
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
SNARE mimic peptide triggered membrane fusion kinetics revealed using single particle techniques†
Guus
van der Borg
 a,
Niek
Crone
b,
Aimee L.
Boyle
c,
Alexander
Kros
b and
Wouter H.
Roos
*a
a,
Niek
Crone
b,
Aimee L.
Boyle
c,
Alexander
Kros
b and
Wouter H.
Roos
*a
aMoleculaire Biofysica, Zernike Instituut, Rijksuniversiteit Groningen, Groningen, The Netherlands. E-mail: w.h.roos@rug.nl
bDepartment of Supramolecular and Biomaterials Chemistry, Leiden Institute of Chemistry, Leiden University, Leiden, The Netherlands
cDepartment of Macromolecular Biochemistry, Leiden Institute of Chemistry, Leiden University, Leiden, The Netherlands
First published on 27th April 2023
Abstract
Membrane fusion is an essential part of the proper functioning of life. As such it is not only important that organisms carefully regulate the process, but also that it is well understood. One way to facilitate and study membrane fusion is to use artificial, minimalist, fusion peptides. In this study the efficiency and kinetics of two fusion peptides, denoted CPE and CPK, were studied using single-particle TIRF microscopy. CPE and CPK are helical peptides which interact with each other, forming a coiled-coil motif. The peptides can be inserted into a lipid membrane using a lipid anchor, and if these peptides are anchored in opposing lipid membranes, then the coiled-coil interaction can provide the mechanical force necessary to overcome the energy barrier to initiate fusion, much in the same way the SNARE complex does. In this study we find that the fusogenic facilitation of CPE and CPK in liposomes is, at least partially, dependent on the size of the particle. In addition, under certain fusogenic conditions such as when using small liposomes of ∼60 nm in diameter, CPK alone is enough to facilitate membrane fusion in both bulk and single-particle studies. We show this using bulk lipid mixing assays utilizing FRET and single-particle TIRF, making use of dequenching fluorophores to indicate fusion. This provides us with new insights into the mechanisms of peptide-mediated membrane fusion and illuminates both challenges as well as opportunities when designing drug delivery systems.
Introduction
Membrane fusion is an integral part of many biological processes that are required for life to function. As such, it is essential that the process is carefully regulated by the organism.1,2 A variety of systems in which membrane fusion is regulated have been described.3–5 Arguably the most studied of these systems are the SNARE (soluble N-ethylmaleimide sensitive factor attachment protein receptor) proteins. The SNARE proteins are a superfamily of proteins generally involved in facilitating the fusion of endosomes with the cellular membrane.6,7 This facilitation of membrane fusion is achieved by the formation of a protein complex which forms a stable four helix coiled-coil bundle and spans both the endosomal membrane and the cell membrane. This functions to stabilize the intermediate states of membrane fusion and lowers the energetic barrier that needs to be overcome to initiate the fusion process in a well-controlled manner.4,8–10To study this process, and possibly to use it for artificially induced membrane fusion, minimalist model systems were developed, one of which is the E/K peptide pair.11–14 This model system relies on two helical peptides, namely peptide ‘K’ (KIAALKE)4 and peptide ‘E’ (EIAALEK)4, which when interacting with each other form a coiled-coil motif.13,15 In order to use this interaction to facilitate membrane fusion the peptides were attached to a membrane anchor, namely cholesterol hemisuccinate, via a flexible, poly(ethylene glycol) (PEG), linker. The resulting SNARE mimics are named ‘CPE’ and ‘CPK’, depending on the attached peptide (Fig. 1A). When two membranes with CPE inserted in one membrane and CPK in the other membrane are in close proximity to one another the E and K peptides zipper in a parallel configuration to form heterodimeric coiled-coil complexes.15 This complex formation first docks the membranes onto each other and then serves to provide the force involved in lowering the energy barrier for fusion, in a manner similar to the SNARE complex.16 In particular, upon contact, CPE and CPK form a heterodimeric complex through a combination of hydrophobic and electrostatic interactions. Following this coiled-coil formation, the liposomes are brought into close proximity and CPK is able to interact with the opposing lipid membrane. The consequences of this peptide–lipid membrane interaction are increased liposome curvature, increased lipid tail protrusion, and phosphatidylethanolamine (PE) accumulation.17,18 All of these factors are thought to increase the likelihood of fusion. Typically, first hemifusion occurs, where only the outer membrane leaflets are fused, and then this state transitions to the formation of a complete fusion pore (Fig. 1B).16,17,19 The E/K peptide pair has been shown to work in multiple systems, such as liposomes, GUVs, planar lipid bilayers, cell membranes, and multilamellar vesicles, both in vitro, and in vivo.16,20–23 This means that it shows promise, not only as a model system for SNARE proteins, but also as a potential drug delivery system. To gain a greater understanding of the system we use both bulk and single particle methods to characterize the efficiency and kinetics of the peptide-induced fusion. Bulk lipid mixing is studied through the fusion of labeled liposomes in a fluorescence spectrometer.24,25 Single-particle studies are performed using Total Internal Reflection Fluorescence (TIRF) microscopy on a planar lipid bilayer in a microfluidic flow cell.26,27 TIRF is a powerful approach to observe liposome fusion at the single-particle level.28,29 Using these techniques, we gain greater insight into both the efficiency and kinetics of the CPE/CPK system. We find that CPE/CPK pairs rapidly induce fusion in membranes both between liposomes and with liposomes and planar bilayers, though there are differences in kinetics. Furthermore, using small vesicles, CPK alone is sufficient to promote membrane fusion.
