Multiphoton breakdown of acetylene; formation of organic building block fragments†
Received
25th September 2022
, Accepted 28th November 2022
First published on 9th December 2022
Abstract
Mass resolved REMPI spectra and electron and ion velocity map images were recorded for REMPI of acetylene in the case of two-photon resonant excitations to low lying 3p and 4p Rydberg states. Combined data analysis of ion signal intensities and electron and ion kinetic energy release distribution revealed multiphoton-fragmentation processes in terms of photodissociation and photoionization channels to form the molecular ion, C2H2+ and the fragment ions H+, C+, CH+, CH2+, C2+ and C2H+. The ratio of fragment ion formation over the parent ion formation increases with excitation energy. To a large extent, multiphoton-fragmentation involves the initial breakdown of the molecule into ground and excited state neutral fragments by two-, three- and four-photon dissociation processes prior to multiphoton ionization. The three-photon dissociation processes via superexcited molecular state(s) are found to be the most important and electronically excited fragment species playing a significant role in the overall multiphoton-fragmentation. Furthermore, the data are indicative of the involvement of secondary photodissociation processes and provide information on fragment energetics as well as state interactions. The question, whether acetylene could be an important source of building block fragments for the formation of organic molecules in interstellar space, is addressed.
I. Introduction
Acetylene (C2H2), the smallest unsaturated organic molecule with a triple bond between two carbon atoms, is of fundamental importance in the fields of organic photochemistry,1,2 astrochemistry2–4 and organic synthesis.5 Synthesis involving acetylene includes the preparation of heterocycles and construction of a number of functionalized molecules with different levels of molecular complexity.5 Photochemical processes following absorption of VUV light have been found to form C2 + H2, C2H + H and metastable acetylene.1 Multiphoton excitation studies have revealed an important involvement of high energy molecular Rydberg states, in photochemical processes,2,6–9 different in nature depending on the states symmetry (u or g) and/or energy. Acetylene has long been known to exist in molecular clouds in interstellar space,3,10 where it could, upon radiation, be the source of fundamental reactive fragment species (radicals and/or ions) as building blocks for bigger organic molecules either via gas-phase photofragmentation and/or surface reactions. Catalytic conversion reactions of acetylene on a solid SiC grain surface have been found to lead to the formation of polycyclic aromatic hydrocarbons (PAHs) and are expected to mimic chemical processes in certain astrophysical environments.1,2,11
Resonance enhanced multiphoton excitation and ionization studies of acetylene reveal broad spectral lines due to transitions via Rydberg states, a growing number of fragment ion formations and weakening of ion signals as the excitation energy is increased.2,6,8,9 These observations can be taken as evidence for opening of increased numbers of predissociation and fragmentation formation channels with excitation energy. Virtually all possible fragment formations (H,2,7,9 H2,2,7 C,9 CH,2 CH2,9 C2,2,6,7,9 and C2H2,7) have been proposed, based on direct or indirect observations. Furthermore, a number of excited C2 fragments (C2*) have been detected.6,7 It has been proposed that fragmentation processes could be competing with an isomerization into the vinylidene radical (H2C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C), which is supposed to be a primary precursor for large polycyclic aromatic hydrocarbons (PAHs).2 Our previous work on mass resolved (2 + n) REMPI of acetylene suggests that the parent molecular ion is the primary ion product for resonant excitations to the lowest grade Rydberg states in the two-photon excitation region of about 72
C), which is supposed to be a primary precursor for large polycyclic aromatic hydrocarbons (PAHs).2 Our previous work on mass resolved (2 + n) REMPI of acetylene suggests that the parent molecular ion is the primary ion product for resonant excitations to the lowest grade Rydberg states in the two-photon excitation region of about 72![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 700–75000 cm−1, whereas a significant amount of both fragments (H, C, CH, C2, and C2H) and parent molecules are ionized via excitations to Rydberg states in the intermediate region of about 75700–76100 cm−1 and mainly fragment species are ionized for transitions to Rydberg states in the 82500–83050 cm−1 region.9
700–75000 cm−1, whereas a significant amount of both fragments (H, C, CH, C2, and C2H) and parent molecules are ionized via excitations to Rydberg states in the intermediate region of about 75700–76100 cm−1 and mainly fragment species are ionized for transitions to Rydberg states in the 82500–83050 cm−1 region.9
Recently we have been emphasizing multiphoton-fragmentation (both photodissociation and photoionization) studies of halogen-containing reagents (hydrogen halides (HX) and methyl halides (CH3X)) by mass-resolved and velocity-map-imaging techniques in association with REMPI for two-photon resonant excitations to molecular Rydberg states.12–17 The studies reveal various photodissociation processes for one-electron excitations of the nonbonding (nb) valence electrons localized on the halogen atoms, depending on the number of photons absorbed. The one-photon excitations, which cause transfer to repulsive valence states, are found to form halogen atoms (X) in its ground electronic state along with H (from HX) and CH3 (from CH3X) prior to multiphoton ionization of the fragments. Two-photon excitations, which cause transfer to Rydberg states, have also been found to form X along with H (from HX)18,19 and CH3 (from CH3X)16,20 by predissociation. An additional absorption of the third photon excites the Rydberg states to metastable superexcited states above the molecular ionization limit followed by molecular autoionization and/or dissociation to form ground or excited state fragments prior to further photoionization. Furthermore, interactions between the intermediate Rydberg states and bound ion-pair/valence states have been found to affect further photon excitations for the hydrogen halides.12–14,18 Indications of ion pair formation for CH3I (i.e. CH3+/I−)17 suggest that this might also be the case for methyl halides.
In the work presented here we move a step further in light of our experience from the work described in the previous paragraph. Here we present multiphoton-fragmentation studies of acetylene by mass-resolved REMPI (MR-REMPI) and the corresponding velocity-map-imaging of electrons and number of fragment ions for two-photon resonant excitations to molecular Rydberg states (VMI-REMPI). The two-photon excitation of the π-electrons in the C–C triple bond excites the molecules directly to the Rydberg states. An additional absorption of the third photon excites the Rydberg states to superexcited states above the molecular ionization limit followed by molecular autoionization and/or various dissociation processes to form the ground or excited state of a number of fragments prior to further photoionization. Analyses of ion mass-resolved spectra, kinetic energy release data for electrons and ions and cross-correlation analysis of the data allow us to derive a picture of the multiphoton breakdown of the molecule into reactive fragments/radicals and ions. Relevant details of the analyses are to be found in Section III, A–D, followed by discussion in Section IV. Readers who wish to skip the details should go directly to Section V (1st paragraph) for a summary.
II. Experimental
The velocity map imaging (VMI) setup used in this work has been described previously and only a brief description will be given here.21,22 A supersonic molecular beam of C2H2 (99.6% purity) was formed by expansion through a homemade piezoelectrically actuated nozzle valve (1 mm orifice) and skimmed (1.5 mm, Beam Dynamics) before entering the interaction region. Typically a stagnation pressure of P0 ∼ 1 bar was used. The photolysis/photofragment ionization laser beam (2.0–2.5 mJ per pulse) was generated by frequency doubling of the output of an excimer laser (Lambda-Physik LPX300, operating with XeCl) pumped Dye laser (LPD3000) using the appropriate dyes (C480, C521, and C540A) and focused (f = 30 cm) on the geometric focal point between single-electrode repeller–extractor plate arrangement, where it intersects the collimated molecular beam at right angles. The apparatus was operated in “VMI mode” and the repeller was always on. Ions traversed a field-free time-of-flight region (45 cm) and a gated, position-sensitive detector (dual, imaging quality Micro-Channel Plates (MCP) array coupled to a phosphor screen) imaged the photofragment sphere. The image frame was recorded asynchronously each second (∼10 laser shots) by a CCD camera and several thousand frames were averaged to form the images. A 2D slice of 3D ion distribution from each final image is extracted by an inverse Abel transformation and integrated from its center over an angle to extract the speeds and over the radius to extract the angular distributions of the photofragments. For photoelectrons, the repeller was negatively charged (−3 kV) and the detector was not gated. To detect high-speed photoelectrons a negative charge of −5 kV was used for recording images no. 6 and 7 (see Table 1).
Table 1 Photoelectron (PE) images and the corresponding velocity map images for fragment ions recorded for C2H2via resonant transitions to specified Rydberg states
| Image no. |
C2H2 Rydberg states; nl; 1Λg, (v1v2v3v4v5)a |
One-photon excitation/nm |
Two-photon excitation/cm−1 |
|
n: principal quantum number for the Rydberg electron. l: Rydberg electron orbital. (v1 v2 v3 v4 v5): vibrational quantum numbers referring to vibrational modes: ν1 (C–H symmetric stretch), ν2 (C–C stretch), ν3 (C–H asymmetric stretch), ν4 and ν5 (bend).
|
| 1 |
3p; 1Δg, (00020) |
270.364 |
73969 |
| 2 |
3p; 1Σg, (00000) |
269.255 |
74279 |
| 3 |
3p; 1Σg, (01000) |
268.262 |
74554 |
| 4 |
3p; 1Δg, (01020) |
263.992 |
75760 |
| 5 |
3p; 1Σg, (01000) |
262.864 |
76085 |
| 6 |
4p; 1Δg, (00000) |
242.245 |
82561 |
| 7 |
4p; 1Σg, (00000) |
240.947 |
83006 |
Mass-resolved REMPI (MR-REMPI) spectra were derived from data which were recorded in a system which has been described before.9 In short, a 20 cm quartz lens was used to focus about 10 ns laser pulses in the energy range of 0.05–0.30 mJ on a molecular beam.
