |||
| C | ν CHn | 2853 (10) | 2860 (17) | 2863 (16) | 2862 (17) | 2860 (10) |
| D | ν CHn | 2872 (7) | 2874 (9) | 2871 (14) | 2870 (16) | 2867 (4) |
| E | ν CHn | 2921 (14) | 2906 (15) | 2911 (13) | 2911 (7) | 2905 (3) |
| F | ν CHn FRc | 2949 (17) | 2943 (18) | 2939 (20) | 2936 (25) | 2933 (23) |
| F |

|
2943 (18) | ||||
| G |

|
2973 (5) | 2977 (11) | |||
| H |
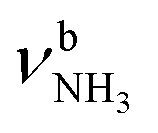
|
2974 (18) | ||||
| I |

|
3009 (39) | 3058 (17) | |||
| J |
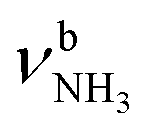
|
3007 (24) | 3069 (39) | 3118 (46) | 3144 (42) | |
| K |

|
3080 (23) | 3127 (24) | 3170 (21) | 3188 (6) | |
| L | 2βNH3 | 3164 (7) | ||||
| M | 2βNH3 | 3185 (5) | 3193 (13) | 3230 (40) | 3268 (30) | 3271 (30) |
| N |

|
3238 (13) | 3279 (17) | 3288 (5) | ||
| O |

|
3315 (12) | 3317 (10) | |||
| P |

|
3317 (21) | 3328 (20) | 3331 (3) | ||
| Q |

|
3344 (7) | ||||
| R |

|
3373 (8) | ||||
| S |

|
3387 (13) | ||||
| T |

|
3403 (39) | 3432 (52) | 3452 (55) | ||
| U | 2βNH2 | 3428 (11) | ||||
| V |
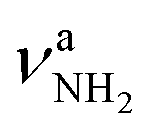
|
3451 (10) | ||||
| W |

|
3552 (11) | 3554 (10) | |||
| X |
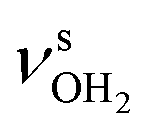
|
3633 (22) | 3638 (22) | 3639 (12) | 3642 (13) | |
| Y |