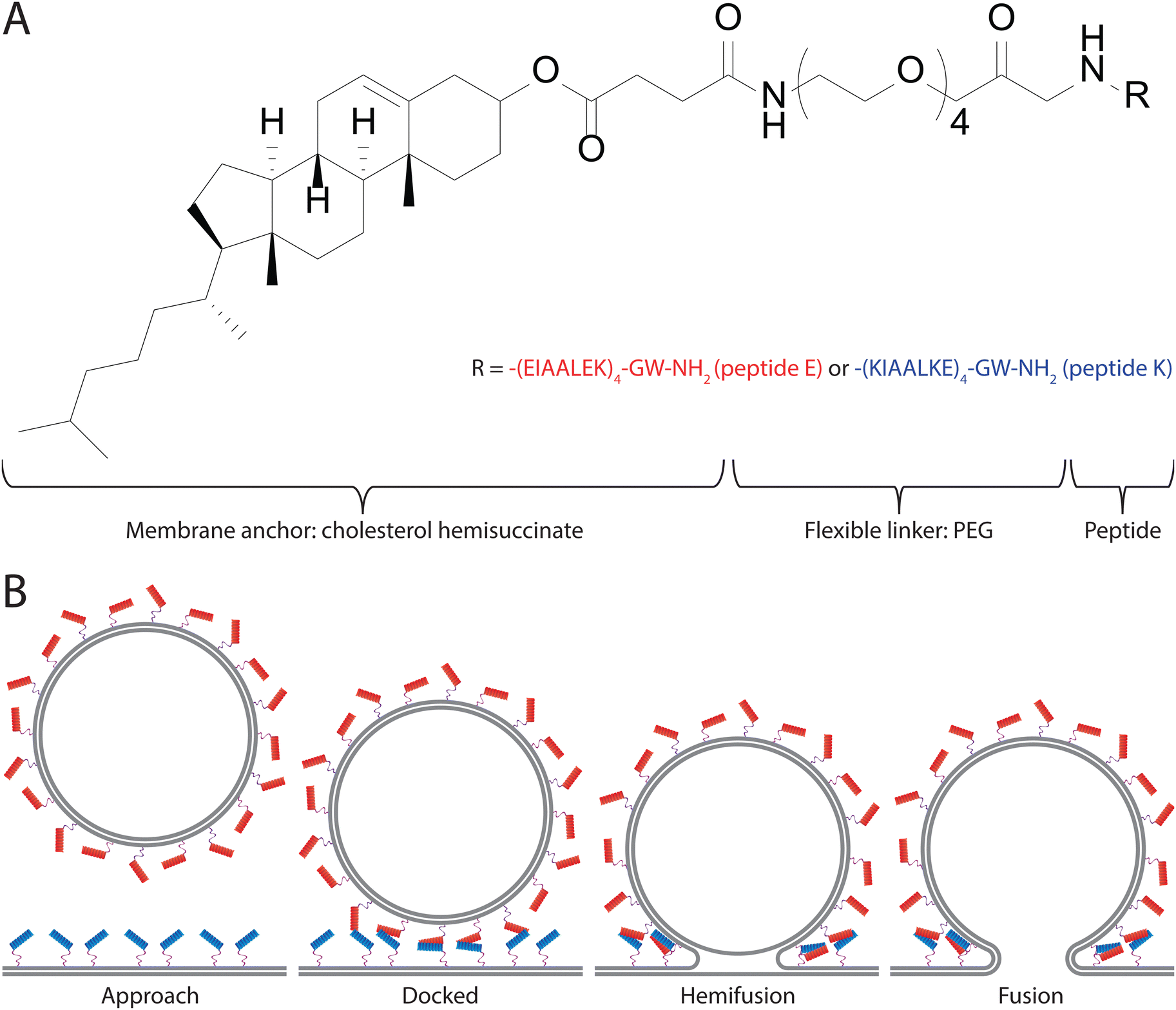 | ||
| Fig. 1 CPE/CPK chemical structure and fusion process. (A) The chemical structure of CPE/CPK. Peptides consist of a cholesterol membrane anchor conjugated via a flexible PEG linker to peptide E or K respectively. (B) A schematic model of the fusion process facilitated by CPE/CPK. Red helices represent the K peptide, blue helices the E peptide, and the purple lines represent the flexible linker. | ||
Experimental
Snare mimic peptide synthesis
Peptides E4 (EIAALEK)4GW and K4 (KIAALKE)4GW were synthesized using a Liberty Blue microwave-assisted peptide synthesizer. A rink-amide resin was used as the solid support. Standard Fmoc chemistry protocols were employed, with 20% piperidine in DMF used to facilitate Fmoc deprotection, and DIC/Oxyma as the activator/base respectively for the coupling steps. After synthesis, Fmoc-PEG4-COOH (1.2 equivalents) was coupled to the N-terminus of the peptides using DIC/Oxyma (3 equivalents each) and left to react overnight. The resin was subsequently washed (DMF, 3×) Fmoc-deprotected, (20% piperidine in DMF, 15 min, 2×) and washed again (DMF 3×) before cholesteryl hemisuccinate (5 equivalents) was coupled using DIC/Oxyma (5 equivalents each) and left to react overnight. The resin was then cleaved (97.5% TFA, 2.5% TIPS, 1 h), precipitated into ice-cold diethyl ether and centrifuged to collect the precipitate. The precipitate was redissolved in H2O/MeCN and freeze-dried.To purify the peptides, reversed-phase high-performance liquid chromatography (RP-HPLC) was employed. A C4 column was used for both lipopeptides. A gradient from 20% to 80% MeCN (with 0.1% TFA) was employed (the other component of the mobile phase was H2O with 0.1% TFA). Fractions were collected, analyzed by Liquid Chromatography-Mass Spectrometry (LCMS) and those deemed to be >95% pure were pooled and freeze-dried. LCMS data was added in the ESI† (Fig. S3–S5).
