
The formation of active phases in Pt-containing catalysts for bicyclohexyl dehydrogenation in hydrogen storage†
Tatiana V.
Bogdan
ab,
Alexander N.
Kalenchuk
ab,
Leonid M.
Kustov
 *ab and
Viktor I.
Bogdan
a
*ab and
Viktor I.
Bogdan
a
aN. D. Zelinsky Institute of Organic Chemistry, Russian Academy of Sciences, 47 Leninsky prosp., Moscow, 119991, Russia. E-mail: lmk@ioc.ac.ru
bChemistry Department, Moscow State University, 1-3 Leninskie Gory, Moscow, 199991, Russia
First published on 30th November 2022
Abstract
The fundamental role of the carbon carrier Sibunit® in the formation of active and selective phases in low-percentage Pt-containing catalysts Pt/C, Pt/Ni/C, Pt/Ni-Cr/C for the complete dehydrogenation of bicyclohexyl into biphenyl (320 °C, 1 atm) is shown. The Pt/Ni-Cr/C catalyst showed the greatest activity and selectivity in the complete dehydrogenation of bicyclohexyl into biphenyl. Detailed analysis of the catalyst surface by XPS, TEM-HR and EDX methods revealed two main processes associated with the high activity and successful course of the reaction of bicyclohexyl dehydrogenation: the formation of an active carbide Pt1−xCx phase and graphitization of the carbon carrier. Both these processes are realized equally in the Pt/Ni-Cr/C catalyst demonstrating the highest activity. The formation of the Pt1−xCx carbide phase at a low extent of graphitization of the catalyst surface is predominant for the Pt/C catalyst. Graphitization of the carbon carrier is most pronounced for Pt/Ni/C, where the yield of biphenyl is significantly reduced. The formation of graphite for the Pt/Ni/C catalyst at the metal–carrier interface leads to the encapsulation of a metal particle in a graphite shell, which apparently determines its low activity in the conversion of bicyclohexyl.
Introduction
Currently, the main task of global energy development is the diversification of the use of fossil raw materials and alternative renewable energy sources. Among alternative energy sources, hydrogen attracts attention as one of the most environmentally friendly fuels. However, the widespread use of hydrogen is impossible without solving the problem of its controlled, safe storage, logistics and on-demand release in places of consumption.1–3 An example of a new approach to solving these problems is the use of liquid organic hydrogen carriers (LOHC) as hydrogen storage materials operating on the basis of catalytic hydrogenation–dehydrogenation reactions.4–9 Unlike all known systems and methods, the gravimetric and volumetric content of hydrogen in LOHC does not depend on the degree of its interaction with the storage medium, which makes it as safe as possible for hydrogen storage and transportation under normal conditions. The key factor in designing an efficient composite catalytic hydrogen storage system is the development of a catalyst that combines high activity with high selectivity toward the final product and is able to minimize undesirable side processes (cracking, hydrogenolysis, etc.), which reduce the number of hydrogenation–dehydrogenation cycles due to the degradation of the substrate. It has been shown10–13 that the biphenyl–bicyclohexyl system is highly efficient in hydrogenation–dehydrogenation processes because it demonstrates the minimum contribution of side reactions (cracking, hydrogenolysis, etc.), which reduce the number of hydrogenation–dehydrogenation cycles due to degradation of the substrate.Noble metal-based catalysts are known to exhibit high catalytic activity in the LOHC hydrogenation, and platinum is the most active metal for dehydrogenation.14 At the same time, due to the high cost of platinum, the strategy of modifying Pt with transition metals is of great importance for reducing the cost of catalysts. The synergistic effect of interaction between platinum and transition metals in composite active clusters can significantly increase the activity and selectivity of catalytic reactions.15–20
Activated carbon or modern carbon materials are usually used as a carrier.21–27 It is known28–30 that metal particles deposited on carbon catalyze various reactions of the carbon carrier with hydrogen and carbon oxides. Therefore, the effect of the carbon support on the activity and selectivity of metal-deposited catalysts in organic reactions at the stages of both pre-activation and in situ reactions is the subject of numerous studies, and, in particular, in the development of new heterogeneous catalytic systems.31–35 Thus, the high efficiency of a new type of Fe-catalyst deposited on plasma-sintered carbon nanotubes in the hydrogenation reaction of supercritical CO2 in Fischer–Tropsch synthesis of hydrocarbons was demonstrated.31 The high stability and activity of catalytic Fe centers decorated with carbon shells of CNTs of increased density, in our opinion, is determined by a high degree of metal–carbon interaction, intensified by increased carbidization and CO activation. Close attention was paid to the interaction of deposited Ni, Pd, and Pt metals with carbon supports in the works.33,34,36–43 It was found that metals catalyze graphitization of amorphous carbon, which increases the resistance of the carbon carrier to gasification reactions.26,36–42 It was found that the interaction of metal particles with the carbon leads to formation of active metal carbides. In particular, it was recently established by in situ HR-TEM that the active phase responsible for the graphitization of carbon is nickel carbide Ni3C, which is formed via the interaction of metallic nickel with amorphous carbon.44,45
In this study we performed catalytic dehydrogenation of bicyclohexyl into biphenyl on Pt–Ni-Cr, Pt–Ni, and Pt catalysts. In order to identify the active phases and the role of the carbon carrier in their formation, a detailed phase analysis of the catalyst surface and at the particle/carrier interface was performed using XPS, HR-TEM, and EDX methods.
Results and discussion
Catalytic experiment
The catalytic dehydrogenation of bicyclohexyl C12H22 proceeds sequentially through cyclohexylbenzene C12H16 with the formation of the final product – biphenyl C12H10: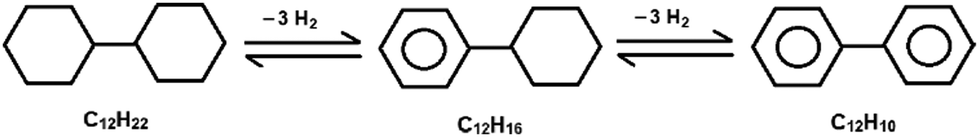
Data on the conversion of bicyclohexyl and the yield of reaction products are given in Table 1 and Fig. 1. The Pt/Ni-Cr/C catalyst showed the best conversion and the highest yield of the target product of complete dehydrogenation. As a result the ratio of cyclohexylbenzene to biphenyl is 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :5. On a monometallic Pt/C catalyst, the ratio of cyclohexylbenzene: biphenyl is 1:2 at a comparable bicyclohexyl conversion. The total conversion of the substrate is reduced by more than 2 times on the Pt/Ni/C catalyst, where the yield of biphenyl is also low, and the products of complete and partial dehydrogenation are formed in approximately equal proportions.
:5. On a monometallic Pt/C catalyst, the ratio of cyclohexylbenzene: biphenyl is 1:2 at a comparable bicyclohexyl conversion. The total conversion of the substrate is reduced by more than 2 times on the Pt/Ni/C catalyst, where the yield of biphenyl is also low, and the products of complete and partial dehydrogenation are formed in approximately equal proportions.
| Catalyst | TOF in the conversion of C12H22 X, mmol g−1 h−1 | Space-time yield Y, mmol g−1 h−1 | |
|---|---|---|---|
| C12H16 | C12H10 | ||
| Pt/C | 8.8 | 2.8 | 6.0 |
| Pt/Ni/C | 3.7 | 2.0 | 1.7 |
| Pt/Ni-Cr/C | 10.5 | 1.7 | 8.8 |
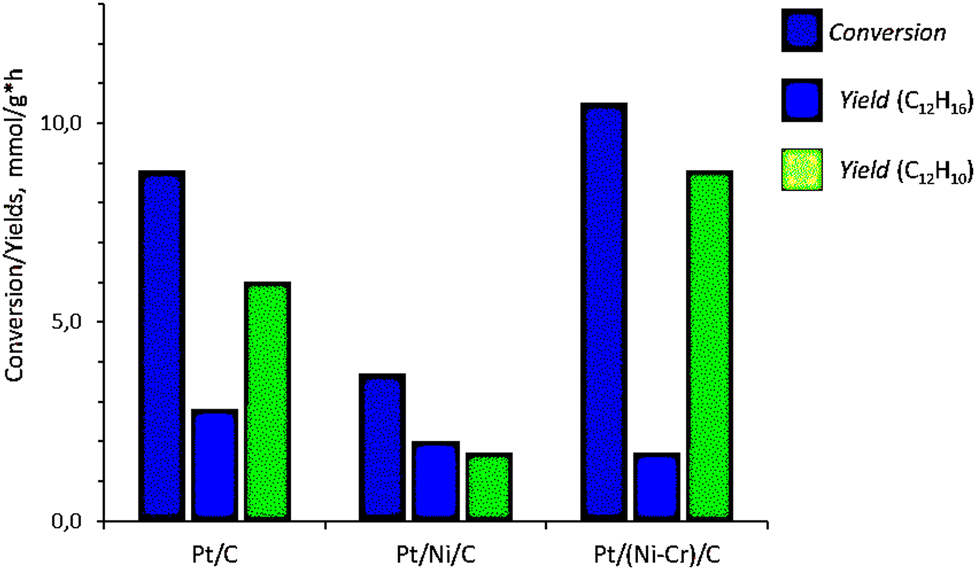 | ||
| Fig. 1 TOF in the conversion of bicyclohexyl C12H22 and space–time yield of products of complete and partial dehydrogenation of bicyclohexyl on Pt/C, Pt/Ni/C, and Pt/Ni-Cr/C catalysts. | ||
Thus, the catalytic phases active in the complete dehydrogenation reaction of bicyclohexyl are formed on the Pt/Ni-Cr/C catalyst to a greater extent than on Pt/C, especially, on Pt/Ni/C. To clarify the nature of the active phases, analysis of the structure of the catalysts was carried out.
