Pyrolysis and oxidation of benzene and cyclopentadiene by NOx: a ReaxFF molecular dynamics study†
Received
22nd September 2022
, Accepted 27th March 2023
First published on 3rd April 2023
Abstract
Benzene (C6H6) and 1,3-cyclopentadiene (c-C5H6) are critical intermediate species in the combustion of fossil fuel and the formation of polycyclic aromatic hydrocarbons (PAHs). This study investigates the underlying mechanisms of pyrolysis and oxidation of C6H6 and c-C5H6 in the presence of O2, NO and NO2, respectively, under combustion conditions via ReaxFF molecular dynamics simulations. The size growth in the pyrolysis system is accompanied by an amorphous nature as well as an increase in the C/H ratio. In the oxidation sytems, NO2 is the most effective in the oxidation of both C6H6 and c-C5H6, followed by NO and O2. In the presence of NOx, O and N radicals generated in the high-temperature decomposition reactions of NO and NO2 are actively involved in the addition and H-abstraction reactions of C6H6 and c-C5H6. Remarkably, the decomposition of NO2 dramatically increases the number of O radicals in the system, which significantly accelerates the ring-opening of C6H6 and c-C5H6 by O-addition and forms linear-C6H6O and C5H6O species, respectively. Afterwards, the formation of –CH2– by H-transfer plays an essential role in the decomposition of linear–C6H6O and –C5H6O. Reaction pathways of O and N radicals with C6H6 and c-C5H6 are reported in detail. The O and N-addition of C6H6 facilitate the decomposition to resonance-stabilized cyclopentadienyl radicals after the restructuring of the C–C bond.
1. Introduction
Concerns about pollutant emissions together with the increasingly stringent emission specification worldwide have motivated research in clean combustion technologies. Soot is mainly exhausted from incomplete fuel combustion in diesel cylinders under high temperatures and pressures.1 Benzene (C6H6), as the “first aromatic ring”, is an important intermediate species in the incomplete combustion of hydrocarbon fuels,2 and it also contributed significantly to the formation of larger polycyclic aromatic hydrocarbons (PAHs) and soot.3 Recently, aromatics with a five-membered ring have drawn much attention in soot studies as PAHs with five-membered rings are observed in experiments via high-resolution atomic force microscopy.4 Meanwhile, the oxidation of six-membered aromatic rings leads to oxyradicals, which subsequently decompose into CO and a five-membered ring at the edge of PAHs such as free-edge, zigzag, and armchair. The in-depth study of C6H6 and 1,3-cyclopentadiene (c-C5H6) is instructive for extending large PAHs since they are the basic units of PAHs.5 Moreover, because of the similarity of the structures, many investigations about PAH have been performed upon monocyclic rings to reduce the computational expense. The pyrolysis of C6H6 and c-C5H6 provides valuable insights into soot formation. Kashiwa et al.6 investigated C6H6 pyrolysis and the formation of particulate matter using a flow reactor. They found that a large amount of particulate matter with a less organic solvable fraction was formed from a C6H6–N2 mixture above 1273 K. Shock tube experiments of C6H6 pyrolysis were conducted by Saggese et al.7 The results show that C6H6 pyrolysis begins with the cleavage of C–H bonds. Experimental studies by Wang et al.8 determined the reaction pathways of c-C5H6 pyrolysis. Kiefer et al.9 studied the c-C5H6 molecule and its radical using quantum simulations and estimated the thermodynamic properties. There is a general agreement that the initial step of c-C5H6 pyrolysis is C–H fission. Kern et al.10 first provided the first quantitative evidence for the H atom shift mechanism for the important secondary reaction c-C5H5 → C3H3 + C2H2. Pitsch et al.11 investigated the H-abstraction and H-addition of c-C5H6 by quantum chemistry calculations. The results indicate that H-abstraction prefers to attack the –CH2– group of c-C5H6. They also found that the reactions of c-C5H6 + H to form bimolecular products are prevalent at high temperatures and low pressures.12