For the synthesis of Atto-488 labelled CPK, the procedure above was followed but the K4 peptide was synthesized with ‘GC’ at the C-terminus in place of ‘GW.’ PEG and cholesteryl hemisuccinate were coupled as described, and the peptide was cleaved from the resin, precipitated and dried before being purified. To couple the Atto-488 dye, the lipopeptide was dissolved in DCM, and 1.1 equivalents of Atto-488 maleimide solution were added. The mixture was left to stir in the dark for one hour, before the solvent was removed by rotary evaporation. The mixture was redissolved in H2O![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :MeCN (4:1) and purified by RP-HPLC as described above. The peptides were designed to form helical structures, and their tertiary structure is further characterized in Rabe et al. (2015).30
:MeCN (4:1) and purified by RP-HPLC as described above. The peptides were designed to form helical structures, and their tertiary structure is further characterized in Rabe et al. (2015).30
Small unilamellar vesicle (SUV) preparation
The intent of the study was to create SUVs of comparable size and comparable size distributions between conditions. However, this turned out to be extremely challenging. The original experimental design called for using the filter extrusion method of creating SUVs, in order to ensure the comparable size. And whilst this worked well for plain SUVs, the process of extrusion removed the CPE and CPK peptides from the lipid membranes. As such it was ultimately decided to use the sonication method of SUV formation, even though this method is sub-optimal for producing a specific size and size distribution of SUV.SUVs were composed of DOPC:DOPE:cholesterol in a ratio of 50:25:25 mol%. Either CPK or CPE lipopeptide was added to the SUVs in a molar ratio of 2 (with respect to above mol%). For the bulk fusion assay 1,2-dioleoyl-sn-glycero-3-phosphoethanolamine-N-(lissamine rhodamine B sulfonyl) (LR-DOPE) and 1,2-dioleoyl-sn-glycero-3-phosphoethanolamine-N-(7-nitro-2-1,3-benzoxadiazol-4-yl) (NBD-DOPE) were added in a molar ratio of either 0.5 or 0.25 for the experimental and ‘50% control’ conditions respectively. And for the TIRF studies, octadecyl rhodamine B (R18) was added with a molar ratio of 5. Lipopeptide solutions were prepared in 1:1 (v/v) chloroform:methanol. A lipid film was then formed by drying an appropriate volume (generally ∼40 μL) of solution in a flat-bottomed, glass, 2 mL vial using a stream of argon. The lipid film was further dried out by placing the vials under low pressure in a desiccator for a minimum of 15 min. The film was rehydrated in phosphate buffered saline (PBS, pH 7.4), vortexed until the lipids were suspended, and then sonicated in a bath sonicator with the water bath at ∼55 °C for 5 min. The liposomes were always used shortly after manufacturing them. At the latest within 4 h of production.
Size determination and bulk mixing assay
The diameter of the SUVs was determined by Nanoparticle Tracking Analysis (NTA) using a NanoSight LM14 (Malvern) system with NTA 3.0 software (Malvern). Measurements were performed several hours after SUV preparation.The bulk lipid mixing assays were conducted with n = 8 on a Tecan Spark multimode microplate reader using a black flat-bottomed 96 well plate. The excitation wavelength was 460 nm, and NBD emission was monitored at 535 nm. 100 μL of 1 mM fluorescent SUVs and 100 μL of 1 mM non-fluorescent SUVs were combined and measured, except for the ‘50% control’ condition where 200 μL of 1 mM fluorescent SUVs were used. For consistent mixing the plate was shaken inside the plate reader for 30 s. Data was collected every 30 s for 1 h at room temperature. The results were normalized, taking the mean fluorescence of the 50% control as 100% lipid mixing and the mean fluorescence of the CPE/plain condition as 0% lipid mixing. As the data points fluctuate around the mean as defined above, also values below 0% and above 100% will be found in the data.
Fluorescence microscopy
TIRF imaging was performed in a microfluidic flow channel. Microfluidic flow channels were produced by pouring polydimethylsiloxane (PDMS) over a mold, followed by a period of curing to harden the PDMS, after which the inlets and outlets were punched into the PDMS using biopsy needles. Lastly, a glass coverslip covered the PDMS to create the channels. Glass coverslips were cleaned in acetone for 30 min in a bath sonicator, followed by 30 min in 96% ethanol in a bath sonicator for 30 min, and lastly 10 min of sonication in 1 M KOH. In between sonications and before drying, the coverslips were rinsed with deionized water three times. Coverslips were dried overnight at 60 °C. Before use, coverslips were cleaned in an oxygen plasma cleaner and attached to the PDMS, forming the microfluidic channels. The flow cell consists of five separate channels with a width and height of 0.5 × 0.2 mm. The setup was placed on a home-built TIRF microscope, using an inverted microscope (IX-71; Olympus) and a high numerical aperture, oil-immersion objective (NA 1.45, 60×; Olympus). Lipid bilayers were formed by filling the microfluidic channels with a 1 mM SUV suspension soon after plasma cleaning the glass and incubating at room temperature for 45 min. For the single-particle fusion experiments the bilayer was formed using non-fluorescent SUVs, after which the planar lipid bilayer was washed to remove excess SUVs using PBS (pH 7.4). A solution of 1 nM fluorescent SUVs was introduced, and docking and fusion events were recorded using a Hamamatsu EM-CCD camera. Videos of 1500 frames of 200 ms each were taken for a total movie length of 5 min (ESI† Video S1). Fluorescent SUVs and planar lipid bilayers were excited using a 561 nm laser. The single-particle fusion data was analyzed using a home-written Python script and ImageJ. Hemifusion time was determined by using the Python script to generate fluorescence over time graphs for every individual particle and then manually designating the time of docking and the time of fusion of each graph. Aggregated particles were identified through unusually high fluorescence values and multiple fusion peaks and were disregarded.For Fluorescence Recovery After Photobleaching (FRAP) experiments the SUVs used to form the bilayer were labeled with either R18 or Atto-488 labeled CPK. R18 labels the membrane by inserting itself into the membrane using an 18-carbon hydrophobic tail. CPK is labeled directly with Atto-488 by covalently binding it to the C terminus of the K peptide via thiol–maleimide coupling. The area of excitation was then reduced to a small area by partially closing a manual aperture in front of the excitation laser (488 or 561 nm; Coherent). This area was then strongly illuminated by increasing the power of the excitation laser, bleaching the fluorophores in the area, after which the laser power was reduced again and the aperture manually opened. After this the recovery of fluorescence was measured over a span of 30 min.
Results
Particle characterization
The lipid films consisting of 50:25:25:2, DOPC:DOPE:Cholesterol:CPE or CPK were processed into a 1 mM solution of SUVs using sonication.16 NTA analysis revealed a modal particle diameter of 54.5 and 61.0 nm for CPK and CPE incorporated SUVs respectively (Fig. 2). These dimensions are below the previously reported ∼100 nm modal diameter.16 The NTA data also revealed the occurrence of SUV aggregation as indicated by the increase in particle size occurrence in multiples of the modal SUV size in the particle size histogram.