XPS data
The XPS spectra of Pt/Ni-Cr/C catalyst before reduction were given in our recent publication.46 This article discusses the XPS of the studied catalysts after treatment under reduction conditions. The high-resolution spectra of the element lines are shown in Fig. 2–5. The fraction of nickel and platinum atoms in different states on the surface of reduced samples according to XPS data are given in Table 2.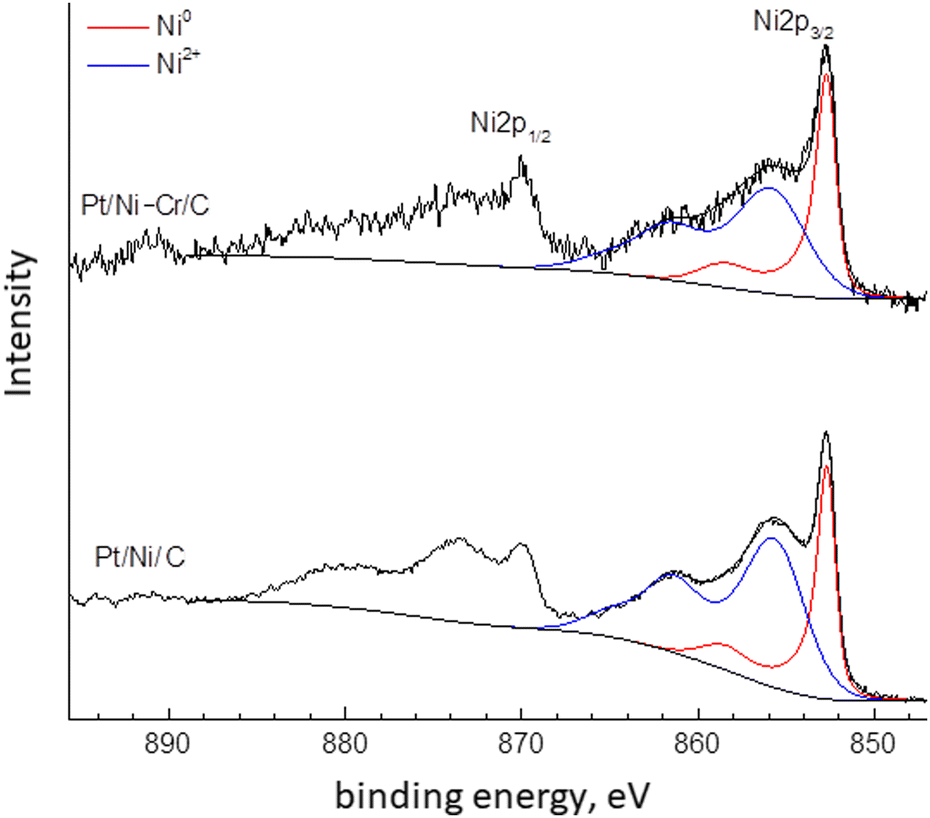 | ||
| Fig. 2 Ni2p XPS spectra of the Pt/Ni/C and Pt/Ni-Cr/C catalysts. | ||
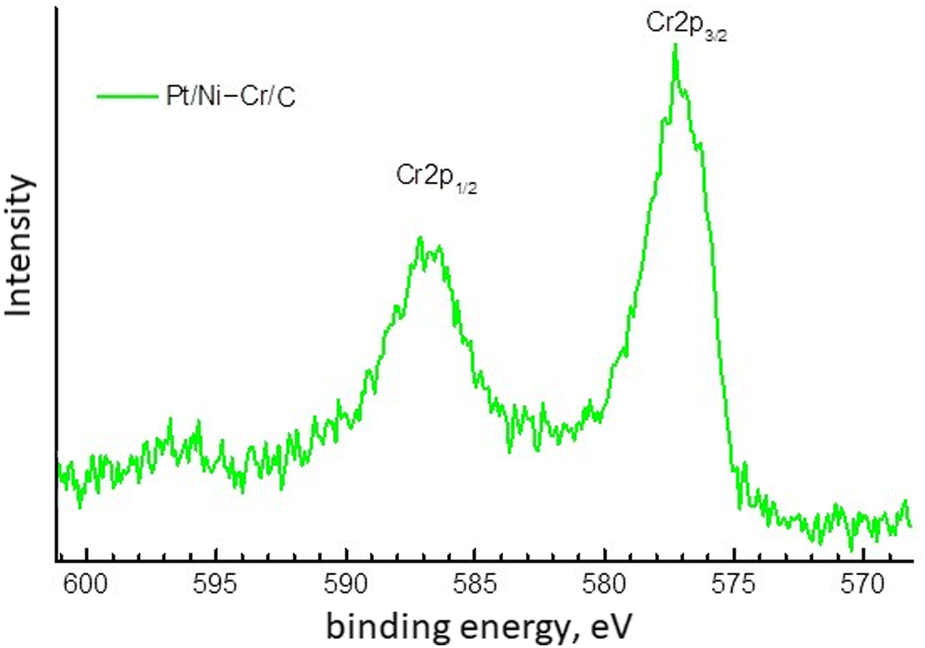 | ||
| Fig. 3 Cr2p XPS spectrum of Pt/Ni-Cr/C catalyst. | ||
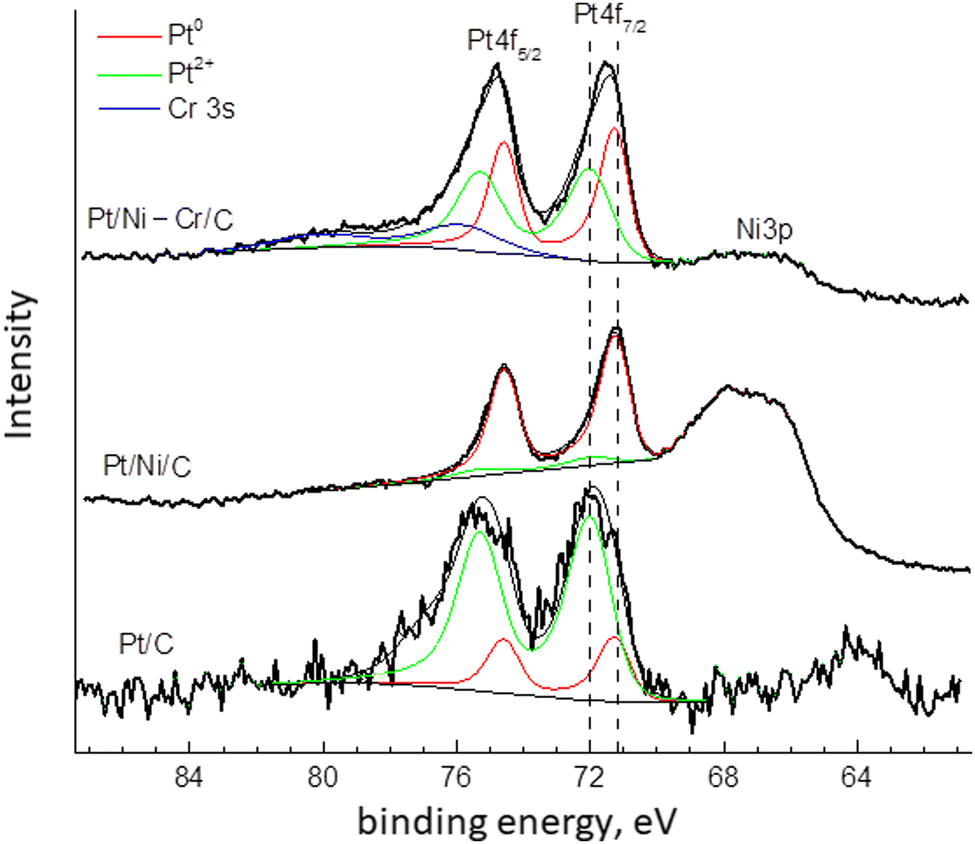 | ||
| Fig. 4 Pt4f, Ni3p η Cr3s XPS spectra of Pt/C, Pt/Ni/C and Pt/Ni-Cr/C catalysts. | ||
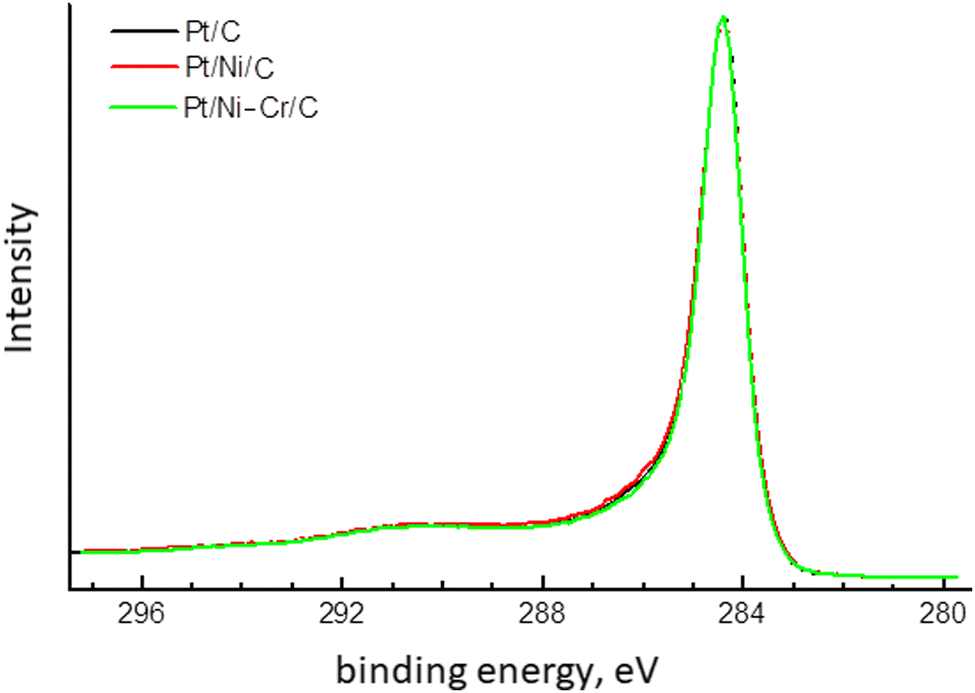 | ||
| Fig. 5 C1s XPS spectra of Pt/C, Pt/Ni/C and Pt/Ni-Cr/C catalysts. | ||
| Catalyst | Ni0 | Ni+2 | Pt0 | Ptδ+ |
|---|---|---|---|---|
| Pt/C | — | — | 21 | 79 |
| Pt/Ni/C | 32 | 68 | 91 | 9 |
| Pt/Ni-Cr/C | 38 | 62 | 49 | 51 |
The Ni2p XPS spectra of Pt/Ni/C and Pt/Ni-Cr/C practically coincide (Fig. 2) and contain a contribution from the metallic nickel Ni2p3/2 component with a binding energy of about 852.7 eV and from the oxidized nickel state to which corresponds the Ni2p3/2 component with a binding energy of about 855.8 eV with an intensive shake-up satellite, which is typical for divalent nickel in various compounds (hydroxide, complex oxides, etc.).20,47 To determine the fraction of metallic nickel in the samples, the spectra were decomposed into components corresponding to the oxidized and metallic forms of nickel. The results of the decomposition are shown in Fig. 2 and Table 2. The ratio of Ni0/Ni+2 in the Pt/Ni/C and Pt/Ni-Cr/C catalysts in the reduced catalysts is close, but the fraction of metallic nickel is higher in Pt/Ni-Cr/C. Thus, the presence of chromium in the trimetallic catalyst contributes to the reduction to metallic nickel.
For the Cr2p component, the XPS spectrum (Fig. 3) is observed as a doublet of broad lines with a poorly defined structure and with a binding energy of the Cr2p3/2 component 577.0 eV, which is typical for trivalent chromium compounds.20,48
To reveal the forms of platinum in the catalysts the spectra were decomposed into two components. The component with a binding energy of 71.2 eV corresponds to metallic platinum Pt0.20,43,49–51 The second component with a binding energy of about 72.0 can be referred to the electron-deficient Ptδ+ (0 < δ ≤ +2).20,51
Fig. 4 and Table 2 show the ratios Ptδ+:Pt0 on the surface of the catalysts Pt/C, Pt/Ni/C, and Pt/Ni-Cr/C after reduction. The electron-deficient platinum species prevail in Pt/C, whereas in Pt/Ni-Cr/C the ratio Ptδ+:Pt0 is approximately equal. Only in the case of Pt/Ni/C, metallic platinum Pt0 predominates on the surface of the catalyst. Note that the catalytic activity of the Pt/Ni/C catalyst in the bicyclohexyl dehydrogenation reaction is low compared to Pt/C and Pt/Ni-Cr/C, which may indicate the important role of the ratio between electron-deficient Ptδ+ and non-oxidized Pt0 species for the reaction of complete dehydrogenation of bicyclohexyl into biphenyl. It is known that the electron work function for nickel is less than for platinum (4.91–5.01 eV versus 5.30–5.55 eV,20 respectively), which apparently prevents the formation of electron-deficient Ptδ+ in the Pt/Ni/C catalyst.