III. Results and analysis
A. MR-REMPI
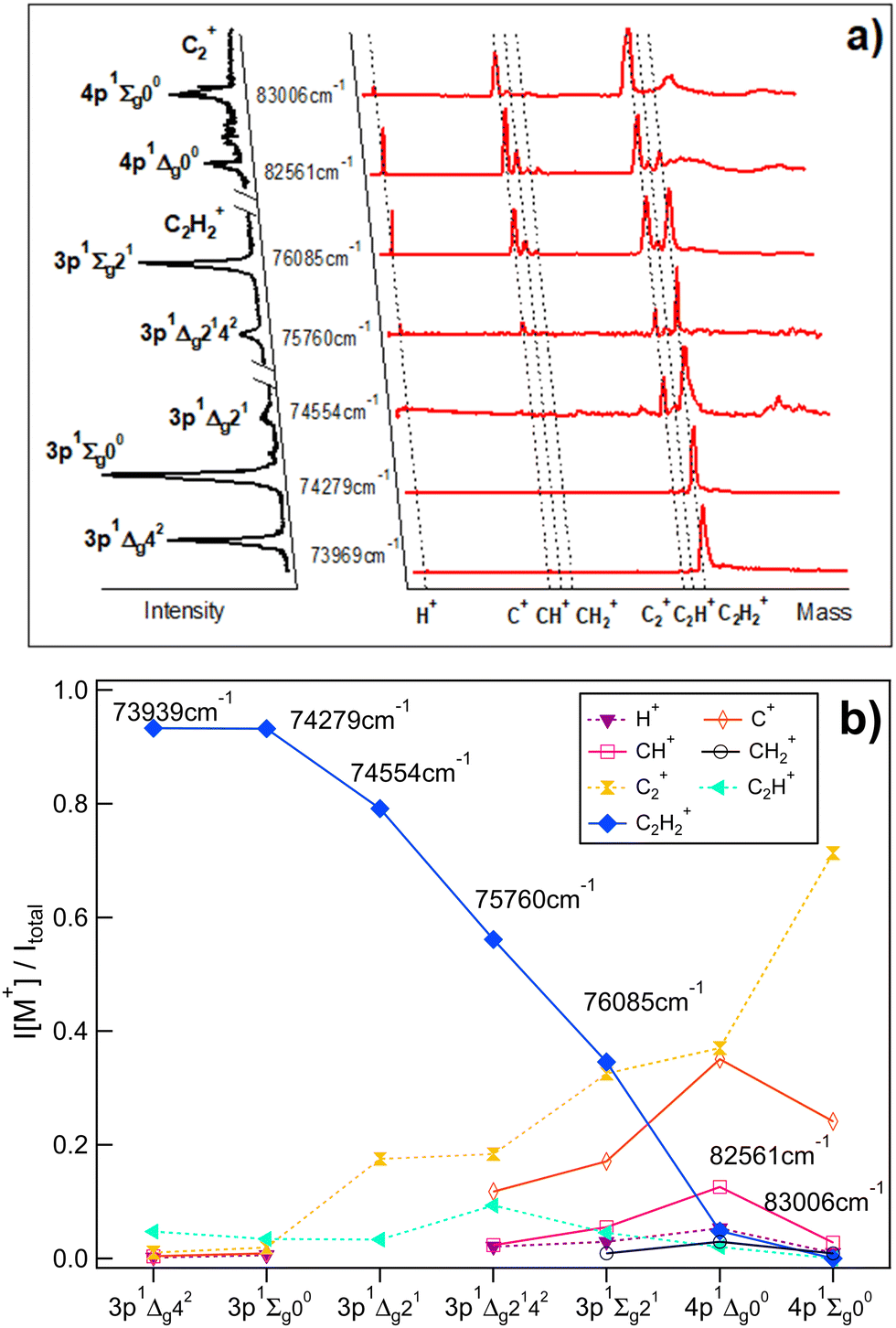
Fig. 1a shows the mass spectra derived for two-photon resonant excitations to a total of seven 3p and 4p molecular Rydberg states in REMPI of acetylene (C2H2) (see also Table 1) along with the derived REMPI spectra. Relative ion signal intensities observed as a function of the excitations are shown in Fig. 1b. Generally, the relative ion signal of the parent molecular ion (C2H2+) is found to decrease with excitation energy, while the relative fragment ion signals (H+, C+, CH+, CH2+, C2+ and C2H+) generally increase with energy. More precisely, the parent molecular ion (C2H2+) signal dominates for the lowest energy excitations and gradually decreases with energy to become virtually negligible for 83006 cm−1. On the contrary the C2+ signal gradually increases with energy, larger in intensity than all the other fragment ion signals and dominates for the highest energy resonance. The H+, C+ and CH+ ion intensities reach maxima for the 82561 cm−1 excitation, while the C2H+ relative ion signal is at a maximum for the 75760 cm−1 excitation. Weak signals for CH2+ were detected for the highest energy excitations only.
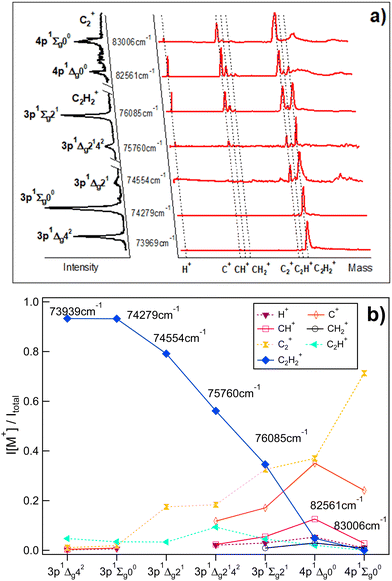 |
| | Fig. 1 Mass-resolved REMPI of C2H2: (a) mass spectra (red) and corresponding REMPI spectra (black, rotated to the left) of C2H2 for two-photon excitations to 3p and 4p molecular Rydberg states (C2H2+ REMPI spectra for the 3p states/C2+ REMPI spectra for the 4p states). The data is partly from ref. 9. (b) Relative ion signal intensities, I[M+]/Itotalvs. excitations for the ions indicated. | |
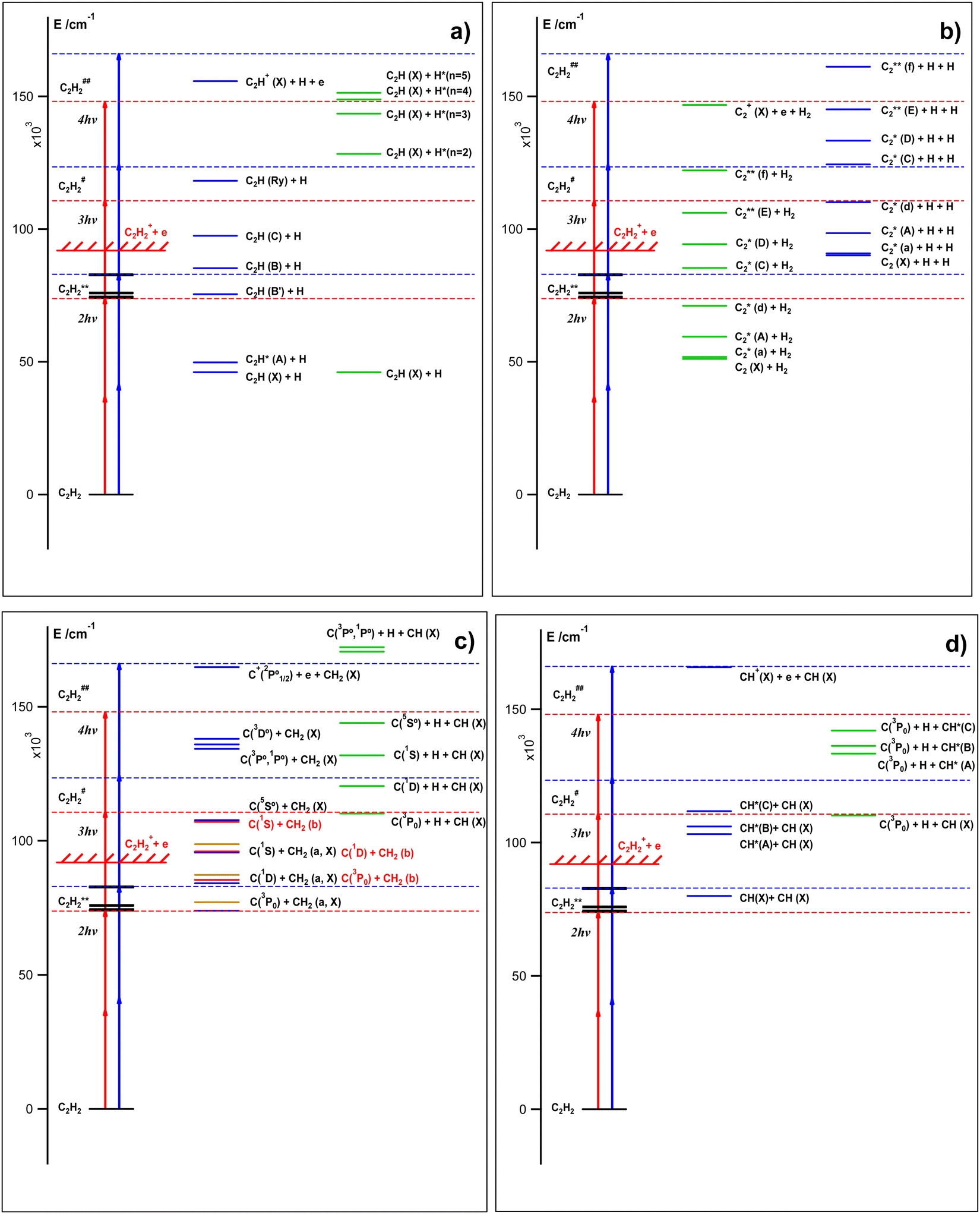
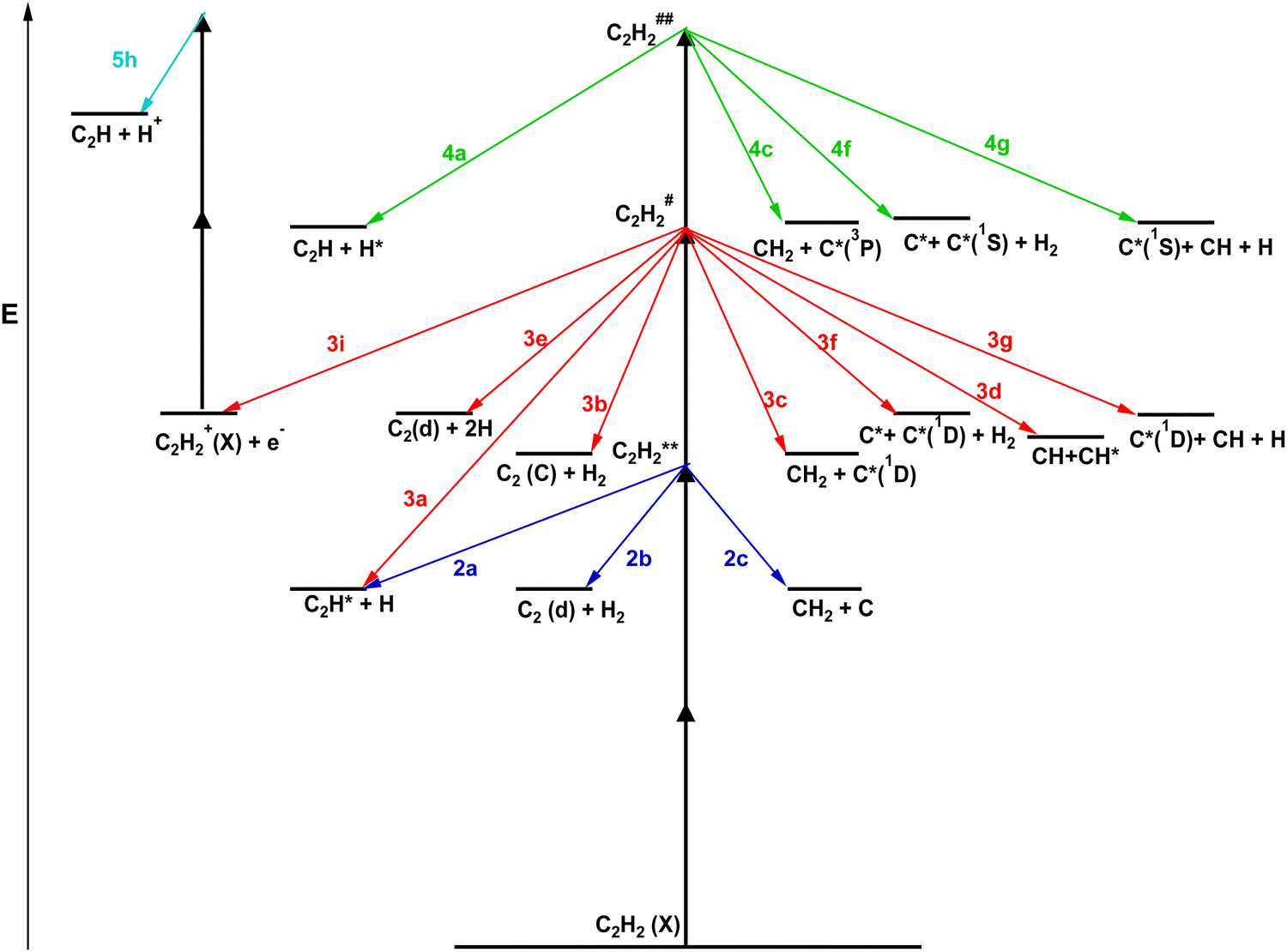
These observations are clear indications of multiphoton excitation and ionization processes. Fig. 2 shows the relevant energetics for the C2H2 molecule and various fragment thresholds as well as the minimum and maximum one- to four-photon excitations of the molecule (see Table 1). The figure gives an indication of the number of photons that may be needed to form the various neutral species directly or by photodissociation in ground and excited states prior to further (photo)ionization. First, one-photon excitation is not sufficient to break any chemical bonds to form fragments and, therefore does not need to be considered here in that respect. The one-photon intermediate excitation step can be assumed to involve a transition to a virtual state (C2H2*) with a character determined by bound valence states close in energy,23
| | | C2H2 + 1hν → C2H2*; one-photon “transition” to a virtual state | (1) |
Second, the two-photon absorption creates the molecular Rydberg states (C
2H
2**; see
Fig. 1a and
Table 1) by resonant excitations and could, energetically, create a number of low energy fragment pairs (F1 + F2/F2*; F1, F2: ground states/F2*: excited state; see also ref.