|
3683 (8) | 3680 (9) | 3683 (8) | ||
| Z |

|
3717 (14) | 3716 (27) | 3712 (42) | 3705 (49) | |
3.3 IRPD of AmaH+Ar
The bands in the AmaH+Ar spectrum have been assigned in detail previously.49 Ar-tagging has little impact on the frequencies of bare AmaH+, and bands marked in black/red are attributed to the ammonium/iminium isomers. Briefly, bands N and P are attributed to the low-frequency NH stretch modes of the ammonium isomer (I), while bands Q and V arise from higher-frequency NH stretch modes of the iminium isomer (II). Although the AmaH+Ar spectrum is strongly dominated by the ammonium isomer formed by simple protonation of Ama at the NH2 group, ∼20–25% of the AmaH+ population is attributed to iminium isomers.3.4 IRPD of AmaH+H2O
The IRPD spectrum of AmaH+H2O shows the distinct free NH stretch signatures of the NH3+ ammonium group by observing the intense bands N and P at 3279 and 3328 cm−1 with widths of 17 and 20 cm−1, respectively. These are close to those of AmaH+Ar ( (N),
(N),  (P)). On the other hand, the intense signatures of the NH2+ amino group of the iminium isomers (Q, V) observed for AmaH+Ar are barely discernible or absent for AmaH+H2O, with the possible exception of the very weak band R at 3373 cm−1. Hence, the AmaH+H2O spectrum is concluded to be predominantly produced by ammonium isomers, while the population of iminium isomers is close to or below the detection limit (<10%). A further major difference between the AmaH+Ar and AmaH+H2O spectra is the appearance of the intense and broad bands J and K at 3007 and 3080 cm−1 with widths of 24 and 23 cm−1 for L = H2O, respectively. These are indicative of a strongly redshifted bonded NH stretch mode, arising from the predominant presence of an H-bonded AmaH+H2O isomer with a strong NH⋯O ionic H-bond between H2O (acceptor) and the acidic NH3+ group (donor). This H-bond is strongly stabilized by the large electrostatic charge–dipole forces between the AmaH+ cation and the H2O dipole which dominate the long-range part of the intermolecular attraction. The strong NH⋯O H-bond causes a large redshift of the bonded
(P)). On the other hand, the intense signatures of the NH2+ amino group of the iminium isomers (Q, V) observed for AmaH+Ar are barely discernible or absent for AmaH+H2O, with the possible exception of the very weak band R at 3373 cm−1. Hence, the AmaH+H2O spectrum is concluded to be predominantly produced by ammonium isomers, while the population of iminium isomers is close to or below the detection limit (<10%). A further major difference between the AmaH+Ar and AmaH+H2O spectra is the appearance of the intense and broad bands J and K at 3007 and 3080 cm−1 with widths of 24 and 23 cm−1 for L = H2O, respectively. These are indicative of a strongly redshifted bonded NH stretch mode, arising from the predominant presence of an H-bonded AmaH+H2O isomer with a strong NH⋯O ionic H-bond between H2O (acceptor) and the acidic NH3+ group (donor). This H-bond is strongly stabilized by the large electrostatic charge–dipole forces between the AmaH+ cation and the H2O dipole which dominate the long-range part of the intermolecular attraction. The strong NH⋯O H-bond causes a large redshift of the bonded  mode down to the spectral range 2800–3200 cm−1, where it couples strongly with NH, OH and CH bend overtones (via anharmonic Fermi resonance (FR)) or with CH stretch fundamentals (via local mode mixing). These interactions result in a broad vibrational multi-band pattern in the IR spectrum. Similar complex patterns arising from NH⋯O ionic H-bonds have been observed previously for a variety of related hydrated aromatic cations such as acetanilide, hydroxyindole, pyrrole, and formanilide.77,78,81,82,103 All IRPD spectra of AmaH+(H2O)n=1–4 show an almost identical CHn stretch pattern of the AmaH+ cage to that observed for AmaH+Ar (C–F). However, because of the redshifted
mode down to the spectral range 2800–3200 cm−1, where it couples strongly with NH, OH and CH bend overtones (via anharmonic Fermi resonance (FR)) or with CH stretch fundamentals (via local mode mixing). These interactions result in a broad vibrational multi-band pattern in the IR spectrum. Similar complex patterns arising from NH⋯O ionic H-bonds have been observed previously for a variety of related hydrated aromatic cations such as acetanilide, hydroxyindole, pyrrole, and formanilide.77,78,81,82,103 All IRPD spectra of AmaH+(H2O)n=1–4 show an almost identical CHn stretch pattern of the AmaH+ cage to that observed for AmaH+Ar (C–F). However, because of the redshifted  mode and the associated strong couplings, their relative integrated intensities are strongly enhanced (when compared to the free NH stretch bands). The CHn stretch pattern consists of one intense doublet C and D at ∼2860 and ∼2870 cm−1 with a total width of ∼45 cm−1 and a more intense and broader doublet E and F at ∼2910 and ∼2940 cm−1 with a total width of ∼60 cm−1. When compared to the AmaH+Ar spectrum, bands C are slightly blueshifted by ∼7 cm−1 while bands E are redshifted by ∼12 cm−1. The distinct blueshifts of the doublet C/D from Ar to H2O may arise from the assignment to different isomers (iminium for Ar and ammonium for H2O). With increasing n, only bands D and F show notable redshifts from 2874 (n = 1) to 2867 cm−1 (n = 4) and from 2943 (n = 1) to 2933 cm−1 (n = 4), respectively. However, in the AmaH+H2O spectrum an additional weak band B is observed at 2821 cm−1 in the CH stretch range. As expected, the AmaH+H2O spectrum reveals two bands in the OH stretch range, which are absent for AmaH+Ar. Bands X and Z at 3633 and 3717 cm−1 are readily assigned to the free symmetric
mode and the associated strong couplings, their relative integrated intensities are strongly enhanced (when compared to the free NH stretch bands). The CHn stretch pattern consists of one intense doublet C and D at ∼2860 and ∼2870 cm−1 with a total width of ∼45 cm−1 and a more intense and broader doublet E and F at ∼2910 and ∼2940 cm−1 with a total width of ∼60 cm−1. When compared to the AmaH+Ar spectrum, bands C are slightly blueshifted by ∼7 cm−1 while bands E are redshifted by ∼12 cm−1. The distinct blueshifts of the doublet C/D from Ar to H2O may arise from the assignment to different isomers (iminium for Ar and ammonium for H2O). With increasing n, only bands D and F show notable redshifts from 2874 (n = 1) to 2867 cm−1 (n = 4) and from 2943 (n = 1) to 2933 cm−1 (n = 4), respectively. However, in the AmaH+H2O spectrum an additional weak band B is observed at 2821 cm−1 in the CH stretch range. As expected, the AmaH+H2O spectrum reveals two bands in the OH stretch range, which are absent for AmaH+Ar. Bands X and Z at 3633 and 3717 cm−1 are readily assigned to the free symmetric  and antisymmetric
and antisymmetric  OH stretch modes of the H2O acceptor in the NH⋯O ionic H-bond. Their modest redshifts of 24 and 39 cm−1 from the transitions of bare H2O
OH stretch modes of the H2O acceptor in the NH⋯O ionic H-bond. Their modest redshifts of 24 and 39 cm−1 from the transitions of bare H2O 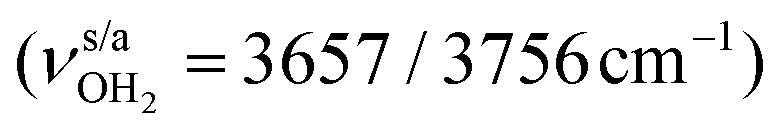 and the strong IR enhancement of
and the strong IR enhancement of 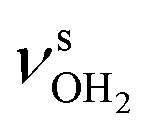 are typical for cation-H2O clusters.57,64,71,83,84,104 Interestingly, band Z is rather broad with a total with of 137 cm−1 and several additional features at 3711, 3759, and 3805 cm−1, which may arise from Q-branches of hindered internal H2O rotation, as has been reported previously for Ada+H2O. However, the spacings between these bands are not regular for AmaH+H2O and not associated directly with rotational constants of bare H2O, indicating that this motion is more complicated than in Ada+H2O. Finally, the weak band M of AmaH+H2O at 3193 cm−1 has a slightly higher frequency than the βNH3 overtone observed for AmaH+Ar (2βNH3 = 3185 cm−1 (M)), consistent with the stronger NH⋯O H-bond providing a larger retarding force by H2O for this mode.
are typical for cation-H2O clusters.57,64,71,83,84,104 Interestingly, band Z is rather broad with a total with of 137 cm−1 and several additional features at 3711, 3759, and 3805 cm−1, which may arise from Q-branches of hindered internal H2O rotation, as has been reported previously for Ada+H2O. However, the spacings between these bands are not regular for AmaH+H2O and not associated directly with rotational constants of bare H2O, indicating that this motion is more complicated than in Ada+H2O. Finally, the weak band M of AmaH+H2O at 3193 cm−1 has a slightly higher frequency than the βNH3 overtone observed for AmaH+Ar (2βNH3 = 3185 cm−1 (M)), consistent with the stronger NH⋯O H-bond providing a larger retarding force by H2O for this mode.
3.5 IRPD of AmaH+(H2O)2
The AmaH+(H2O)2 spectrum shows significant changes compared to n = 1, except for the CH stretch bands (C–F). Significantly, instead of two intense free NH stretch bands (N, P), only one free NH stretch band O at 3315 cm−1 dominates that spectral range, indicating the predominant presence of a n = 2 isomer, in which two H2O molecules bind separately to the NH3+ group via individual NH⋯O H-bonds while one N–H bond remains free (interior ion solvation motif). Band O occurs roughly midway between N and P, as expected for the change from two coupled free NH stretch oscillators to a single uncoupled one. The resulting bound NH stretch bands J and K are blueshifted by 62 and 47 cm−1 to 3069 and 3127 cm−1 compared to n = 1, respectively, and gain significantly in IR intensity. Because interior ion solvation is slightly noncooperative (due to enhanced delocalization of the positive charge), the NH⋯O H-bonds in n = 2 are somewhat weaker than in n = 1, resulting in stronger N–H donor bonds and smaller redshifts in the bonded NH stretch modes. As a result, the CH stretch band G observed in AmaH+Ar (νCH2 = 3756 cm−1) is for n = 2 no longer obscured and detected at 2977 cm−1 in the shoulder of band F. The free OH stretch bands X and Z remain at nearly the same frequencies of 3638 and 3716 cm−1 and are again attributed to and
and  of the individual uncoupled H2O ligands, respectively. The observed increase in relative IR intensity of both bands is attributed to the larger number of ligands. Interestingly, the number of sub-peaks of band Z increased even further at n = 2, with additional reproducible peaks at 3683, 3705, 3737, and 3772 cm−1, but again no simple internal rotation pattern with equidistant lines can be recognized.
of the individual uncoupled H2O ligands, respectively. The observed increase in relative IR intensity of both bands is attributed to the larger number of ligands. Interestingly, the number of sub-peaks of band Z increased even further at n = 2, with additional reproducible peaks at 3683, 3705, 3737, and 3772 cm−1, but again no simple internal rotation pattern with equidistant lines can be recognized.
Significantly, the OH stretch range provides clear evidence for the minor presence of a second isomer, in which a H-bonded (H2O)2 dimer with an OH⋯O H bond is attached to one of the NH proton donors of the NH3+ group (H-bonded solvent network motif). The broader band band T at 3403 cm−1 with a width of 39 cm−1 is observed in the spectral range of bound OH stretch modes  . Compared to bare (H2O)2 (3601 cm−1),105 the
. Compared to bare (H2O)2 (3601 cm−1),105 the  frequency is drastically reduced, which is due to the large cooperativity of the H-bonded network resulting from the strong polarization forces induced by the nearby positive charge of AmaH+. The associated free OH stretch mode
frequency is drastically reduced, which is due to the large cooperativity of the H-bonded network resulting from the strong polarization forces induced by the nearby positive charge of AmaH+. The associated free OH stretch mode  of the H2O donor can be identified at 3683 cm−1 (Y), which is also redshifted compared to bare (H2O)2 (3735 cm−1).105 The two free NH stretch bands of this isomer are expected close to bands N and P of n = 1. The signal in the n = 2 spectrum in this range is rather small (but visible), indicating that the population of this isomer is relatively small compared to the predominant interior ion solvation isomer.
of the H2O donor can be identified at 3683 cm−1 (Y), which is also redshifted compared to bare (H2O)2 (3735 cm−1).105 The two free NH stretch bands of this isomer are expected close to bands N and P of n = 1. The signal in the n = 2 spectrum in this range is rather small (but visible), indicating that the population of this isomer is relatively small compared to the predominant interior ion solvation isomer.
Finally, band M gains width and intensity and shifts somewhat to the blue by 37 cm−1 to 3230 cm−1 due to further retarding forces of the H2O ligands on the βNH3 mode. In addition, a broad but weak band A is observed at 2754 cm−1, while the weak band B disappears. However, the true intensity of band A is difficult to estimate because the laser intensity decreases rapidly in the lower frequency range.
3.6 IRPD of AmaH+(H2O)3
In the n = 3 spectrum, bands J and K are further blueshifted to 3118 and 3170 cm−1 compared to n = 2 and an additional band I appears at 3009 cm−1 with a width of 39 cm−1. All three bands are attributed to the modes of an isomer, in which all NH bonds are singly hydrated by H2O ligands. The slightly blueshifted peak O (3317 cm−1) is still visible but weak, indicative of a minor isomer with a single free NH bond of the NH3+ group. The associated band T, attributed to
modes of an isomer, in which all NH bonds are singly hydrated by H2O ligands. The slightly blueshifted peak O (3317 cm−1) is still visible but weak, indicative of a minor isomer with a single free NH bond of the NH3+ group. The associated band T, attributed to  of a (H2O)2 dimer unit, is also blueshifted to 3432 cm−1 and gains intensity, indicating the abundance increase of isomeric structures with (H2O)2 binding to the NH3+ group. Moreover, an additional weak band W appears at 3552 cm−1 with a width of 11 cm−1 in the weakly H-bonded OH stretch range, which can be attributed to a
of a (H2O)2 dimer unit, is also blueshifted to 3432 cm−1 and gains intensity, indicating the abundance increase of isomeric structures with (H2O)2 binding to the NH3+ group. Moreover, an additional weak band W appears at 3552 cm−1 with a width of 11 cm−1 in the weakly H-bonded OH stretch range, which can be attributed to a  mode of a (H2O)3 water ring structure attached to the NH3+ group. Compared to bare cyclic (H2O)3 (3533 cm−1),105 the frequency is slightly blueshifted by 19 cm−1. The bands X (3639 cm−1), Y (3680 cm−1), Z (3712 cm−1) associated with the free OH stretch modes have almost the same frequencies as for n = 2, although band Z broadens to 42 cm−1. The broad band M assigned to 2βNH3 is probably composed of several sharper transitions and shifted further to the blue by 38 cm−1 to 3268 cm−1 due to increased retarding forces. Transition A gains slightly in intensity and redshifts down to 2712 cm−1.
mode of a (H2O)3 water ring structure attached to the NH3+ group. Compared to bare cyclic (H2O)3 (3533 cm−1),105 the frequency is slightly blueshifted by 19 cm−1. The bands X (3639 cm−1), Y (3680 cm−1), Z (3712 cm−1) associated with the free OH stretch modes have almost the same frequencies as for n = 2, although band Z broadens to 42 cm−1. The broad band M assigned to 2βNH3 is probably composed of several sharper transitions and shifted further to the blue by 38 cm−1 to 3268 cm−1 due to increased retarding forces. Transition A gains slightly in intensity and redshifts down to 2712 cm−1.