Soot oxidation represents an essential and complex process in soot evolution, competing with surface growth, thus hampering particle emissions.13 High-temperature combustion takes place in the flame after PAH formation. NOx is inherently produced when the temperature exceeds 2200 K.14,15 Experimental studies indicate that NOx, especially NO2, are more effective oxidizers than O2 for soot oxidation.16,17 Oxidation and reburn types are two mechanisms responsible for the reduction of soot and NOx.18 In the actual in-cylinder combustion, the oxidation of soot by NOx inevitably occurs at the flame surface. By applying the flue gas recirculation technique, the presence of a given nitrogen oxide in the recirculated mixture can affect the emissions of soot, which could cause reburn type reactions of soot and NOx. Therefore, the coexistence of soot and NOx is worth studying. In the study of Setiabudi et al.19 by analyzing the soot combustion characteristic from room temperature to 900 K, NO2 is first involved in the soot oxidation to produce surface complexes that are reactive to O2. Shrivastava et al.20 studied the kinetics of soot oxidation by NO2 using the online aerosol technique, indicating that NO2 decomposes to NO at temperatures higher than 773.15 K. They also found that the addition of NO2 significantly reduces the activation energy of oxidation compared to the oxidation with air. Sander et al.21 investigated the reaction mechanism of the non-catalytic soot reduction by NO, which was validated using density functional theory. However, the subsequent pathway of the surface oxygen desorbing from the soot as CO was not elaborated. In the study of Abián et al.18 the effects of the presence and concentration of NO, NO2 and N2O on soot formation during ethylene pyrolysis in a quartz flow reactor have been determined. The experimental results indicate that the lowest soot tendency has been achieved in the presence of NO2, followed by NO and N2O. In addition, N radical is one of the products of the high-temperature reaction of NO.22 However, few studies have been performed on the chemistry of N radicals under conditions associated with combustion applications. In addition, the mixed combustion of ammonia and fossil fuels was proposed recently to meet the needs of ‘carbon neutrality’. Therefore, the high-temperature reaction of N with PAHs has attracted more attention. Therefore, the oxidation mechanism with NOx still needs to be theoretically studied for refinement.
In this paper, the ReaxFF MD method was utilized to investigate the reaction of C6H6 and c-C5H6 under high-temperature and pressure conditions including both the pyrolysis and the oxidation processes at the atomic level. In particular, the pyrolysis system focuses on the growth routes from reactants to large species. The oxidation by NO and NO2 emphasizes the detailed pathways of O and N radicals in the reaction. A detailed understanding of the key reactive events related to pyrolysis and oxidation of monocyclic hydrocarbon will not only facilitate our understanding of combustion chemistry but also assist in developing comprehensive kinetic models.
2. Methodology
2.1 Simulation method
ReaxFF molecular dynamics (MD) is a novel simulation method that retains nearly the accuracy of quantum mechanics but allows MD for computational costs nearly as low as for classical force fields. ReaxFF MD can simulate large-scale systems with complex chemical reactions, such as pyrolysis and combustion of hydrocarbons.23
The ReaxFF MD is a general bond-order dependent potential method that can smoothly describe the bond breakage and formation among molecules in a system. The connectivity-dependent terms containing bond, angle and torsion are calculated from interatomic distances updated every MD step. Non-bonded interactions, including van der Waals and Coulomb, are calculated between every pair of atoms as they are independent of the bonded interactions. A geometry-dependent charge calculation scheme and Electronegative Equalization Method (EEM) are used to calculate atomic charges. ReaxFF force field includes the following energy components:
| | | Esystem = Ebond + Eover + Elp + Eval + EvdWaals + ECoulomb | (1) |
where terms on the right-hand side of the equation represent bond energy, over-coordination energy penalty, under-coordination stability, lone pair energy, valence angle energy, torsion angle energy, van der Waals energy, and Coulomb energy, respectively. A more detailed description of the ReaxFF force field can be found in the work by van Duin
et al.