24) as,
| | | C2H2 + 2hν → C2H2**; two-photon resonant excitation | (2) |
| | | C2H2** → H + C2H/C2H*; dissociation to form fragment pairs | (2a) |
| | | C2H2** → H2 + C2/C2*; dissociation to form fragment pairs | (2b) |
| | | C2H2** → C + CH2/CH2*; dissociation to form fragment pairs | (2c) |
| | | C2H2** → CH + CH; dissociation to form fragment pairs | (2d) |
Third, three-photon absorption could excite the molecule beyond its ionization limit to form metastable/superexcited state(s) (C
2H
2#). There will be a competition between autoionization of short lived C
2H
2# to form C
2H
2+ and dissociation to form fragments. The fragment pairs, mentioned above (F1 + F2/F2*; see
(2a)–(2d)), of the same or higher energy, could be formed as well as species from dissociation leading to three fragments (F1 + F2 + F3/F3*) (see also ref.
24),
| | | C2H2 + 3hν → C2H2#; three-photon (2 + 1) REMP-excitation via C2H2** | (3) |
| | | C2H2# → C2H2+ + e−; autoionization | (3i) |
| | | C2H2# → H + C2H/C2H*; dissociation to form fragment pairs | (3a) |
| | | C2H2# → H2 + C2/C2*; dissociation to form fragment pairs | (3b) |
| | | C2H2# → C/C* + CH2/CH2*; dissociation to form fragment pairs | (3c) |
| | | C2H2# → CH + CH*; dissociation to form fragment pairs | (3d) |
| | | C2H2# → H + H + C2/C2*; dissociation to form three fragments | (3e) |
| | | C2H2# → H2 + C/C* + C/C*; dissociation to form three fragments | (3f) |
| | | C2H2# → H + CH + C/C*; dissociation to form three fragments | (3g) |
The overall, inverse relationship between the ion signals for the parent molecular ion (C
2H
2+) and the fragment ions (Fi
+) with excitation energy (
Fig. 1b), fits a decreasing branching into autoionizations
vs. increasing branching into dissociation with excitation energy. This strongly suggests that the processes
(3) could be of major importance in the multiphoton-ionization of C
2H
2. Observed maxima and fall in relative ion intensities as a function of the excitation energy could be an indication of a varying competition between the different dissociation processes
(3a)–(3g) with energy and/or due to a variation in the dissociative branching with energy at the two-photon excitation step.
 |
| | Fig. 2 C2H2 energetics and excitations: schematic energy diagrams and two-, three- and four-photon molecular excitations to the Rydberg states (C2H2**), first superexcited states (C2H2#) and second superexcited states (C2H2##), respectively (see the main text). Energy levels for the C2H2** states (see no. 1–7 in Table 1) and various fragment formation thresholds are shown. The red and blue vertical arrows correspond to the two-photon resonant transitions of 73969 cm−1 (no. 1; Table 1) and 83006 cm−1 (no. 7), respectively. | |
Due to the metastable/short lifetime nature of the superexcited molecular state(s), C2H2#, formed by three-photon excitation (3) and the wealth of possible exit channels (3i), (3a)–(3g), there is a reason to believe that further molecular photoexcitation is not important under our experimental conditions used to record the MR-REMPI (Section II). However, ion formations due to one- (or few-) photon ionizations of highly excited fragment species formed upon photodissociation by four photons cannot be ruled out. Thus, for example, the H+ signal could be formed by one-photon excitation of H* (n = 2), after four-photon excitation of the molecule (to form C2H2##) rather than by three-photon ionization of H (n = 1) following two- or three-photon dissociation (see also ref. 24), etc.
| | | C2H2 + 4hν → C2H2##; four-photon (2 + 2) REMP-excitation via C2H2** | (4) |
| | | C2H2## → C2H + H*(n = 2); dissociation to form fragment pairs | (4a) |
| | | C2H2## → H2 + C2/C2*; dissociation to form fragment pairs | (4b) |
| | | C2H2## → C/C* + CH2/CH2*; dissociation to form fragment pairs | (4c) |
| | | C2H2## → CH + CH*; dissociation to form fragment pairs | (4d) |
| | | C2H2## → H + H + C2/C2*; dissociation to form three fragments | (4e) |
| | | C2H2## → H2 + C/C* + C/C*; dissociation to form three fragments | (4f) |
| | | C2H2## → H + CH/CH* + C/C*; dissociation to form three fragments | (4g) |
Notice that
eqn (1)–(4), above, are marked as (
nx) where
n is the number of photons required for each step and x are suitable letters.
The minimum number of photons required to ionize the fragment species (mmin) formed according to the dissociation channels (2) to (4) (see Fig. 2) range between 1 to 3,24
| | | H/H* + mhν → H+ + e−; mmin = 3/1 for H/H* | (5a) |
| | | C/C* + mhν → C+ + e−; mmin = 3/1, 2, 3 for C/C* | (5b) |
| | | CH/CH* + mhν → CH+ + e−; mmin = 3/2 for CH/CH* | (5c) |
| | | CH2/CH2* + mhν → CH2+ + e−; mmin = 3/1, 2, 3 for CH2/CH2* | (5d) |
| | | C2/C2* + mhν → C2+ + e−; mmin = 3/1, 2, 3 for C2/C2* | (5e) |
| | | C2H/C2H* + mhν → C2+ + e−; mmin = 3/1, 2, 3 for C2H/C2H* | (5f) |
| | | H/H* (n = 2) + mhν → H+ + e−; mmin = 3/1, 2, 3 for H/H* | (5g) |
In addition to the photoionization channels
(5a)–(5g) photodissociation of the parent molecular ion (C
2H
2+(X)) to form H
+ and/or C
2H
+ also needs to be considered,
| | | C2H2+(X) + 2hν → C2H + H+; ion photodissociation | (5h) |
| | | C2H2+(X) + 2hν → C2H+ + H; ion photodissociation | (5i) |
Furthermore, autoionization and photodissociation of fragment species (Fi = H, C, CH,…) according to,
| | | Fi/Fi* + nhν → Fi#/Fi# → Fi+ + e−; photoexcitation/autoionization | (5j) |
| | | Fi/Fi* + nhν → Fi**/Fi** → Fi1 + Fi2; photodissociation | (5k) |
also need to be considered.
In the subsections below (B–E) we present and describe data and relevant analysis based on velocity map images of electrons and ions (H+, C+, CH+, C2+, C2H+) formed by the multiphoton excitations of concern (Table 1 and Fig. 1). These are suitable to characterize what photofragmentation processes (within eqn (2)–(5)) are involved.
B. Photoelectrons; e-KERs
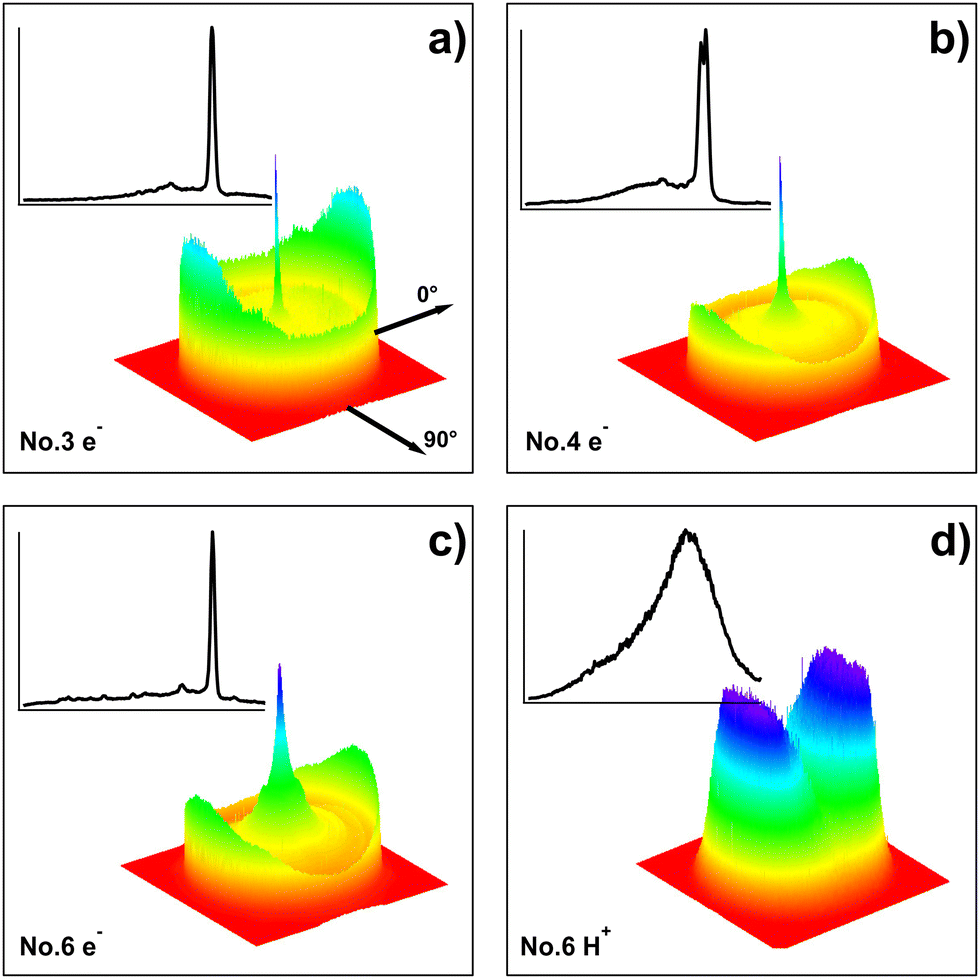
Photoelectron images were recorded for two-photon resonant excitation of C2H2 to all the molecular Rydberg states listed in Table 1 and marked in Fig. 1a. Electron kinetic energy release spectra (e-KERs)/photoelectron spectra (PES) were derived from the images. Selected images, as three dimensional (3D) contour diagrams, and the corresponding e-KERs are shown in Fig. 3(a–c) (see also ref. 24). Fig. 4 shows all the e-KERs/PES. All the spectra (images) show clear peaks (rings) varying in number and positions (KER) depending on the excitation energy. The spectra in Fig. 4 have been shifted by one- (a), two- (b) and three- (c) photon energy differences, (Δ(1hv) = |1hvi − 1hv0|, (a); Δ(2hv) = |2hvi − 2hv0|, (b); Δ(3hv) = |3hvi − 3hv0|, (c); i refers to different spectra) with respect to the spectrum derived for the highest energy excitation (i.e. the “reference spectrum”; subscript 0) for the one-, two- and three-photon excitations of no. 7 (Table 1). Thus, spectral features due to one- (a), two- (b) and three- (c) photon ionization of the same species will align. Calculated threshold energies (i.e. maxima of kinetic energy release) for the various ionization processes of relevance to our interpretation in the following paragraphs are also marked in the figures.