3.7 IRPD of AmaH+(H2O)4
The n = 4 spectrum exhibits a somewhat poorer signal-to-noise ratio due to the reduced abundance of AmaH+(H2O)4 clusters produced in the ion source. Nonetheless, at least 16 peaks can still be identified. As mentioned above, the four CH stretch bands (C–F) have almost the same positions as in the n = 1–3 spectra, although bands D and E are narrower with widths of 4 and 3 cm−1, respectively. Bands I, J, and K attributed to the modes are again slightly blueshifted to 3058, 3144, and 3188 cm−1, respectively, and an additional band H appears at 2974 cm−1 with a width of 18 cm−1. In addition, band O disappears, indicating that at this cluster size all N–H bonds of the NH3+ group are fully solvated and that isomers with a free N–H bond are below the detection limit. The bands T and W at 3452 and 3554 cm−1 associated with
modes are again slightly blueshifted to 3058, 3144, and 3188 cm−1, respectively, and an additional band H appears at 2974 cm−1 with a width of 18 cm−1. In addition, band O disappears, indicating that at this cluster size all N–H bonds of the NH3+ group are fully solvated and that isomers with a free N–H bond are below the detection limit. The bands T and W at 3452 and 3554 cm−1 associated with  modes are also blueshifted. Band W exhibits increased intensity, indicating an increasing abundance of isomers with cyclic water ring structures bound to the NH3+ group. Moreover, an additional band S is observed at 3387 cm−1 with a width of 13 cm−1 in the range of
modes are also blueshifted. Band W exhibits increased intensity, indicating an increasing abundance of isomers with cyclic water ring structures bound to the NH3+ group. Moreover, an additional band S is observed at 3387 cm−1 with a width of 13 cm−1 in the range of  modes. The free OH stretch bands X–Z are hardly shifted, with frequencies of 3642, 3683, and 3705 cm−1. The broad band M remains almost at the same frequency at 3271 cm−1, although it is more difficult to analyze because of the lower signal. Band A is again further redshifted by 32 cm−1 down to 2680 cm−1.
modes. The free OH stretch bands X–Z are hardly shifted, with frequencies of 3642, 3683, and 3705 cm−1. The broad band M remains almost at the same frequency at 3271 cm−1, although it is more difficult to analyze because of the lower signal. Band A is again further redshifted by 32 cm−1 down to 2680 cm−1.
In summary, the IRPD spectra of AmaH+(H2O)n with n = 1–4 can be interpreted with a predominant sequential cluster growth in which the three NH protons of the NH3+ group of the predominant N-protonated ammonium isomer of AmaH+ are gradually solvated with H2O molecules. This view is justified by the observation of the multi-band pattern F, J, and K, which is typical for NH⋯O ionic H-bonds, as well as by the progressive disappearance of the free N–H stretch bands (N, P, O). As the number of H2O ligands increases, the presence of solvent network isomers also increases, as evidenced by the increased intensity of bands T and W. The population of open-cage iminium isomers is very low, because the signal strength in the range of the characteristic iminium NH stretch transitions of the NH2+ group (Q, R, V) is very low.
4. Computational results and assignments
4.1 Ama, Ama+, AmaH+
To determine the AmaH+(H2O)n=1–4 structures responsible for the recorded IRPD spectra, it is required to first consider the relevant monomer isomers of Ama, Ama+(I–III), and AmaH+(I–IV), which have been recently studied in detail by the same computational and spectroscopic approach.47–49 Their structures and NBO charge distributions are shown in ESI† (Fig. S4, ESI†), and we briefly review the information relevant for the present work. Neutral Ama (1A1, Cs), with its NH2 group attached to the adamantyl cage (C10H15) in pyramidal configuration (sp3 hybridisation of N), has short N–H bonds leading to relatively high NH stretch frequencies with rather low IR intensity (Ia/s = 3/0.2 km mol−1). The structure and IR spectrum calculated for Ama agree well with previous experimental and computational data.51–54
with rather low IR intensity (Ia/s = 3/0.2 km mol−1). The structure and IR spectrum calculated for Ama agree well with previous experimental and computational data.51–54
The energetically favourable protonation site of Ama is the basic NH2 group with a calculated proton affinity of PA = 957 kJ mol−1, which is in good agreement with the accepted experimental value (PA = 949 kJ mol−1).91 The resulting ammonium ion (I, C2v, 1A1) is the global minimum on the ground state potential (E0 = 0 kJ mol−1). N-protonation leads to a drastic elongation of the N–H bonds by 7 mÅ to 1.022 Å, resulting in massive redshift of the  modes of ∼70/60 cm−1
modes of ∼70/60 cm−1 and a drastic increase in their IR intensities (by a factor of ∼10/500) due to the increased charge on the NH protons (qH = 0.429 e). The NH2 angles increase slightly to 108.1°, while preserving the pyramidal configuration. Due to the higher positive charge of the NH3+ group (qNH3 = 0.605 e) and its electron-withdrawing character, the neighbouring C–N bond is elongated by 77 mÅ to 1.544 Å and the adjacent C–C bonds contract compared to Ama (rC1C2 = 1.530 vs. 1.538 Å, rC1C3 = 1.530 vs. 1.544 Å), while the C–C bonds parallel to the C3 symmetry axis are slightly stretched (rC4C5 = 1.544 vs. 1.538 Å). Most C–H bonds contract (ΔrCH = 2 mÅ), resulting in small blueshifts of the νCH modes (νCH = 2890 vs. 2899 cm−1, νCH = 2917 vs. 2956 cm−1, Table S1, ESI†).
and a drastic increase in their IR intensities (by a factor of ∼10/500) due to the increased charge on the NH protons (qH = 0.429 e). The NH2 angles increase slightly to 108.1°, while preserving the pyramidal configuration. Due to the higher positive charge of the NH3+ group (qNH3 = 0.605 e) and its electron-withdrawing character, the neighbouring C–N bond is elongated by 77 mÅ to 1.544 Å and the adjacent C–C bonds contract compared to Ama (rC1C2 = 1.530 vs. 1.538 Å, rC1C3 = 1.530 vs. 1.544 Å), while the C–C bonds parallel to the C3 symmetry axis are slightly stretched (rC4C5 = 1.544 vs. 1.538 Å). Most C–H bonds contract (ΔrCH = 2 mÅ), resulting in small blueshifts of the νCH modes (νCH = 2890 vs. 2899 cm−1, νCH = 2917 vs. 2956 cm−1, Table S1, ESI†).
Although protonation of Ama at a C atom is energetically very unfavourable (as it would lead to a nonclassical fivefold coordinated C atom), at least one isomer with an iminium group (NH2+) instead of an ammonium group (NH3+) has been identified by Ar-tagging IRPD, AmaH+(II), which must therefore also be taken into account when studying microhydration of AmaH+.49 This open-cage isomer II is likely formed by addition of an H radical to the open-cage Ama+ radical cation isomer, Ama+(III),49 which is formed by EI of Ama in addition to the canonical Ama+(I) cage isomer.47,48AmaH+(II) (1A’, Cs, E0 = 3.0 kJ mol−1) has also an open-cage structure with a C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N double bond (1.297 Å), but with a C9H group instead of the CR3˙ radical centre and a much wider open cage (θC1C9C3 = 155.3° vs. 106.1°, rC1⋯C3 = 4.47 vs. 3.35 Å, Fig. 1). The C9–H bond is oriented toward the nearly planar CNH2+ group (rC9H⋯C1 = 2.450 Å) and the C–C bonds at the C9 atom are drastically stretched compared to Ama+(III) (rC3C9 = 1.482 vs. 1.529 Å, rC9C8 = 1.492 vs. 1.536 Å). The newly formed C9–H bond (rC9H = 1.098 Å) is the longest C–H bond (rCH = 1.090–1.098 Å) and its intense νCH mode (νCH = 2848 cm−1) is therefore significantly redshifted from the other νCHn modes. The N–H bonds are much shorter compared to those of the NH3+ group of AmaH+(I) (rNH = 1.012 vs. 1.022 Å), resulting in νNH2 modes at
N double bond (1.297 Å), but with a C9H group instead of the CR3˙ radical centre and a much wider open cage (θC1C9C3 = 155.3° vs. 106.1°, rC1⋯C3 = 4.47 vs. 3.35 Å, Fig. 1). The C9–H bond is oriented toward the nearly planar CNH2+ group (rC9H⋯C1 = 2.450 Å) and the C–C bonds at the C9 atom are drastically stretched compared to Ama+(III) (rC3C9 = 1.482 vs. 1.529 Å, rC9C8 = 1.492 vs. 1.536 Å). The newly formed C9–H bond (rC9H = 1.098 Å) is the longest C–H bond (rCH = 1.090–1.098 Å) and its intense νCH mode (νCH = 2848 cm−1) is therefore significantly redshifted from the other νCHn modes. The N–H bonds are much shorter compared to those of the NH3+ group of AmaH+(I) (rNH = 1.012 vs. 1.022 Å), resulting in νNH2 modes at  well separated (by 100 cm−1) from those of the NH3+ group. Although the positive charge of the NH2+ group (qNH2 = 0.210 e) is much lower than that of the NH3+ group of Ama+(I) (qNH3 = 0.604 e), the NH2+ group is still very attractive for hydration, because of similar positive charges of the protons (qH = 0.419 vs. 0.427 e). However, the high positive charge of the adjacent C1 atom (qC1 = 0.538 e) also influences the attachment of H2O ligands. In addition to isomer II, two further open-cage structures, AmaH+(III) and AmaH+(IV), are calculated (Fig. S4, ESI†), which differ structurally only slightly from isomer II.49 Both isomers show similar solvation behaviour as II with nearly identical IR spectra and can thus experimentally not be distinguished (Fig. S5, ESI†). For this reason, these structures and their hydrated clusters will not be discussed in detail here. This procedure may be justified by their much higher relative energy (E0 > 27 kJ mol−1). For completeness, the structures, IR spectra, and energies of all considered AmaH+(I–IV)(H2O)n=1–4 isomers are available in ESI† (Fig. S6–S25 and Tables S2–S6), along with their NBO charge distribution (Fig. S26–S29, ESI†).
well separated (by 100 cm−1) from those of the NH3+ group. Although the positive charge of the NH2+ group (qNH2 = 0.210 e) is much lower than that of the NH3+ group of Ama+(I) (qNH3 = 0.604 e), the NH2+ group is still very attractive for hydration, because of similar positive charges of the protons (qH = 0.419 vs. 0.427 e). However, the high positive charge of the adjacent C1 atom (qC1 = 0.538 e) also influences the attachment of H2O ligands. In addition to isomer II, two further open-cage structures, AmaH+(III) and AmaH+(IV), are calculated (Fig. S4, ESI†), which differ structurally only slightly from isomer II.49 Both isomers show similar solvation behaviour as II with nearly identical IR spectra and can thus experimentally not be distinguished (Fig. S5, ESI†). For this reason, these structures and their hydrated clusters will not be discussed in detail here. This procedure may be justified by their much higher relative energy (E0 > 27 kJ mol−1). For completeness, the structures, IR spectra, and energies of all considered AmaH+(I–IV)(H2O)n=1–4 isomers are available in ESI† (Fig. S6–S25 and Tables S2–S6), along with their NBO charge distribution (Fig. S26–S29, ESI†).
In our previous work, we provide a detailed vibrational and isomer-specific assignment of the bands C–G, L, M, N, P, Q, U, and V of the AmaH+Ar spectrum.49 Briefly, the influence of Ar ligands is negligible justifying a direct assignment of the AmaH+Ar bands to the ammonium (AmaH+(I)) and iminium (AmaH+(II)) isomers. Most of the intense bands are readily assigned to the most stable AmaH+(I) isomer (Table S7, ESI†). Bands N (3238 cm−1) and P (3317 cm−1) are attributed to  of the NH3+ group (3214 and 3301 cm−1), bands D (2872 cm−1) and F (2949 cm−1) to the convoluted peaks of the νCHn modes (2899 and 2956 cm−1), and the small peaks M (3185 cm−1) and L (3164 cm−1) to 2βNH3 overtones (3222 cm−1). On the other hand, the bands Q, U, and V cannot be explained by AmaH+(I) but arise from the iminium isomer AmaH+(II). Specifically, bands Q (3344 cm−1) and V (3451 cm−1) agree well with
of the NH3+ group (3214 and 3301 cm−1), bands D (2872 cm−1) and F (2949 cm−1) to the convoluted peaks of the νCHn modes (2899 and 2956 cm−1), and the small peaks M (3185 cm−1) and L (3164 cm−1) to 2βNH3 overtones (3222 cm−1). On the other hand, the bands Q, U, and V cannot be explained by AmaH+(I) but arise from the iminium isomer AmaH+(II). Specifically, bands Q (3344 cm−1) and V (3451 cm−1) agree well with  of the NH2+ group of AmaH+(II) (3331 and 3435 cm−1), while band U (3428 cm−1) is assigned to 2βNH2 (3328 cm−1). Finally, bands C (2853 cm−1), E (2921 cm−1), F (2949 cm−1), and G (2973 cm−1) are attributed to the convoluted peaks of the νCHn modes of AmaH+(II) (2848, 2913, 2952, 2977 cm−1). The population ratio for AmaH+(I) and AmaH+(II) is estimated as 4:1 or 3:1 from computed and measured relative band intensities.
of the NH2+ group of AmaH+(II) (3331 and 3435 cm−1), while band U (3428 cm−1) is assigned to 2βNH2 (3328 cm−1). Finally, bands C (2853 cm−1), E (2921 cm−1), F (2949 cm−1), and G (2973 cm−1) are attributed to the convoluted peaks of the νCHn modes of AmaH+(II) (2848, 2913, 2952, 2977 cm−1). The population ratio for AmaH+(I) and AmaH+(II) is estimated as 4:1 or 3:1 from computed and measured relative band intensities.
4.2 AmaH+H2O
Starting from the AmaH+ isomers I–IV (Fig. S4 and Table S2, ESI†),49 stable AmaH+(H2O)n structures are constructed by adding H2O ligands to the acidic N–H bonds or to the less positively charged adamantyl cage (C10H15). All obtained monohydrated structures exhibit a favorable charge–dipole configuration, with the electronegative O atom of H2O directed toward AmaH+. Two I-H2O and four II-H2O structures are found (Table S3 and Fig. S6, S7, ESI†). The two most stable isomers in each case are compared to Ama, AmaH+(I), and AmaH+(II) in Fig. 1, while their computed IR spectra are compared with the experimental IRPD spectrum in Fig. 3. The position and widths of the transitions observed in the IRPD spectrum of AmaH+H2O are listed in Table 2, along with their vibrational and isomer assignment. In the most stable isomers I-H2O(I) and II-H2O(II), H2O forms a strong NH⋯O ionic H-bond to the NH2+ or NH3+ group, while in the others it is bound to the adamantyl cage by weaker charge–dipole forces, resulting in much higher relative energies (E0 > 30 kJ mol−1). | ||
| Fig. 3 IRPD spectrum of AmaH+H2O compared to linear IR absorption spectra of AmaH+(I,II)(H2O)(I,II) calculated at the B3LYP-D3/cc-pVTZ level. The positions of the transition observed in the IRPD spectrum of AmaH+H2O and their vibrational assignment are listed in Table 2. Differences in relative energy (E0, G) are given in kJ mol−1. Note the different IR intensity scale for the computed IR spectra. The bands that can only be assigned to the iminium isomer II-H2O(I) are highlighted in red. | ||
| AmaH+H2O | Modeb | I-H2O(I) | II-H2O(I) | H2O C 2v |
|---|---|---|---|---|
a IR intensities in km mol−1 are given in parentheses.
b Stretching (ν), bending (β).
c Coupled 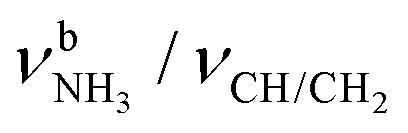 modes.
d FR = Fermi resonance with modes.
d FR = Fermi resonance with  .
e Coupled βNH3/βOH2. .
e Coupled βNH3/βOH2.
|
||||
| B 2821 (8) | ν CH | 2867 (49) | ||
| C 2860 (20) | ν CH/CH2 | 2890 (12) | 2886 (3) | |
| 2895 (19) | 2892 (11) | |||
| 2900 (44) | 2905 (19) | |||
| D 2874 (9) | ν CH/CH2 | 2913 (27) | 2907 (7) | |
| 2914 (27) | 2913 (41) | |||
| 2915 (1) | 2914 (10) | |||
| E 2906 (19) | ν CH/CH2 | 2930 (22) | 2939 (12) | |
| F 2943 (18) | ν CH/CH2, FRd | 2940 (14) | 2942 (25) | |
| 2942 (4) | 2948 (20) | |||
| 2948 (44) | 2949 (16) | |||
| 2948 (44) | 2952 (17) | |||
| 2949 (27) | 2962 (19) | |||
| 2951 (20) | 2972 (3) | |||
| 2955 (2) | 2974 (30) | |||
| 2955 (22) | 2978 (20) | |||