Note that for the catalyst Pt/C before reduction,46 the binding energy of the Pt 4f7/2 component was equal to 71.9 eV, which is corresponds to electron-deficient Ptδ+ (0 ≤ δ ≤ +2). In addition, the XPS spectrum before reduction contained satellites with a binding energy of about 74.3 eV, which corresponds to oxidized forms of platinum with binding energies close to Pt4+.20,51 Thus, platinum on the catalyst surface before reduction was represented both as lectron-deficient Ptδ+ and oxidized Pt4+ states,46 and component Pt+4 was absent after reduction.
C1s XPS spectra (Fig. 5) are identical for all investigated samples and are observed as a narrow asymmetric line with a maximum at 284.4 eV, which is typical for sp2-carbon.52–54
Thus, under reduction conditions nickel and platinum exist in the metallic and electron-deficient or oxidized states on the surface of catalysts, and chromium is present in the oxidized state. The phase composition of the catalyst surface was clarified using high-resolution TEM.
High resolution TEM
The TEM image of the Pt/C catalyst before reduction shows the high dispersity of the supported metal (Fig. 6) and separate nanoparticles. EDX spectra for nanoparticles in the Pt/C catalyst are presented in Fig. S1, ESI.† The interplane distances determined from the FFT image (Fig. 6) fall into the intervals corresponding to that for graphite, as well as metallic platinum (100) and its carbide or oxides (ESI,† Table S1). However, we believe that mainly platinum carbides Pt1−xCx rather than platinum oxides are formed on the surface of the Pt/C catalyst. The reasons are as follows. First, according to the XPS data (see Table 2), electron-deficient Ptδ+ with the binding energy 72.0 eV predominates on the catalyst surface, but the binding energy for Pt+2 in PtO is 73.8–74.2 eV.51 Second, we take into account the data36 about the appearance of superstructural reflections 100 for metallic platinum deposited on a carbon carrier explained by the formation of carbides Pt1−xCx of the type of substitution solid solutions.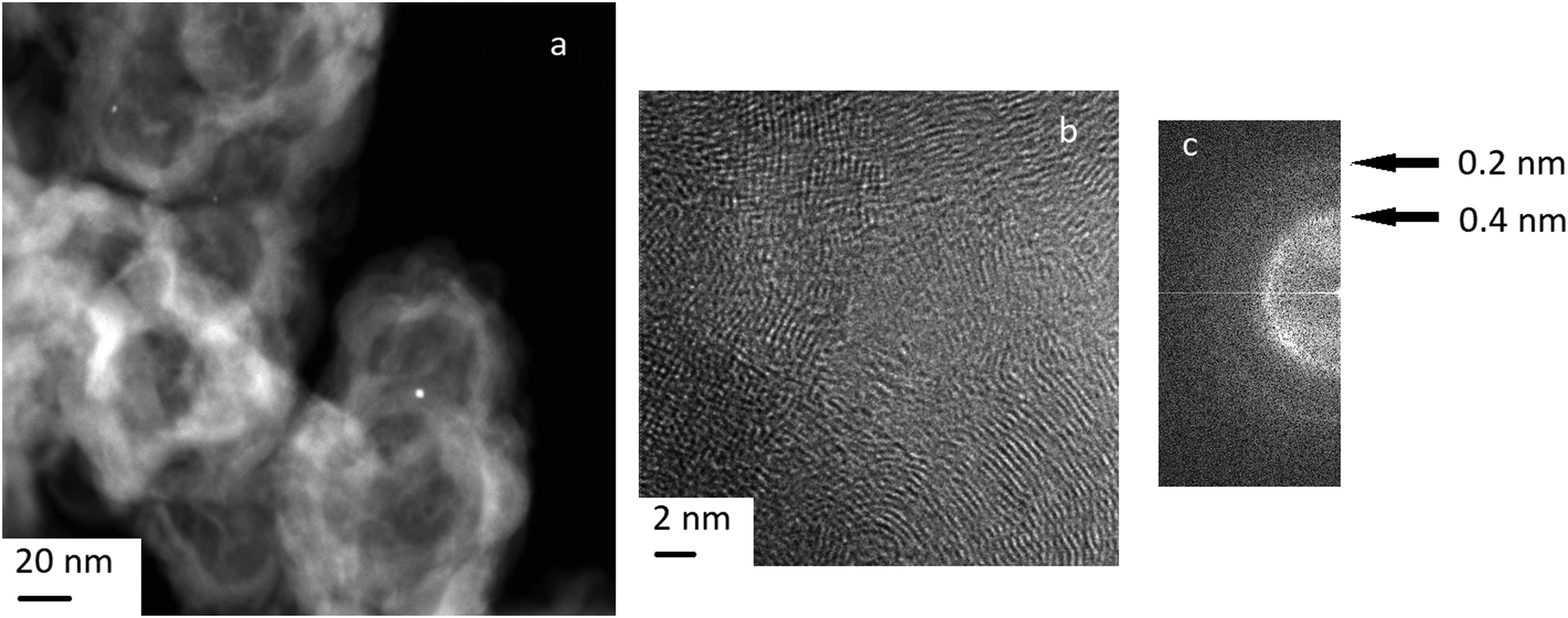 | ||
| Fig. 6 TEM image of the Pt/C catalyst surface. (a) – low-magnitude dark-field micrograph, (b) – high-magnitude micrograph and its FFT (c) (A) is a graphite-like region; (B) is a region of finely dispersed metal deposition with the formation of platinum carbide/oxides. | ||
When platinum interacts with a carbon carrier, a partial electron transfer from Pt to carbon atoms occurs due to the difference in the electronegativity of the elements. As a result of the charge transfer, the effective radius of a carbon atom increases and the carbon atoms can substitute platinum in the structure with formation of a carbide as a substitution solid solution. Third, the electron-deficient Ptδ+ state is formed as a result of the charge transfer and carbide formation. It is known that electron-deficient platinum Ptδ+ demonstrates a high activity in the reaction of dehydrogenation and dehydrocyclization of paraffins, as well as high dispersion, absence of bonds between platinum atoms, and high resistance to sintering.55,56 The indirect proof of the formation of electron-deficient platinum on the Pt/C catalyst is its high activity in the bicyclohexyl dehydrogenation under consideration.