 |
| | Fig. 3 Images and KERs: three-dimensional (3D) contour diagrams of electron (a–c) and ion (H+, (d)) images (right, bottom) and corresponding kinetic energy release spectra (e-KERs (a–c) and Ion-KERs (d); left top) derived from the images, for the two-photon resonant excitations of 74554 cm−1 (no. 3 in Table 1) (a), 75760 cm−1 (no. 4) (b), 82561 cm−1 (no. 6) (c) and 74554 cm−1 (no. 3) (d). The KERs are normalized to the strongest peak in each spectrum. The laser polarization direction is indicated by the axis labelled 0°. | |
 |
| | Fig. 4 e-KERs: electron kinetic energy release spectra (e-KERs) derived from images no. 1–7 (Table 1) plotted as a function of the kinetic energy release (KER) for no. 7 (KER0), shifted by one- (a), two- (b) and three- (c) photon energy differences, (Δ(1hν) (a); Δ(2hν) (b); Δ(3hν) (c)) with respect to the “reference spectrum”, no. 7 (see the main text). KER thresholds for the ionization of fragments, as specified, are indicated by sticks above the spectra (a–c). KER thresholds for the ionization of C2H2 to form the ground state ion C2H2+ in different vibrational states (C2H2+(X, v+1, v+2, v+3, v+4)) are indicated in (c). The inset in (c) shows an expanded part of the spectrum no. 6 at high KER. The spectra are normalized to the strongest peak in each spectrum, whereas the spectrum for no. 6 has been expanded by a factor of 7 in (a) and (b). | |
C2H2 three-photon autoionization.
In the e-KERs for images no. 1–7 relatively sharp peaks due to autoionization of C2H2#, following three-photon excitation of C2H2(X) via resonant excitation to all the Rydberg states (3p and 4p), are identified (Fig. 4c). These signals dominate for images no. 1–5. These correspond to the formation of C2H2+(X) by eqn (3) and (3i), above for different vibrational states (v+1, v+2, v+3, v+4) as indicated in Fig. 4c. The threshold energies (Ethr(v+1, v+2, v+3, v+4)) shown in Fig. 4c are the energy differences between the three-photon excitations (3hν) and the ionization energies of C2H2 in the ground electronic state and the lowest vibrational energy, C2H2(X; 0,0,0,0) to form C2H2+(X; v+1,v+2,v+3,v+4) (i.e. IE(v+1,v+2,v+3,v+4)),25| | | Ethr(v+1,v+2,v+3,v+4) = 3hν − IE(v+1,v+2,v+3,v+4) | (6) |
The peaks closely resemble those observed in the photoelectron spectra by Ashfold et al.,8 whereas our vibrational assignments differ somewhat for the excitations/spectra no. 3–7.
Fragment (Fi) ionizations.
The major signals in the images/e-KERs no. 1–5 for the 3p Rydberg states are due to the three-photon autoionization of the parent molecule via resonant excitations to the Rydberg states. The importance of signals due to ionization of fragment species on the other hand increases for the resonant excitations to the 4p Rydberg states and dominates in the image/e-KER no. 7. Furthermore, some broad features and weak spectral peaks in the e-KERs no. 4–5 are also attributed to fragment ionizations. Some relevant threshold energies for ionization of various fragment species (Ethr(Fi/Fi*)) are marked in Fig. 4a–c. These correspond to the energy difference between photon excitations (mhν) and the ionization energies of the fragments (IE(Fi/Fi*)),25| | | Ethr(Fi/Fi*) = mhν − IE(Fi/Fi*); m = 1, 2, 3 | (7) |
The thresholds are arranged in Fig. 4a–c by the number of photons (m) needed for the ionization as m = 1 in Fig. 4a, m = 2 in Fig. 4b and m = 3 in Fig. 4c. Judging from the comparison of thresholds and spectral peaks or features the major observations are as follows.
H*.
Weak, but significant peaks are seen in the e-KERs no. 4, 5 and 6 at KER0 = 1.75 eV in Fig. 4a, which fits a one-photon ionization (m = 1) of H* (n = 2),
| (5a): H* (n = 2) + 1hν → H+ + e−; e-KERs no. 4, 5 |
H* (n = 2) could be formed by a minimum of four- (n = 4; via C2H2##) photon dissociation of C2H2 (eqn (4) and (4a)).
C*.
A number of peaks associated with two- (m = 2) and one- (m = 1) photon ionization of excited carbon atoms (C*) (C*(2p2;1D), C*(2p2;1S) and C*(2s2p3;5S) for m = 2 and C*(3s;3P), C*(3s;1P) and C*(2s2p3;3D) for m = 1) were identified in the e-KERs no. 6 and 7 (Fig. 4a and b),
| (5b): C*(1D, 1S, 5S) + 2hν → C+ + e−; e-KERs no. 6, 7 |
| (5b): C*(3P, 1P, 3D) + 1hν → C+ + e−; e-KERs no. 6, 7 |
These could be formed by a minimum of three- (n = 3; via C2H2#) and four- (n = 4; via C2H2##) photon dissociation of C2H2, to form the fragment pairs C* and CH2(X) according to eqn (3c) and (4c), respectively.
CH*.
A number of peaks associated with two- (m = 2) photon ionization of the excited methylidyne radical (CH*(A, B, C)) were identified in the e-KERs no. 4–7 as, weak peaks at KER0 = 2.51 eV in spectra no. 4–7 (CH*(A)), strong to moderate peaks at KER0 = 2.84 eV in the spectra no. 6 and 7, respectively (CH*(B)) and weak peak at KER0 = 3.60 eV in the spectrum no. 7 (CH*(C); see Fig. 4b),
| (5c): CH*(A) + 2hν → CH+ + e−; e-KERs no. 4, 5, 6, 7 |
| (5c): CH*(B) + 2hν → CH+ + e−; e-KERs no. 6, 7 |
| (5c): CH*(C) + 2hν → CH+ + e−; e-KERs no. 7 |
These could be formed by a minimum of three- (n = 3) photon dissociation of C2H2, (via C2H2#) to form the fragment pairs CH* and CH(X) according to eqn (3d).
CH2.
A weak but significant peak corresponding to three- (m = 3) photon ionization of the methylene ground state radical (CH2(X)) was identified in the e-KERs of no. 6 at KER0 = 5.03 eV (see Fig. 4c and ref. 24),
| (5d): CH2 + 3hν → CH2+ + e−; e-KERs no. 6 |
These could be formed by a minimum of two- (n = 2) photon dissociation of C2H2, (via C2H2**) to form the fragment pairs C and CH2(X) according to eqn (2c).
C2*.
A clear vibrational structure due to autoionization of superexcited C2# formed by two-photon excitation of C2*(d) is seen in the low KER part of the e-KERs for no. 6 and 7 at KER0 < 0.88 eV (Fig. 4b). Analyses reveal that the structure corresponds to two-photon transitions from C2*(d, v′ = 0, 1) to C2# followed by autoionization to form C2+(X; v+ = 0–6) according to eqn (5j) for n = 2 and Fi* = C2*(d) (see ref. 24). Furthermore, weak peaks in no. 1–3 (KER0 = 2.45 eV) are also indicative of autoionization of C2# following two-photon excitation of C2*(C),
| (5j): C2*(d, v′ = 0, 1) + 2hν → C2#/C2# → C2+(X, v+ = 0–6) + e−; e-KERs no. 6, 7 |
| (5j): C2*(C, v′ = 0) + 2hν → C2#/C2# → C2+(X, v+ = 0–6) + e−; e-KERs no. 1, 2, 3 |
A minimum of two- (n = 2) and three- (n = 3) photon dissociation processes to form the C2*(d) and C2*(C) species along with H2, respectively, could explain these observations.
C2H*.
Broad spectral features are seen in the low KER part of the e-KERs no. 4 and 5. These match on the “Δ(2hν) shift scale” in the region below KER0 of about 2.3 eV (Fig. 4b), which corresponds to the threshold for two-photon ionization of C2H*(B′).28 This suggests that the broad feature is due to autoionization of C2H# to form C2H+(X) after two-photon excitation of C2H*(B′), by analogy to the observation mentioned in the previous paragraph concerning C2*(d)/C2#/C2+(X),
| (5j): C2H*(B′) + 2hν → C2H#/C2H# → C2H+(X) + e−; e-KERs no. 4, 5 |
A closer look at the broad feature of the e-KERs for no. 424 reveals an overlapping vibrational structure of a frequency close to that of the e CC-vibrational stretching mode (ν3) for C2H+(X; v3).23 A minimum of two- (n = 2) photon dissociation processes to form the C2H*(B′) along with H could explain these observations.