|
2921 (682) | |||
| J 3007 (24) |

|
2921 (682) | ||
| ν CH/CH2 , FRd | 2961 (204) | |||
| K 3080 (23) |

|
3044 (1047) | ||
| M 3193 (13) | 2βNH3e | 3198 | ||
| 2βOH2 | 3204 | |||
| N 3279 (17) |

|
3258 (43) | ||
| P 3328 (20) |

|
3312 (38) | ||
| R 3373 (8) |

|
3391 (87) | ||
| X 3633 (22) |

|
3650 (55) | 3649 (48) | 3662 (3) |
| Z 3717 (14) |

|
3736 (116) | 3736 (113) | 3759 (41) |
In the I-H2O(I) global minimum, H2O forms a strong and nearly linear NH⋯O H-bond (175.7°) to the NH3+ group with a bond length of 1.742 Å and a high binding energy of 70.9 kJ mol−1. Upon hydration, the N–H donor bond stretches drastically by 21 mÅ to 1.043 Å and the NH2 angle increases slightly to 108.5°, resulting in a massive redshift of the  mode by 380 cm−1 to 2921 cm−1, associated with a huge increase (by a factor of 13) in IR intensity (I = 682 km mol−1). These structural and spectroscopic effects are typical for NH⋯O ionic H-bonds in RNH3+-H2O dimers.79,106,107 The two remaining free N–H bonds contract slightly by 2 mÅ to 1.020 Å, resulting in modest
mode by 380 cm−1 to 2921 cm−1, associated with a huge increase (by a factor of 13) in IR intensity (I = 682 km mol−1). These structural and spectroscopic effects are typical for NH⋯O ionic H-bonds in RNH3+-H2O dimers.79,106,107 The two remaining free N–H bonds contract slightly by 2 mÅ to 1.020 Å, resulting in modest  blueshifts of 44/11 cm−1 to 3258/3312 cm−1, respectively. The C–N bond is shortened by 14 mÅ to 1.530 Å compared to I, while the other C–C and C–H bonds change only slightly (ΔrCC < 3 mÅ, ΔrCH < 2 mÅ). The NBO analysis reveals a partial charge transfer of 42 me from AmaH+ to H2O and E(2) = 75.2 kJ mol−1 for the donor–acceptor interaction between the lone pairs of O and the antibonding σ* orbital of the N–H donor bond involved in the NH⋯O ionic H-bond (Fig. S26, ESI†). As a result, the O–H bonds slightly elongate by 2 mÅ to 0.963 Å and the OH2 bond angle opens from 104.5° to 106.0°, leading to a rise in IR intensity of the νOH2 modes (by a factor of 18/3) and a redshift of their frequencies by 12/23 cm−1 down to
blueshifts of 44/11 cm−1 to 3258/3312 cm−1, respectively. The C–N bond is shortened by 14 mÅ to 1.530 Å compared to I, while the other C–C and C–H bonds change only slightly (ΔrCC < 3 mÅ, ΔrCH < 2 mÅ). The NBO analysis reveals a partial charge transfer of 42 me from AmaH+ to H2O and E(2) = 75.2 kJ mol−1 for the donor–acceptor interaction between the lone pairs of O and the antibonding σ* orbital of the N–H donor bond involved in the NH⋯O ionic H-bond (Fig. S26, ESI†). As a result, the O–H bonds slightly elongate by 2 mÅ to 0.963 Å and the OH2 bond angle opens from 104.5° to 106.0°, leading to a rise in IR intensity of the νOH2 modes (by a factor of 18/3) and a redshift of their frequencies by 12/23 cm−1 down to  , respectively.
, respectively.
In the high-energy I-H2O(II) isomer (E0 = 44.8 kJ mol−1), H2O binds with a low binding energy of 26.1 kJ mol−1 to CH(2) groups opposite to the NH3+ group of the adamantyl cage of Ivia three weak CH⋯O contacts based mostly on charge–dipole forces. Due to the weak and strongly nonlinear CH⋯O contacts with intermolecular distances greater than 2.5 Å, the charge transfer from I to H2O (Δq = 6 me) and the impact of hydration on the monomer structure are negligible, except for the slight contractions of the involved C–H bonds (ΔrCH = 2 mÅ). As a result, the IR spectrum of I-H2O(II) is essentially a combination of the spectra of I and bare H2O.
The most stable iminium monohydrate II-H2O(I) is 8.3 kJ mol−1 above the most stable ammonium hydrate I-H2O(I). Thus, monohydration increases the ammonium–iminium gap by 5 kJ mol−1 because the NH2+ group is less acidic than the NH3+ group leading to weaker NH⋯O H-bonds. In II-H2O(I), H2O binds to NH2+ group via a strong and nearly linear NH⋯O H-bond (172.0°) with a binding energy of 65.6 kJ mol−1. As a result, the NH⋯O H-bond (1.770 Å) is slightly longer and the N–H bond (1.031 Å) is less elongated compared to I-H2O(I) (ΔrNH = 19 vs. 21 mÅ), resulting in a less pronounced redshift of the 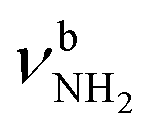 mode to 3044 cm−1 (287 vs. 380 cm−1) and a slightly smaller increase in IR intensity (factor 7). However, the absolute IR activity of the
mode to 3044 cm−1 (287 vs. 380 cm−1) and a slightly smaller increase in IR intensity (factor 7). However, the absolute IR activity of the 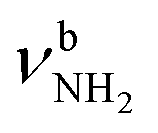 mode is significantly higher than that of the
mode is significantly higher than that of the  mode (I = 1047 vs. 682 km mol−1). The charge transfer to H2O and orbital interaction energy are slightly smaller than in I-H2O(I) (Δq = 39 vs. 42 me, E(2)= 68.3 vs. 75.2 kJ mol−1, Fig. S26, ESI†). The NH2 bond angle increases slightly by 1.1° to 118.0° and the length of the free N–H bond remains at 1.012 Å, resulting in
mode (I = 1047 vs. 682 km mol−1). The charge transfer to H2O and orbital interaction energy are slightly smaller than in I-H2O(I) (Δq = 39 vs. 42 me, E(2)= 68.3 vs. 75.2 kJ mol−1, Fig. S26, ESI†). The NH2 bond angle increases slightly by 1.1° to 118.0° and the length of the free N–H bond remains at 1.012 Å, resulting in  at 3391 cm−1. The OH2 bond angle opens slightly from 104.5° to 105.8° and the O–H bonds are slightly stretched, leading to the same redshifts in νOH2 as for I-H2O(I),
at 3391 cm−1. The OH2 bond angle opens slightly from 104.5° to 105.8° and the O–H bonds are slightly stretched, leading to the same redshifts in νOH2 as for I-H2O(I),  ). Upon hydration, the CN double bond of II is slightly shortened by 4 mÅ to 1.293 Å and the adjacent C–C bonds are somewhat elongated (rC1C2/C4 = 1.488/1.489 vs. 1.486 Å). The θC1C9C3 angle opens slightly from 155.3° to 155.7° and the C9–H bond contracts slightly, leading to a small blueshift of the associated νCH mode closer to the range of the other νCH(2) modes (νCH = 2867 vs. 2848 cm−1). Most of the other C–C and C–H bonds do not change significantly.
). Upon hydration, the CN double bond of II is slightly shortened by 4 mÅ to 1.293 Å and the adjacent C–C bonds are somewhat elongated (rC1C2/C4 = 1.488/1.489 vs. 1.486 Å). The θC1C9C3 angle opens slightly from 155.3° to 155.7° and the C9–H bond contracts slightly, leading to a small blueshift of the associated νCH mode closer to the range of the other νCH(2) modes (νCH = 2867 vs. 2848 cm−1). Most of the other C–C and C–H bonds do not change significantly.
In the second iminium monohydrate, II-H2O(II) at E0 = 30.7 kJ mol−1, H2O binds to the adamantyl cage via weak CH⋯O contacts, which are substantially shorter (2.39 Å) and stronger (43.2 kJ mol−1) than for I-H2O(II). The reason is probably the high positive charge on C1 (qC1 = 0.556 e), to which H2O is attracted at a distance of 3.27 Å. Nevertheless, charge transfer to H2O is again quite small (Δq = 9 me) and the effects on monomer structure and IR spectrum of II are negligible. The structures and IR spectra of the less favourable iminium isomers II-H2O(III,IV) are very similar to those of II-H2O(II) (Fig. S7, ESI†) and not discussed because of their relatively high energies (E0 > 43 kJ mol−1).
The IRPD spectrum of AmaH+H2O is compared in Fig. 3 to linear IR absorption spectra calculated for the four considered I/II-H2O(I,II) isomers. At first glance, most bands of the IRPD spectrum can readily be assigned to the most stable I-H2O(I) isomer (Table 2). The typical signature of a NH⋯O ionic H-bond is observed as a broad multi-band pattern (F, J, K) that can be associated with the redshifted 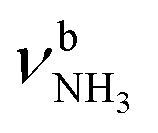 mode of I-H2O(I) and its coupled modes (FR and/or local mode mixing).77,78,81,82,103 Thus, an assignment of the intense bands F, J, and K to specific modes of I-H2O(I) is not trivial. Since bands C–F were already observed in the AmaH+Ar spectrum and assigned to νCH(2) modes, they are again attributed to νCH(2) modes here, while band J at 3007 cm−1 is mostly associated with the
mode of I-H2O(I) and its coupled modes (FR and/or local mode mixing).77,78,81,82,103 Thus, an assignment of the intense bands F, J, and K to specific modes of I-H2O(I) is not trivial. Since bands C–F were already observed in the AmaH+Ar spectrum and assigned to νCH(2) modes, they are again attributed to νCH(2) modes here, while band J at 3007 cm−1 is mostly associated with the  mode predicted at 2921 cm−1. Band K at 3080 cm−1 is then explained by a FR. Bands N and P (3279 and 3328 cm−1) are unambiguously assigned to the two remaining free
mode predicted at 2921 cm−1. Band K at 3080 cm−1 is then explained by a FR. Bands N and P (3279 and 3328 cm−1) are unambiguously assigned to the two remaining free  modes of I-H2O(I) predicted at
modes of I-H2O(I) predicted at 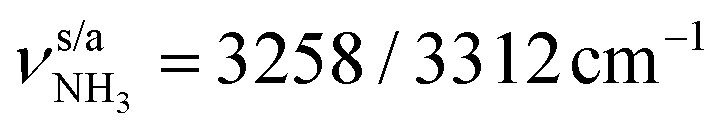 with deviations of 21/16 cm−1, respectively. Moreover, bands X and Z observed at 3633 and 3717 cm−1 agree well with the free
with deviations of 21/16 cm−1, respectively. Moreover, bands X and Z observed at 3633 and 3717 cm−1 agree well with the free  modes of I-H2O(I) computed at 3650/3736 cm−1. As for AmaH+Ar,49 band M at 3193 cm−1 is attributed to the 2βNH3 overtone of I-H2O(I) estimated at 3198 cm−1, which is slightly blueshifted by Ar → H2O substitution consistent with the larger retarding force arising from the stronger H-bond to H2O. Hence, with the exception of the weak bands B and R, the IRPD spectrum of AmaH+H2O can be fully explained by the I-H2O(I) global minimum. As bands B and R are weak, they may also be attributed to overtone or combination bands, or in the case of band B even to a CH stretch fundamental of I-H2O(I), although the latter assignment gives a large discrepancy between calculation and experiment (69 cm−1). Thus, in an alternative and strongly favoured second scenario, these bands are assigned to transitions of a minor population of the II-H2O(I) iminium isomer, whose monomer structure was also detected in the AmaH+Ar spectrum.49 Specifically, band R at 3373 cm−1 is associated with the
modes of I-H2O(I) computed at 3650/3736 cm−1. As for AmaH+Ar,49 band M at 3193 cm−1 is attributed to the 2βNH3 overtone of I-H2O(I) estimated at 3198 cm−1, which is slightly blueshifted by Ar → H2O substitution consistent with the larger retarding force arising from the stronger H-bond to H2O. Hence, with the exception of the weak bands B and R, the IRPD spectrum of AmaH+H2O can be fully explained by the I-H2O(I) global minimum. As bands B and R are weak, they may also be attributed to overtone or combination bands, or in the case of band B even to a CH stretch fundamental of I-H2O(I), although the latter assignment gives a large discrepancy between calculation and experiment (69 cm−1). Thus, in an alternative and strongly favoured second scenario, these bands are assigned to transitions of a minor population of the II-H2O(I) iminium isomer, whose monomer structure was also detected in the AmaH+Ar spectrum.49 Specifically, band R at 3373 cm−1 is associated with the  mode (predicted at 3391 cm−1) of II-H2O(I) and then the redshifted
mode (predicted at 3391 cm−1) of II-H2O(I) and then the redshifted  mode (predicted at 3044 cm−1) is overlapping with band K at 3080 cm−1 with deviations of 18 and 36 cm−1, respectively. Moreover, band B at 2821 cm−1 arises from the intense and low-frequency νCH mode of the long C9–H bond of II-H2O(I) predicted at 2867 cm−1. Following this second scenario, the relative population of II-H2O(I) can roughly be estimated as <10% of I-H2O(I) by considering the computed and observed integrated peak intensities of the isolated peaks N, P, and R. The two much less stable isomers I-H2O(II) and I-H2O(II) can be excluded as noticeable carrier of the IRPD spectrum, because their free νNH2/3 modes are not observed and have a predicted IR intensity ratio in large disagreement with experiment. In summary, the IRPD spectrum clearly shows the predominant presence of I-H2O(I) and (at most) only a small population of II-H2O(I). In both dimers, H2O binds to the AmaH+ isomers via strong ionic NH⋯O H-bonds, while there is no spectroscopic evidence for isomers with H2O attached to the adamantyl cage.
mode (predicted at 3044 cm−1) is overlapping with band K at 3080 cm−1 with deviations of 18 and 36 cm−1, respectively. Moreover, band B at 2821 cm−1 arises from the intense and low-frequency νCH mode of the long C9–H bond of II-H2O(I) predicted at 2867 cm−1. Following this second scenario, the relative population of II-H2O(I) can roughly be estimated as <10% of I-H2O(I) by considering the computed and observed integrated peak intensities of the isolated peaks N, P, and R. The two much less stable isomers I-H2O(II) and I-H2O(II) can be excluded as noticeable carrier of the IRPD spectrum, because their free νNH2/3 modes are not observed and have a predicted IR intensity ratio in large disagreement with experiment. In summary, the IRPD spectrum clearly shows the predominant presence of I-H2O(I) and (at most) only a small population of II-H2O(I). In both dimers, H2O binds to the AmaH+ isomers via strong ionic NH⋯O H-bonds, while there is no spectroscopic evidence for isomers with H2O attached to the adamantyl cage.
4.3 AmaH+(H2O)2
For the AmaH+(H2O)2 dihydrate, seven isomers are considered for both I and II (Fig. S10, S11 and Table S4, ESI†). In the following, only I-(H2O)2(I–III) and II-(H2O)2(I,II) are discussed, because the other isomers have an energy difference of >30 kJ mol−1 to the global minimum and are not necessary for explaining the IRPD spectrum. These most stable structures are shown in Fig. 4, while their computed IR spectra are compared in Fig. 5 with the IRPD spectrum. | ||
| Fig. 4 Calculated equilibrium structures (in Å and degrees) of (H2O)2, AmaH+(I)(H2O)2(I–III) and AmaH+(II)(H2O)2(I,II) in their ground electronic state (B3LYP-D3/cc-pVTZ). All bond lengths are shown in ESI† (Fig. S23). | ||
 | ||
| Fig. 5 IRPD spectrum of AmaH+(H2O)2 compared to linear IR absorption spectra of AmaH+(I)(H2O)2(I–III) and AmaH+(II)(H2O)2(I,II) calculated at the B3LYP-D3/cc-pVTZ level. The positions of the transition observed in the IRPD spectrum of AmaH+(H2O)2 and their vibrational assignment are listed in Table S8 (ESI†). Differences in relative energy (E0, G) are given in kJ mol−1. Note the different IR intensity scale for the computed IR spectra. | ||
In the I-(H2O)2(I) global minimum (Cs), both H2O molecules are symmetrically bound to the NH3+ group via equivalent N–H⋯O H-bonds (1.794 Å, 173.5°) and a total binding energy of 133.1 kJ mol−1, yielding 66.6 kJ mol−1 per H-bond. This binding motif corresponds to interior ion solvation. The NH2 angles decrease slightly to 108.0°, while the N–H donor bonds are stretched from 1.022 to 1.036 Å. Compared to I-H2O(I), the H-bonds are somewhat weaker and the resulting perturbation of the monomer properties are correspondingly smaller, because of the noncooperative effect typical for internal ion solvation. As a result, the  modes are less redshifted to 3007 and 3033 cm−1
modes are less redshifted to 3007 and 3033 cm−1 . The single remaining free N–H bond contracts slightly by 3 mÅ to 1.019 Å, resulting in a blueshift of
. The single remaining free N–H bond contracts slightly by 3 mÅ to 1.019 Å, resulting in a blueshift of  by 83 cm−1 to 3297 cm−1. Due to the two H2O ligands, the C–N bond contracts even more by 22 mÅ to 1.522 Å compared to I, while the other C–C bonds and C–H bonds change only slightly (ΔrCC < 3 mÅ, ΔrCH < 2 mÅ). The NBO analysis yields a total charge transfer from AmaH+ to the H2O ligands of 70 me and E(2) = 60.8 kJ mol−1 per H-bond (Fig. S27, ESI†). As a result, the O–H bonds are slightly elongated by 2 mÅ to 0.963 Å and the OH2 bond angle opens from 104.5° to 105.8°, leading to a redshift of their coupled free OH stretch modes to νOH2s = 3653/4 cm−1 and νOH2a = 3740/1 cm−1, respectively, along with an increase in IR intensity.
by 83 cm−1 to 3297 cm−1. Due to the two H2O ligands, the C–N bond contracts even more by 22 mÅ to 1.522 Å compared to I, while the other C–C bonds and C–H bonds change only slightly (ΔrCC < 3 mÅ, ΔrCH < 2 mÅ). The NBO analysis yields a total charge transfer from AmaH+ to the H2O ligands of 70 me and E(2) = 60.8 kJ mol−1 per H-bond (Fig. S27, ESI†). As a result, the O–H bonds are slightly elongated by 2 mÅ to 0.963 Å and the OH2 bond angle opens from 104.5° to 105.8°, leading to a redshift of their coupled free OH stretch modes to νOH2s = 3653/4 cm−1 and νOH2a = 3740/1 cm−1, respectively, along with an increase in IR intensity.
In the only slightly less stable isomer I-(H2O)2(II) with E0 = 4.9 kJ mol−1, a H-bonded (H2O)2 dimer is attached to the NH3+ group via a single NH⋯O ionic H-bond (D0 = 128.5 kJ mol−1). This binding motif corresponds to formation of a H-bonded solvent network. The (H2O)2 dimer is oriented toward the adamantyl cage, because of weak CH⋯O contacts. Due to the larger proton affinity of (H2O)2 compared to H2O (808 vs. 691 kJ mol−1),91,92 the NH⋯O H-bond is much stronger and shorter than in I-H2O(I) (1.653 vs. 1.742 Å). The affected N–H bond is drastically stretched by 31 mÅ to 1.053 Å due to the cooperative effects of the water solvent network, while the two remaining N–H bonds are slightly shortened to 1.020 Å. As a result, the  mode is strongly redshifted by 556 cm−1 down to 2745 cm−1 while