At the same time, we noted the formation of graphite-like structures on the surface of the catalyst (Fig. 6). Other authors also observed graphitization of the carbon carrier in the presence of platinum.36,57 Graphitization contributed to a sharp drop in the specific surface area and the enlargement of Pt particles. The reduction of the surface area during graphitization complicates the distribution of Pt in a finely dispersed state and contributes to the formation of large agglomerates. It should be reminded that our results about the high dispersion of Pt were obtained for the very low weight concentration of platinum −0.1%. An increase in the concentration of platinum results in noticeable sintering of platinum nanoparticles and increase in their size.58,59
The formation of the graphite phase was found36 to be accompanied by the disappearance of a superstructural reflexes 100 for Pt0 (see above). It is indicated that graphitization occurs due to the destruction of platinum carbide Pt1−xCx. The disappearance of super-structural reflections also observed after Pt/C treatment with hydrogen or heating above 1000 °C. Thus, treatment of the Pt/C catalyst under reducing conditions as well as exposure to hydrogen under dehydrogenation promotes the destruction of the carbide phase and leads to partial graphitization of the carrier.
The structure of the Pt/Ni/C catalyst was considered both before reduction and after treatment under reducing conditions. The TEM image and EDX mapping patterns are shown in Fig. 7. EDX spectra are presented in Fig. S2 (ESI†). Prior to reduction, the surface of the catalyst is represented by deposited metal particles with a size of 5–10 nm and finely dispersed metals. The particles mainly consist of nickel atoms, and platinum atoms are dispersed over the surface of the catalyst. Oxygen atoms are located over the whole surface of the catalyst, indicating partial functionalization/oxidation of the carbon carrier surface. At the same time, the amount of oxygen on the metal particles is enriched compared to the surface of the carrier. This is consistent with the XPS data showing that nickel is predominantly represented as oxides on the surface of the catalyst.
 | ||
| Fig. 7 Pt/Ni/C catalyst before reduction: low-magnitude TEM image in the dark-field mode (a) and EDX mapping pattern: platinum (b); nickel (c); and oxygen (d). | ||
HR-TEM image of the Pt/Ni/C before reduction and its FFT are shown in Fig. 8. The FFT image is similar to those for Pt/C (see Fig. 6). The analysis of FFT indicates that the bright points corresponding to d = 0.21 nm and in accordance with XPS data might refer to NiO (111) (space group Fm![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m). At the particle/carrier boundary, a phase with interplanar spacing in the range of 0.20–0.22 nm was formed (see Fig. 8c and d), which can be attributed carbides of nickel Ni3C (d113 = 0.20 nm, d006 = 0.22 nm) or/and platinum Pt1−xCx,(d111 = 0.22 nm, d200 = 0.20 nm) as a result of interaction with a carbon carrier. It should be noted the distances from FFT fall within the same interval as for the Pt/C catalyst, where this could only be due to the formation of platinum compounds.
m). At the particle/carrier boundary, a phase with interplanar spacing in the range of 0.20–0.22 nm was formed (see Fig. 8c and d), which can be attributed carbides of nickel Ni3C (d113 = 0.20 nm, d006 = 0.22 nm) or/and platinum Pt1−xCx,(d111 = 0.22 nm, d200 = 0.20 nm) as a result of interaction with a carbon carrier. It should be noted the distances from FFT fall within the same interval as for the Pt/C catalyst, where this could only be due to the formation of platinum compounds.
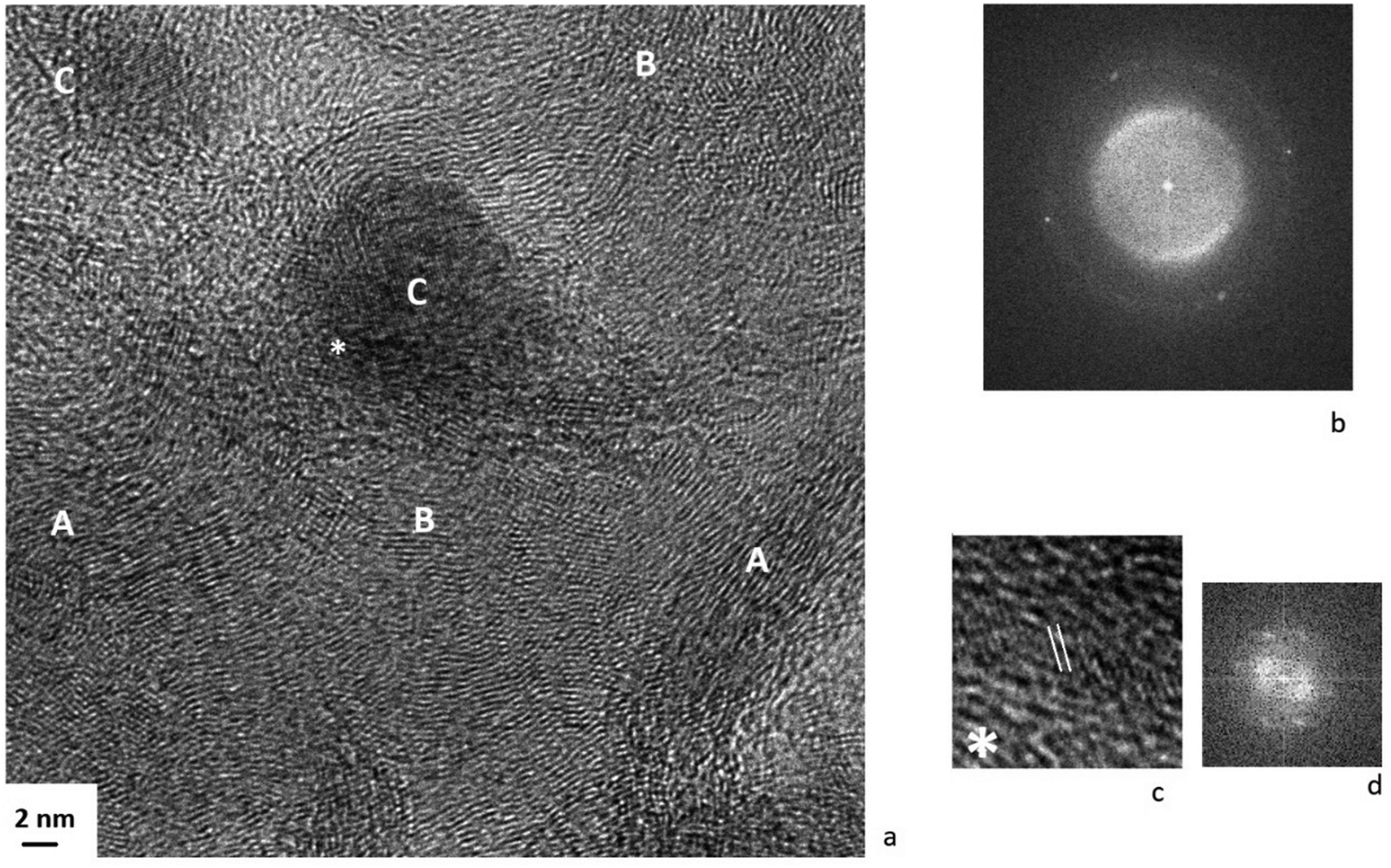 | ||
| Fig. 8 TEM image of the Pt/Ni/C catalyst before reduction (a) and its FFT (b): (A) – the area of the graphitized carrier; (B) – the area of finely dispersed particles of metals: nickel and platinum, as well as their carbides and oxides; (C) – a metal/metal oxide particle. TEM image of the catalyst surface at the metal/carrier boundary (c) and its FFT (d). | ||
Two features can be distinguished on the micrograph of the surface of the Pt/Ni/C catalyst after the reduction treatment (Fig. 9): an increase in the size of metal particles and noticeable graphitization of the carrier surface. In addition, metal particles are partially or completely covered by a graphite shell. The dark-field TEM image of the catalyst surface (Fig. S3, ESI†) shows that the increase in the particle size occurs due to “spreading” metal over the carrier. Perhaps processes spreading and enlargement of metal particles in the presence of Pt are connected mainly with well dispersed platinum (see EDX spectra in Fig. S3, ESI†).