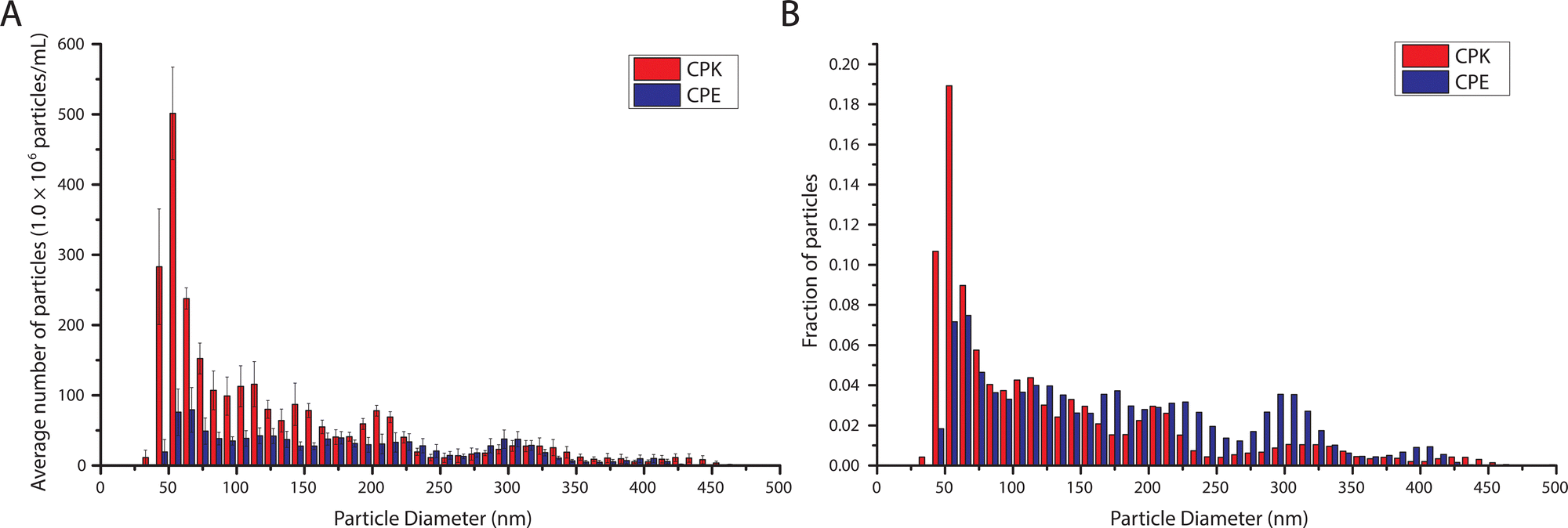 | ||
| Fig. 2 Vesicle diameter determination by NTA. Histograms show the absolute and relative number of particles of a certain range of diameter. Red bars are CPK incorporated SUVs and blue bars are CPE incorporated SUVs. Bin size is 10 nm. (A) The absolute concentrations of particles of a certain diameter. Error bars depict the standard error of the mean. (B) Relative concentration of particles as a function of diameter. | ||
Bulk lipid mixing assay
To observe whether these smaller SUVs behaved similarly to the ∼100 nm SUVs previously described by Marsden et al., a bulk lipid mixing assay was performed.16 Using different combinations of SUVs, the ability of SUV-SUV fusion was tested. The lipid mixing assay, a FRET based approach with one unlabeled SUV batch and one FRET donor and acceptor, (NBD-DOPE and LR-DOPE respectively), fluorescently labeled batch, was performed. If lipid mixing occurs through the fusion of a labeled SUV with an unlabeled SUV the local concentration of the FRET pairs is lowered. This concentration decrease, in turn, increases the stochastic distance between the donor and acceptor, and thus decreases the FRET efficiency (Fig. 3A). This is recorded as an increase in FRET donor emission. Four different types of SUVs were used in this assay. SUVs with incorporated CPK (CPK SUVs), SUVs with incorporated CPE (CPE SUVs), ‘Plain’ SUVs, which are SUVs without incorporated fusion peptides, and ‘50% control’ SUVs, which are SUVs with only half of the fluorophore concentration when compared to the other fluorescent assay SUVs. As this assay is FRET based and it depends on the local concentration of the fluorophores influencing FRET efficiency, the ‘50% control’ SUVs function as a 100% lipid mixing control, as they contain the same local fluorophore concentration as a fully fused florescent/non-fluorescent SUV pair and where no change in donor emission is expected upon fusion (Fig. 3A). The tested conditions were CPE/CPK, CPE/Plain, CPK/Plain, with the conditions all being denoted as fluorescent/non-fluorescent, and the 50% control where all SUVs were labeled. As hypothesized, CPE/CPK showed notable lipid mixing. However, contrary to our expectations, CPK/Plain also showed notable lipid mixing (Fig. 3B). Also notable is that, unlike bulk studies previously done with larger liposomes (LUVs),14 the lipid mixing occurred rapidly, with the majority of the mixing having already taken place before the first measurement could be taken.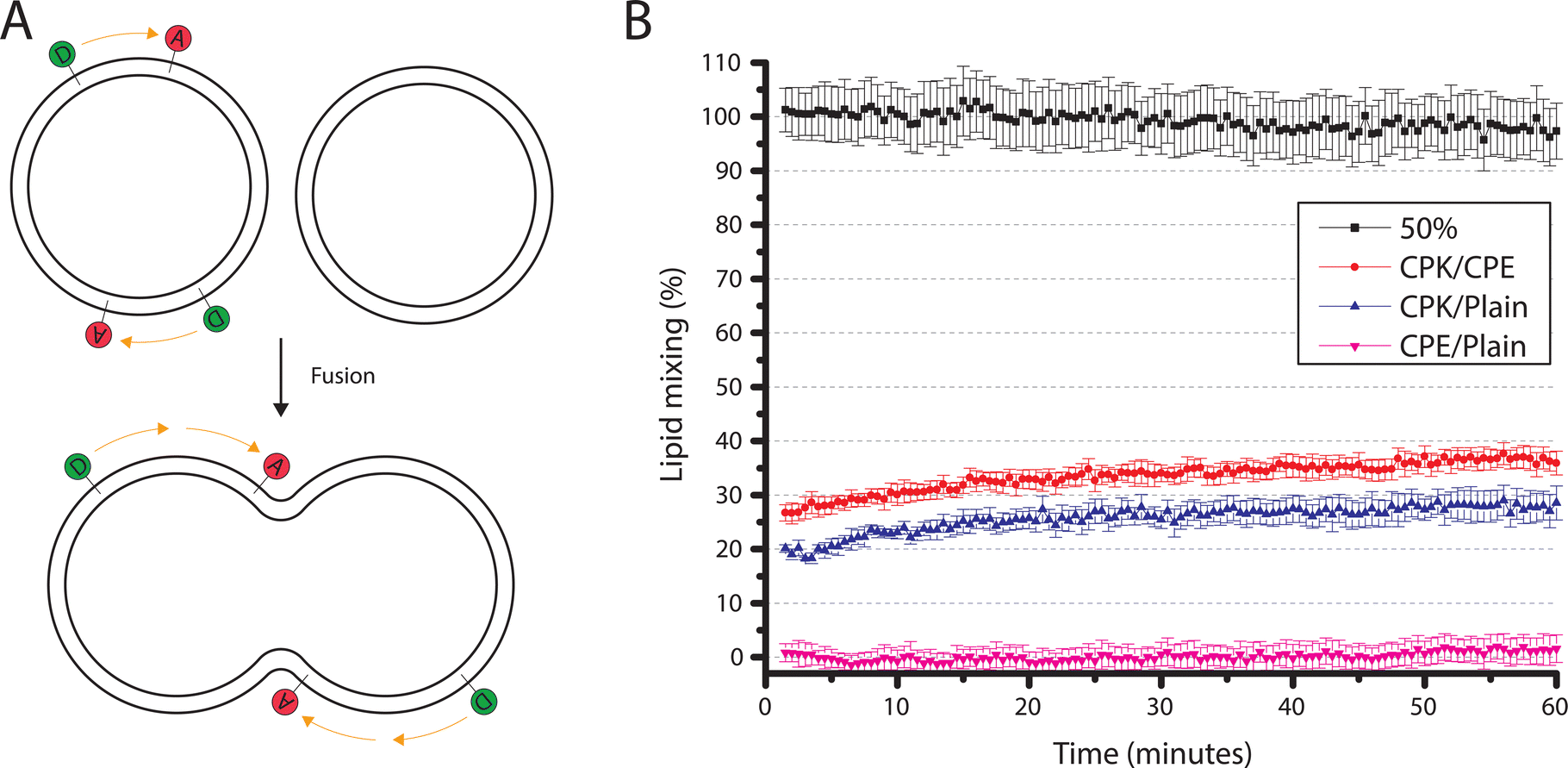 | ||
| Fig. 3 Bulk lipid mixing assay (A) A schematic representation of the lipid mixing assay. In this assay a suspension of SUVs labeled with both a FRET acceptor and donor is mixed with a suspension of unlabeled SUVs. If lipid mixing occurs through fusion then the effective concentration of the fluorophores will lessen, thus stochastically increasing the distance between the acceptor and donor pair, and thus decreasing the FRET efficiency and increasing donor emission. (B) Results of the bulk lipid mixing assay. In this assay labeled and unlabeled SUVs were mixed with the tested condition being denoted as labeled/unlabeled. Conditions tried were CPE/CPK, CPE/Plain, CPK/Plain, and the 50% control. The 50% condition (black line) serves as a control for 100% lipid mixing and only contains SUVs labeled with a lowered fluorescence concentration of 50% when compared to the other conditions, mimicking full lipid mixing. The values were normalized with the 50% control being taken as 100% mixing and CPE/Plain as 0% lipid mixing. Whiskers denote the standard error of the mean. | ||
Single particle fusion microscopy