mode is strongly redshifted by 556 cm−1 down to 2745 cm−1 while 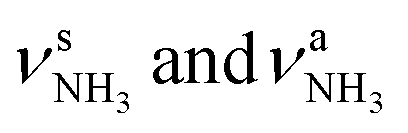 are slightly blueshifted to 3262 and 3320 cm−1, respectively. The total charge transfer is increased from 42 to 59 me and E(2) increases from 75.2 to 107.9 kJ mol−1 compared to I-H2O(I) (Fig. S27, ESI†). The affected NH2 angle increases to 108.7°, while the other NH2 angle decreases slightly to 107.9°. The C–N bond is shortened by 19 mÅ to 1.525 Å, while the other C–C bonds and C–H bonds are hardly affected by (H2O)2 dimer attachment (ΔrCC < 4 mÅ, ΔrCH < 3 mÅ). However, the C–H bonds pointing toward (H2O)2 are shortened due to the weak CH⋯O contacts, while the other C–H bonds maintain their length. The OH⋯O H-bond in I-(H2O)2(II) is much stronger and shorter than in bare (H2O)2 (1.772 vs. 1.946 Å), because of the strong cooperativity of the nonadditive three-body polarization forces caused by the AmaH+ charge. In addition, the binding energy of the terminal H2O is much higher compared to bare (H2O)2 (D0 = 57.4 vs.19.7 kJ mol−1) as is the E(2) energy of the OH⋯H H-bond (E(2) = 62.0 vs. 34.0 kJ mol−1). The O–H donor bond is stretched by 18 mÅ to 0.979 Å and the remaining free O–H bond is slightly shortened to 0.961 Å compared to I-H2O(I). As a result,
are slightly blueshifted to 3262 and 3320 cm−1, respectively. The total charge transfer is increased from 42 to 59 me and E(2) increases from 75.2 to 107.9 kJ mol−1 compared to I-H2O(I) (Fig. S27, ESI†). The affected NH2 angle increases to 108.7°, while the other NH2 angle decreases slightly to 107.9°. The C–N bond is shortened by 19 mÅ to 1.525 Å, while the other C–C bonds and C–H bonds are hardly affected by (H2O)2 dimer attachment (ΔrCC < 4 mÅ, ΔrCH < 3 mÅ). However, the C–H bonds pointing toward (H2O)2 are shortened due to the weak CH⋯O contacts, while the other C–H bonds maintain their length. The OH⋯O H-bond in I-(H2O)2(II) is much stronger and shorter than in bare (H2O)2 (1.772 vs. 1.946 Å), because of the strong cooperativity of the nonadditive three-body polarization forces caused by the AmaH+ charge. In addition, the binding energy of the terminal H2O is much higher compared to bare (H2O)2 (D0 = 57.4 vs.19.7 kJ mol−1) as is the E(2) energy of the OH⋯H H-bond (E(2) = 62.0 vs. 34.0 kJ mol−1). The O–H donor bond is stretched by 18 mÅ to 0.979 Å and the remaining free O–H bond is slightly shortened to 0.961 Å compared to I-H2O(I). As a result,  is strongly redshifted down to 3379 cm−1, consistent with a large increase in its IR intensity (factor ∼13), while
is strongly redshifted down to 3379 cm−1, consistent with a large increase in its IR intensity (factor ∼13), while  is slightly blueshifted to 3725 cm−1 and modes of the terminal H2O at 3648 and 3735 cm−1 nearly do not change.
is slightly blueshifted to 3725 cm−1 and modes of the terminal H2O at 3648 and 3735 cm−1 nearly do not change.
In I-(H2O)2(III) at E0 = 7.0 kJ mol−1, the two nonequivalent H2O ligands are attached to an N–H bond via NH⋯O H-bonds of different strength and connected to each other by a strongly bent and thus weak neutral OH⋯O H-bond. This binding motif corresponds to the formation of a cyclic H-bonded solvent network. The stronger and more linear NH⋯O H-bond has a bond length of 1.747 Å and a bond angle of 155.9°, while the weaker less linear one has 2.075 Å and 138.5°. The corresponding N–H bonds are stretched by 19 and 4 mÅ to 1.041 and 1.026 Å, respectively, resulting in redshifts of the corresponding  modes to 2953 and 3188 cm−1. The total charge transfer to the H2O ligands is 56 me (35 and 21 me) and the E(2) energies are 70.2 and 18.6 kJ mol−1 (Fig. S27, ESI†) for the strong and weak NH⋯O H-bond, respectively. The rather weak OH⋯O H-bond is strongly nonlinear (130.1°) and much longer (2.213 Å) than in bare (H2O)2, as also indicated by the low E(2) energy of 7.4 kJ mol−1. The involved O–H donor bond is only slightly more elongated by 5 mÅ to 0.966 Å than the other free O–H bonds with 0.962 and 0.964 Å. As a result, the strongly coupled νOH2 modes are slightly redshifted to
modes to 2953 and 3188 cm−1. The total charge transfer to the H2O ligands is 56 me (35 and 21 me) and the E(2) energies are 70.2 and 18.6 kJ mol−1 (Fig. S27, ESI†) for the strong and weak NH⋯O H-bond, respectively. The rather weak OH⋯O H-bond is strongly nonlinear (130.1°) and much longer (2.213 Å) than in bare (H2O)2, as also indicated by the low E(2) energy of 7.4 kJ mol−1. The involved O–H donor bond is only slightly more elongated by 5 mÅ to 0.966 Å than the other free O–H bonds with 0.962 and 0.964 Å. As a result, the strongly coupled νOH2 modes are slightly redshifted to  and
and 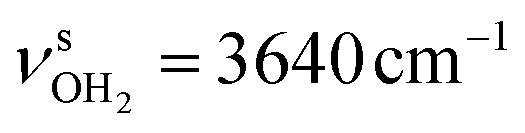 and to
and to  and
and  for the first and second H2O molecule. The effects on the adamantyl cage are again negligible, except for the contraction of the C–N bond from 1.544 Å to 1.520 Å.
for the first and second H2O molecule. The effects on the adamantyl cage are again negligible, except for the contraction of the C–N bond from 1.544 Å to 1.520 Å.
The most stable dihydrated iminium isomer, II-(H2O)2(I), lies 13.5 kJ mol−1 above the global minimum, I-(H2O)2(I), and has also Cs symmetry because both H2O ligands are symmetrically bound to the NH2+ group via separate NH⋯O H-bonds (1.823 Å, 170.5°) with a total binding energy of 122.7 kJ mol−1. As the NH2+ group is less acidic than the NH3+ group, dihydration increases the energy gap between ammonium and iminium isomers further. The total charge transfer of 64 me and E(2) energy per H-bond (55.3 kJ mol−1) are correspondingly smaller than in I-(H2O)2(I) (Fig. S27, ESI†). The N–H bonds elongate by 14 mÅ to 1.026 Å, resulting in  redshifts to 3088 and 3178 cm−1, along with an increase in IR intensity. The NH2 angle increases by 2.1° to 119.0°. The OH2 bond angle rises to 105.7° and the O–H bonds slightly elongate to 0.963 Å, resulting in small redshifts to
redshifts to 3088 and 3178 cm−1, along with an increase in IR intensity. The NH2 angle increases by 2.1° to 119.0°. The OH2 bond angle rises to 105.7° and the O–H bonds slightly elongate to 0.963 Å, resulting in small redshifts to  and
and  . H2O attachment slightly shortens the CN double bond by 7 mÅ to 1.290 Å, while the adjacent C–C bonds are stretched by 5 mÅ to 1.491 Å. However, the effect of the H2O attachment on the adamantyl cage is again negligible (ΔrCH < 1 mÅ, ΔrCC < 5 mÅ).
. H2O attachment slightly shortens the CN double bond by 7 mÅ to 1.290 Å, while the adjacent C–C bonds are stretched by 5 mÅ to 1.491 Å. However, the effect of the H2O attachment on the adamantyl cage is again negligible (ΔrCH < 1 mÅ, ΔrCC < 5 mÅ).
In II-(H2O)2(II) at E0 = 18.6 kJ mol−1, a H-bonded (H2O)2 dimer is attached to the NH2+ group of IIvia a NH⋯O ionic H-bond (D0 = 117.6 kJ mol−1, 1.700 Å, 164.2°, Δq = 56 me, E(2) = 89.8 kJ mol−1). The N–H donor bond is drastically stretched by 27 mÅ to 1.039 Å due to the cooperative effect of the H-bonded solvent network, while the free N–H bond slightly contracts by 1 mÅ to 1.011 Å. As a result,  is strongly redshifted by 521 cm−1 to 2914 cm−1, while
is strongly redshifted by 521 cm−1 to 2914 cm−1, while  is slightly blueshifted to 3399 cm−1, associated with an increase in IR intensities by a factor of 13 and 4, respectively. The OH⋯O H-bond of the attached (H2O)2 is longer and less linear than in I-(H2O)2(II) (1.794 vs. 1.772 Å, 165.7° vs. 168.4°). The involved O–H donor bond is stretched by 17 mÅ to 0.978 Å and the remaining free O–H bond slightly shortens to 0.961 Å compared to II-H2O(I). As a result,
is slightly blueshifted to 3399 cm−1, associated with an increase in IR intensities by a factor of 13 and 4, respectively. The OH⋯O H-bond of the attached (H2O)2 is longer and less linear than in I-(H2O)2(II) (1.794 vs. 1.772 Å, 165.7° vs. 168.4°). The involved O–H donor bond is stretched by 17 mÅ to 0.978 Å and the remaining free O–H bond slightly shortens to 0.961 Å compared to II-H2O(I). As a result,  is redshifted to 3399 cm−1, accompanied with an increase in intensity (factor ∼5) and
is redshifted to 3399 cm−1, accompanied with an increase in intensity (factor ∼5) and  is slightly blueshifted to 3725 cm−1, while
is slightly blueshifted to 3725 cm−1, while  of the terminal water remain at
of the terminal water remain at  and
and  . The CN double bond is slightly shortened by 6 mÅ to 1.291 Å, while the other C–C and C–H bonds are hardly affected (ΔrCC < 3 mÅ, ΔrCH < 3 mÅ).
. The CN double bond is slightly shortened by 6 mÅ to 1.291 Å, while the other C–C and C–H bonds are hardly affected (ΔrCC < 3 mÅ, ΔrCH < 3 mÅ).
Comparison of the measured IRPD spectrum of AmaH+(H2O)2 with the IR spectra computed for I-(H2O)2(I-III) and II-(H2O)2(I,II) in Fig. 5 (Table S8, ESI†) clearly shows the signature of the I-(H2O)2(I) global minimum. The isolated band O at 3315 cm−1 is a clear-cut sign of a single free NH stretch oscillator of an ammonium ion and agrees well with the predicted frequency 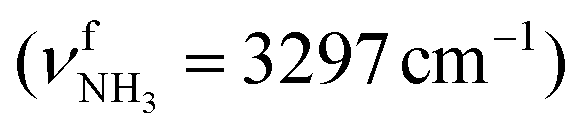 . Similar to AmaH+H2O, a specific assignment of the two
. Similar to AmaH+H2O, a specific assignment of the two  modes is difficult due to the FR. Tentatively, the broad band J (3069 cm−1) is predominantly assigned to the two
modes is difficult due to the FR. Tentatively, the broad band J (3069 cm−1) is predominantly assigned to the two  modes of I-(H2O)2(I) computed at 3007 and 3033 cm−1 with deviations of 62 and 36 cm−1, respectively. As for n = 1, band K (3127 cm−1) is associated with a FR. The corresponding νOH2 modes
modes of I-(H2O)2(I) computed at 3007 and 3033 cm−1 with deviations of 62 and 36 cm−1, respectively. As for n = 1, band K (3127 cm−1) is associated with a FR. The corresponding νOH2 modes  are attributed to bands X and Z at 3638 and 3716 cm−1. Band M at 3230 cm−1 with a large width is again assigned to 2βNH3 (3293 cm−1) and may also have contributions 2βOH2 (3200 and 3208 cm−1). As for n = 1, bands C-F can arise from νCH(2) of all present isomers, due to the small influence of hydration on the adamantyl cage. However, bands A and T at 2754 and 3403 cm−1 are not explainable by I-(H2O)2(I) and can, according to Fig. 5, only be attributed to I-(H2O)2(II). This isomer with an attached (H2O)2 has a strongly redshifted
are attributed to bands X and Z at 3638 and 3716 cm−1. Band M at 3230 cm−1 with a large width is again assigned to 2βNH3 (3293 cm−1) and may also have contributions 2βOH2 (3200 and 3208 cm−1). As for n = 1, bands C-F can arise from νCH(2) of all present isomers, due to the small influence of hydration on the adamantyl cage. However, bands A and T at 2754 and 3403 cm−1 are not explainable by I-(H2O)2(I) and can, according to Fig. 5, only be attributed to I-(H2O)2(II). This isomer with an attached (H2O)2 has a strongly redshifted  mode at 2745 cm−1 and a redshifted
mode at 2745 cm−1 and a redshifted  mode at 3379 cm−1 which both agree well with the observed bands A and T, with minor deviations of 9 and 24 cm−1, respectively. Band A is rather broad as expected for a strongly redshifted proton donor band and its experimental intensity may largely be underestimated by the low laser intensity in this spectral range. Moreover, band Y (3683 cm−1) can be assigned to
mode at 3379 cm−1 which both agree well with the observed bands A and T, with minor deviations of 9 and 24 cm−1, respectively. Band A is rather broad as expected for a strongly redshifted proton donor band and its experimental intensity may largely be underestimated by the low laser intensity in this spectral range. Moreover, band Y (3683 cm−1) can be assigned to  at 3725 cm−1 of I-(H2O)2(II) and the two free
at 3725 cm−1 of I-(H2O)2(II) and the two free 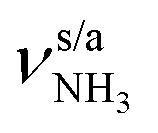 modes at 3262 and 3320 cm−1 are probably weakly present at N and P at 3288 and 3331 cm−1. Thus, the IRPD spectrum of AmaH+(H2O)2 can be fully explained by the major contribution of the most stable I-(H2O)2(I) isomer and a minor contribution of the next stable I-(H2O)2(II) isomer, whereby the population of the latter is estimated to be of the order of 20% from the observed and computed IR intensities of the free NH stretch modes. There is no need to invoke other higher-energy isomer isomers, although their minor presence cannot be ruled out completely. In particular, weak signal near 3600 cm−1 may indicate a small population of the cyclic I-(H2O)2(III) isomer. A significant population of dihydrated iminium isomers, II-(H2O)2, is not expected based on their high relative energy and their small population for n = 1. As the energy difference between iminium and ammonium isomers increases with n (3.0 < 8.3 < 13.5 < 21.5 kJ mol−1 for n = 0–3), we do not consider herein hydrated iminium isomers in detail for n ≥ 3 but focus only on I-(H2O)n clusters.
modes at 3262 and 3320 cm−1 are probably weakly present at N and P at 3288 and 3331 cm−1. Thus, the IRPD spectrum of AmaH+(H2O)2 can be fully explained by the major contribution of the most stable I-(H2O)2(I) isomer and a minor contribution of the next stable I-(H2O)2(II) isomer, whereby the population of the latter is estimated to be of the order of 20% from the observed and computed IR intensities of the free NH stretch modes. There is no need to invoke other higher-energy isomer isomers, although their minor presence cannot be ruled out completely. In particular, weak signal near 3600 cm−1 may indicate a small population of the cyclic I-(H2O)2(III) isomer. A significant population of dihydrated iminium isomers, II-(H2O)2, is not expected based on their high relative energy and their small population for n = 1. As the energy difference between iminium and ammonium isomers increases with n (3.0 < 8.3 < 13.5 < 21.5 kJ mol−1 for n = 0–3), we do not consider herein hydrated iminium isomers in detail for n ≥ 3 but focus only on I-(H2O)n clusters.
4.4 AmaH+(H2O)3
For trihydrated I, we find nine isomers (Fig. S14, S15 and Table S5, ESI†) but discuss in more detail only the structures and IR spectra of the four most stable isomers, I-(H2O)3(I–IV) in Fig. 6 and 7, because the other isomers are higher in energy (E0 > 12 kJ mol−1) and not necessary to explain the IRPD spectrum. In addition, for brevity we keep the discussion on the structures and IR spectra for the n = 3 and also n = 4 isomers shorter than for n = 0–2 and refer to ESI† for more details. In general, the structural, energetic, vibrational, and charge transfer trends, arising from cooperativity of the formation of H-bonded networks and noncooperativity of interior ion solvation discussed for n = 1–2 are also apparent for n = 3 and 4. The main change for the larger clusters with n ≥ 3 is that entropic effects have a decisive role on their relative energies. Hence, we discuss also free energies, which favor isomers with flexible hydration structures (e.g., single ligands) over rigid ones (e.g., cyclic rings or longer solvent chains interacting with the adamantyl cage). For example, while the energetic order for the four isomers in Fig. 6 is I-IV, with E0 = 0, 2.5, 4.4, and 11.9 kJ mol−1, their free energies vary as G0 = 0, −8,8, −1.2, and 12.8 kJ mol−1, i.e., II and III become more stable than I at elevated temperature due to entropy. This temperature effect may become an issue as cooling in the supersonic plasma expansion may be incomplete and not equilibrated (the effective temperature is difficult to estimate) and kinetic trapping may occur during the expansion. | ||
| Fig. 6 Calculated equilibrium structures (in Å and degrees) of AmaH+(I)(H2O)3(I-IV) in their ground electronic state (B3LYP-D3/cc-pVTZ). All bond lengths are shown in ESI† (Fig. S24). | ||
 | ||
| Fig. 7 IRPD spectrum of AmaH+(H2O)3 compared to linear IR absorption spectra of AmaH+(I)(H2O)3(I–IV) calculated at the B3LYP-D3/cc-pVTZ level. The positions of the transition observed in the IRPD spectrum of AmaH+(H2O)3 and their vibrational assignment are listed in Table S9 (ESI†). Differences in relative energy (E0, G) are given in kJ mol−1. Note the different IR intensity scale for the computed IR spectra. | ||
In the most stable I-(H2O)3(I) with Cs symmetry (E0/G = 0/0) and a total binding energy of 190.4 kJ mol−1, the three H2O ligands and the NH3+ group form a cyclic eight-membered ring with two equivalent O–H⋯O H-bonds (1.932 Å, 162.1°) and two equivalent but stronger N–H⋯O ionic H-bonds (1.765 Å, 161.5°). Two H2O ligands act as single-donor single-acceptor, while the terminal H2O molecule closing the ring acts as a double-acceptor ligand. To close the ring, the two H2O ligands involved in the NH⋯O H-bonds have to rotate into the ring plane but due to cooperativity of the formed ring, they are stronger and shorter than in I-(H2O)2(I). Due to higher strain and resulting larger nonlinearity as compared to I-(H2O)2(II) with a (H2O)2 chain, the OH⋯O H-bonds are weaker and longer. Compared to cyclic I-(H2O)2(III) with a cyclic six-membered ring, the addition of one ligand into the ring decreases strain and thus the OH⋯O H-bonds are stronger and more linear. The strengths of the H-bonds correlate with the charge transfer and E(2) energies and have direct impact on the perturbation of the intramolecular O–H and N–H donor bonds and resulting frequency shifts. The IR spectrum of I-(H2O)3(I) is characterized by two intense and moderately redshifted bound OH stretch bands at  , two intense and strongly redshifted bound NH stretch bands at
, two intense and strongly redshifted bound NH stretch bands at  , and a weaker and nearly unshifted free NH stretch band at