 | ||
| Fig. 9 Multi-magnitude TEM images of the Pt/Ni/C catalyst after treatment under reduction conditions. | ||
The behavior of metallic nickel particles deposited on an amorphous carbon carrier was studied.37,39,41,42,44,45 As it was observed,37,45 the size of nickel particles increased after heating. An increase in temperature and/or heating time was accompanied by a change in the structure of the carrier – the amorphous carbon of the support is graphitized. It was established by in situ TEM that the carbide phase Ni3C is responsible for the growth of nickel particles and the growth of graphite layers around the metal particle.44,45 The authors37 also note that nickel particles migrate along the surface of the carrier, catalyzing the transformation of amorphous carbon into graphite. The same process of carbon nanofiber growth on a nickel catalyst on a silica–alumina carrier was observed under conditions of the reaction atmosphere H2/CH4 by in situ TEM methods.44 The reducing hydrogen atmosphere promotes such particle migration, whereas in a vacuum the graphitization process slows down due to the complete or partial encapsulation of nickel particles in the graphite shell.37,44,45
In the present work, we believe there are the same reasons for increasing the size of the metal particles and graphitization of the carrier. The interaction of metal atoms with a carbon support leads to the formation of carbides. In the next stage, carbides are destroyed, which is accompanied by graphitization of the carbon carrier. In the case of the co-presence of nickel and platinum carbides, nickel carbide is destroyed first. Graphitization of the carrier increases significantly when the Pt/Ni/C catalyst was treated in hydrogen. Graphitization around the metal particle promotes its encapsulation. Thus, under the conditions of the dehydrogenation reaction, we can assume the intensification of graphitization processes for the Pt/Ni/C catalyst and its deactivation.
Fig. 10 shows a TEM image of the Pt/Ni-Cr/C catalyst before reduction. The surface phases are represented by finely dispersed metal particles, carbides, oxides and graphitized regions of the carbon carrier. The FFT image of fragment 1 (Fig. 10, right) repeats the FFT for the Pt/C catalyst; FFT image of fragment 2 (Fig. 10, right) is as Pt/Ni/C catalyst and corresponds a metal particle in an oxide shell. FFT image of fragment 3 (Fig. 10, right) corresponds to the graphitized carrier. Formation of graphite-like structures takes place from metal carbides.
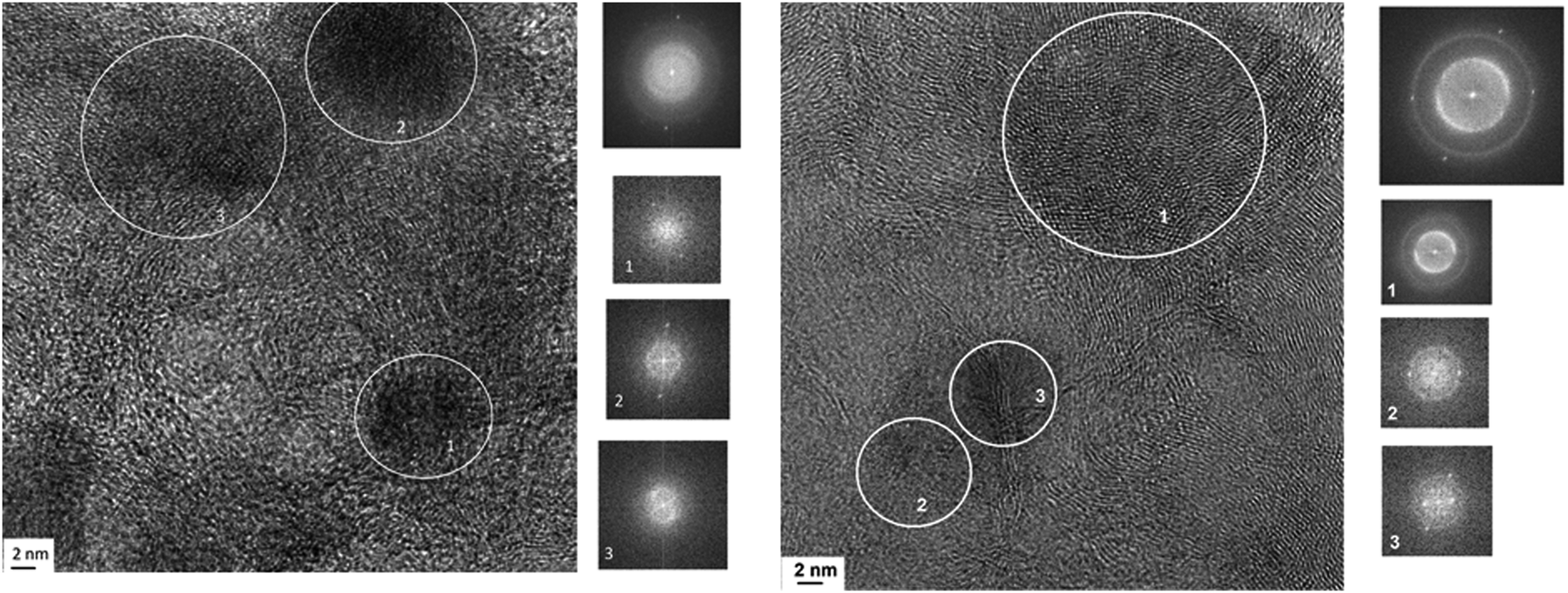 | ||
| Fig. 10 Micrographs of the Pt/Ni-Cr/C catalyst surface before reduction in hydrogen and FFT images for marked regions. | ||
Fig. 11 shows a micrograph of the surface of the Pt/Ni-Cr/C catalyst after reduction. The Pt/Ni-Cr/C catalyst after reduction contains metallic nickel, in accordance with the XPS data. The Pt/Ni-Cr/C catalyst surface after reduction is represented by the dispersed metal phase on the carrier with metal particles encapsulating and spreading over the surface. In FFT images on Fig. 11, the intensity of the bright points corresponding to the metal particle decreases (see Fig. 11a) or its disappears completely (see Fig. 11b). It should be noted that, according to EDX data (Fig. S4, ESI†), the amount of oxygen on the carrier surface decreases after reduction, which is more suggestive of carbides rather than metal oxides. On Fig. 11a, the fragments of the catalyst surface are shown: a large metal particle (B), graphitized carrier (C), carbides (D).