C. Ions; ion-KERs
H+, C+, CH+, C2+ and C2H+ ion images were recorded for the two-photon resonant excitation of C2H2 to the molecular Rydberg states no. 1–2 and 4–7 as listed in Table 1 and marked in Fig. 1a. Ion kinetic energy release spectra (ion-KERs) were derived from the images. A selected image, as a three-dimensional (3D) contour diagram and the corresponding ion-KERs is shown in Fig. 3d (see also ref. 24). None of the images show sharp rings and all the ion-KERs typically consist of one or two broad peaks, peaking at low kinetic energy release (KER) and tailing towards a high kinetic energy release. In cases where the ions are formed by photoionization of fragments this structure is indicative of an energy redistribution among the molecule's internal degrees of freedom prior to dissociation.16,17 The ion-KERs were displayed as shifted by two-, three- and four-photon energy differences (Δ(2hv) = |2hvi − 2hv0|; Δ(3hv) = |3hvi − 3hv0|; Δ(4hv) = |4hvi − 4hv0|; i refers to different spectra) with respect to the spectrum derived for the highest energy excitation (i.e. the “reference spectrum”, no. 7; subscript 0) on the energy scale for the “reference spectrum” (KER0; eV) to allow comparison of common spectral features due to two-, three- and four-photon dissociation processes, respectively.12–17 Threshold kinetic energies (Ethr), corresponding to the energy difference between the photoexcitations (nhν) and the energies of the various fragments (Fi) formed (E(Fi)) according to eqn (2)–(4) above were evaluated,| | | Ethr = nhν − E(Fi); n = 2, 3, 4 | (8) |
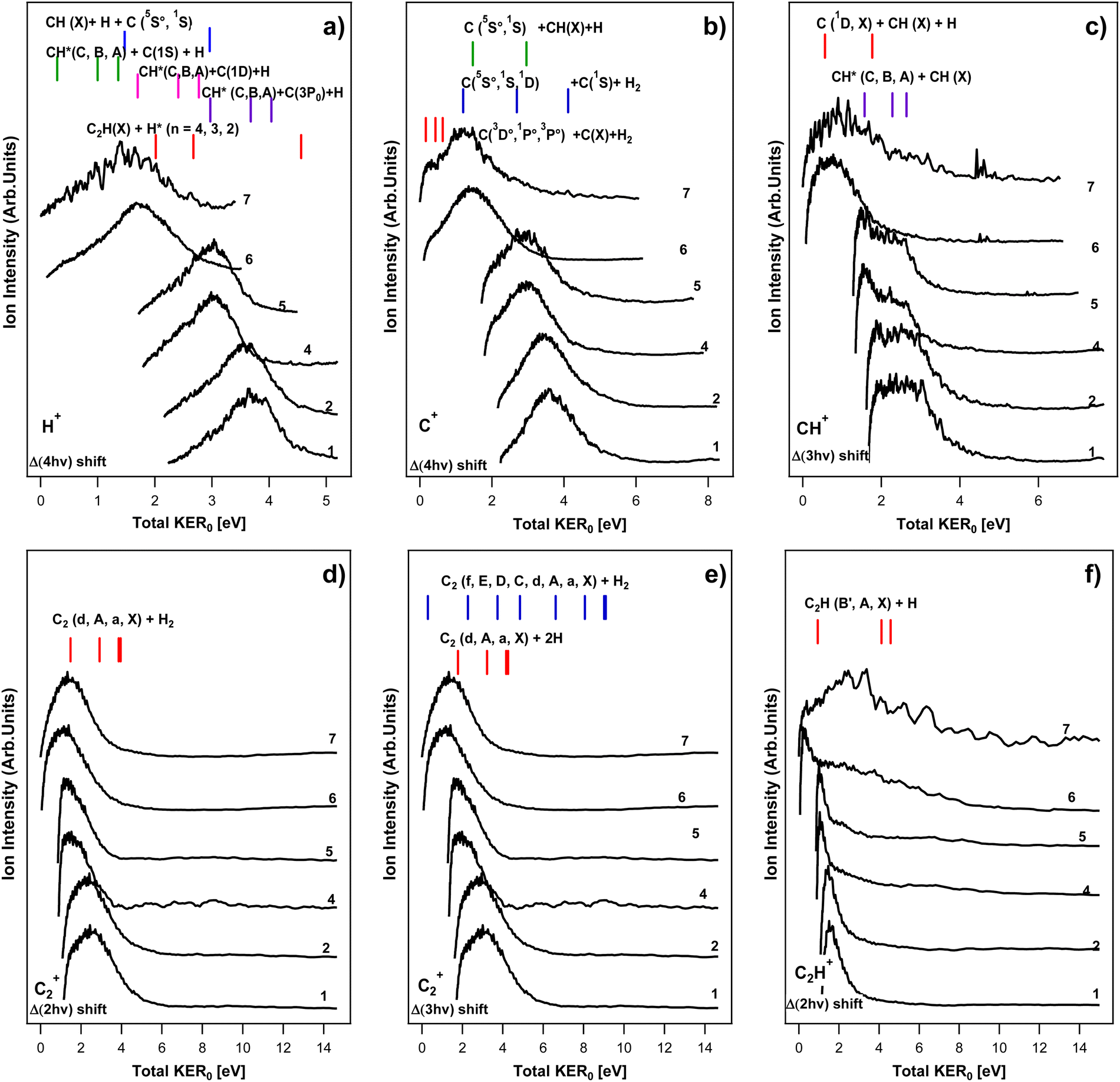
and compared with the spectra to explore the signals origin. The broad spectral features with tails towards the high kinetic energy release, lower than the threshold, could, in principle, all correspond to processes marked by the thresholds. In the following subsections we collect possible fragmentation channels, within eqn (2)–(4), which could be responsible for the ion-KERs signals, based on these criteria. More relevant figures are to be found in the supporting material,24 whereas only a limited number (see Fig. 5) are presented here, mainly for demonstration purposes.
 |
| | Fig. 5 Ion-KERs: ion kinetic energy release spectra (ion-KERs) derived from images no. 1, 2, 4–7 (see Table 1). The spectra are plotted as a function of the total kinetic energy release for no. 7 (total KER0) and shifted by n-photon energy differences (Δ(nhv)) with respect to the “reference spectrum”, no. 7 (see the main text) as, Δ(4hv) for H+ (a), Δ(4hv) for C+ (b), Δ(3hv) for CH+ (c), Δ(2hv) for C2+ (d), Δ(3hv) for C2+ (e) and Δ(2hv) for C2H+ (f). KER thresholds for fragment formations by n-photon-dissociation (n = 2, 3 and 4 for photodissociation via the C2H2**, C2H2# and C2H2## intermediate species), as specified, are indicated by sticks above the spectra. The spectra are normalized to the strongest peak in each spectrum. | |
H+ KERs.
The H+ signals show broad peaks, possibly with two overlapping contributions in some instances, ranging typically over about 3 eV on the total KER scale (see Fig. 5a and ref. 24). The analyses revealed that the following fragmentation channels (nx) could, in principle, be responsible for H/H* formations, prior to ionization:
| (2a): C2H2** → H + C2H*(B′); Ion-KERs no. 6, 7, low KER |
| (2a): C2H2** → H + C2H(X)/C2H*(A); All ion-KERs |
| (3a): C2H2# → H + C2H*(Ry); Ion-KERs no. 6, 7, low KER |
| (3a): C2H2# → H + C2H*(C, B); All ion-KERs |
| (3e): C2H2# → H + H + C2*(d); Ion-KERs no. 4, 5, 6, 7, low KER |
| (3e): C2H2# → H + H + C2/C2*(A, a); All ion-KERs |
| (3g): C2H2# → CH + H + C*(1D); Ion-KERs no. 6, 7, low KER |
| (3g): C2H2# → CH + H + C*(3P); Ion-KERs no. 4, 5, 6, 7, low KER |
| (4a): C2H2## → C2H + H*(n > 2); All ion-KERs |
Furthermore, a molecular ion photodissociation could also be responsible for the H+ formation:
| (3i): C2H2# → C2H2+ + e−; followed by, |
| (5h): C2H2+(X) + 2hν → C2H + H+; All ion-KERs |
C+ KERs.
The C+ signals show broad peaks with overlapping contributions in all spectra, ranging typically over about 4–5 eV on the total KER scale (see Fig. 5b and ref. 24). The analyses revealed that the following fragmentation channels could, in principle, be responsible for C/C* formations, prior to ionization:
| (2c): C2H2** → C + CH2/CH2*(a); Ion-KERs no. 1–7, low KER |
| (3c): C2H2# → C/C*(5S,1S,1D) + CH2/CH2*(b, a); All ion-KERs |
| (3f): C2H2# → H2 + C/C*(1D) + C/C*(1S,1D); All ion-KERs |
| (3g): C2H2# → H + CH + C/C*(1D); Ion-KERs no. 6–7 |
| (4c): C2H2## → C*(3D,1P,3P) + CH2/CH2*(b, a); Ion-KERs no. 1–5 |
| (4f): C2H2## → H2 + C/C*(1S) + C/C*(many); Ion-KERs no. 6–7, low KER |
| (4g): C2H2## → H + CH + C*(5S,1S); All ion-KERs |
CH+ KERs.
The CH+ signals show broad peaks with clear overlapping contributions in no. 1–5, ranging typically over about 3–4 eV on the total KER scale (see Fig. 5c and ref. 24). The analyses revealed that the following fragmentation channels could, in principle, be responsible for CH/CH* formations prior to ionization:
| (2d): C2H2** → CH(X) + CH(X); Ion-KERs no. 6, 7, low KER |
| (3d): C2H2# → CH + CH*(C,B,A); Ion-KERs no. 1–5 |
| (3g): C2H2# → CH + H + C*(1D); Ion-KERs no. 6, 7, low KER |
| (3g): C2H2# → CH + H + C; All ion-KERs, low KER |
| (4g): C2H2## → H + CH/CH*(C,B,A) + C/C*(1D,1S); Ion-KERs no. 1–7 |
C2+ KERs.
The C2+ signals show broad peaks with some overlapping contributions, ranging typically over about 4–5 eV on the total KER scale (see Fig. 5d and ref. 24). The analyses revealed that the following fragmentation channels could, in principle, be responsible for C2/C2* formations prior to ionization (see above):
| (2b): C2H2** → H2 + C2/C2*(d, A, a); All ion-KERs |
| (3b): C2H2# → H2 + C2/C2*(f, E, D, C); All ion-KERs |
| (3e): C2H2# → H + H + C2/C2*(d, A, a); All ion-KERs |
C2H+ KERs.
The C2H+ signals show relatively narrow peaks at low KER for no. 1, 2, 4–6, ranging typically over about 1–2 eV on the total KER scale and a broad high KER contribution for all the images/spectra. The analyses revealed that the following fragmentation channels could, in principle, be responsible for the low KER contributions to the C2H/C2H* formations prior to ionization:
| (2a): C2H2** → H + C2H/C2H*(B′, A); All ion-KERs, low KER |
| (3a): C2H2# → H + C2H*(Ry); Ion-KERs no. 6, 7, low KER |
| (3a): C2H2# → H + C2H/C2H*(C, B, B′, A); All ion-KERs |
Furthermore, a molecular ion photodissociation could also be responsible for the C2H+ formation:
| (3i): C2H2# → C2H2+ + e−; followed by |
| (5i): C2H2+(X) + 2hν → C2H+(X) + H; All ion-KERs, low KER |
D. Correlation analyses (A, B, C combined)
Based on the analyses described in Sections A (MR-REMPI), B (e-KERs) and C (ion-KERs), above concerning possible origins of the ion (A, C) and corresponding electron (B) signals the following results, relevant to the ion formations, are derived (correlation analyses) (see also Fig. 1b, 4, 5 and Table 1):
H+.
H+ signals were detected for the excitations no. 4–7 in MR-REMPI with a maximum relative ion signal for no. 6 (A) (Fig. 1b). e-KERs of images no. 4–5 suggest that one- (m = 1) photon ionization of H* (n = 2) is occurring (B) (Fig. 4a). Judging from the ion-KERs H+ ions could, in principle, be formed after four- (n = 4) photon dissociation of C2H2, via C2H2##, to form H* (n = 2) along with C2H(X) (C) (see Fig. 5a and ref. 24). In combination we conclude that the following multiphoton-fragmentation processes are important:
| (4a): C2H2## → C2H + H* (n = 2); All excitations |
| (5a): H* (n = 2) + 1hν → H+ + e−; e-KERs no. 4, 5 |
C+.