, and a weaker and nearly unshifted free NH stretch band at  .
.
The second most stable I-(H2O)3(II) isomer on the potential energy surface (E0/G = 2.5/−8.8 kJ mol−1) becomes the global minimum on the free energy surface. It has three single H2O ligands attached to the NH3+ group via three equivalent NH⋯O ionic H-bonds (1.840 Å, 172.4°), leading to a structure with (near) C3v symmetry and a total binding energy of D0 = 187.9 kJ mol−1. Due to noncooperativity of interior ion solvation, the three H-bonds of the fully solvated NH3+ group are weaker than those of the corresponding n = 2 and 1 isomers (1.742 < 1.794 < 1.840 Å for n = 1–3), as also seen in the corresponding E(2) energies (75.2 > 60.8 > 50.3 kJ mol−1) and the smaller redshifts in  . Due to its high symmetry, the IR spectrum of this isomer is rather simple and characterized by three intense redshifted
. Due to its high symmetry, the IR spectrum of this isomer is rather simple and characterized by three intense redshifted  bands near 3100 cm−1 (two of which are degenerate in C3v), two free OH stretch bands
bands near 3100 cm−1 (two of which are degenerate in C3v), two free OH stretch bands 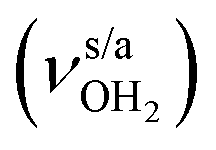 , and missing free NH and bound OH stretch bands
, and missing free NH and bound OH stretch bands  .
.
In the I-(H2O)3(III) isomer (E0/G = 4.4/-1.2 kJ mol−1) with a total binding energy of D0 = 186.0 kJ mol−1, two N–H bonds of the NH3+ group are solvated by a single H2O ligand and a (H2O)2 dimer. The NH⋯O H-bond to H2O (1.807 Å, 173.1°) is weaker than that to (H2O)2 (1.708 Å, 172.6°), as also indicated by the lower E(2) energy (57.6 and 88.0 kJ mol−1) and the smaller redshift in the bound NH stretch mode  . Overall, attachment of the single H2O ligand slightly weakens the NH⋯O and OH⋯O H-bonds of the (H2O)2 ligand via noncooperativity. The convoluted IR spectrum of this isomer is characterized by two strongly redshifted and intense
. Overall, attachment of the single H2O ligand slightly weakens the NH⋯O and OH⋯O H-bonds of the (H2O)2 ligand via noncooperativity. The convoluted IR spectrum of this isomer is characterized by two strongly redshifted and intense 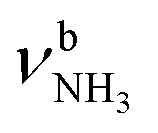 bands, one weak free
bands, one weak free  band, one intense and strongly redshifted νOHb band, and two free OH stretch bands.
band, one intense and strongly redshifted νOHb band, and two free OH stretch bands.
In the I-(H2O)3(III) isomer (E0/G = 12.8/11.9 kJ mol−1), a linear H-bonded (H2O)3 trimer chain forms a rather strong and nearly linear NH⋯O H-bond to the NH3+ group (1.625 Å, 173.6°, E(2) = 123.5 kJ mol−1) supported by weaker CH⋯O contacts, resulting in a total binding energy of D0 = 178.5 kJ mol−1. As the PA of (H2O)n clusters increases with n, the NH⋯O H-bond is stronger and shorter than those of the related isomers of n = 1 (isomer I) and 2 (isomer II) (1.742 > 1.700 > 1.625 Å for n = 1–3), resulting in a larger redshift in the bound NH stretch mode ( =2921 > 2745 > 2652 cm−1) and increase in IR intensity (I = 682 < 1139 < 1268 km mol−1), arising from the stronger elongation of the N–H donor bond (rNH = 1.031 > 1.053 > 1.059 Å). The stronger NH⋯O H-bond is also visible in the increasing charge transfer to solvent (Δq = 42 < 59 < 75 me) and the corresponding orbital interaction energy (E(2) = 75.2 < 107.9 < 123.5 kJ mol−1). Due to closer proximity to the positive charge, the first O–H⋯O H-bond (1.702 Å, 168.5°, E(2) = 82.7 kJ mol−1) is much shorter and more linear than the second one (1.794 Å, 164.2°, E(2)= 55.6 kJ mol−1), resulting in correspondingly larger and smaller redshifts in their
=2921 > 2745 > 2652 cm−1) and increase in IR intensity (I = 682 < 1139 < 1268 km mol−1), arising from the stronger elongation of the N–H donor bond (rNH = 1.031 > 1.053 > 1.059 Å). The stronger NH⋯O H-bond is also visible in the increasing charge transfer to solvent (Δq = 42 < 59 < 75 me) and the corresponding orbital interaction energy (E(2) = 75.2 < 107.9 < 123.5 kJ mol−1). Due to closer proximity to the positive charge, the first O–H⋯O H-bond (1.702 Å, 168.5°, E(2) = 82.7 kJ mol−1) is much shorter and more linear than the second one (1.794 Å, 164.2°, E(2)= 55.6 kJ mol−1), resulting in correspondingly larger and smaller redshifts in their 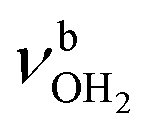 mode (3243 vs. 3412 cm−1). The convoluted IR spectrum of this isomer is characterized by one very strongly redshifted and intense
mode (3243 vs. 3412 cm−1). The convoluted IR spectrum of this isomer is characterized by one very strongly redshifted and intense  band, two weak and coupled free
band, two weak and coupled free  bands, two intense, strongly redshifted, and well-separated νOHb bands, and two free OH stretch bands.
bands, two intense, strongly redshifted, and well-separated νOHb bands, and two free OH stretch bands.
Comparison of the IRPD spectrum of AmaH+(H2O)3 to the IR spectra calculated for I-(H2O)3(I–IV) in Fig. 7 (Table S9, ESI†) suggests the presence of at least three isomers based on the observation of the four redshifted NH3 bands (A, I, J, K). The global minimum I-(H2O)3(I) is identified by band W at 3552 cm−1 assigned to  modes (3509/3533 cm−1) and band O at 3317 cm−1 assigned to
modes (3509/3533 cm−1) and band O at 3317 cm−1 assigned to  at 3307 cm−1. However,
at 3307 cm−1. However,  modes could be also assigned to band T at 3432 cm−1. Moreover, its
modes could be also assigned to band T at 3432 cm−1. Moreover, its  modes (2982/2984 cm−1) agree well with band I at 3009 cm−1, with only minor deviations of 25/27 cm−1. Band M (3268 cm−1) can be assigned to 2βOH2 (3237 cm−1) and/or 2βNH3 (3244 cm−1). The calculated
modes (2982/2984 cm−1) agree well with band I at 3009 cm−1, with only minor deviations of 25/27 cm−1. Band M (3268 cm−1) can be assigned to 2βOH2 (3237 cm−1) and/or 2βNH3 (3244 cm−1). The calculated  modes at 3637 and 3721 cm−1 agree with the observed OH2 bands X and Z at 3639 and 3712 cm−1. Due to negligible influence of hydration on the adamantyl cage, bands C-F are consistent with the νCH(2) modes of I-(H2O)3(I) but also all other isomers. Indeed, the observed bands A, B, J, K, and T imply the presence of further isomers. Bands J and K (at 3118 and 3170 cm−1) can be assigned to three redshifted
modes at 3637 and 3721 cm−1 agree with the observed OH2 bands X and Z at 3639 and 3712 cm−1. Due to negligible influence of hydration on the adamantyl cage, bands C-F are consistent with the νCH(2) modes of I-(H2O)3(I) but also all other isomers. Indeed, the observed bands A, B, J, K, and T imply the presence of further isomers. Bands J and K (at 3118 and 3170 cm−1) can be assigned to three redshifted  modes of the fully solvated NH3+ group of I-(H2O)3(II), with again relatively large deviations of 57 and 65 cm−1. However, the experimental blueshifts of J
modes of the fully solvated NH3+ group of I-(H2O)3(II), with again relatively large deviations of 57 and 65 cm−1. However, the experimental blueshifts of J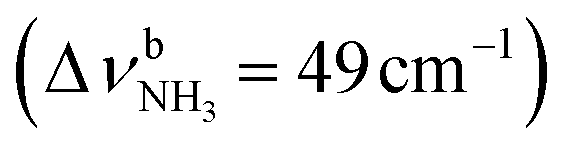 and K
and K agree well with the predicted ones
agree well with the predicted ones  upon further solvation of the NH3+ group of I-(H2O)2(I). The broad transition T (3432 cm−1) can only be attributed to redshifted
upon further solvation of the NH3+ group of I-(H2O)2(I). The broad transition T (3432 cm−1) can only be attributed to redshifted  modes of isomers with an attached (H2O)2,3 cluster and is therefore assigned to
modes of isomers with an attached (H2O)2,3 cluster and is therefore assigned to  of I-(H2O)3(III) (3408 cm−1) and/or
of I-(H2O)3(III) (3408 cm−1) and/or  of I-(H2O)3(IV) (3412 cm−1). Moreover, band O (3317 cm−1) contains to
of I-(H2O)3(IV) (3412 cm−1). Moreover, band O (3317 cm−1) contains to  (νNH3f = 3304 cm−1) and band J (3118 cm−1) the redshifted
(νNH3f = 3304 cm−1) and band J (3118 cm−1) the redshifted  mode of I-(H2O)3(III) (
mode of I-(H2O)3(III) ( = 3045 cm−1) associated with the H-bond to a single H2O. The further redshifted
= 3045 cm−1) associated with the H-bond to a single H2O. The further redshifted 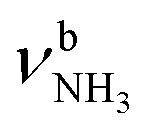 mode I-(H2O)3(III) (
mode I-(H2O)3(III) ( = 2872 cm−1) associated with the H-bond to (H2O)2 agrees well with band B (2823 cm−1), even though the intensity in the IRPD spectrum is too low (possibly for reasons of low laser intensity). Furthermore,
= 2872 cm−1) associated with the H-bond to (H2O)2 agrees well with band B (2823 cm−1), even though the intensity in the IRPD spectrum is too low (possibly for reasons of low laser intensity). Furthermore,  and
and  of I-(H2O)3(IV) can also be attributed to M (3268 cm−1) and O (3317 cm−1), with minor deviations of 11 and 5 cm−1. Its strongly redshifted
of I-(H2O)3(IV) can also be attributed to M (3268 cm−1) and O (3317 cm−1), with minor deviations of 11 and 5 cm−1. Its strongly redshifted 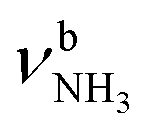 mode (2652 cm−1) due to the attached (H2O)3 is also observed with band A at 2712 cm−1, with a deviation of 60 cm−1. Assuming that only isomers I-(H2O)3(I–IV) are detected and bands A, B, K, and W can each be assigned to only one isomer, the relative populations of I–IV can be roughly estimated from the calculated and observed peak intensities (peak area) as 1:4:4:1, respectively.
mode (2652 cm−1) due to the attached (H2O)3 is also observed with band A at 2712 cm−1, with a deviation of 60 cm−1. Assuming that only isomers I-(H2O)3(I–IV) are detected and bands A, B, K, and W can each be assigned to only one isomer, the relative populations of I–IV can be roughly estimated from the calculated and observed peak intensities (peak area) as 1:4:4:1, respectively.
4.5 AmaH+(H2O)4
As the number of H2O ligands increases, the number of possible low-energy isomers grows drastically. In total, we identified ten low-energy AmaH+(H2O)4 isomers for each monomer (I–IV) (Fig. S18–S21 and Table S6, ESI†). Due to the large number of observed peaks in the AmaH+(H2O)4 spectrum, almost no isomer can be excluded by spectroscopy. Hence, we discuss herein only the most stable isomers I-(H2O)4(I–VI) of monomer I (Fig. 8 and Fig. S26, ESI†), which are sufficient to explain the IRPD spectrum (Fig. 9). Isomers I–III and V are derived by hydrating the most stable cyclic I-(H2O)3(I) trihydrated cluster at different positions of the eight-membered solvation ring or the third N–H bond of the NH3+ unit, while IV is obtained by starting the second hydration shell of the I-(H2O)3(II) isomer with a fully hydrated NH3+ group and VI by hydrating I-(H2O)3(II) to form a Cs symmetric structure with two N–H⋯O H-bonds to two (H2O)2 units. Isomers II and IV have a fully hydrated NH3+ group, while I, III, and VI have one free NH group, illustrating the strong competition between interior ion solvation and the formation of a H-bonded solvent network. As with n = 3, the energetic order of these isomers I-VI of n = 4 (E0 = 0, 0.6, 1.7, 4.9, 7.1, 7.2 kJ mol−1) change drastically with inclusion of entropy (G = 0, −7.0, −2.9, −13.2, 3.0, −7.7 kJ mol−1), so that the energetically most stable isomer I becomes the fifth stable at room temperature, while the energetically forth stable isomer IV becomes the global minimum on the free energy surface (Table S6, ESI†). Indeed, IV has only one OH⋯O H-bond, leading to a flexible and thus entropically favoured structure, while the other low-energy isomers have larger constrain due to two (II, VI) or three OH⋯O H-bonds (I, III, V). As with n = 3, we refer for a detailed discussion on the structures and IR spectra for the n = 4 isomers to ESI.† In general, the structural, energetic, vibrational, and charge transfer trends, arising from cooperativity of the formation of H-bonded networks and noncooperativity of interior ion solvation discussed for n = 1–3 are also observed for n = 4. | ||
| Fig. 8 Calculated equilibrium structures (in Å and degrees) of AmaH+(I)(H2O)4(I,II) and AmaH+(I)(H2O)4(IV–VI) in their ground electronic state (B3LYP-D3/cc-pVTZ). All bond lengths are shown in ESI† (Fig. S25). | ||
 | ||
| Fig. 9 IRPD spectrum of AmaH+(H2O)4 compared to linear IR absorption spectra of AmaH+(I)(H2O)4(I–VI) calculated at the B3LYP-D3/cc-pVTZ level. The positions of the transition observed in the IRPD spectrum of AmaH+(H2O)4 and their vibrational assignment are listed in Table S10 (ESI†). Differences in relative energy (E0, G) are given in kJ mol−1. Note the different IR intensity scale for the computed IR spectra. | ||
Comparison of the IRPD spectrum of AmaH+(H2O)4 to calculated IR spectra of I-(H2O)4(I–V) in Fig. 9 (Table S10, ESI†) provides evidence for the presence of at least three isomers, because five observed bands (A, H, I, J, K) are assigned to  modes. The large number of peaks and the several similar frequencies calculated for different isomers prevent a definitive isomer assignment. Nonetheless, it is possible to explain the IRPD spectrum with the six most stable isomers I-(H2O)4(I–VI) with E0 < 8 kJ mol−1. As before, bands C–F can be assigned to the νCH/CH2 modes of all possible isomers I-(H2O)4(I–VI), as their C–H bonds remain almost unchanged upon hydration. Moreover, the free OH stretch bands X–Z are consistent with their
modes. The large number of peaks and the several similar frequencies calculated for different isomers prevent a definitive isomer assignment. Nonetheless, it is possible to explain the IRPD spectrum with the six most stable isomers I-(H2O)4(I–VI) with E0 < 8 kJ mol−1. As before, bands C–F can be assigned to the νCH/CH2 modes of all possible isomers I-(H2O)4(I–VI), as their C–H bonds remain almost unchanged upon hydration. Moreover, the free OH stretch bands X–Z are consistent with their  modes. For example, the
modes. For example, the  ,
, 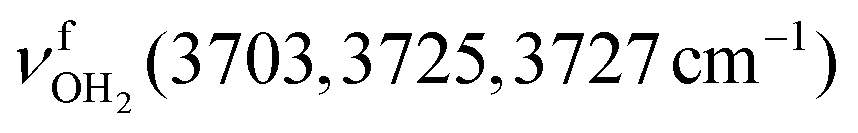 , and
, and  modes of isomer I agree well with bands X (3642 cm−1), Y (3683 cm−1), and Z (3705 cm−1), respectively, with maximum, mean, and summed deviations of 40, 20, and 23 cm−1. On the other hand, peak A at 2680 cm−1, can uniquely be identified with the intense and strongly redshifted
modes of isomer I agree well with bands X (3642 cm−1), Y (3683 cm−1), and Z (3705 cm−1), respectively, with maximum, mean, and summed deviations of 40, 20, and 23 cm−1. On the other hand, peak A at 2680 cm−1, can uniquely be identified with the intense and strongly redshifted  mode (2776 cm−1) of V. The isomer assignment for the other bands (H–W) is less clear. For example, the bound NH stretch peak H (2974 cm−1) may be attributed I
mode (2776 cm−1) of V. The isomer assignment for the other bands (H–W) is less clear. For example, the bound NH stretch peak H (2974 cm−1) may be attributed I , III (2937/2961 cm−1), IV (2979 cm−1), and/or VI (2929 cm−1). The three
, III (2937/2961 cm−1), IV (2979 cm−1), and/or VI (2929 cm−1). The three  modes of II predicted at 3049, 3068, and 3123 cm−1 correlate with bands I, J, and K (3058, 3144, 3188 cm−1) with larger differences (maximum, mean, and summed deviations of 76, 65, and 50 cm−1), as expected for proton-donor stretch modes of strong H-bonds. However, band I also fits to
modes of II predicted at 3049, 3068, and 3123 cm−1 correlate with bands I, J, and K (3058, 3144, 3188 cm−1) with larger differences (maximum, mean, and summed deviations of 76, 65, and 50 cm−1), as expected for proton-donor stretch modes of strong H-bonds. However, band I also fits to  of V (3008 cm−1), and bands J and K to the
of V (3008 cm−1), and bands J and K to the  modes of IV (3095/3126 cm−1) with similar large maximum, mean, and summed deviations of 62, 50, and 54 cm−1. Although the differences between calculated and observed
modes of IV (3095/3126 cm−1) with similar large maximum, mean, and summed deviations of 62, 50, and 54 cm−1. Although the differences between calculated and observed  bands are relatively large, they are of the same order of magnitude as for n = 1-3, and the observed blueshifts of the peaks upon hydration (n = 3 → 4) is also predicted by the calculations. For example, the averaged incremental
bands are relatively large, they are of the same order of magnitude as for n = 1-3, and the observed blueshifts of the peaks upon hydration (n = 3 → 4) is also predicted by the calculations. For example, the averaged incremental  blueshifts calculated for II, IV, and V agree well with the observed ones (ΔI = 45 vs. 49 cm−1, ΔJ = 21 vs. 26 cm−1, ΔK = 18 vs.18 cm−1), supporting the suggested assignments. Band M (3271 cm−1) is again probably 2βOH2 and/or 2βNH3, but the
blueshifts calculated for II, IV, and V agree well with the observed ones (ΔI = 45 vs. 49 cm−1, ΔJ = 21 vs. 26 cm−1, ΔK = 18 vs.18 cm−1), supporting the suggested assignments. Band M (3271 cm−1) is again probably 2βOH2 and/or 2βNH3, but the  modes of I, III, and V (all at 3311 cm−1) may also contribute to this broad band. The bonded O–H modes of the low-energy isomers I and III at
modes of I, III, and V (all at 3311 cm−1) may also contribute to this broad band. The bonded O–H modes of the low-energy isomers I and III at  = 3421/3404 and 3466/3455 cm−1 may be attributed to bands S (3387 cm−1) and T (3452 cm−1). Their third and remaining intense