For the single-particle studies a fluid planar lipid bilayer is formed on the surface as a target membrane. These planar lipid bilayers were formed by incubating SUVs on a glass surface made hydrophilic by oxygen plasma cleaning. To prove that this planar lipid bilayer was formed properly FRAP was performed. FRAP experiments showed that full recovery had taken place after 10 min. Next, to test the mobility of the fusion peptides in the planar lipid membrane, a second FRAP experiment was performed using Atto-488 labeled CPK peptides. In these experiments, after 30 min, recovery plateaued at 51% ± 3% recovery, meaning that approximately half of the CPK was immobile (ESI,† Fig. S1). As the planar lipid bilayer is formed using SUVs that have CPK incorporated in both leaflets of the membrane there is CPK on both sides of the planar lipid membrane as well. In addition, as the glass is slightly negatively charged due to the oxidation caused by the plasma cleaning, it is likely that the net positively charged K peptides that are located on the glass side electrostatically bind to the surface, thus explaining the ∼50% immobile fraction of CPK. This indicates that the peptides involved in membrane fusion, i.e. the peptides on the other side of the membrane, are fully mobile. Single particle fusion assays were performed using TIRF microscopy where, the fusing event was defined as a spike in fluorescence intensity followed by a sharp decrease in signal (Fig. 4). These assays were performed for the following conditions, with the fluorescently labeled SUVs always mentioned first: CPE/CPK, Plain/CPK, CPK/CPK, CPE/Plain, Plain/Plain. CPE/CPE was omitted as no planar lipid bilayer could be made with CPE integrated liposomes. The inability of CPE incorporated membranes to form planar lipid membranes in the microfluidic channel could be explained by the electrostatic repulsion of the net negatively charged E peptide from the negatively charged oxidized glass. Of the tested conditions, Plain/Plain and CPE/Plain particles showed little to no docking or fusion. All conditions involving CPK showed docking and fusion, however. Fusion efficiency was measured by dividing the number of particles that underwent membrane fusion by the total amount of particles docked onto the planar lipid bilayer. Aggregate particles, identified by multiple fusion peaks and/or excessively high base fluorescence (a factor of >2 higher than the average particle in the experiment), were identified and disregarded. The CPE/CPK, Plain/CPK, and CPK/CPK conditions showed a similar mean fusion efficiency of 26%, 17% and 21% respectively (Fig. 5). Indeed, the CPK/CPK condition was not significantly different from either of the other conditions at the α < 0.05 level when tested with a two sample t-test. Next, we looked at the time necessary to go from docking to full fusion to obtain an indication of the kinetics involved in the CPE/CPK mediated fusion. As the fusing velocity is not normally distributed and strongly skewed towards shorter timeframes, we look at the median value to get an idea of the fusion speed. Median fusion times of the CPE/CPK, plain/CPK, and CPK/CPK conditions were 2.6 s, 0 s, and 1.8 s respectively. With ‘0 s’ meaning that, with the available temporal resolution, no distinction could be made between time of docking and time of fusion. All median fusion times were significantly different at the α < 0.05 level when tested with the Mood's median test. When plotting the fusion times of the particles as a histogram it was found that the data follows a bi-exponential decay function (Fig. 6, ESI† Fig. S2). With the function fitted being| y = Ae−k1t + Be−k2t |
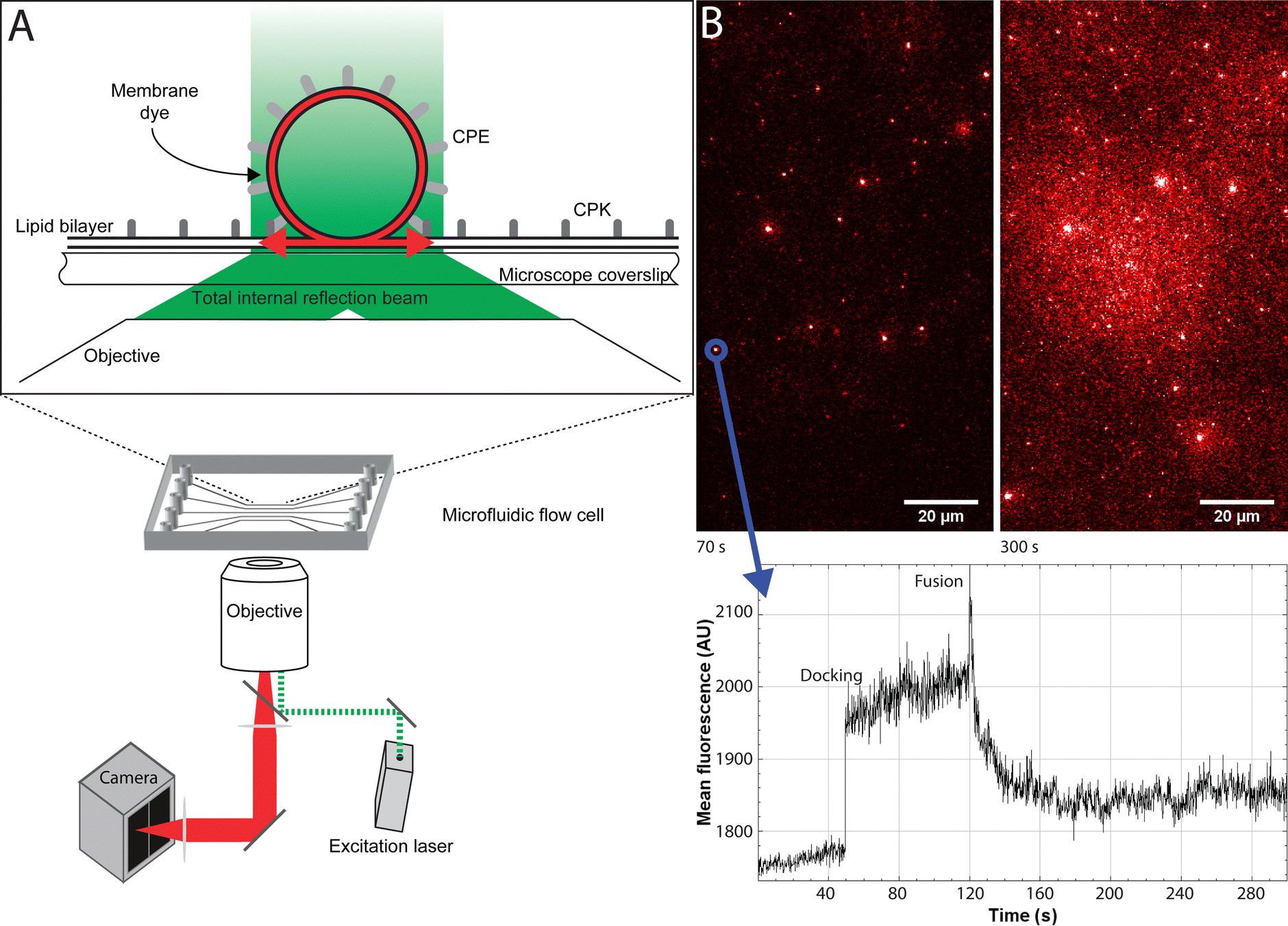 | ||