24
2.2 Computational details
The ReaxFF MD simulation is carried out with the REAXC package in the Large-scale Atomic/Molecular Massively Parallel Simulation (LAMMPS).25 The CHON-2019 reactive force field developed by Kowalik et al.26 is utilized for MD simulations in the current work. The bond/angle/dihedral parameters that involve any N atoms are trained against available experimental and quantum mechanics-based density functional theory data, which were validated in our previous work27 and in the ESI.† The setups of the ReaxFF MD simulations of C6H6 and c-C5H6 are shown in Tables 1 and 2, respectively. There are 100 reactants in the pyrolysis systems. The system of C6H6 + O2 contains 100 C6H6 and 375 O2 molecules with an initial equivalence ratio of 2. Similarly, 100 c-C5H6 and 325 O2 are set in the c-C5H6 + O2 system. To investigate the effect of NOx, O2 is replaced by the same number of NO and NO2. Ar is added as the bath gas to maintain the same initial mole fraction of C6H6 or c-C5H6 in pyrolysis and oxidation systems. The interaction between Ar and other species is via the van der Waals interaction. The periodic boundary conditions were applied in three directions. The bond order cutoff is set to be 0.2, which is related to species analysis and does not affect the actual simulation. Firstly, energy minimization is performed for every system using a conjugate gradient algorithm to eliminate the artificial effects on the initial geometric configuration. The MD simulations are carried out through the isothermal–isobaric ensemble (NPT) with constant atom number, pressure, and temperature. NPT-MD simulations are performed at 50 atm, which on the one hand is high enough to ensure reactions occur at the ReaxFF MD simulation timescale,28 and on the other hand, is of practical interest for high-pressure combustion engines. The system temperature is controlled by the Nosé–Hoover thermostat with a damping constant of 50 fs. A Nose–Hoover barostat is used to relax the pressure with a damping constant of 1000 timesteps. The starting configurations are equilibrated via low-temperature (300 K) ReaxFF simulations (time step of 0.1 fs for 200 ps) to prevent chemical reactions from occurring during equilibration. Then, the MD simulations are carried out for 5000 ps with a time step of 0.2 fs.28 Note that each case is calculated three times with the same starting configurations.
Table 1 Specific simulation conditions of C6H6 systems
| System |
Molecules |
Temperature (K) |
Pressure (atm) |
| C6H6 |
100C6H6 + 750Ar |
2000, 2500 |
50 |
| C6H6 + O2 |
100C6H6 + 375O2 + 375 Ar |
| C6H6 + NO |
100C6H6 + 375NO + 375Ar |
| C6H6 + NO2 |
100C6H6 + 375NO2 + 375Ar |
Table 2 Specific simulation conditions of c-C5H6 systems
| System |
Molecules |
Temperature (K) |
Pressure (atm) |
| c-C5H6 |
100c-C5H6 + 750Ar |
2000, 2500 |
50 |
| c-C5H6 + O2 |
100c-C5H6 + 325O2 + 425 Ar |
| c-C5H6 + NO |
100c-C5H6 + 325NO + 425Ar |
| c-C5H6 + NO2 |
100c-C5H6 + 325NO2 + 425Ar |
3. Results and discussion
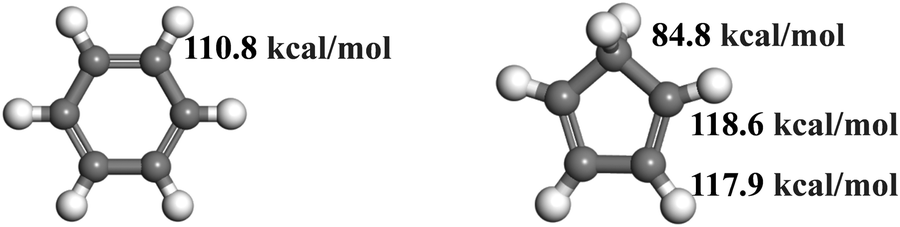