= 3421/3404 and 3466/3455 cm−1 may be attributed to bands S (3387 cm−1) and T (3452 cm−1). Their third and remaining intense  mode agrees well with band M, but their high predicted IR intensity (I = 800/1460 km mol−1) is not reflected in the measured spectrum, which makes this assignment obsolete and could be taken as an exclusion criterion for I and III. In fact, their free energies G move them backwards in the stability order. On the other hand, it is well known that proton-donor stretch bands move to the blue at elevated temperature, because intermolecular excitation weakens the H-bond.108–110 Following this line of argument, these intense modes may indeed be observed by bands S and T. The latter bands also agree with the
mode agrees well with band M, but their high predicted IR intensity (I = 800/1460 km mol−1) is not reflected in the measured spectrum, which makes this assignment obsolete and could be taken as an exclusion criterion for I and III. In fact, their free energies G move them backwards in the stability order. On the other hand, it is well known that proton-donor stretch bands move to the blue at elevated temperature, because intermolecular excitation weakens the H-bond.108–110 Following this line of argument, these intense modes may indeed be observed by bands S and T. The latter bands also agree with the  modes of the bound (H2O)2 dimers of IV (3456 cm−1) and V (3427/3418 cm−1), which have the lowest free energies. Even if the ring isomers I and III were excluded, it is still considered certain that ring isomers are detected, since the signature of such water ring structures, band W, is observed at 3554 cm−1 and agrees well with
modes of the bound (H2O)2 dimers of IV (3456 cm−1) and V (3427/3418 cm−1), which have the lowest free energies. Even if the ring isomers I and III were excluded, it is still considered certain that ring isomers are detected, since the signature of such water ring structures, band W, is observed at 3554 cm−1 and agrees well with  modes of II (3519/3542 cm−1) and/or V (3598 cm−1).
modes of II (3519/3542 cm−1) and/or V (3598 cm−1).
5. Further discussion
The analysis of the IRPD spectra of AmaH+(H2O)n with B3LYP-D3 calculations clearly reveals that mainly hydrates of the ammonium ion (I) of AmaH+ are detected, indicating that the formation of the iminium isomers of AmaH+ (II–IV) in a hydration environment is substantially suppressed compared to an Ar solvation environment. Indeed the population of II-H2O is estimated as <10% of I-H2O, while the corresponding II-Ar population is ∼30% of I-Ar.49 This result is consistent with the observed isomer distribution of the Ama+(I–III) radical cation, for which the population of the iminium ion Ama+(III) is also lower in water solvation than in Ar solvation (10% vs. 35%).48 In the latter case, computations show that H2O attachment substantially increases the barrier for cage-opening required for forming iminium ions.48 Since it is assumed that isomer II of AmaH+ is formed by H-radical addition to the distonic Ama+(III) ion and its population is much smaller in a water environment, the population of II-H2O is thus also reduced.In addition to the identity of the AmaH+ core ion, the analysis of the IRPD spectra provides a consistent picture of the microhydration process of the predominant AmaH+(I) ion, which gradually shifts from interior ion solvation to the formation of a H-bonded solvent network. Except for n = 1, where a small population of II-H2O(I) is detected, all main bands of the AmaH+(H2O)1–4 spectra are assigned to the most stable microhydrates of the ammonium ion I. In these clusters, the H2O ligands form a growing hydration network attached to the acidic NH3+ group, while the spectra contain no signature of much less stable isomers with H2O binding to the aliphatic adamantyl cage. For n ≤ 2, the H2O ligands bind preferentially to the acidic protons of the NH3+ group via strong and almost linear NH⋯O ionic H-bonds. However, an isomer with a H-bonded (H2O)2 dimer attached to the NH3+ group is also observed for n = 2 (with an estimated population of 20%), which becomes competitive due to the strong cooperative effects of the H-bonded solvent network (E0 = 4.9 kJ mol−1). For n = 3, more types of hydration are observed. In addition to the isomer with a fully hydrated NH3+ group and an isomer with H2O and (H2O)2 separately attached to two NH protons, an isomer with a linear (H2O)3 trimer attached to one NH group and the adamantyl cage and an isomer with a cyclic eight-member solvation ring including part of the NH3+ group also occur. Although the latter ring isomer is the global minimum on the potential energy surface of I-(H2O)3 at 0 K, the isomer with the fully solvated NH3+ group (ΔG = −8.8 kJ mol−1) and the isomer with a dangling (H2O)2 ligand (ΔG = −1.2 kJ mol−1) become more stable on the free energy surface at 298.15 K. The latter effect explains the estimated observed population distribution of I-(H2O)3(I–IV) of 10, 40, 40, and 10%, respectively. At n = 4, the formation of the H-bonded solvation network already dominates over internal ion solvation, because the completely saturated NH3+ group offers only three acidic NH protons and bifurcated NH⋯O H-bonds of one NH donor to two H2O ligands are less stable and indeed no such minima are found on the potential energy surface. Furthermore, the isomers with cyclic solvation rings dominate at n = 4, so that among the five energetically best isomers, four isomers have such rings, which benefit from cooperative three-body forces. When considering the free energies, the energetic order changes such that isomer IV with a fully saturated NH3+ group (bonded (H2O)2 dimer and two H2O ligands, ΔG = −13.2 kJ mol−1) and the local minimum VI with two bonded (H2O)2 dimers (ΔG = −7.7 kJ mol−1) are most favourable, but closely followed by isomer II with a ring structure (ΔG = −7.0 kJ mol−1).
Overall, H-bonded isomers are energetically preferred over other isomers in which the adamantyl cage is gradually solvated by individual H2O ligands via cation-dipole forces supported by weak CH⋯O contacts. Interior ion solvation dominates over H-bonded solvent network until the NH3+ group is completely saturated with H2O (n ≤ 3), although isomers with H-bonded solvent network are also detected in somewhat lower populations, which become competitive due to the strong cooperative effects as n increases. In general, the hydration energies calculated for n = 1–4 (71–51 kJ mol−1) are higher than the absorbed IR photon energy (hν < 48 kJ mol−1 ∼4000 cm−1), indicating that, under single-photon absorption conditions with an unfocused IR laser beam, only cluster ions with substantial internal energy can undergo the IRPD process. This result also explains the widths of the bands and the entropy contribution required to evaluate the energy ordering of the competing isomers. The calculated binding energies for the observed AmaH+(I)(H2O)n clusters decrease as D0 = 71 > (62–55) > (57–50) > (53–46) kJ mol−1 for n = 1–4 and linear extrapolation to larger cluster sizes suggests that around n = 10 internal ion solvation becomes again more favourable than extending the solvation network (Fig. S31, ESI†). However, this prediction ignores the observed enthalpy of sublimation of bulk ice of 51.0 kJ mol−1,111 and thus larger AmaH+(I)(H2O)n clusters will also favour structures in which the hydrophobic adamantyl cage is located at the surface of the H-bonded water solvent network, along with increasing charge delocalization into the solvent.
The detailed evolution of the properties of the N–H bonds (rNH, E(2),  ,
,  ) of the observed AmaH+(I)(H2O)n clusters as a function of n is visualized in Fig. 10. To distinguish the different N–H bonds and to deduce correlations, a colour code is used according to the ligand attached to the N–H bond: free N–H bond (blue); single H2O (red, black); (H2O)2 dimer (magenta); (H2O)3 trimer (cyan); water ring (green); extended ring (orange). In general, the calculated and measured frequencies of the νNH modes agree very well with respect to the incremental changes, even though in some cases the absolute values differ somewhat, mainly due to improper evaluation of anharmonicity of the proton donor stretch modes involved in the NH⋯O ionic H-bonds. For clusters with single H2O ligands (red), the N–H bonds elongate at n = 1, accompanied by a large redshift of the corresponding νNH3 mode, due to the E(2) energy for the donor–acceptor interaction between the lone pairs of O and the antibonding σ* orbital of the N–H donor bond. As n increases, the H-bonds become somewhat weaker (E(2) decreases) due to noncooperative effects typical for interior ion solvation, which in turn leads to a contraction of the N–H bonds and associated incremental blueshifts in νNH3. For the N–H bonds to which a single water is added in a second step (black), the behaviour is similar, although logically the N–H bond does not elongate until n = 2 and E(2) is lower due to the first H2O already bound to an adjacent N–H bond (interior ion solvation), resulting in a weaker redshift in νNH3. The situation is different for N–H bonds to which a (H2O)2 dimer (magenta) or a (H2O)3 trimer (cyan) are attached, resulting in much stronger NH⋯O ionic H-bonds at n = 2 and n = 3, respectively. Due to the higher E(2) energies, the N–H bonds are more stretched, leading to larger redshifts in νNH3. For the (H2O)2 dimer, E(2) becomes lower again at n > 3 due to further internal ion solvation, which in turn leads to blueshifts of the νNH3 modes. The first intermolecular NH⋯O H-bonds forming a ring with H2O appear at n = 3 (green) and are stronger than those from a single bonded H2O, but weaker than those from a bonded (H2O)2 or (H2O)3 cluster, resulting in a moderate redshift of the corresponding νNH3 modes. When at n = 4 the remaining free N–H bond is hydrated by another H2O, E(2) decreases again, the N–H bonds contract, and the νNH3 modes become slightly blueshifted. When the solvation network of the ring is extended (orange), the intermolecular bonds become stronger (E(2) increases) and the N–H bonds elongate, pushing the associated νNH3 modes further to the red.
) of the observed AmaH+(I)(H2O)n clusters as a function of n is visualized in Fig. 10. To distinguish the different N–H bonds and to deduce correlations, a colour code is used according to the ligand attached to the N–H bond: free N–H bond (blue); single H2O (red, black); (H2O)2 dimer (magenta); (H2O)3 trimer (cyan); water ring (green); extended ring (orange). In general, the calculated and measured frequencies of the νNH modes agree very well with respect to the incremental changes, even though in some cases the absolute values differ somewhat, mainly due to improper evaluation of anharmonicity of the proton donor stretch modes involved in the NH⋯O ionic H-bonds. For clusters with single H2O ligands (red), the N–H bonds elongate at n = 1, accompanied by a large redshift of the corresponding νNH3 mode, due to the E(2) energy for the donor–acceptor interaction between the lone pairs of O and the antibonding σ* orbital of the N–H donor bond. As n increases, the H-bonds become somewhat weaker (E(2) decreases) due to noncooperative effects typical for interior ion solvation, which in turn leads to a contraction of the N–H bonds and associated incremental blueshifts in νNH3. For the N–H bonds to which a single water is added in a second step (black), the behaviour is similar, although logically the N–H bond does not elongate until n = 2 and E(2) is lower due to the first H2O already bound to an adjacent N–H bond (interior ion solvation), resulting in a weaker redshift in νNH3. The situation is different for N–H bonds to which a (H2O)2 dimer (magenta) or a (H2O)3 trimer (cyan) are attached, resulting in much stronger NH⋯O ionic H-bonds at n = 2 and n = 3, respectively. Due to the higher E(2) energies, the N–H bonds are more stretched, leading to larger redshifts in νNH3. For the (H2O)2 dimer, E(2) becomes lower again at n > 3 due to further internal ion solvation, which in turn leads to blueshifts of the νNH3 modes. The first intermolecular NH⋯O H-bonds forming a ring with H2O appear at n = 3 (green) and are stronger than those from a single bonded H2O, but weaker than those from a bonded (H2O)2 or (H2O)3 cluster, resulting in a moderate redshift of the corresponding νNH3 modes. When at n = 4 the remaining free N–H bond is hydrated by another H2O, E(2) decreases again, the N–H bonds contract, and the νNH3 modes become slightly blueshifted. When the solvation network of the ring is extended (orange), the intermolecular bonds become stronger (E(2) increases) and the N–H bonds elongate, pushing the associated νNH3 modes further to the red.
 | ||
| Fig. 10 Plots of various calculated and experimental properties of the N–H bonds of the most stable Ama+(H2O)n clusters as a function of the cluster size n: calculated N–H bond lengths (rN–H); calculated second-order perturbation energies (E(2)) of donor–acceptor orbital interactions involved in the H-bonds; calculated and experimental NH stretch frequencies (νNH3). Symbols denote individual data points, while the connected points correspond to the values of the assigned isomers. The colour code indicates the type of ligand attached to the N–H bonds: free N–H bond (blue); first single H2O (red); second single H2O (black); (H2O)2 dimer (magenta); (H2O)3 trimer (cyan); cyclic water ring (green); extended ring (orange). Experimentally observed peaks associated with νNH3 modes are labeled (A, B, H, I, J, K, N, O, P). | ||
The detailed evolution of the properties of the water O–H bonds (rOH, E(2),  ,
,  ) of the observed AmaH+(I)(H2O)n clusters as a function of n is shown in ESI† (Fig. S32). Similar to the N–H bonds, it also shows good agreement of the incremental changes of calculated and measured νOH2 frequencies as well as consistent correlations with the O–H bond lengths and orbital interaction energies E(2). Overall, the neutral OH⋯O H-bonds of the hydration network are weaker than the ionic NH⋯O H-bonds of H2O to the charged NH3+ group, resulting in smaller perturbations of the O–H donor bonds and correspondingly smaller redshifts in
) of the observed AmaH+(I)(H2O)n clusters as a function of n is shown in ESI† (Fig. S32). Similar to the N–H bonds, it also shows good agreement of the incremental changes of calculated and measured νOH2 frequencies as well as consistent correlations with the O–H bond lengths and orbital interaction energies E(2). Overall, the neutral OH⋯O H-bonds of the hydration network are weaker than the ionic NH⋯O H-bonds of H2O to the charged NH3+ group, resulting in smaller perturbations of the O–H donor bonds and correspondingly smaller redshifts in  .
.
Although there is increasing partial charge transfer from AmaH+ to the (H2O)n solvent cluster with increasing n, no proton transfer to solvent is observed up to n = 4 and most of the positive charge remains with AmaH+. This result is somewhat expected because the proton affinity of Ama (PA = 948.8 kJ mol−1) is substantially higher than that of (H2O)n clusters in the considered size range (PA = 691, 808, 862, 900, 904, 908 kJ mol−1 for n = 1–6),91–95 and the large difference in PA cannot be compensated by differences in the solvation energies of clusters of the type AmaH+(H2O)n and Ama(H2O)nH+. Although no complete proton transfer is observed, the tendency for NH3+ protons to shift from AmaH+ to (H2O)n gradually increases with n. It is possible to derive a lower limit for the cluster size n for proton transfer from the calculated C–N bond lengths and the partial charges of the adamantyl cage (C10H15) for the identified AmaH+(I)(H2O)n clusters, which decrease continuously for n = 1–4 and approach the values of neutral Ama. Although linear extrapolations to larger clusters suggest that proton transfer may occur at n = 11–13, the trends will certainly be nonlinear and thus this cluster size is a safe lower limit (Fig. S33, ESI†). Although we could not find any study reporting the degree of protonation of Ama in liquid water, most Ama molecules occur in their N-protonated form under physiological cell conditions. Under conditions of pharmaceutical action, AmaH+ clearly occurs in its N-protonated form and the acidic protons of the NH3+ group interact strongly with various bio-active molecular binding sites via H-bonding or cation–π interactions (docking).112
It is instructive to compare microhydration in AmaH+(I)(H2O)n with that in Ama+(I)(H2O)n radical cation clusters to evaluate the impact of the additional proton attached to the NH2 group (NH3+/NH2+) on the structure of the hydration shell as well as on the strength the NH⋯O ionic H-bonds. Microhydration of AmaH+(I) shows the same preferential cluster growth as Ama+(I)(H2O)n, in which the N–H bonds are first hydrated and then a H2O solvent network is formed.57 Furthermore, populations of clusters with solvent networks are already detected for both core ions starting from n = 2, and this binding motif becomes more and more competitive with increasing n due to the cooperative effects. However, the detection of isomers with a cyclic solvation ring in AmaH+(I)(H2O)n for n ≥ 3 clearly shows a difference in cluster growth from that in Ama+(I)(H2O)n, where no such ring structures with three H2O molecules are detected. It appears that the additional acidic N–H bond of the NH3+ group in AmaH+(I) favours the formation of small cyclic solvation rings. This observation is probably due to the fact that the NH2 bond angle of the two neighbouring N–H bonds involved in the ring is much smaller in the NH3+ group of AmaH+(I) than in the NH2+ group of Ama+(I) (108.1° vs. 116.8°). Thus, the NH⋯O H-bonds of the cyclic solvation ring in AmaH+(I)(H2O)n can be more linear (161.5° vs. 154.8°), shorter (1.765 vs. 1.787 Å) and thus more stable (E(2) = 67.3 vs. 30.3 kJ mol−1) than in Ama+(I)(H2O)3(I).57 In related clusters such as aniline+(H2O)n,88,89 these small cyclic solvation rings with only three bound H2O molecules cannot be detected, while for n > 4 populations of isomers with larger hydration rings consisting of four water molecules are observed. Another major difference is revealed for the NH⋯O H-bonds, which are much stronger, more linear, and shorter in AmaH+(I)(H2O)n than in Ama+(I)(H2O)n, as seen from the NH⋯O H-bond parameters of AmaH+(I)(H2O)(I) and Ama+(I)(H2O)(I) (1.742 vs. 1.768 Å, 175.7° vs. 170.5°, E(2) = 75.2 vs. 33.9 kJ mol−1). The reason for this is that the positive charge on the NH2+ group of Ama+(I) is lower by a factor of 2 compared to the NH3 group+ of AmaH+(I) (313 vs. 604 me), resulting in less acidic N–H bonds. For example, the redshifted NH stretch bands occur at  for the monohydrates of AmaH+(I) and Ama+(I).