| Fig. 4 Experimental setup of the single particle measurements using TIRF (A) A schematic depiction of the TIRF setup. (top) A schematic depiction of a fusion event with CPE incorporated liposomes and a CPK incorporated planar lipid bilayer. After fusion the fluorescent label (in red) in the liposome membrane is free to diffuse through the planar lipid bilayer. (bottom) A schematic view of the microscope setup. (B) A representative image and graph of a single particle fusion experiment. (top) Images of, respectively, 70 s and 300 s into the experiment. After 300 s the background is significantly brighter due to the increasing amount of fluorescent dyes diffused into the planar lipid bilayer. (bottom) A fluorescence intensity graph over a region of interest, namely a particle. It clearly shows the time of docking, through the sudden increase of fluorescence as a plateau, and the fusing event, as a spike in fluorescence intensity followed by a sharp decrease to baseline intensity. | ||
 | ||
| Fig. 5 A boxplot of the fusion efficiency of the different conditions. The datapoints describe individual experiments, the box shows the standard error of the mean, the dotted line is the median, and the small open square marks the mean value. * denotes a statistically significant difference at the α < 0.05 level as tested by a two-sample t-test. | ||
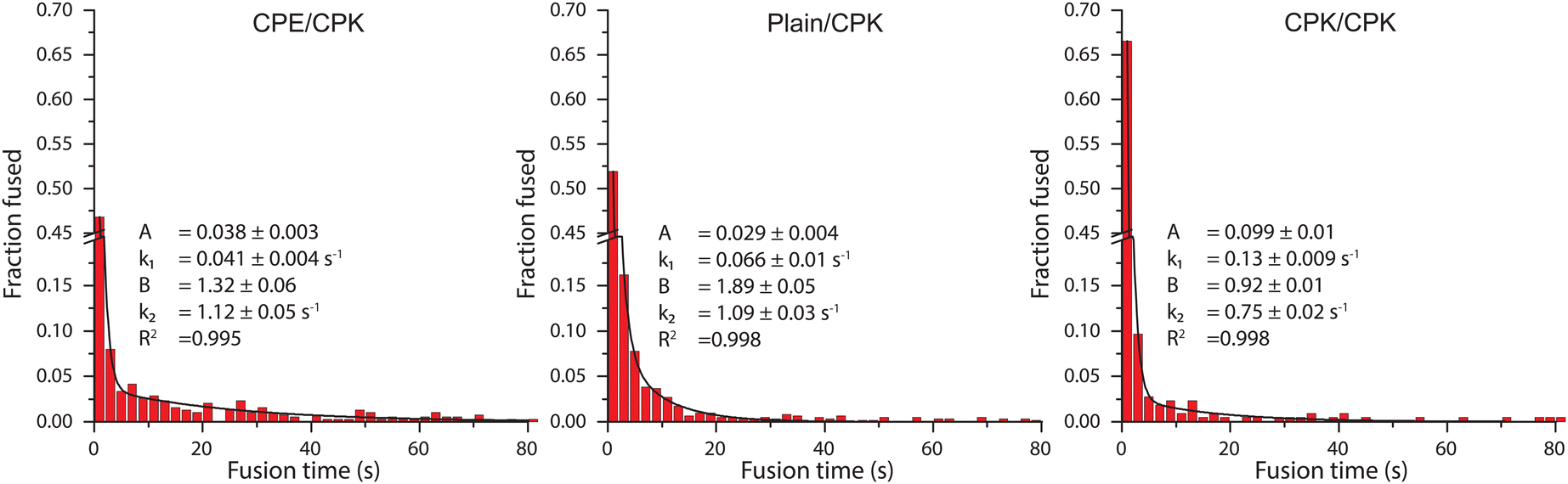 | ||
| Fig. 6 A histogram of the fusion time of individual particles. The histograms were fitted with a bi-exponential decay function. The bi-exponential decay function is fit with 4 variables: with A and B being the amplitudes and k1 and k2 being the decay constants, with the range indicating the standard error. R2 shows the coefficient of determination of the fit. Scalebars were truncated (y-axis) or cut (x-axis) to increase clarity. The full histogram can be found as ESI† Fig. S2. | ||
Discussion
We have shown that peptide induced fusion can be studied at the single particle level using a TIRF microscopy imaging approach. Next to the expected fusion between CPE–CPK pairs, we also observed, under certain conditions, that CPK alone can promote membrane fusion. The observation that CPK affects the lipid membrane in a way that promotes membrane fusion has been reported before, however, this had not been investigated in detail.17,31–33 As CPK induced fusion is both observed in the bulk lipid mixing assays as in the single particle TIRF studies, it is likely that the size of the SUV strongly influences fusion. Compared to the Marsden et al. publication the vesicles that were produced in the current study were notably smaller, with a modal size of ∼60 nm as opposed to ∼100 nm.16 In other words, we now study SUVs whereas previously LUVs were studied. It is likely that this change in size did not directly affect the fusogenic capabilities of the CPK and/or CPE fusion peptides, but that it had an effect on the membrane properties which lowered the ‘baseline’ energy barrier preventing fusion. Depending on the lipid shape, it is expected that the membranes of the vesicles with the same lipid composition, yet smaller diameter, are under notably higher bending stress when compared to the larger vesicles.34,35 It is known that a membrane under higher bending stress fuses more readily due to the energy barrier needed to overcome to initiate fusion being smaller.36 If this energy barrier is lowered then it will require a less robust fusion promotor to facilitate fusion. Which could possibly explain why CPK alone could facilitate fusion in a 60 nm SUV whilst being unable to do so in a 100 nm sized LUV. The folding properties of the peptides do not seem responsible for the observed phenomena, as can be concluded from circular dichroism spectra [Daudey et al., Chem. Sci., 2021 doi: https://doi.org/10.1039/D0SC06635D, and