Firstly, a global analysis of the pyrolysis and oxidation of (a) C6H6 and (b) c-C5H6 is conducted. Fig. 1 shows the time evolution of the molecular number of C6H6 and c-C5H6 under the pyrolysis and oxidation conditions with different oxidizers (O2, NO, NO2) at 2000 K and 2500 K, respectively. Compared with the results at 2000 K, the number of the reactant decreases quicker at 2500 K. It is even significant for C6H6 as it is difficult to react at 2000 K under both the pyrolysis and the oxidation by O2 conditions, whereas c-C5H6 is relatively easier to react at this temperature. In addition, at both 2000 K and 2500 K, systems containing C6H6 react more slowly than c-C5H6 with the same oxidizer. These phenomena can be explained by the lower C–H bond dissociation energy (BDEs) of c-C5H6 than that of C6H6. The molecular structures and C–H BDEs of C6H6 and c-C5H6 are shown in Fig. 2. The c-C5H6 molecule has a symmetry number of two, and three different types of C–H bonds. The C–H bond dissociation energy of –CH2– in c-C5H6 is 84.8 kcal mol−1,12 about 28.2 kcal mol−1 lower than the C–H bond dissociation energy of C6H6.29 The effects of different oxidizers on the five- and six-membered rings have similar trends. O2 is the weakest in the oxidation of C6H6 and c-C5H6 compared to NO and NO2. NO2 is the most significant for the oxidation of both c-C5H6 and C6H6. However, the rate of elimination of c-C5H6 by NO essentially overlaps with that of NO2, which is caused by the superior H-abstraction by NO on –CH2–. NO is abundant at the beginning in the system of c-C5H6 + NO. Moreover, NO prefers to react with c-C5H6 by H-abstractions instead of direct decomposition, which is significantly different from NO2 as it is hard to be involved in the reaction directly. In the system of c-C5H6 + NO2, NO2 is first decomposed to NO and O. The detailed mechanisms of pyrolysis and oxidation by different oxidizers will be discussed later.
 |
| | Fig. 1 Time evolution of the number of C6H6 and C5H6 molecules under different conditions at (a) 2000 K and (b) 2500 K, respectively. | |
 |
| | Fig. 2 Molecular structures and C–H BDEs of C6H612 and c-C5H6.29 | |
3.1 Pyrolysis of C6H6 and c-C5H6
The first-step reaction of monocyclic hydrocarbons is of great significance to understanding and controlling PAH growth. Fig. 3 shows the proportion of different reaction types that trigger the first-step reaction of C6H6 (blue columns) and c-C5H6 (red columns) in the pyrolysis systems. The upper and lower parts represent the ratio of forward and reverse reactions to the total net reaction. 35% of c-C5H6 molecules start by breaking the C–H bonds in –CH2–, which is 30.4% higher than the percentage of dehydrogenation of C6H6, attributed to the weaker C–H bond in –CH2– of c-C5H6. The C–C bond cleavage is observed during the pyrolysis of c-C5H6. However, the delocalized π bond of C6H6 is difficult to destroy directly, which is much stronger than the C–C bond in c-C5H6.7,30 One pathway for C6H6 pyrolysis begins with the C–H bond breaking. Among them, the probability of the H-addition reaction is slightly higher than that of H-abstraction. According to the potential energy surfaces, the energy barrier for the H-addition of C6H6 is 8.9 kcal mol−1,31 while that of the H-abstraction by H is 16 kcal mol−1.32 Another vital pathway of C6H6 pyrolysis is the addition of CxHy species generated by the decomposition of C6H6.