C+ signals were detected for the excitations no. 4–7 in MR-REMPI with a maximum relative ion signal for excitations no. 6 (A) (Fig. 1b). e-KERs of images no. 6–7 suggest that two- (m = 2) photon ionization of C*(1D, 1S, 5S) and one- (m = 1) photon ionization of C*(3P, 1P, 3D) is occurring (B) (Fig. 4a and b). Judging from the ion-KERs C+ ions could, in principle, be formed after three- (n = 3) photon dissociation of C2H2, via C2H2#, to form C*(5S,1S,1D) along with CH2/CH2*, H2 and H + CH and after four- (n = 4) photon dissociation of C2H2, via C2H2##, to form C*(3D,1P,3P) along with CH2/CH2* and H2 as well as C*(5S,1S) along with H and CH (C) (see Fig. 5b and ref. 24). In combination we conclude that the following multiphoton-fragmentation processes could be important:
| (3c): C2H2# → C*(5S,1S,1D) + CH2/CH2*(b,a); All ion-KERs |
| (3f): C2H2# → H2 + C*(1D) + C*(1S,1D); All ion-KERs |
| (3g): C2H2# → H + CH + C*(1D); Ion-KERs no. 6–7 |
| (4c): C2H2## → C*(3D,1P,3P) + CH2/CH2*(b, a); Ion-KERs no. 1–5 |
| (4f): C2H2## → H2 + C*(1S) + C*(many); Ion-KERs no. 6–7, low KER |
| (4g): C2H2## → H + CH + C*(5S,1S); Ion-KERs no. 6–7, low KER |
| (5b): C*(1D, 1S, 5S) + 2hν → C+ + e−; e-KERs no. 6, 7 |
| (5b): C*(3P, 1P, 3D) + 1hν → C+ + e−; e-KERs no. 6, 7 |
CH+.
Maximum relative ion signals of CH+ are observed in MR-REMPI for excitation no. 6 (A) (Fig. 1b). e-KERs of images no. 4–7 suggest that two- (m = 2) photon ionization of a number of CH* species (CH*(A, B, C)) is occurring (B) (Fig. 4b). Judging from the ion-KERs CH+ ions could, in principle, be formed after three- (n = 3) photon dissociation of C2H2, via C2H2#, to form CH*(C,B,A) along with CH and after four- (n = 4) photon dissociation of C2H2, via C2H2##, to form CH*(C,B,A) along with C/C* (C) (see Fig. 5c and ref. 24). In combination we conclude that the following multiphoton-fragmentation processes are important:
| (3d): C2H2# → CH + CH*(C); Excitations no. 4–7, low ion-KER |
| (3d): C2H2# → CH + CH*(B, A); All excitations, low ion-KER |
| (4g): C2H2## → H + CH*(C, B, A) + C*(1S); Excitations no. 6–7 |
| (4g): C2H2## → H + CH*(C, B, A) + C/C*(1D); All excitations |
| (5c): CH*(C, B, A) + 2hν → CH+ + e−; e-KERs no. 4, 5, 6, 7 |
CH2+.
The relative ion signals of low CH2+ intensities observed in MR-REMPI reaches maximum for no. 6 (A) (Fig. 1b). The e-KER of image no. 6 suggests that three-(m = 3) photon ionization of the ground state CH2(X) is occurring (B) (Fig. 4c). Energetically CH2(X) could be formed along with C by two- (n = 2) photon dissociation, via C2H2** and along with a number of C* atoms as well as C by three- (n = 3) photon dissociation, via C2H2# (Fig. 2c). In combination, we conclude that the following multiphoton-fragmentation processes could be important:
| (2c): C2H2** → C + CH2; All excitations |
| (3c): C2H2# → C/C* + CH2; All excitations |
| (5d): CH2 + 3hν → CH2+ + e−; e-KER no. 6 |
C2+.
The relative ion signals of C2+ observed in MR-REMPI are found to increase gradually with excitation (A) (Fig. 1b). e-KERs of images no. 6 and 7 suggest that two-photon ionization of C2*(d; v′ = 0, 1) is occurring (B) (Fig. 4b). e-KERs of no. 1–3 suggest that two-photon ionization of C2*(C) is occurring (B) (Fig. 4b). Judging from the ion-KERs C2+ ions could, in principle, be formed after two- (n = 2) and three- (n = 3) photon dissociation of C2H2, via C2H2** and C2H2#, respectively to form C2*(d) along with H2 (n = 2, 3) and 2H (n = 3) for all excitations (C) (see Fig. 5d, e and ref. 24). C2+ ions could also be formed after three- (n = 3) photon dissociation of C2H2 to form C2*(C) along with H2 for all excitations (C) (Fig. 5d). In combination, we conclude that the following multiphoton-fragmentation processes are important:
| (2b): C2H2** → H2 + C2*(d); All excitations, low ion-KER |
| (3b): C2H2# → H2 + C2*(C); All excitations |
| (3e): C2H2# → H + H + C2*(d); All excitations, low ion-KER |
| (5j): C2*(d, v′ = 0, 1) + 2hν → C2#/C2# → C2+(X, v+ = 0–6) + e−; e-KERs no. 6, 7 |
| (5j): C2*(C, v′ = 0) + 2hν → C2#/C2# → C2+(X, v+ = 0) + e−; e-KERs no. 1, 2, 3 |
C2H+.
Maximum relative ion signals of C2H+ are observed in MR-REMPI for excitations no. 4 and 5 (A) (Fig. 1b). e-KERs of images no. 4 and 5 suggest that two- (m = 2) photon ionization of C2H*(B′) is occurring (B) (Fig. 4b). Judging from the ion-KERs C2H+ ions could, in principle, be formed after two- (n = 2) and three- (n = 3) photon dissociation of C2H2, via C2H2** and C2H2#, respectively to form C2H*(B′) along with H for all the excitations (C) (see Fig. 5f and ref. 24). In combination we conclude that the following multiphoton-fragmentation processes are important:
| (2a): C2H2** → H + C2H*(B′); All excitations, low ion-KER |
| (3a): C2H2# → H + C2H*(B′); All excitations, high ion-KER |
| (5j): C2H*(B′) + 2hν → C2H#/C2H# → C2H+(X) + e−; e-KERs no. 4, 5 |
E. Angular distributions
All the ion images recorded display shapes corresponding to either parallel or isotropic distributions depending on the ions and the kinetic energy released (see Fig. 3d and ref. 24). Given the number of photons and the multiple channels/pathways involved in the production of each ion and electron angular distribution recorded, a detailed fitting and analysis will not provide easily interpretable results as the extracted anisotropy parameters would be the average of multiple processes. However, to obtain an insight on the general trend, we fitted selected KER ranges with the one-step (non-resonant) expression for angular distribution,15| |  | (9) |
to derive anisotropy parameters β2n (n = 1–3), where n is the number of photons involved in the photolysis, P2n is the 2n-th order Legendre polynomial and A is a scaling factor. β2n parameters derived from the data are to be found in ref. 24. β2, which is a convenient parameter to indicate the degree of anisotropy of the transitions involved, ranges from +2 (purely parallel transition) to −1 (purely perpendicular transition), via 0 (isotropic). These were found to be in the range of +1.3 (H+) to −0.2 (C2H+). There is an overall tendency toward a decreasing β2 value with ion mass. Furthermore, a tendency of a gradual decrease in β2 with the excitation energy is observed for all the fragment ions except C2H+, which shows a drop in the value from about +0.75 ± 0.25 (for excitations no. 1, 2, 4) to about 0 ± 0.15 (for no. 5, 6, 7). An attempt was made to derive β2 parameters for separate channels from images which appear to display bi- (or multi-) modal contributions by evaluating β2 for different KER ranges (low and high ion-KER). Significantly different β2 values were obtained for high and low ion-KER contributions of the C+ and CH+ images (see ref. 24). No further interpretations of the angular distribution data24 in connection with the fragmentation processes are attempted here.
IV. Discussion
C2H*(B′)/C2H+
Judging from our data (see Section III, D) low KER peaks seen in the C2H+ ion-KERs no. 4 and 5 (Fig. 5f) are likely to be due to two-photon ionization of C2H*(B′) after a “near-resonant” energy transfer from the corresponding Rydberg states (C2H2**) (Table 1) to form C2H*(B′) and H after two-photon excitation of C2H2 (eqn (2a)). This could be used to estimate the energy of the C2H*(B′) species, which is the subject of an uncertainty: The value of the energy for the C2H*(B′) state (E(C2H*(B′))) reported in NIST25 of 3.640 eV (29360 cm−1) is determined from an absorption spectrum recorded after photolysis of C2H2 trapped in a solid argon matrix.26 In addition, calculated values of 3.81 eV (thermodynamic calculation)27 and 3.716 eV (ab initio calculation)28 have been reported for E(C2H*(B′)). An alternative estimate of E(C2H*(B′)), based on our data (gas phase condition), is as follows.
Conservation of the energy in the two-photon excitation step gives,
| | | 2hν = D0(C2H–H) + E(C2H*(B′)) + Ethr | (10a) |
where
D0(C
2H–H) is the bond energy of C
2H
2(X) to form C
2H(X) and H
29 and
Ethr is the threshold energy for C
2H*(B′) + H (
eqn (8)). This gives,
| | | E(C2H*(B′)) = 2hν − D0(C2H–H) − Ethr | (10b) |
The threshold energies (
Ethr) refer to the upper limits of the low ion kinetic energy release peaks for no. 4 and 5, which cannot be determined due to overlap of spectral contributions (see
Fig. 5f) Using the low ion-KERs peak positions (
Epeak) as an approximation for
Ethr, however, allows estimates of the upper limit for
E(C
2H*(B′)),
| | | E(C2H*(B′)) ≤ 2hν − D0(C2H–H) − Epeak | (10c) |
of about 3.54 ± 0.01 eV (see ref.
24).
C2*/C2+
In what follows we will discuss the probable fragmentation channels prior to C2+ formation according to our observations (see Section III, D) in the context of other observations.
C2*(d)/C2+.
According to our data, C2+ could be formed by autoionization of a superexcited state C2# formed by two-photon ionization of C2*(d, v′ = 0, 1) (eqn (5j)) for the excitations no. 6 and 7 (Table 1). It also shows that C2*(d, v′ = 0, 1) could be formed by two- (n = 2) photon excitation of C2H2 (via C2H2**) along with H2 (eqn (2b)) and/or by three- (n = 3) photon excitation of C2H2 (via C2H2#) along with 2H (eqn (3e)) for all the excitations (see Section III, D). The following observations/arguments favor formation of C2*(d) along with 2H by three-photon dissociation over the formation of C2*(d) along with H2 by two-photon dissociation:
(i) Fluorescence studies based on the C2*(d) → C2*(a) transition (the Swan band6) reveal no C2*(d) formation for C2H2 for one-photon excitation corresponding to the two-photon excitation region of concern here (i.e. 73900–83100 cm−1),30 whereas a significant C2*(d) fluorescence is detected for excitation to the region of 110850–124650 cm−1, which corresponds to the three-photon excitation here.30 This suggests that three-photons are required for the C2*(d) formation here.