for the monohydrates of AmaH+(I) and Ama+(I).
6. Concluding remarks
IRPD spectroscopy of size-selected AmaH+(H2O)n cluster with n = 1–4 recorded in the sensitive OH, NH, and CH stretch range are analysed with dispersion-corrected DFT calculations at the B3LYP-D3/cc-pVTZ level to reveal the first steps in the microhydration process of this important pharmaceutical molecular drug. Significantly, these spectra provide the first spectroscopic impression of the intermolecular interaction between the AmaH+ cation and water solvent molecules at the molecular level. The major results may be summarized as follows. The population of open-cage iminium ions of AmaH+ (II–IV) in the EI expansion of Ama compared to the canonical N-protonated ammonium ion (I), the pharmaceutically active structure, is significantly lower in H2O environment than in Ar solvation. This result is consistent with the decreased population of distonic iminium ions of the Ama+ radical cation in H2O environment which is considered to be the precursor of AmaH+ in the employed EI source. Microhydration of the predominant isomer I of AmaH+ can be summarized as follows. Considering free energies, the spectra of the n = 1–4 clusters are dominated by the most stable isomers predicted by the DFT calculations, although energetically higher isomers with somewhat lower populations are also detected. Microhydration of AmaH+(I) occurs by solvating the acidic NH protons via strong NH⋯O ionic H-bonds until the NH3+ group is completely hydrated. Due to noncooperative effects of this interior ion solvation process, the strength of the NH⋯O H-bonds decreases slightly with n for n = 1–3. However, in addition to isomers with interior ion solvation motifs, isomers with a H-bonded solvent network are also detected with somewhat lower population, which become competitive due to the strong cooperative effects arising from nonadditive induction forces. In particular, for n ≥ 3 isomers are observed in which the H2O ligands form an eight-membered cyclic ring involving the NH3+ group via four H-bonds. In contrast to interior ion solvation, the formation of such H-bonded networks is strongly cooperative and the strength of NH⋯O H-bonding increases with n because the proton affinity of (H2O)n clusters rises with size. At n = 4, the preferred cluster growth continues with further extension of the H-bonded solvent network by formation of (H2O)2 dimers or by attachment of a further H2O ligand to the cyclic hydration ring via OH⋯O H-bonds. However, interior ion solvation is still competitive due to the saturation of a remaining free N–H bond not involved in a water ring. It is clear that isomers in which H2O ligands bind to the adamantyl cage via weak CH⋯O contacts (mostly stabilized by charge–dipole forces) are much less stable than those with solvation of the NH3+ group due to its higher positive partial charge. The charge transfer from AmaH+ to (H2O)n increases with n, but remains below 0.1 e in the size range n ≤ 4. Similarly, no proton transfer is observed in this size range, consistent with the difference in proton affinity of Ama and (H2O)n, thus justifying the notation of AmaH+⋯(H2O)n. In general, the IRPD spectra and calculations show a solvation process similar to that previously observed for Ama+(H2O)n and related RNH2+(H2O)n clusters, in which the formation of a H-bonded solvation network is favoured over internal ion solvation after saturation of the acidic NH2/3+ group.57,75,88,89,106,107 However, the cluster growth of AmaH+(H2O)n differs by the formation of small cyclic solvation rings involving only three water molecules for n ≥ 3 enabled by the additional N–H bond of the NH3+ group. Moreover, the NH protons of the NH3+ group are much more acidic than those of the NH2+ group, leading to much stronger NH⋯O ionic H-bonds. In the future, it will be interesting to investigate the interaction of AmaH+ with amino acids or small peptides using the same experimental and computational strategy to explore the biological activity of this drug at the molecular level.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
This study was supported by Deutsche Forschungsgemeinschaft (grant DO 729/8).References
- J. Kirschbaum, in Analytical Profiles of Drug Substances, ed. K. Florey, Academic Press, 1983, vol. 12, pp. 1–36 Search PubMed.
- S. Landa and V. Machacek, Collect. Czech. Chem. Commun., 1933, 5, 1 CrossRef CAS.
- R. C. Fort and P. V. R. Schleyer, Chem. Rev., 1964, 64, 277–300 CrossRef CAS.
- J. E. Dahl, S. G. Liu and R. M. K. Carlson, Science, 2003, 299, 96–99 CrossRef CAS PubMed.
- P. V. R. Schleyer, J. Am. Chem. Soc., 1957, 79, 3292 CrossRef CAS.
- J. E. P. Dahl, J. M. Moldowan, Z. Wei, P. A. Lipton, P. Denisevich, R. Gat, S. Liu, P. R. Schreiner and R. M. K. Carlson, Angew. Chem., Int. Ed., 2010, 49, 9881–9885 CrossRef CAS PubMed.
- H. Schwertfeger, A. A. Fokin and P. R. Schreiner, Angew. Chem., Int. Ed., 2008, 47, 1022–1036 CrossRef CAS PubMed.
- P. R. Schreiner, L. V. Chernish, P. A. Gunchenko, E. Y. Tikhonchuk, H. Hausmann, M. Serafin, S. Schlecht, J. E. P. Dahl, R. M. K. Carlson and A. A. Fokin, Nature, 2011, 477, 308 CrossRef CAS PubMed.
- W. L. Yang, J. D. Fabbri, T. M. Willey, J. R. I. Lee, J. E. Dahl, R. M. K. Carlson, P. R. Schreiner, A. A. Fokin, B. A. Tkachenko and N. A. Fokina, Science, 2007, 316, 1460–1462 CrossRef CAS PubMed.
- A. A. Fokin and P. R. Schreiner, Chem. Rev., 2002, 102, 1551–1594 CrossRef CAS PubMed.
- L. Wanka, K. Iqbal and P. R. Schreiner, Chem. Rev., 2013, 113, 3516–3604 CrossRef CAS PubMed.
- D. F. Blake, F. Freund, K. F. M. Krishnan, C. J. Echer, R. Shipp, T. E. Bunch, A. G. Tielens, R. J. Lipari, C. J. D. Hetherington and S. Chang, Nature, 1988, 332, 611 CrossRef CAS PubMed.
- R. S. Lewis, E. Anders and B. T. Draine, Nature, 1989, 339, 117 CrossRef CAS.
- O. Pirali, M. Vervloet, J. E. Dahl, R. M. K. Carlson, A. Tielens and J. Oomens, Astrophys. J., 2007, 661, 919 CrossRef CAS.
- J.-Y. Raty and G. Galli, Nat. Mater., 2003, 2, 792 CrossRef CAS PubMed.
- P. R. Schreiner, N. A. Fokina, B. A. Tkachenko, H. Hausmann, M. Serafin, J. E. P. Dahl, S. Liu, R. M. K. Carlson and A. A. Fokin, J. Org. Chem., 2006, 71, 6709–6720 CrossRef CAS PubMed.
- L. Landt, K. Klünder, J. E. Dahl, R. M. K. Carlson, T. Möller and C. Bostedt, Phys. Rev. Lett., 2009, 103, 47402 CrossRef PubMed.
- T. Henning and F. Salama, Science, 1998, 282, 2204–2210 CrossRef CAS PubMed.
- P. B. Crandall, D. Müller, J. Leroux, M. Förstel and O. Dopfer, Astrophys. J., 2020, 900, L20 CrossRef CAS.
- J. Cernicharo, B. Tercero, A. Fuente, J. L. Domenech, M. Cueto, E. Carrasco, V. J. Herrero, I. Tanarro, N. Marcelino and E. Roueff, Astrophys. J., Lett., 2013, 771, L10 CrossRef.
- E. E. Etim, P. Gorai, A. Das and E. Arunan, Adv. Space Res., 2017, 60, 709–721 CrossRef CAS.
- L. J. Allamandola, S. A. Sandford, A. G. Tielens and T. M. Herbst, Science, 1993, 260, 64–66 CrossRef CAS PubMed.
- O. Guillois, G. Ledoux and C. Reynaud, Astrophys. J., Lett., 1999, 521, L133 CrossRef CAS.
- M. Steglich, F. Huisken, J. E. Dahl, R. M. K. Carlson and T. Henning, Astrophys. J., 2011, 729, 91 CrossRef.
- A. Patzer, M. Schütz, T. Möller and O. Dopfer, Angew. Chem., Int. Ed., 2012, 51, 4925–4929 CrossRef CAS PubMed.
- B. A. McGuire, Astrophys. J., Suppl. Ser., 2022, 259, 30 CrossRef.
- W. L. Davies, R. R. Grunert, R. F. Haff, J. W. McGahen, E. M. Neumayer, M. Paulshock, J. C. Watts, T. R. Wood, E. C. Hermann and C. E. Hoffmann, Science, 1964, 144, 862–863 CrossRef CAS PubMed.
- R. S. Schwab, A. C. England, D. C. Poskanzer and R. R. Young, JAMA, 1969, 208, 1168–1170 CrossRef CAS PubMed.
- G. Hubsher, M. Haider and M. S. Okun, Neurology, 2012, 78, 1096–1099 CrossRef CAS PubMed.
- K. Spilovska, F. Zemek, J. Korabecny, E. Nepovimova, O. Soukup, M. Windisch and K. Kuca, Curr. Med. Chem., 2016, 23, 3245–3266 CrossRef CAS PubMed.
- J. S. Oxford and A. Galbraith, Pharmacol. Ther., 1980, 11, 181–262 CrossRef CAS PubMed.
- J. Hu, R. Fu and T. A. Cross, Biophys. J., 2007, 93, 276–283 CrossRef CAS PubMed.
- I. V. Chizhmakov, F. M. Geraghty, D. C. Ogden, A. Hayhurst, M. Antoniou and A. J. Hay, J. Physiol., 1996, 494, 329–336 CrossRef PubMed.
- WHO, 2010–2011. Retrieved 2019.
- A. S. Monto and N. H. Arden, Clin. Infect. Dis., 1992, 15, 362–367 CrossRef CAS PubMed.
- R. P. Grelak, R. Clark, J. M. Stump and V. G. Vernier, Science, 1970, 169, 203–204 CrossRef CAS PubMed.
- L.-O. Farnebo, K. Fuxe, M. Goldstein, B. Hamberger and U. Ungerstedt, Eur. J. Pharmacol., 1971, 16, 27–38 CrossRef CAS PubMed.
- P. F. V. Voigtlander and K. E. Moore, Science, 1971, 174, 408–410 CrossRef PubMed.
- A. A. Spasov, T. V. Khamidova, L. I. Bugaeva and I. S. Morozov, Pharm. Chem. J., 2000, 34, 1–7 CrossRef CAS.
- J. Kornhuber, M. Weller, K. Schoppmeyer and P. Riederer, J. Neural Transm., 1994, 43, 91–104 CAS.
- T. J. Murray, Can. J. Neurol. Sci., 1985, 12, 251–254 CrossRef CAS PubMed.
- K. M. Kampman, J. R. Volpicelli, A. I. Alterman, J. Cornish and C. P. O’Brien, Am. J. Psychiatry, 2000, 157, 2052–2054 CrossRef CAS PubMed.
- J. Kornhuber, W. Retz and P. Riederer, J. Neural Transm., Suppl., 1995, 46, 315–324 CAS.
- J. W. Johnson and S. E. Kotermanski, Curr. Opin. Pharmacol., 2006, 6, 61–67 CrossRef CAS PubMed.
- C. G. Parsons, A. Stöffler and W. Danysz, Neuropharmacology, 2007, 53, 699–723 CrossRef CAS PubMed.
- F. Weber and S. Gottfried, Nachr. Chem., 2014, 62, 997 CrossRef.
- M. A. R. George and O. Dopfer, J. Phys. Chem. Lett., 2022, 13, 449–454 CrossRef PubMed.
- M. A. R. George and O. Dopfer, Chem. – Eur. J., 2022, 28, e202200577 CrossRef CAS PubMed.
- M. A. R. George and O. Dopfer, Phys. Chem. Chem. Phys., 2022, 24, 16101–16111 RSC.
- L. Rivas, S. Sanchez-Cortes, J. Stanicova, J. V. García-Ramos and P. Miskovsky, Vib. Spectrosc., 1999, 20, 179–188 CrossRef CAS.
- I. V. Litvinyuk, Y. Zheng and C. E. Brion, Chem. Phys., 2000, 261, 289–300 CrossRef CAS.
- P. J. Linstrom and W. G. Mallard, NIST Chemistry WebBook, NIST Standards and Technology, Gaithersburg, MD, 20889, https://webbook.nist.gov, accessed 2022-09-01 Search PubMed.
- P. C. Yates, T. J. Hill and M. Kaur, THEOCHEM, 1992, 258, 299–313 CrossRef.
- C. Parlak and Ö. Alver, Chem. Phys. Lett., 2017, 678, 85–90 CrossRef CAS.
- J. C. Garcia, J. F. Justo, W. V. M. Machado and L. V. C. Assali, J. Phys. Chem. A, 2010, 114, 11977–11983 CrossRef CAS PubMed.
- J. Staničová, G. Fabriciová, L. Chinsky, V. Šutiak and P. Miškovský, J. Mol. Struct., 1999, 478, 129–138 CrossRef.
- M. A. R. George, F. Buttenberg, M. Förstel and O. Dopfer, Phys. Chem. Chem. Phys., 2020, 22, 28123–28139 RSC.
- M. A. R. George, M. Förstel and O. Dopfer, Angew. Chem., Int. Ed., 2020, 12098–12104 CrossRef CAS PubMed.
- M. Štícha, R. L. Chayrov and I. G. Stankova, Anal. Lett., 2019, 52, 2069–2076 CrossRef.
- J. W. Turczan and T. Medwick, J. Pharm. Sci., 1974, 63, 425–427 CrossRef CAS PubMed.
- H. A. Omara and A. S. Amin, Arab. J. Chem., 2011, 4, 287–292 CrossRef CAS.
- A. M. Mahmoud, N. Y. Khalil, I. A. Darwish and T. Aboul-Fadl, Int. J. Anal. Chem., 2009, 2009, 810104 Search PubMed.
- Y. Dou, Y. Sun, Y. Ren, P. Ju and Y. Ren, J. Pharm. Biomed., 2005, 37, 543–549 CrossRef CAS PubMed.
- O. Dopfer, Z. Phys. Chem., 2005, 219, 125–168 CrossRef CAS.
- O. Dopfer and M. Fujii, Chem. Rev., 2016, 116, 5432–5463 CrossRef CAS PubMed.
- N. Solcà and O. Dopfer, Chem. Phys. Lett., 2001, 347, 59–64 CrossRef.
- N. Solcà and O. Dopfer, Chem. – Eur. J., 2003, 9, 3154–3161 CrossRef.
- O. Dopfer, N. Solcà, J. Lemaire, P. Maitre, M.-E. Crestoni and S. Fornarini, J. Phys. Chem. A, 2005, 109, 7881–7887 CrossRef CAS PubMed.
- H.-S. Andrei, N. Solcà and O. Dopfer, Chem. Phys. Chem., 2006, 7, 107–110 CrossRef CAS PubMed.
- S. Chakraborty, A. Patzer, A. Lagutschenkov, J. Langer and O. Dopfer, Chem. Phys. Lett., 2010, 485, 49–55 CrossRef CAS.
- K. Chatterjee and O. Dopfer, J. Chem. Phys. A, 2020, 124, 1134–1151 CrossRef CAS PubMed.
- K. Tanabe, M. Miyazaki, M. Schmies, A. Patzer, M. Schütz, H. Sekiya, M. Sakai, O. Dopfer and M. Fujii, Angew. Chem., Int. Ed., 2012, 124, 6708–6711 CrossRef.
- I. Alata, M. Broquier, C. Dedonder-Lardeux, C. Jouvet, M. Kim, W. Y. Sohn, S.-S. Kim, H. Kang, M. Schütz and A. Patzer, J. Chem. Phys., 2011, 134, 74307 CrossRef PubMed.
- J. Klyne, M. Schmies, M. Miyazaki, M. Fujii and O. Dopfer, Phys. Chem. Chem. Phys., 2018, 20, 3148–3164 RSC.
- M. Schmies, M. Miyazaki, M. Fujii and O. Dopfer, J. Chem. Phys., 2014, 141, 214301 CrossRef PubMed.
- O. Dopfer, A. Patzer, S. Chakraborty, I. Alata, R. Omidyan, M. Broquier, C. Dedonder and C. Jouvet, J. Chem. Phys., 2014, 140, 124314 CrossRef PubMed.
- M. Schütz, Y. Matsumoto, A. Bouchet, M. Öztürk and O. Dopfer, Phys. Chem. Chem. Phys., 2017, 19, 3970–3986 RSC.
- J. Klyne, M. Schmies, M. Fujii and O. Dopfer, J. Phys. Chem. B, 2015, 119, 1388–1406 CrossRef CAS PubMed.
- A. Bouchet, M. Schütz and O. Dopfer, Chem. Phys. Chem., 2016, 17, 232–243 CrossRef CAS PubMed.
- M. Schütz, K. Sakota, R. Moritz, M. Schmies, T. Ikeda, H. Sekiya and O. Dopfer, J. Phys. Chem. A, 2015, 119, 10035–10051 CrossRef PubMed.
- J. Klyne, M. Miyazaki, M. Fujii and O. Dopfer, Phys. Chem. Chem. Phys., 2018, 20, 3092–3108 RSC.
- J. Klyne and O. Dopfer, Phys. Chem. Chem. Phys., 2019, 21, 2706–2718 RSC.
- K. Chatterjee and O. Dopfer, Phys. Chem. Chem. Phys., 2017, 19, 32262–32271 RSC.
- K. Chatterjee and O. Dopfer, Chem. Sci., 2018, 9, 2301–2318 RSC.
- K. Chatterjee and O. Dopfer, J. Chem. Phys., 2018, 149, 174315 CrossRef PubMed.
- K. Chatterjee and O. Dopfer, J. Phys. Chem. A, 2019, 123, 7637–7650 CrossRef CAS PubMed.
- K. Chatterjee and O. Dopfer, Phys. Chem. Chem. Phys., 2020, 13092–13107 RSC.
- Y. Inokuchi, K. Ohashi, Y. Honkawa, N. Yamamoto, H. Sekiya and N. Nishi, J. Phys. Chem. A, 2003, 107, 4230–4237 CrossRef CAS.
- M. Alauddin, J. K. Song and S. M. Park, Int. J. Mass Spectrom., 2012, 314, 49–56 CrossRef CAS.
- S.-K. Jiang, D. Yang, X.-T. Kong, C. Wang, X.-Y. Zang, H.-J. Zheng, G. Li, H. Xie, W.-Q. Zhang and X.-M. Yang, Chin. J. Chem. Phys., 2020, 33, 31–36 CrossRef CAS.
- E. P. L. Hunter and S. G. Lias, J. Phys. Chem. Ref. Data, 1998, 27, 413–656 CrossRef CAS.
- D. J. Goebbert and P. G. Wenthold, Eur. J. Mass Spectrom., 2004, 10, 837–845 CrossRef CAS PubMed.
- A. Courty, M. Mons, J. Le Calvé, F. Piuzzi and I. Dimicoli, J. Phys. Chem. A, 1997, 101, 1445–1450 CrossRef CAS.
- M. Miyazaki, A. Fujii, T. Ebata and N. Mikami, Chem. Phys. Lett., 2004, 399, 412–416 CrossRef CAS.
- R. Knochenmuss, Chem. Phys. Lett., 1988, 144, 317 CrossRef CAS.
- O. Dopfer, Int. Rev. Phys. Chem., 2003, 22, 437–495 Search PubMed.
- T. Oka, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 12235–12242 CrossRef CAS PubMed.
- O. Dopfer, D. Roth, R. V. Olkhov and J. P. Maier, J. Chem. Phys., 1999, 110, 11911–11917 CrossRef CAS.
- F. M. Pasker, N. Solcà and O. Dopfer, J. Phys. Chem. A, 2006, 110, 12793–12804 CrossRef CAS PubMed.
- M. J. Frisch, et al., Gaussion 09, Rev. D.01, Gaussian, Inc., Wallingford, CT, 2009.
- E. D. Glendening, J. K. Badenhoop, A. E. Reed, J. E. Carpenter, J. A. Bohmann, C. M. Morales, C. R. Landis and F. Weinhold, NBO 6.0, Theoretical Chemistry Institute, University of Wisconsin, Madison, 2013 Search PubMed.
- A. E. Reed, L. A. Curtiss and F. Weinhold, Chem. Rev., 1988, 88, 899–926 CrossRef CAS.
- J. Klyne, M. Schmies, M. Miyazaki, M. Fujii and O. Dopfer, Phys. Chem. Chem. Phys., 2018, 20, 3148–3164 RSC.
- K. Chatterjee and O. Dopfer, Phys. Chem. Chem. Phys., 2019, 21, 25226–25246 RSC.
- F. Huisken, M. Kaloudis and A. Kulcke, J. Chem. Phys., 1996, 104, 17–25 CrossRef CAS.
- T. M. Chang, J. S. Prell, E. R. Warrick and E. R. Williams, J. Am. Chem. Soc., 2012, 134, 15805–15813 CrossRef CAS PubMed.
- K. Hirata, F. Haddad, O. Dopfer, S.-I. Ishiuchi and M. Fujii, Phys. Chem. Chem. Phys., 2022, 24, 5774–5779 RSC.
- R. V. Olkhov, S. A. Nizkorodov and O. Dopfer, Chem. Phys., 1998, 239, 393–407 CrossRef CAS.
- S. A. Nizkorodov, D. Roth, R. V. Olkhov, J. P. Maier and O. Dopfer, Chem. Phys. Lett., 1997, 278, 26–30 CrossRef CAS.
- M. Fujii and O. Dopfer, Int. Rev. Phys. Chem., 2012, 31, 131–173 Search PubMed.
- S. Heiles, R. J. Cooper, M. J. DiTucci and E. R. Williams, Chem. Sci., 2017, 8, 2973–2982 RSC.
- Q.-S. Du, S.-Q. Wang, D. Chen, J.-Z. Meng and R.-B. Huang, PLoS One, 2014, 9, e93613 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2cp04556g |
| This journal is © the Owner Societies 2023 |