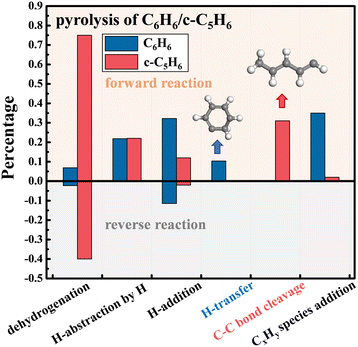 |
| | Fig. 3 Proportion of different first-step reactions of C6H6 (blue columns) and c-C5H6 (red columns) in the pyrolysis systems at 2500 K. | |
As shown in Fig. 4 and 5, the time evolution of the number of species with different carbon numbers was monitored to understand the soot formation. C# represents the number of all species containing carbon atoms of #. For C6H6 pyrolysis, the H-transfer of C6H6 is first observed at 705.6 ps, as shown in Fig. 4(f). Then, C6H6 is dehydrogenated at 876.4 ps. The hydrogen-related reactions including dehydrogenation, H-abstraction and H-addition have a lower percentage in the pyrolysis of C6H6, as reflected by the consistently low amount of C6 (except C6H6). C6 species are mainly decomposed into C2 and C4, as shown in the shaded boxes in Fig. 4(b) and (d), with C2 and C4 growing simultaneously from 1500 ps to 3500 ps, which provide resources for species growth. C7 and heavier species are formed due to the successive CxHy addition after 1463.6 ps. The largest species, C22H6, is observed at 4792.4 ps. Fig. 5 shows that the pyrolysis of c-C5H6 starts with dehydrogenation at 144.0 ps. The C–C bond cleavage of c-C5H6 occurs almost simultaneously with dehydrogenation. Note that the pyrolysis of c-C5H6 begins much earlier than C6H6 because of the weaker C–H bond in c-C5H6. Since dehydrogenation accounts for a large proportion of the first-step reaction, the conversion between C5H6 and C5 (except C5H6) is frequent, as shown in Fig. 5(e). The production of C2 and C3 by the C–C bond fission of C5 is the most critical decomposition pathway, as illustrated by the rapid growth from 500 ps to 2500 ps by shaded boxes in Fig. 5(b) and (c). C6–C9 species are first observed at 1200.0 ps. C21H3, formed at 4826.8 ps, is the largest species in the pyrolysis system of c-C5H6. Notably, the pyrolysis of C6H6 produces significantly more C10–19 species than that in the pyrolysis system of c-C5H6, indicating that the addition reaction of graphitization in C6H6 pyrolysis is preferred. The study of Ossler et al.33,34 has demonstrated that high-temperature combustion generates more curved, more amorphous and less ordered structures. Notably, the carbon-to-hydrogen (C/H) ratio shows a considerable difference between the largest species (C22H6/C21H3) and reactants (C6H6/C6H5), indicating that the size growth is accompanied by an increase in mass as well as an increase in the C/H ratio.35,36
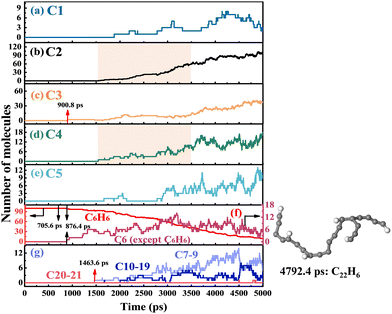 |
| | Fig. 4 Time evolution of the number of species with different carbon numbers in the system of C6H6 pyrolysis. In (a)–(g), C# represents the number of all species containing carbon atoms of #. | |
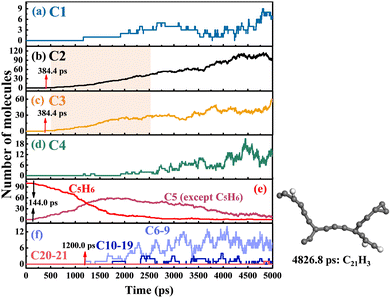 |
| | Fig. 5 Time evolution of the number of species with different carbon numbers in the system of c-C5H6 pyrolysis. In (a)–(f), C# represents the number of all species containing carbon atoms of #. | |
The ring opening and decomposition of C6H6 and c-C5H6 at the atomic level are essential for understanding the growth mechanism of PAHs. Fig. 6 shows the reaction pathway of pyrolysis of C6H6 at 2500 K. The number of reactions is indicated in red. The addition of CxHy species to C6H6 plays a vital role in the initial step of C6H6 pyrolysis. C2 species, especially C2H and C2, participate in the growth of aromatics. C4 species are also frequently involved in the addition of C6H6, which further verified that C2 and C4 species are the main decomposition products of C6H6. Four elementary reactions related to hydrogen are proposed for the initial step of C6H6: H-addition, H-transfer, H-abstraction and dehydrogenation, forming C6H7 (S1), isomer-C6H6 (S4) and C6H5 (S5), respectively. However, subsequent reactions of S1 have not been explored in-depth in previous studies. After isomerization to methylcyclopentadienyl radicals (S2), three of them react to fulvene by breaking one of the C–H bonds of the methyl (–CH3) group and then isomerize to S3, as shown by pathway (1). In addition, we have found that an S2 radical experiences bond cleavage to form methyl radicals and c-C5H4 (pathway (2)). Pathway (3) shows that five S5 species are directly converted to linear-C6H5via C–C β-scission. At last, C–C β-scission after H-transfer is also a vital route of destroying the ring structure of S1 (pathway (4)).