(ii) A close correlation is found between the signals of REMPI spectra derived for the molecular parent ion (C2H2+)8,9 and a fluorescence excitation spectrum due to C2*(d) → C2*(a)6 derived for multiphoton excitations via the Rydberg states no. 1–6 (see Fig. 6). This strongly suggests that the formations of C2H2+ and C2*(d) are of the same origin, i.e. from the superexcited state(s) C2H2#, formed by three-photon excitation.
 |
| | Fig. 6 Fragment fluorescence and acetylene spectra. (a) Fluorescence quantum yield for C2H*(B′), due to the C2H*(B′) → C2H(X) + hν emission transition in one-photon-dissociation of C2H2.27 (b) Fluorescence excitation spectrum of C2H2 for the C2*(d) → C2*(a) + hν (Swan band) emission transition following two-photon excitation to C2H2** Rydberg states.6 (c) C2H2+ REMPI spectra for two-photon resonant excitations to C2H2** Rydberg states, from ref. 9 (black), 8 (brown) and 6 (purple). (d) Absorption spectrum of C2H2.27 | |
(iii) Dissociation of C2H2** to form C2*(d) and H2 (eqn (2b)) will involve a transition via a cis-conformation geometry.9 Calculations reveal an energy maximum for the lowest energy singlet state cis-conformer of about 76000 ± 1000 cm−1, which excludes a corresponding dissociation of C2H2** for no. 1–3 and limits the corresponding transition/dissociation probabilities for no. 4–5 (energies 75760 cm−1 and 76085 cm−1, respectively). Furthermore, all the Rydberg states of concern (C2H2**) are, to a first approximation, considered to be linear in structure,8 which limits the possibility of transformation to a cis-geometry without additional state transfer processes.
We, therefore, conclude that C2*(d) is formed by the three-photon dissociation mechanism (3e), rather than by the two-photon dissociation mechanism (2b), for all the excitations. While C2*(d) is formed for all the excitations according to the fluorescence data,6 C2+ signals from the ionization of C2*(d) (Fig. 4) are only observed for the excitations no. 6 and 7, for energetic reasons, since only two-photons are required to ionize C2*(d) in no. 6 and 7 compared to three-photons for no. 1–5. A striking difference, however, is seen in the C2+ signal intensities for ionization of C2*(d) for no. 6 and 7 as (no. 7) ≫ (no. 6) (see Fig. 4). Most likely this is due to a significant difference in the branching fractions for the C2*(d) formations (Γ(C2*(d))) via channel (3e) for no. 6 and 7, as (Γ(C2*(d)) for no. 7) ≫ (Γ(C2*(d)) for no. 6). This is further evident from the striking difference in the relative intensities of the C2H2+ REMPI signal and the C2*(d) fluorescence signal as seen in Fig. 6 for no. 7 as well as the relatively small C2H2+ signal in the e-KERs for no. 7 (Fig. 4(c)).
While (3e) is likely to be the dominant channel for the C2*(d) formation, a contribution to the formation of C2*(d) from the photodissociation of C2H*(B′) (see above) is worth an investigation. As mentioned above (subsection “C2H*(B′)/C2H+”) our data suggests that C2H*(B′) is formed after a two-photon excitation (via C2H2**) (channel (2a)). C2H*(B′) was found to be a long lived species31 formed to an increasing extent with excitation energy in one-photon excitation27 in the region of concern, reaching a quantum yield of about 6.7% for the excitation to no. 7 (see Fig. 6). Further photoexcitation of C2H*(B′) (in no. 6 and 7) could, energetically, form excited states (Rydberg32 and/or valence states33), which are known to exist in that energy region and which could predissociate to form C2*(d) and H (channel (5k)).33 This could be an important additional contribution to the C2*(d) formation, for no. 6 and 7 in particular.
C2*(C)/C2+.
According to our data, C2+ could also be formed by autoionization of a superexcited state C2# formed by two-photon ionization of C2*(C) (eqn (5j)) for no. 1, 2 and 3 (Table 1). It also shows that C2*(C) could be formed by three- (n = 3) photon excitation of C2H2 (via C2H2#) along with H2 (eqn (3b)) for all the excitations (see Section III, D). This matches the result of fluorescence studies by Han et al. who observed C2*(C) fluorescence due to the C2*(C) → C2*(A) transition for one-photon excitation of C2H2, which corresponds to an excitation within our three-photon excitation region, above the threshold for C2*(C) + H2 and below the threshold for C2*(C) + 2H (see Fig. 2b).30
C2*(B′, v′ = 3)/C2+.
A sharp peak sticks out in the e-KERs for no. 6 on the low KER side of the threshold for the C2*(d, v′ = 0) ionization (see Fig. 4b). A corresponding weak peak, which matches on the “Δ(2hν) shift scale” was also found for no. 7, depending on the image recording conditions.24 This suggests that these peaks are of the same origin for two-photon ionization. These could not be assigned to any ionizations of relevant atom fragments or molecular fragments for zero vibrational states. Instead we assign these peaks to ionizations of C2*(B′, v′ = 3). This is based on the calculated potential energy curves for C2*34,35 and spectroscopic parameters for C2*(B′)34 and C2*(d),25 which suggest that the peak positions match an ionization of C2*(B′, v′ = 3), which is close in energy to C2*(d, v′ = 0) (ΔE ≈ 0.1 eV) (see also ref. 24). The appearance of peaks due to ionization of the v′ = 3 level only and an absence of peaks due to ionization of other v′ levels of C2*(B′) could be associated with mixing between the two states, which will depend on the Franck-Condon overlap of the vibrational wave functions and be inversely proportional to the energy gap between the v′ levels for the two states,12–14,18 to exaggerate the ionization process for C2*(B′, v′ = 3) due to near-degenerate mixing with C2*(d, v′ = 0), i.e. an “intensity borrowing” effect.
CH*(C,B,A)/CH+
Judging from our data (see Section III, D) CH+(X) could be formed by two- (m = 2) photon ionization of (CH*(C,B,A) (eqn (5c)) (possibly via autoionization of superexcited CH# (eqn (5j)) for no. 4–7 and that CH*(C,B,A) could be formed by three- (n = 3) and/or four- (n = 4) photon dissociation processes along with CH (channel (3d)) and C* (4g), respectively. Our prediction of the former photodissociation channels (3d) matches observations in fluorescence studies by Han et al., who detected CH*(C,B,A) fluorescence due to transitions from CH*(C,B,A) to CH(X) in the case of one-photon excitation of C2H2 in the excitation region corresponding to our three-photon excitations (see Fig. 2b).30 The latter channel (4g), however, cannot be ruled out. Thus, the broad range of the ion-KERs for CH+ for no. 7, in particular (see Fig. 5c) and observation of strong C*(1S) and CH*(B) two- (m = 2) photon ionization signals in the e-KER for no. 6 (Fig. 4b) suggest its involvement.
C*/C+
Based on our data (see Section III, D) C+ could be formed by two- (m = 2) and one- (m = 1) photon ionization of lower (C*(1D, 1S, 5S)) and higher (C*(3P, 1P, 3D)) energy C* atoms, respectively, for no. 6 and 7. The lower energy carbon atoms (C*(1D, 1S, 5S)) could be formed by three-photon dissociation channels ((3c), (3f) and (3g)), whereas the higher energy atoms (C*(3P, 1P, 3D)) would be formed by four-photon dissociation channels ((4c), (4f) and (4g)). In both cases the total number of photons (n + m) would be 5. The byproducts are either CH2/CH2* (channels (3c) and (4c)), H2 (channels (3f) and (4f)) or H and CH (channels (3g) and (4g)). The observation of a CH2(X) ionization according to the e-KERs for no. 6 supports the existence of channels (3c) and/or (4c), as the source for the lower energy (C*) species. An additional contribution to the source of C*(1D) could be a one-photon dissociation of CH*(A) (channel (5k)), which is found to be formed for no. 6 and 7 (see above). It has been shown by calculations36 and experimentally37,38 that high lying Rydberg states of CH (CH**), which could be accessed by one-photon excitation of CH*(A) in the excitation energy region of concern, could be predissociated by a repulsive 2Π state to form C*(1D) + H. Thus, for example, CH*(A) could be formed by three-photon dissociation of C2H2 (along with CH(X)) followed by one-photon excitation of CH*(A) and predissociation to form C*(1D) (i.e. total number of photons in the excitation of n = 3) prior to two- (m = 2) photon ionization of C*(1D) (i.e. n + m = 5).
V. Summary, conclusions and closing remarks
Data of mass-resolved (MR) REMPI (Section III, A) and velocity map images (VMI) of photoelectron and ion products following two-photon resonant excitations of C2H2 to seven Rydberg states (VMI-REMPI) (Section III, B and C) (Table 1) were recorded and analyzed. The analyses involved determination of relative ion intensities, derivation of electron- and ion KER spectra (e-KERs and ion-KERs) and angular distributions for a number of ions formed as a function of the excitation. Threshold energies corresponding to the maxima of kinetic energy release for electrons formed in multiphoton ionization as well as fragments formed in multiphoton dissociation processes for various species (both in ground and excited states) and dissociation channels were determined (Fig. 4 and 5). These were compared systematically with electron- and ion-KERs signals/spectral features in our attempt to explore the signal origin. In combination, the analyses revealed multiphoton-fragmentation (photodissociation and/or photoionization) processes relevant to the formation of the parent molecular ion (C2H2+) and the fragment ions H+, C+, CH+, CH2+, C2+ and C2H+ (Section III, D). Whereas, the parent molecular ion formation dominated for the lower energy excitations, the fragment ion formation gradually increases with energy and was found to dominate for the excitation no. 7 (Table 1). The major photodissociation channels observed are summarized schematically in Fig. 7 and collected in Table 2. First, C2H2+ is formed by autoionization of superexcited molecules (C2H2#) formed by three-photon excitation for all the cases (no. 1–7; Table 1). This appears to be the major channel for no. 1–5. Second, various neutral fragments are formed either in ground or excited states by two-, three- or four-photon dissociation processes via the Rydberg states (C2H2**) or superexcited states (C2H2# and C2H2##), respectively (marked as nx, where n = 2, 3, 4/number of photons in the excitations and x = a–g in Fig. 7 and Table 2), prior to photoionization. All in all, the three-photon dissociation processes, via the first superexcited state(s) (C2H2#), are found to play the biggest role in the overall multiphoton-fragmentation process. Although all the photoexcited molecular states (C2H2**, C2H2# and C2H2##) are found to be involved in multiphoton-fragmentation, their involvement and importance will vary, depending on the photon density. The first superexcited state(s) (C2H2#), being metastable, hence short lived, with respect to both autoionization and dissociation, is an important gateway for fragment formation in competition with molecular autoionization. Further excitation to the second superexcited state(s) (C2H2##), will compete with these fast fragmentation and autoionization channels and, therefore, largely diminish with decreasing photon density. The resonant excitation to the Rydberg states, to a large extent, acts as an enhancement step for further transitions to the C2H2# state(s). Thus, in combination, it makes sense that the “intermediate” excited state/first superexcited state (C2H2#) plays the most important role as a gateway towards the fragment formation, prior to ionization under our experimental conditions. Thus, for example, the formation of C2*(d) along with two hydrogen atoms (2H) by three-photon dissociation is found to be favored over two-photon dissociation to form C2*(d) along with H2. Overall, the involvement of ionization of electronically excited state fragment species (Fi*) in the multiphoton-fragmentation processes is found to be important (Table 2) and the e-KER spectra reveal a characteristic vibrational structure for the C2*(d) ionization indicative of autoionization processes of superexcited C2#. In addition to the major processes detected and shown in Fig. 7 and Table 2, further photofragmentation processes involving secondary photodissociation channels (i.e. photodissociation of fragment species C2H*(B′) and CH*(A); eqn (5k)) followed by photoionization processes (eqn (5x)) are also proposed (see Section IV). Furthermore, the data allowed evaluation relevant to the energetics of the C2H*(B′) state, which is the subject of uncertainty in the literature26–28 (see Section IV, “C2H*(B′)/C2H+”) and revealed a state interaction in C2* between C2*(d, v′ = 0) and C2*(B′, v′ = 3)34,35 (see Section IV, “C2*(B′, v′ = 3)/C2+”).
 |
| | Fig. 7 Multiphoton-fragmentation processes: schematic energy diagram for multiphoton-excitation processes of C2H2 according to this work. Vertical black arrows denote photon absorption. Blue, red and green arrows denote fragmentation processes after the two-, three- and four-photon excitations, respectively. The fragmentation processes are marked by the number of photons (n) required for the excitations and letters (nx) (see Section III, A). C2H2(X), C2H2**, C2H2# and C2H2## are the ground-, Rydberg- and first and second superexcited states, respectively. | |
Table 2 Fragment ion (Fi+) formation processes detected
| Fi+ |
Photofragmentation |
Photoionization |
| (nx)a |
No. excitationsb |
m
|
Species ionized |
No. excitationsb |
|
(nx) refers to the eqn numbers given in Section III, A; n = number of photons in photon-fragmentation; x = letter, a, b,….