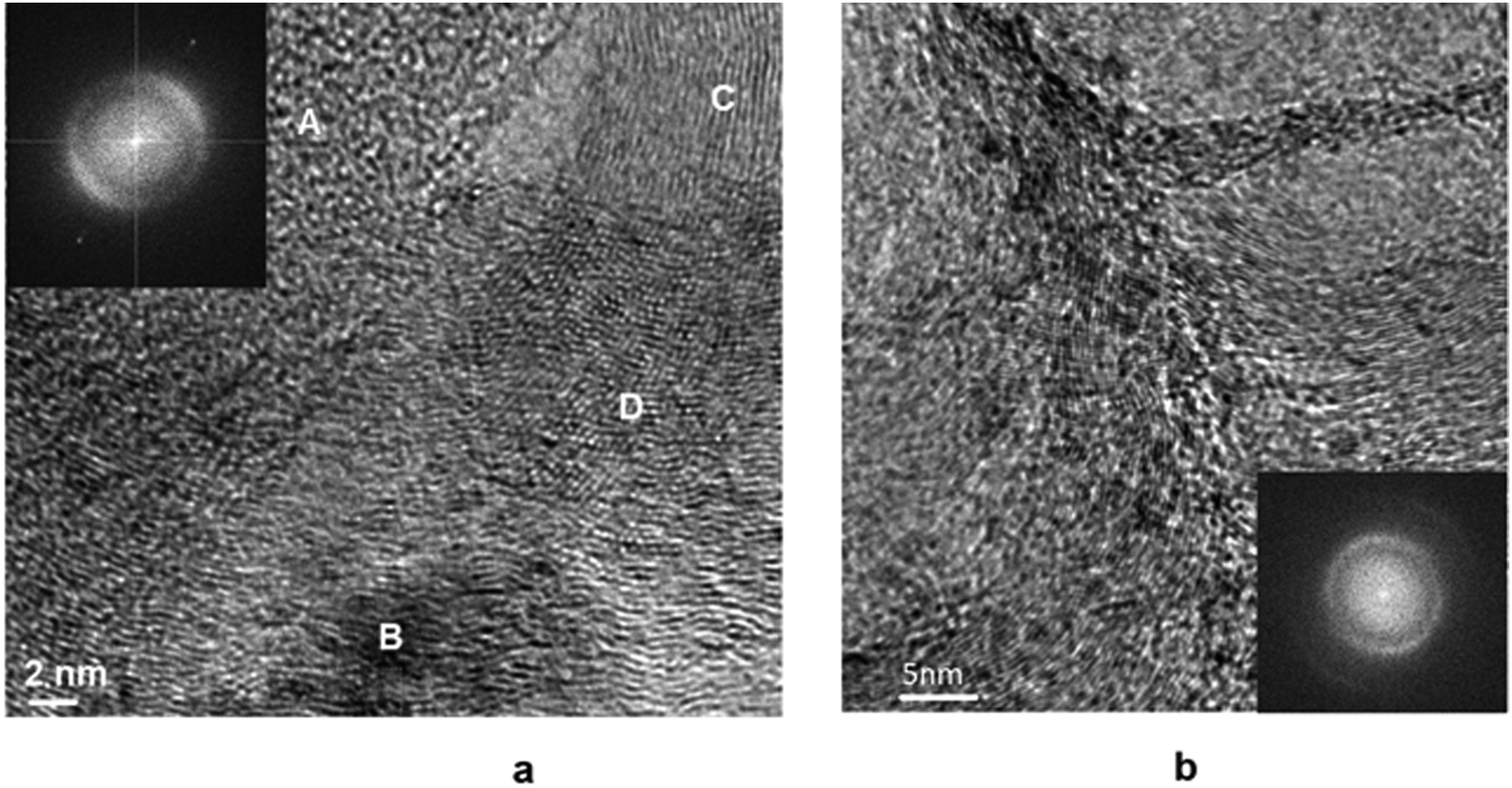 | ||
| Fig. 11 TEM images of the Pt/Ni-Cr/C catalyst after reduction by hydrogen and FFTs. (A) – the region of an amorphous carrier; (B) – a metal particle partially coated with a carrier; (C) – graphitized carbon, (D) – the area of metal carbides. | ||
At the same time, we note another feature of the structure of the catalyst after reduction, namely, the formation of spherical composite particles on the surface of the carrier (Fig. 12). The formation of large spherical metal particles deposited on a non-metallic carrier was noted in the literature.38,58 It was pointed out that carbides are formed on large particles of metallic palladium deposited on a carbon carrier, which, when destroyed, form a graphite phase.38 It is also indicated that at high temperatures, metal particles can be encapsulated in an inert graphite shell under the action of gases released during the gasification of the carbon carrier.
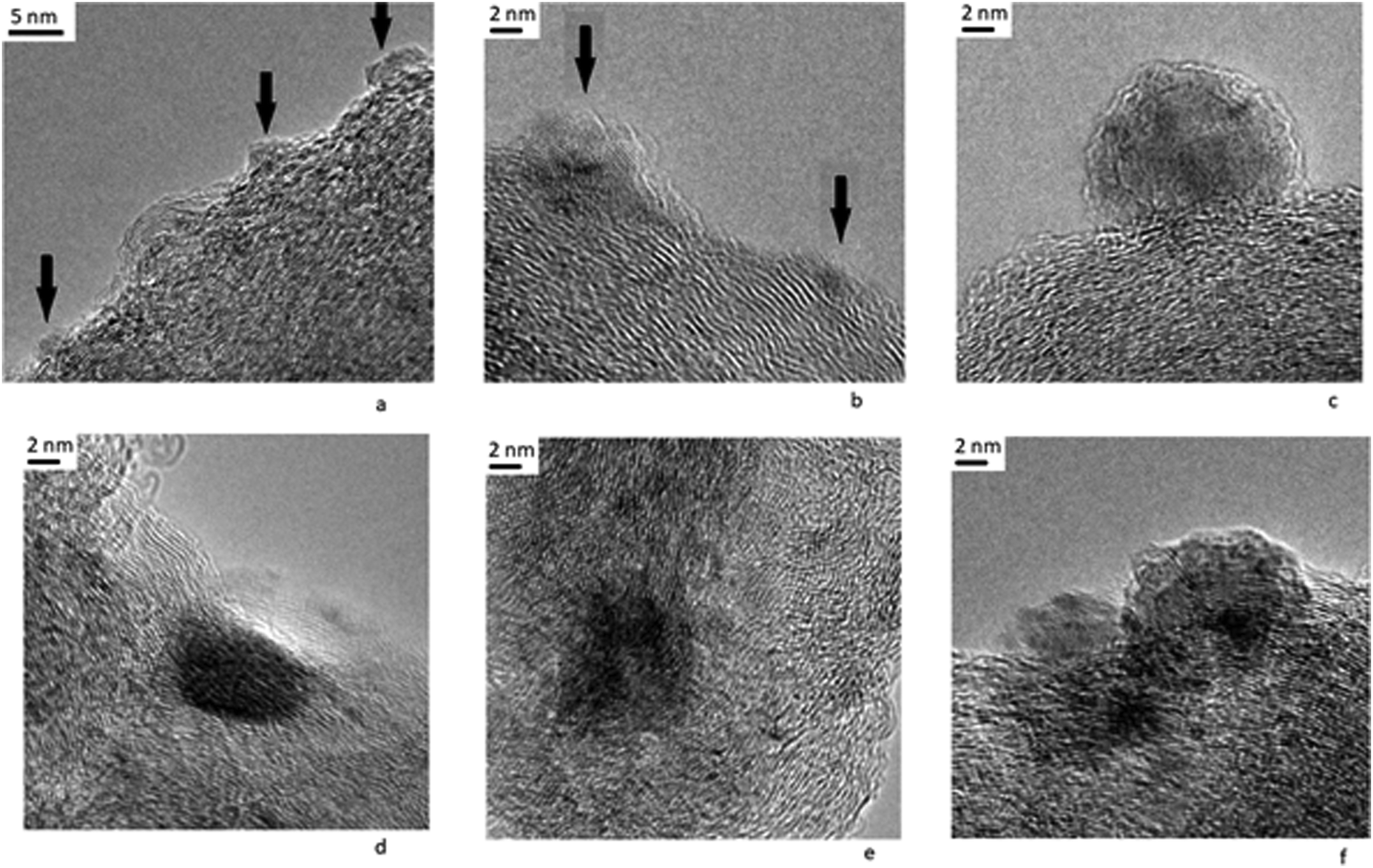 | ||
| Fig. 12 Micrographs of the Pt/Ni-Cr/C: interaction metal with the carrier: (a–c) formation of spherical particles on the surface of amorphous carbon; (d–e) formation of a graphite shell on top of composite Pt-Ni-Cr particles; and (f) both processes are presented. | ||
Thus, in the case of the Pt/Ni-Cr/C catalyst we distinguish the following processes occurring during interaction of metal particles with a carrier under reducing conditions: (1) the formation of an active carbide phase; (2) metal-catalyzed graphitization of the carrier, leading to the formation of an inert graphite shell on the surface of a metal particle; (3) formation of spherical particles in an inert shell on the surface of the carrier.
Conclusions
The fundamental role of the carbon carrier – Sibunit® in the formation of active and selective phases in low-percentage Pt-containing catalysts for the complete dehydrogenation of bicyclohexyl into biphenyl (320 °C, 1 atm) is shown. Detailed analysis of Pt/C, Pt/Ni/C, Pt/Ni-Cr/C catalyst surfaces by XPS, TEM-HR, EDX methods revealed interesting phenomena occurring on the carbon carrier. The dynamics of transformations include (i) graphitization of amorphous carbon carrier, (ii) encapsulation of metal particles in the graphite shell, (iii) formation and destruction of metal carbides on the carrier surface, (iv) changes in the phase composition of composite particles, and (v) enlargement of catalyst particles, including due to “spreading” of particles on the surface of the carbon support.But two main processes were associated with activity and successful course of the reaction of bicyclohexyl dehydrogenation: the formation of platinum carbide Pt1−xCx and graphitization of the carbon carrier. The Pt/Ni-Cr/C catalyst showed the greatest activity and selectivity in complete dehydrogenation of bicyclohexyl into biphenyl, and both processes reach the equilibrium. The surface of the monometallic catalyst Pt/C is mainly represented by finely dispersed deposited metallic platinum and its carbide with minor formation graphite-like region. In a bimetallic Pt/Ni catalyst the formation of significant graphite areas occurs under mild thermodynamic conditions.