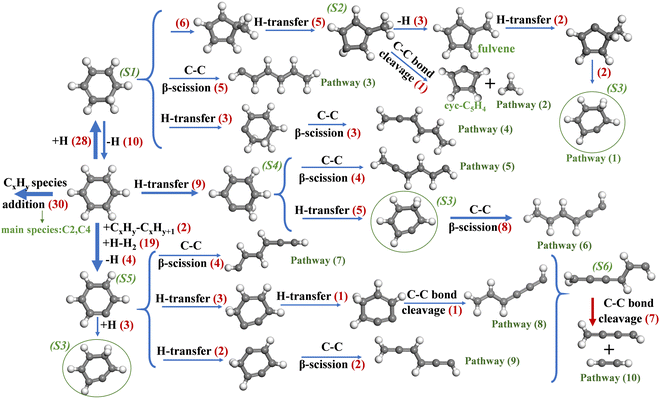 |
| | Fig. 6 Major reaction pathways of C6H6 pyrolysis at 2500 K. The number of reactions is indicated in red. | |
Pyrolysis at extremely high pressures and temperatures probably leads to the emergence of reactions rarely observed at medium and low temperatures, which should be considered. H-transfer of C6H6 to form S4 greatly facilitates the ring opening of C6H6 in the present work. Pathways (5) and (6) show the C–C β-scission of two types of C6H6 isomers (S3 and S4), respectively. C6H6 is converted to phenyl radicals (S5) via dehydrogenation and H-abstraction.7 Species that trigger H-abstraction include H radicals and CxHy, such as C2H and C2. One of the first steps of c-C6H5 conversion is the C–C bond breaking. As shown by Pathways (7), c-C6H5 isomerizes to a linear open-chain species, which agrees with the calculated results by Madden37 and experimental results.38,39 Pathways (8) and (9) show the ring opening after H-transfer to form linear-C6H5 radicals. The way in which H-transfer leads to the ring opening of c-C6H5 has not been found in previous studies, which should be of concern in high-temperature reactions. Linear-C6H5 generated by Pathways 7–9 undergoes isomerization to form S6 and eventually decomposes into C4H3 and C2H2, which agrees with previous studies.7,40
Fig. 7 shows the main pathways of c-C5H6 pyrolysis. The carbon in –CH2– of c-C5H6 is defined as carbon (a). The ortho- and para-positions of carbon (a) are defined as carbon (b) and carbon (c), respectively. The elimination of hydrogen from carbon (a) to form cyclopentadienyl accounts for the largest proportion. Meanwhile, a great number of cyclopentadienyl radicals are produced by H-abstraction via H radicals. However, the abstraction of hydrogen from carbon (c) to produce S8 plays a minor role. The difference in probability between the two abstraction routes is attributed to the low barrier height of the H-abstraction at carbon (a), as calculated by the theoretical study by Mao et al.12 Further H-abstraction of cyclopentadienyl radicals results in the formation of c-C5H4. The unsaturation degree rises with hydrogen removal, promoting the addition reactions. Carbon not connected to hydrogen in c-C5H4 is denoted as carbon (d). Except for the H-addition to carbon (d) to regenerate cyclopentadienyl radicals, few H-addition reactions take place at ortho- and para-positions to carbon (d), forming S7 and S8, respectively. As shown by pathway (11), S7 is converted into linear C5H5 radicals via C–C β-scission, which agrees well with the computational result obtained by Roy et al.41 S8 experiences ring-opening via C–C β-scission, as presented by pathway (12), which has however been neglected in the previous studies.