See Table 1.
m = number of photons in photoionization.
C*(low energy): C*(5S,1S,1D); C*(high energy): C*(3D,1P,3P).
Channel (3e) is favoured over (2b): see Section IV, “C2*/C2+”.
CH*(A, B, C): CH valence states A2Δ, B2Σ−, C2Σ+; CH2(X): CH2 ground state X2B1; C2*(d, C): C2 excited states d2Πg, C2Πg; C2H*(B′): C2H B′2A1.
|
| H+ |
4a |
1–7 |
1 |
H* (n = 2) |
4, 5 |
| C+ |
3c, 3f |
1–7 |
2 |
C*(low energy)d |
6, 7 |
| C+ |
3g |
6, 7 |
2 |
C*(low energy)d |
6, 7 |
| C+ |
4c |
1–5 |
1 |
C*(high energy)d |
6, 7 |
| C+ |
4f, 4g |
6, 7 |
1 |
C*(high energy)d |
6, 7 |
| CH+ |
3d, 4g |
1–7 |
2 |
CH*(A, B, C)f |
4–7 |
| CH2+ |
2c, 3c |
1–7 |
3 |
CH2(X)f |
6 |
| C2+ |
3ee |
1–7 |
2 |
C2*(d)f |
6, 7 |
| C2+ |
3b |
1–7 |
2 |
C2*(C)f |
1–3 |
| C2H+ |
2a, 3a |
1–7 |
2 |
C2H*(B′)f |
4, 5 |
The acetylene molecule (C2H2), which is the smallest organic molecule in terms of the number of atoms, has been observed in interstellar space.3,10 The question arises whether it could be an important source of reactive organic intermediate species (radicals and ions) such as those observed in this work, upon UV radiation in space. Thus, it could act as a source of important building blocks for further creation of bigger organic molecules formed by radical and/or ion reactions either on surfaces or in space. The triple bond of the molecule is an underlying reason for the large number of possible transitions by photoexcitation of the π valence electrons to Rydberg orbitals, hence the formation of Rydberg states. These belong to Rydberg series which either converge to the ground ionic state (i.e. C2H2**) or excited ionic states (i.e. superexcited states C2H2#), the latter of which are found to be important gateways for radical and excited state fragment formations. Thus, it can be argued that the “unsaturated nature” of acetylene makes it particularly suitable for the formation of small organic radicals and ion species upon UV radiation corresponding to excitation to superexcited Rydberg states beyond its ionization limit (11.4 eV), corresponding to λ < 109 nm, one-photon excitation. Although our excitation scheme involves three-photon excitation of C2H2 to C2H2#via C2H2** rather than one-photon excitation directly to C2H2# the dominant selection rule of g ↔ u per photon excitation (hence g ↔ u for three- and odd number-photon excitations) for C2H2 ensures similarity for both one- and three-photon excitations to the superexcited state(s) (C2H2#) region. We, therefore, feel that our observations are of important relevance to understanding one-photon UV excitation of acetylene as well as for (multi)photon-fragmentation processes in molecules in general.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
The financial support from the University Research Fund, University of Iceland and the Icelandic Research Fund (Grant No. 184693-053) is gratefully acknowledged. Aðalsteinn Kristjánsson's memorial fund for the promotion of natural sciences and chemistry is also acknowledged. The imaging part of the reported results was carried out at the Ultraviolet Laser Facility at IESL-FORTH, supported in part by the European Union's Horizon 2020 Research and Innovation Programme LASERLAB-EUROPE (Grant Agreement No. 871124).
References
- H. Okabe, Can. J. Chem., 1983, 61, 850–855 CrossRef CAS.
- M. S. Child, R. W. Field, D. Gauyacq, J. K. G. Watson, N. Shafizadeh, J.-H. Fillion, D. Gauyacq and S. Couris, Philos. Trans. R. Soc. A, 1997, 355, 1637–1658 CrossRef.
- J. H. Lacy, N. J. Evans, II, J. M. Achtermann, D. E. Bruce, J. F. Arens and J. S. Carr, Astrophys. J., 1989, 342, L43 CrossRef CAS.
- T. Q. Zhao, Q. Li, B. S. Liu, R. K. E. Gover, P. J. Sarre and A. S. C. Cheung, Phys. Chem. Chem. Phys., 2016, 18, 3489–3496 RSC.
- V. V. Voronin, M. S. Ledovskaya, A. S. Bogachenkov, K. S. Rodygin and V. P. Ananikov, Molecules, 2018, 23, 2442 CrossRef PubMed.
- K. Tsuji, N. Arakawa, A. Kawai and K. Shibuya, J. Phys. Chem. A, 2002, 106, 747–753 CrossRef CAS.
- Y. C. Hsu, M. S. Lin and C. P. Hsu, J. Chem. Phys., 1991, 94, 7832–7841 CrossRef CAS.
- M. N. R. Ashfold, B. Tutcher, B. Yang, Z. K. Jin and S. L. Anderson, J. Chem. Phys., 1987, 87, 5105–5115 CrossRef CAS.
- K. Matthíasson, H. Wang and Á. Kvaran, Chem. Phys. Lett., 2008, 458, 58–63 CrossRef.
- S. T. Ridgway, D. N. B. Hall, S. G. Kleinmann, D. A. Weinberger and R. S. Wojslaw, Nature, 1976, 264, 345–346 CrossRef CAS.
- M. Frenklach and E. D. Feigelson, Astrophys. J., 1989, 341, 372 CrossRef CAS.
- D. Zaouris, A. Kartakoullis, P. Glodic, P. C. Samartzis, H. Rafn Hróðmarsson and Á. Kvaran, Phys. Chem. Chem. Phys., 2015, 17, 10468–10477 RSC.
- P. Glodic, D. Zaouris, P. C. Samartzis, A. Hafliðason and Á. Kvaran, Phys. Chem. Chem. Phys., 2016, 18, 26291–26299 RSC.
- H. R. Hróðmarsson, A. Kartakoullis, D. Zaouris, P. Glodic, H. Wang, P. C. Samartzis and Á. Kvaran, Phys. Chem. Chem. Phys., 2017, 19, 11354–11365 RSC.
- A. Hafliðason, P. Glodic, G. Koumarianou, P. C. Samartzis and Á. Kvaran, Phys. Chem. Chem. Phys., 2018, 20, 17423–17433 RSC.
- A. Hafliðason, P. Glodic, G. Koumarianou, P. C. Samartzis and Á. Kvaran, Phys. Chem. Chem. Phys., 2019, 21, 10391–10401 RSC.
- K. Matthíasson, G. Koumarianou, M.-X. Jiang, P. Glodic, P. C. Samartzis and Á. Kvaran, Phys. Chem. Chem. Phys., 2020, 22, 4984–4992 RSC.
- J. Long, H. Wang and Á. Kvaran, J. Chem. Phys., 2013, 138, 044308 CrossRef PubMed.
- J. Long, H. R. Hróðmarsson, H. Wang and Á. Kvaran, J. Chem. Phys., 2013, 138, 044308 CrossRef PubMed.
- F. Wang, M. L. Lipciuc, X. Yang and T. N. Kitsopoulos, Phys. Chem. Chem. Phys., 2009, 11, 2234–2240 RSC.
- C. R. Gebhardt, T. P. Rakitzis, P. C. Samartzis, V. Ladopoulos and T. N. Kitsopoulos, Rev. Sci. Instrum., 2001, 72, 3848–3853 CrossRef CAS.
- V. Papadakis and T. N. Kitsopoulos, Rev. Sci. Instrum., 2006, 77, 083101 CrossRef.
- K. Malsch, R. Rebentisch, P. Swiderek and G. Hohlneicher, Theor. Chem. Acc., 1998, 100, 171–182 Search PubMed.
- See ESI†.
- NIST Chemistry WebBook – (National Institute of Standards and Technology) https://webbook.nist.gov/chemistry/name-ser/.
- K. W. Chang and W. R. M. Graham, J. Chem. Phys., 1982, 76, 5238–5244 CrossRef CAS.
- M. Suto and L. C. Lee, J. Chem. Phys., 1984, 80, 4824–4831 CrossRef CAS.
- S. Boyé, A. Campos, S. Douin, C. Fellows, D. Gauyacq, N. Shafizadeh, P. Halvick and M. Boggio-Pasqua, J. Chem. Phys., 2002, 116, 8843–8855 CrossRef.
- D. H. Mordaunt and M. N. R. Ashfold, J. Chem. Phys., 1994, 101, 2630–2631 CrossRef CAS.
- J. C. Han, C. Ye, M. Suto and L. C. Lee, J. Chem. Phys., 1989, 90, 4000–4007 CrossRef CAS.
- J. Jiang, A. K. Muthike, T. J. Erickson and R. W. Field, J. Mol. Spectrosc., 2019, 361, 24–33 CrossRef CAS.
- T. A. Cool and P. M. Goodwin, J. Chem. Phys., 1991, 94, 6978–6988 CrossRef CAS.
- D. Duflot, J. M. Robbe and J. P. Flament, J. Chem. Phys., 1994, 100, 1236–1246 CrossRef CAS.
- M. Martin, J. Photochem. Photobiol., A, 1992, 66, 263–289 CrossRef CAS.
- J. F. Babb, R. T. Smyth and B. M. McLaughlin, Astrophys. J., 2019, 876, 38 CrossRef CAS.
- G. J. Vázquez, J. M. Amero, H. P. Liebermann, R. J. Buenker and H. Lefebvre-Brion, J. Chem. Phys., 2007, 126, 164302 CrossRef PubMed.
- A. Hafliðason, H. Wang and Á. Kvaran, Phys. Chem. Chem. Phys., 2016, 18, 1797–1806 RSC.
- J. Long, H. Wang and Á. Kvaran, J. Phys. Chem. A, 2014, 118, 182 CrossRef PubMed.
|
| This journal is © the Owner Societies 2023 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  a,
Ioannis C.
Giannakidis
a,
Ioannis C.
Giannakidis