Rabe et al., Langmuir, 2015, 31, 9953–9964]. However, there are a few indicators as to how CPK alone can serve as a fusion facilitator. For one, the increased docking capability of the plain particles on the CPK integrated bilayer as opposed to plain particles on a plain bilayer indicates that CPK interacts with the target lipid membrane even in the absence of a complimentary peptide. Interaction of peptide K with both the host lipid membrane and the opposing lipid bilayer consisting of a 2:1:1 DOPC:DOPE:cholesterol ratio has been reported before.31,32 In fact, CPK has been known to affect the membrane properties of the opposing membrane in a way that promotes membrane fusion as well.17,32,33 Furthermore, the interaction with the opposing membrane would bring the liposome and planar lipid bilayer in close proximity, thereby imposing a certain force on the system, increasing the odds of fusion occurring. It is of interest to note that these interactions are not seen with CPE, and that the study described in Rabe et al. (2014) suggests that CPE, unlike CPK, does not interact with the lipid membrane in a significant way. Which would explain it's lack of fusogenic properties without a complementary peptide.32 This may also explain why the fusion of the Plain/CPK condition seems to be even faster than that of the CPE/CPK condition. It is known that CPK, when the complimentary peptide is present, preferentially binds to CPE over interacting with the membrane.33 This non-specific interaction may also explain the bi-exponential decay characteristic of the fusion time data. A bi-exponential decay function implies that there are two major pathways of membrane fusion occurring in the system. The presence of a fusion mechanism unmediated by peptide binding, accomplished merely through the effects peptide K has on the membrane it is incorporated in, would explain one of the mechanisms. The other could then be associated with peptide K interacting with the opposing membrane. This could provide not only a binding force but also local alterations in the opposing planar lipid membrane which would facilitate membrane fusion. As the median fusion times of the CPE/CPK and CPK/CPK SUV interaction pairs is higher than for Plain/CPK, steric effects could play a role for these conditions.
Conclusions
This study shows that the effect CPK has on the membrane is large enough to promote fusion on its own, provided that the energy barrier for fusion has already been lowered by other factors. The small sized SUVs, which are under notable membrane tension, are sufficiently prone to fusion that the full CPE and CPK pair is not needed. This has important implications for further use of the SNARE mimic peptides as we now have a better description of the different modes in which these peptides can promote fusion. The fact that, under certain conditions, fusogenic molecules on only one of the two membranes is needed, creates many more possibilities for targeted fusion approaches. This can fuel innovations in controlled membrane fusion. Finally, the reported new insights also reveal that the mechanism through which these peptides promote fusion is not completely understood yet. Follow-up studies investigating how SUV size as well as membrane tension affects fusion is likely to reveal new insights on the mechanism behind peptide induced fusion events.Conflicts of interest
There are no conflicts to declare.Acknowledgements
WR thanks Fundamenteel Onderzoek der Materie (FOM) for funding via a projectruimte grant.Notes and references
- S. Ferro-Novick and R. Jahn, Nature, 1994, 370, 191–193 CrossRef CAS PubMed.
- M. K. Bennett and R. H. Scheller, Proc. Natl. Acad. Sci. U. S. A., 1993, 90, 2559–2563 CrossRef CAS PubMed.