 |
| | Fig. 7 Major reaction pathways of c-C5H6 pyrolysis at 2500 K. | |
The decomposition of c-C5H6 to C2 and C3 species follows pathways (13) and (14). Pathway (13) shows that c-C5H6 isomerizes by C–C bond cleavage and then experiences H-transfer to form a –CH2 group in 1,2,4-pentatriene, which eventually decomposes to C3H3 and C2H3. Pathway (13) is crucial for the accurate prediction of c-C5H6 consumption at 2500 K, which however has been ignored in the previous studies. Pathway (14) begins with the H-addition to carbon (b) to form S10, followed by isomerization via C–C β-scission and H-transfer, and eventually decomposition to C2H3 and C3H4. Notably, a small number of H radicals is added to the carbon (c) of c-C5H6 to form S9. After that, all S9 species isomerize to S10 by H-transfer as the resonance-stabilized S10 is more stable.12
3.2 Oxidation of C6H6 and c-C5H6 by NOx
Oxygen is one of the most significant oxidizers in combustion. Oxidation of soot by NOx is more efficient than O2, obtained by the global analysis from Fig. 1. In the presence of O2, O2 is rarely involved in the oxidation of C6H6 or c-C5H6 directly. The oxidation rate is limited by the branching reaction: O2 + H = O + OH. More details of C6H6 and c-C5H6 oxidation by O2 are shown in the ESI.† In the presence of NO, the largest species in C6H6 and c-C5H6 systems are C12H10O and C9H5N2, respectively. Furthermore, C7H6O2N2 and C7H7O are the largest species observed in the systems of C6H6 + NO2 and c-C5H6 + NO2, indicating that NOx, especially NO2, effectively inhibits the growth of soot precursors. Fig. 8 shows the time evolution of the main species in the pure NO and pure NO2 systems. The pure NO system has 375 NO molecules and 475 Ar atoms. Similarly, 375 NO2 and 475 Ar are involved in the pure NO2 system. Both systems are equilibrated at the 300 K for 200 ps, which subsequently performed high-temperature simulation at 2500 K for 5000 ps. Details of the settings are the same as for C6H6 + NO/C6H6 + NO2 in section 2.2. Fig. 9 shows the main species in the reaction of C6H6/c-C5H6 with NO and NO2, respectively. The arrows on the right side present the number of O, N2, NO and N in the pure NO/NO2 system. The oxidation of C6H6 and c-C5H6 by NOx is a chain reaction initiated at high temperatures. The mechanism of C6H6 oxidized by NO is as follows:Once initiated, the chain propagation proceeds:
 |
| | Fig. 8 Time evolution of the number of key species in the (a) pure NO system and (b) pure NO2 system at 2500 K. | |
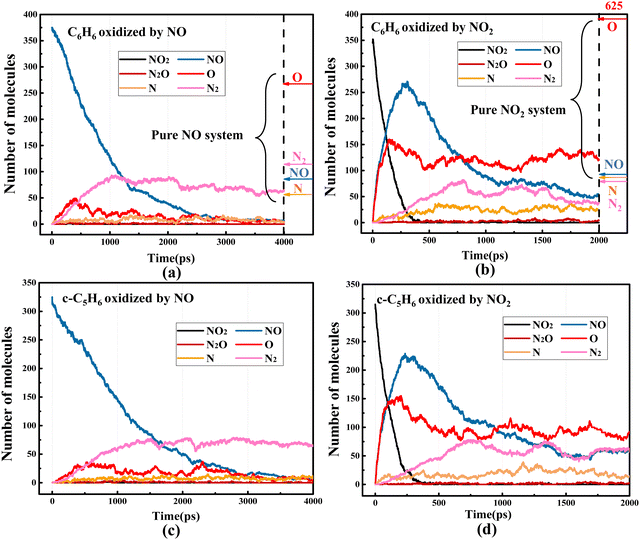 |
| | Fig. 9 Time evolution of the number of key species in the system of (a) C6H6 and NO, (b) C6H6 and NO2, (c) c-C5H6 and NO, and (d) c-C5H6 and NO2 at 2500 K. The arrows on the right side of (a) and (b) present the number of O, N2, NO and N in the pure NO and pure NO2 systems, respectively. | |
The production of N2 in (R2) is consistent with that observed in the experiment.42 Notably, the consumption of NO in the presence of C6H6 and C5H6 is slower than that in the pure NO system. This is because a portion of N radicals produced by (R1) is involved in the reaction with CxHy species. Therefore, (R2) reacts slowly due to the reduction of N radicals.