We assume that effective interaction of the catalysts with the aromatic substrate is ensured by the presence of graphene fragments on the carrier surface. Activity of the Pt/C catalyst during bicyclohexyl dehydrogenation is lower than that of the Pt/Ni-Cr/C catalyst, and Pt/C catalyst is the least prone to graphitization. Graphitization of the carbon carrier is most pronounced for Pt/Ni/C, where the yield of biphenyl, the target product of complete dehydrogenation of bicyclohexyl, is significantly reduced. And the formation of graphite for the Pt/Ni/C catalyst at the metal–carrier interface leads to the encapsulation of a metal particle in a graphite shell, which apparently determines its low activity in the conversion of bicyclohexyl.
The formation of carbides is facilitated by the deposition of finely dispersed metal particles on a carbon carrier. Platinum carbide Pt1−xCx is the formula for the existence of active electron deficient Ptδ+. The formation of active carbide phases is a result of metal spreading on the carbon carrier surface. The formation of platinum carbide Pt1−xCx is observed for all the studied catalytic systems, but it is most pronounced for Pt/C and Pt/Ni-Cr/C systems, where the yield of the target product of complete dehydrogenation of bicyclohexyl is noticeably higher than on the Pt/Ni/C catalyst.
Experimental
To carry out catalytic experiments, a carbon carrier Sibunit® (referred to as C), producing by the Omsk division of Boreskov Institute of Catalysis, was used in spherical granules with diameters 1.5–2.0 mm and following textural characteristics: specific surface area 243 m2 g−1; average pore size 4.8 nm; and pore volume0.45 cm3 g−1.60 The textural characteristics of both the initial carbon carrier Sibunit® and the catalysts prepared on its basis were determined by the BET method. The surface area, porosity of ready metal-loaded catalysts do not differ from the Sibunit® characteristics. Also, the studies of catalysts by CO chemisorption showed that after supporting the metals onto the carbon carrier, the surface area and average pore size did not change compared to the carrier.The catalysts were prepared by impregnating the carrier with aqueous salt solutions using the incipient wetness procedure. The composition of the metal phase in the prepared catalysts in mass percentages was as follows: 0.1% Pt, 1.5% Cr, 3% Ni. In the process of catalyst preparation, we calculated the mass amount of metals for the declared catalyst composition based on the mass of the carrier. Then we recalculated the necessary amount of salts and the corresponding volume of aqueous solutions of a given concentration for supporting the metals onto the surface of the carrier.
Before supporting the active components, the surface of the carrier was oxidized by boiling Sibunit® granules in concentrated nitric acid.61 Before the impregnation of salt solutions, the pre-oxidized carrier was calcined in air for 2 hours at T = 105–110 °C and then cooled to room temperature.
Pt/C
An aqueous solution of H2PtCl6 6H2O (Khimmed, Russia, chemical purity grade) was applied dropwise to the freshly calcined carrier at room temperature. Then the sample was dried for 24 hours in air at room temperature and then was kept in a desiccator for 4 hours at 130 °C. The dried catalysts were additionally calcined for 2 hours in a stream of N2 (99.9%, 50 mL min−1) at 350 °C.Pt/Ni/C
At the 1st stage of the Pt/Ni/C synthesis, the Ni/C sample was prepared as follows. A solution of Ni(NO3)2 × 6H2O (Khimmed, Russia, chemical purity grade) was applied to the surface of a freshly calcined carrier. The impregnated samples were dried for 24 hours in air at room temperature and then were kept in a desiccator for 4 hours at 130 °C. Then the sample was additionally calcined for 2 hours in a flow of N2 (99.9%, 50 mL min−1) at 500 °C. At the 2nd stage, platinum was supported onto the Ni/C sample according to the procedure described above for Pt/C (impregnation, drying, and calcination).Pt/Ni-Cr/C
In step 1 of Pt/Ni-Cr/C preparation, the Ni-Cr/C sample was prepared as follows. A solution of Cr(NO3)3 × 9H2O (Acros Organics, chemical purity grade) and Ni(NO3)2 × 6H2O (Khimmed, Russia, chemical purity grade) salts was applied dropwise to the prepared carrier. After that, the (Ni-Cr)/C sample was dried for 24 hours in air at room temperature, then it was kept in a desiccator for 4 hours at 130 °C and then calcined for 2 hours in a flow of N2 (99.9%, 50 mL min−1) at 500 °C. At step 2, platinum salts were supported onto the prepared (Ni-Cr)/C catalyst using the procedure described above for Pt/C (impregnation, drying, calcination).Treatment of the catalysts in a reducing medium
The prepared catalysts were treated in a flow reactor according to the following procedure: (1) heating to 105 °C in a flow of N2 (99.9%, 50 mL min−1); (2) maintaining at these conditions for 1 h; (3) heating to 150 °C in a flow of N2 (99.9%, 50 mL min−1); (4) heating to 500 °C in a flow of H2 (99.9%, 30 mL min−1); (5) keeping at these conditions for 2 h; (6) cooling in a flow of H2 (99.9%, 30 mL min−1) to the reaction temperature.The bicyclohexyl (99%, Acros Organics®) dehydrogenation reaction was carried out in a flow reactor at a temperature of 320 °C, a pressure of 1 atm and a weight hourly space velocity WHSV = 2.8 h−1 for 4 hours. The catalyst loading (1.85 g) was activated immediately before the reaction in a steel reactor (ID 10 mm) for 2 hours at a temperature of 320 °C in a flow of H2 with a linear feed rate of 30 mL min−1. Then the substrate was fed into the reactor at a rate of 6 mL h−1 (density 0.864 g mL−1) by a high-pressure pump HPR 5001. The reaction products were analyzed on a Crystallux-4000M chromatograph (Russia) using a ZB-5 capillary column (Zebron, USA). The analysis was performed in a programmable temperature mode of 70–220 °C at a heating rate of 6 °C min−1. The purity of hydrogen released was determined by gas chromatography with a thermal conductivity detector on a packed column with Porapak Q. The conversion (X) was calculated as the ratio of the amount of bicyclohexyl after the reaction to its initial amount. The selectivity (S) of the biphenyl production reaction was determined as the ratio of the amount of biphenyl to the total amount of reaction products.
The charge state of metals and the composition of the catalyst surface were determined by X-ray photoelectron spectroscopy (XPS) using a Kratos Axis Ultra DLD device (Great Britain) with monochromatic radiation Al Ka (hν = 1486.6 eV, 150 W). The standard energy of the analyzer for high-resolution spectra was 160 eV and 40 eV.
The composition and structure of Pt/C, Pt/Ni/C, Pt/Ni-Cr/C catalyst surface was determined using high-resolution transmission electron microscopy (HR-TEM) with a JEOL-2100F (Japan) electron microscope in light and dark field modes at an accelerating voltage of 200 kV, resolution at points of 0.1 nm, along the lines of 0.08 nm. The phase identification of crystallites was performed by analysing experimental FFT images and reference diffraction data.62 Also the element distribution on the catalyst surface was analyzed by the method of energy dispersive X-ray spectroscopy (EDX).
Author contributions
T. V. Bogdan: treatment of structural data, analysis, visualization, writing & editing; A. N. Kalenchuk: investigation, catalytic experiment, analysis, writing; L. M. Kustov: hydrogen project administration, editing, substantive translation; V. I. Bogdan: conceptualization, editing, supervision, hydrogen and catalysis project administration.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors are grateful to S. V. Savilov, K. I. Maslakov and S. V. Maksimov (Chemistry Department, Moscow State University) for XPS, TEM, EDX measurements performed at the MSU “Nanochemistry and Nanomaterials” equipment center acting under the MSU Program of Development.References
- Z. Abdin, A. Zafaranloo, A. Rafiee, W. Mérida, W. Lipiński and K. Khalilpour, Energy Rev., 2020, 120, 109620 Search PubMed.
- J. Abe, A. Popoola, E. Ajenifuja and O. M. Popoola, Int. J. Hydrogen Energy, 2019, 44, 15072 CrossRef CAS.
- P. Preuster, A. Alekseev and P. Wassersche, Annu. Rev. Chem. Biomol. Eng., 2017, 8, 445 CrossRef CAS.
- P. Modisha, C. Ouma, R. Garidzirai, P. Wasserscheid and D. Bessarabov, Energy Fuels, 2019, 33, 2778 CrossRef CAS.
- P. Aakko-Saksa, C. Cook, J. Kiviaho and T. Repo, J. Power Sources, 2018, 396, 803 CrossRef CAS.
- P. Preuster, C. Papp and P. Wasserscheid, Acc. Chem. Res., 2017, 50, 74 CrossRef CAS.
- E. Gianotti, M. Taillades-Jacquin, J. Rozière and D. Jones, ACS Catal., 2018, 8, 4660 CrossRef CAS.
- M. Markiewicz, Y. Zhang, A. Bösmann, N. Brückner, J. Thöming, P. Wasserscheid and S. Stolte, Energy Environ. Sci., 2015, 8, 1035 RSC.