- J. M. Hernandez and B. Podbilewicz, Development, 2017, 144, 4481–4495 CrossRef CAS PubMed.
- B. J. Nichols and H. R. Pelham, Biochim. Biophys. Acta, 1998, 1404, 9–31 CrossRef CAS PubMed.
- S. Boonstra, J. S. Blijleven, W. H. Roos, P. R. Onck, E. van der Giessen and A. M. van Oijen, Annu. Rev. Biophys., 2018, 47, 153–173 CrossRef CAS PubMed.
- A. Kadkova, J. Radecke and J. B. Sorensen, Neuroscience, 2019, 420, 50–71 CrossRef CAS PubMed.
- T. C. Sudhof and J. E. Rothman, Science, 2009, 323, 474–477 CrossRef PubMed.
- T. Y. Yoon and M. Munson, Curr. Biol., 2018, 28, R397–R401 CrossRef CAS PubMed.
- R. W. Baker and F. M. Hughson, Nat. Rev. Mol. Cell Biol., 2016, 17, 465–479 CrossRef CAS PubMed.
- M. P. Madrigal, A. Portales, M. P. SanJuan and S. Jurado, Neuroscience, 2019, 420, 12–21 CrossRef CAS PubMed.
- B. van Lengerich, R. J. Rawle, P. M. Bendix and S. G. Boxer, Biophys. J., 2013, 105, 409–419 CrossRef CAS PubMed.
- B. E. Hubrich, P. Kumar, H. Neitz, M. Grunwald, T. Grothe, P. J. Walla, R. Jahn and U. Diederichsen, Angew. Chem., Int. Ed., 2018, 57, 14932–14936 CrossRef CAS PubMed.
- H. Robson Marsden, N. A. Elbers, P. H. Bomans, N. A. Sommerdijk and A. Kros, Angew. Chem., Int. Ed., 2009, 48, 2330–2333 CrossRef PubMed.
- H. R. Marsden, I. Tomatsu and A. Kros, Chem. Soc. Rev., 2011, 40, 1572–1585 RSC.
- T. Zheng, A. Boyle, H. Robson Marsden, D. Valdink, G. Martelli, J. Raap and A. Kros, Org. Biomol. Chem., 2015, 13, 1159–1168 RSC.
- H. Robson Marsden, A. V. Korobko, T. Zheng, J. Voskuhl and A. Kros, Biomater. Sci., 2013, 1, 1046–1054 RSC.
- M. Rabe, C. Aisenbrey, K. Pluhackova, V. de Wert, A. L. Boyle, D. F. Bruggeman, S. A. Kirsch, R. A. Bockmann, A. Kros, J. Raap and B. Bechinger, Biophys J., 2016, 111, 2162–2175.F CrossRef CAS PubMed.
- D. N. Woolfson, Adv. Protein Chem., 2005, 70, 79–112 CrossRef CAS PubMed.
- F. Versluis, J. Voskuhl, B. van Kolck, H. Zope, M. Bremmer, T. Albregtse and A. Kros, J. Am. Chem. Soc., 2013, 135, 8057–8062 CrossRef CAS PubMed.
- J. Yang, Y. Shimada, R. C. Olsthoorn, B. E. Snaar-Jagalska, H. P. Spaink and A. Kros, ACS Nano, 2016, 10, 7428–7435 CrossRef CAS PubMed.
- L. L. Schwenen, R. Hubrich, D. Milovanovic, B. Geil, J. Yang, A. Kros, R. Jahn and C. Steinem, Sci. Rep., 2015, 5, 12006 CrossRef PubMed.
- Z. Chen, J. Wang, W. Sun, E. Archibong, A. R. Kahkoska, X. Zhang, Y. Lu, F. S. Ligler, J. B. Buse and Z. Gu, Nat. Chem. Biol., 2018, 14, 86–93 CrossRef CAS PubMed.
- L. Kong, Q. Chen, F. Campbell, E. Snaar-Jagalska and A. Kros, Adv. Healthc. Mater., 2020, 9, e1901489 CrossRef PubMed.
- D. K. Struck, D. Hoekstra and R. E. Pagano, Biochemistry, 1981, 20, 4093–4099 CrossRef CAS PubMed.
- P. S. Uster, Methods Enzymol., 1993, 221, 239–246 CAS.
- G. van der Borg, S. Braddock, J. S. Blijleven, A. M. van Oijen and W. H. Roos, J. Phys.: Condens. Matter, 2018, 30, 204005 CrossRef PubMed.
- M. K. S. van Duijl-Richter, J. S. Blijleven, A. M. van Oijen and J. M. Smit, J. Gen. Virol., 2015, 96, 2122–2132 CrossRef CAS PubMed.
- M. G. Malle, P. M. G. Loffler, S. S. Bohr, M. B. Sletfjerding, N. A. Risgaard, S. B. Jensen, M. Zhang, P. Hedegard, S. Vogel and N. S. Hatzakis, Nat. Chem., 2022, 14, 558–565 CrossRef CAS PubMed.
- J. Otterstrom and A. M. van Oijen, Biochemistry, 2013, 52, 1654–1668 CrossRef CAS PubMed.
- M. Rabe, H. R. Zope and A. Kros, Langmuir, 2015, 31, 9953–9964 CrossRef CAS PubMed.
- N. L. Mora, A. L. Boyle, B. J. V. Kolck, A. Rossen, S. Pokorna, A. Koukalova, R. Sachl, M. Hof and A. Kros, Sci. Rep., 2020, 10, 3087 CrossRef CAS PubMed.
- M. Rabe, C. Schwieger, H. R. Zope, F. Versluis and A. Kros, Langmuir, 2014, 30, 7724–7735 CrossRef CAS PubMed.
- A. Koukalova, S. Pokorna, A. L. Boyle, N. Lopez Mora, A. Kros, M. Hof and R. Sachl, Nanoscale, 2018, 10, 19064–19073 RSC.
- L. G. Toscano-Flores, D. Jacinto-Mendez and M. D. Carbajal-Tinoco, J. Phys. Chem. B, 2016, 120, 5655–5661 CrossRef CAS PubMed.
- L. Tayebi, D. Vashaee and A. N. Parikh, Chem. Phys. Chem., 2012, 13, 314–322 CrossRef CAS PubMed.
- L. V. Chernomordik and M. M. Kozlov, Nat. Struct. Mol. Biol., 2008, 15, 675–683 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2cp04448j |
| This journal is © the Owner Societies 2023 |