- L. Kustov, A. Kalenchuk and V. Bogdan, Rus. Chem. Rev., 2020, 89, 897 CrossRef.
- D. Zaitsau, V. Emel’yanenko, A. Pimerzin and S. Verevkin, J. Chem. Thermodyn., 2018, 122, 1 CrossRef.
- D. Han, Y. Jo, B. Shin, M. Jang, J. Kang, J. Han, S. Nam and C. Yoon, Energy Technol., 2019, 7, 113 CrossRef.
- A. Kalenchuk, V. Bogdan, S. Dunaev and L. Kustov, Fuel, 2020, 280, 118625 CrossRef.
- A. Kalenchuk and L. Kustov, Molecules, 2022, 27, 2236 CrossRef PubMed.
- J. Cho, H. Kim, J. Oh and B. Park, Catalysts, 2021, 11, 1497 CrossRef CAS.
- U. Rayhan, Z. Kowser, M. N. Islam, C. Redshaw and T. Yamato, Top. Catal., 2018, 61, 560 CrossRef CAS.
- V. Bogdan, A. Kalenchuk, P. Chernavsky, T. Bogdan, I. Mishanin and L. Kustov, Int. J. Hydrogen Energy, 2021, 46, 14532 CrossRef CAS.
- V. Baglio, A. Stassi, A. Di Blasi, C. D’Urso, V. Antonucci and A. Aricò, Electrochim. Acta, 2007, 53, 1360 CrossRef CAS.
- L. Xiong and A. Manthiram, J. Electrochem. Soc., 2005, 152, 697 CrossRef.
- T. Bogdan, A. Kalenchuk, S. Maksimov and V. Bogdan, Rus. J. Phys. Chem. A, 2021, 95, 523 CrossRef CAS.
- D. Kaewsai and M. Hunsom, Nanomaterials, 2018, 8, 299 CrossRef.
- E. Antolini, J. Mater. Sci., 2003, 38, 2995 CrossRef CAS.
- M. Román-Martínez, D. Cazorla-Amorós, A. Linares-Solano and C. Salinas-Martínez, Appl. Catal. A Gen., 1994, 116, 187 CrossRef.
- J. Du, C. Song, J. Song, J. Zhao and Z. Zhu, J. Fuel Chem. Tech., 2009, 37, 468 CrossRef.
- V. Babar, S. Jaiswal and V. Kumar, Chem. Phys. Lett., 2013, 560, 42 CrossRef.
- E. Auer, A. Freund, J. Pietsch and T. Tacke, Appl. Catal. A Gen., 1998, 173, 259 CrossRef.
- K. Iost, V. Borisov, V. Temerev, Y. Surovikin, P. Pavluchenko, M. Trenikhin, A. Arbuzov, D. Shlyapin, P. Tsyrulnikov and A. Vedyagin, Reac. Kinet. Mech. Cat., 2019, 127, 103 CrossRef.
- E. Lam and J. Luong, ACS Catal., 2014, 4, 3393 CrossRef.
- A. Koklin, N. Bobrova, T. Bogdan, I. Mishanin and V. Bogdan, Molecules, 2022, 27(5), 1494 CrossRef PubMed.
- Y. Pokusaeva, A. Koklin, V. Lunin and V. Bogdan, Mendeleev Commun., 2019, 29(4), 382 CrossRef.
- Y. Pokusaeva, A. E. Koklin, O. L. Eliseev, R. V. Kazantsev and V. Bogdan, Rus. Chem. Bul. Int. Ed., 2020, 69(2), 237 CrossRef.
- S. Chernyak, A. Ivanov, D. Stolbov, S. Maksimov, K. Maslakov, P. Chernavskii, Y. Pokusaeva, A. Koklin, V. Bogdan and S. Savilov, Carbon, 2020, 168, 475 CrossRef.
- Z. Tian, C. Wang, J. Yue, X. Zhang and L. Ma, Catal. Sci. Technol., 2019, 9, 2728 RSC.
- E. Bus and J. Bokhoven, J. Phys. Chem. C, 2007, 111, 9761 CrossRef.
- A. Bugaev, A. Guda, A. Lazzarini, K. Lomachenko, E. Groppo, R. Pellegrini, A. Piovano, H. Emerich, A. Soldatov, L. Bugaev, V. Dmitriev, J. Bokhoven and C. Lamberti, Catal. Today, 2017, 283, 119 CrossRef.
- I. Mishanin, T. Bogdan, A. Koklin and V. Bogdan, Chem. Eng. J., 2022, 446, 137184 CrossRef.
- R. Lamber and N. Jaeger, Surface Sci., 1993, 289, 247 CrossRef.
- R. Lamber, N. Jaeger and G. Schulz-Ekloff, Surface Sci., 1988, 197, 402 CrossRef.
- R. Lamber, N. Jaeger and G. Schulz-Ekloff, Surface Sci., 1990, 227, 15 CrossRef.
- Y. Chen, J. Wang, P. Schützendübe, Z. Wang and E. J. Mittemeijer, Carbon, 2020, 159, 37 CrossRef.
- M. Zheng, K. Takei, B. Hsia, H. Fang, X. Zhang, N. Ferralis, H. Ko, Y. Chueh, Y. Zhang, R. Maboudian and A. Javey, Appl. Phys. Lett., 2010, 96, 063110 CrossRef.
- Y. Bleu, V. Barnier, F. Christien, F. Bourquard, A. Loir, F. Garrelie and C. Donnet, Carbon, 2019, 155, 410 CrossRef.
- Q. Yu, J. Lian, S. Siriponglert, H. Li, Y. Chen and S. Pei, Appl. Phys. Lett., 2008, 93, 113103 CrossRef.
- E. Paparazzo, Carbon, 2013, 63, 562 CrossRef.
- Y. Lyu, P. Wang, D. Liu, F. Zhang, T. Senftle, G. Zhang, Z. Zhang, J. Wang and W. Liu, Small Methods, 2022, 6, 2200235 CrossRef PubMed.
- M. Plodinec, H. Nerl, T. Lunkenbein and R. Schlögl, Microsc. Microanal., 2022, 28(S1), 146 CrossRef.
- A. Kalenchuk, K. Maslakov, T. Bogdan, P. Chernavskiy and V. Bogdan, Rus. Chem. Bul. Int. Ed., 2021, 70, 323 CrossRef.
- M. Biesinger, L. Lau, A. Gerson and R. Smart, Phys. Chem. Chem. Phys., 2012, 14, 2434 RSC.
- C. Biesinger, C. Brown, J. Mycroft, R. Davidson and N. McIntyre, Surf. Interface Anal., 2004, 36, 1550 CrossRef.
- V. Saveleva, V. Papaefthimiou, M. Daletou, W. Doh, C. Ulhaq-Bouillet, M. Diebold, S. Zafeiratos and E. Savinova, J. Phys. Chem. C, 2016, 120, 15930 CrossRef.
- R. Mom, L. Frevel, J. Velasco-Velez, M. Plodinec, A. Knop-Gericke and R. Schlogl, J. Am. Chem. Soc., 2019, 141, 6537 CrossRef.
- J. F. Moulder, W. F. Stickle, P. E. Sobol and K. D. Bomben, Handbook of X-ray photoelectron spectroscopy, ed. J. Chastain, PerkinElmer Corp., 1992. p. 261 Search PubMed.
- T. Susi, T. Pichler and P. Ayala, Beilstein J. Nanotechnol., 2015, 6, 177 CrossRef.
- S. Suzuki, Y. Watanabe and S. Heun, Curr. Opin. Solid State Mater. Sci., 2006, 10, 53 CrossRef.
- G. Speranza, Materials, 2022, 15, 4434 CrossRef.
- A. A. Shukla, P. V. Gosavi, J. V. Pande, V. P. Kumar, K. V. R. Chary and R. B. Biniwale, Int. J. Hydrogen Energy, 2010, 35, 4020 CrossRef.
- Y. Jo, T. W. Kim, J. Oh, D. Kim and Y.-W. Suh, J. Catal., 2022, 413, 127 CrossRef.
- G. C. Gazzadi and S. Frabboni, Appl. Phys. Lett., 2009, 94, 173112 CrossRef.
- J. Harris, E. Boyes and J. Cairns, J. Catalysis, 1983, 82, 127 CrossRef.
- Q. Liu, P. Rzepka, H. Frey, J. Tripp, A. Beck, L. Artiglia, M. Ranocchiari and J. A. van Bokhoven, Mater. Today Nano, 2022, 20, 100273 CrossRef.
- Y. Yermakov, V. Surovikin, G. Plaksin, V. Semikolenov, V. Likholobov, L. Chuvilin and S. Bogdanov, React. Kinet. Catal. Lett., 1987, 33, 435 CrossRef.
- A. Kalenchuk, V. Bogdan, S. Dunaev and L. Kustov, Int. J. Hydrogen Energy, 2018, 43, 6191 CrossRef.
- ICDD (2018). The Powder Diffraction File. International Centre for Diffraction Data, Newtown Square, Pennsylvania, USA.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2cp04457a |
| This journal is © the Owner Societies 2023 |