In the presence of NO2, the main reaction route of NO2 acts as a rich source producing O radicals via:
Subsequent NO reactions are the same as (R1) and (R2). NO can also be formed by the reaction of NO2 and H radicals:
which provides OH radicals in the system. Some OH radicals react with H radicals and N radicals:
In addition, OH experiences OH addition and H-abstraction by OH with CxHy species, including:
| | | C3H4 + HO = H2O + C3H3 | (R12) |
The mechanism of reaction at high temperatures is dominated by OH addition with H-abstraction being a minor contributor.43 The OH radicals can also provide O for CxHy species:
In the system of C6H6 + NO2 (Fig. 9(b)), the number of O and N radicals is lower than those in the pure NO2 system. In particular, the reduction of O radicals is more noticeable than that in the pure NO2 system, with a difference of approximately 500. Similarly, in the system where c-C5H6 reacts with NO or NO2, a major role is played by O and N radicals in the c-C5H6 oxidation.
The increase in O radicals and N radicals significantly altered the types and distribution of the initial reaction. Fig. 10(a) shows the proportion of different reaction types that trigger the first-step reaction of C6H6 and c-C5H6 in the presence of NO. The percentage of reactions that take place in the pyrolysis system drops drastically. Notably, as seen from the red column, NO can abstract hydrogen from the –CH2– of c-C5H6, with a percentage of about 65%. Xu et al.44 found that NO is effective for H-abstraction of HCO. For the more stable C–H bond in –CH2– of c-C5H6, NO still has an excellent ability to abstract hydrogen. However, for the C–H bond in C6H6, H-abstraction by NO is not dominant, which only accounts for about 4.4%. In the system of C6H6 + NO, more than half of the C6H6 molecules are activated by O and N radicals. Among them, reaction with O radicals accounts for more than 30%, which accounts for the highest percentage of all initial steps. O and N radicals participate in the reaction via H-abstraction and addition, represented by shaded and non-shaded parts, respectively. The probability of O-addition reactions is much higher than H-abstraction by O for both reactants of C6H6 and c-C5H6. However, nearly half of the reactions with N are achieved by H-abstraction in both systems. It is also worth noting that c-C5H6 is more likely to undergo H-abstraction by O or N radicals than C6H6. In the presence of NO2 shown in Fig. 10(b), reactions in the pyrolysis system account for less than fifteen percent. Abundant O radicals greatly increase the probability of reaction with O radicals in both systems of C6H6 and c-C5H6, in proportions of 63.8% and 47.6%, respectively. NO2 is rarely involved in the reaction with C6H6/c-C5H6 directly. NO, O and N produced during the decomposition of NO2 are actively involved in the first-step reaction. NO has a strong capacity for H-abstraction. However, O and N radicals, especially O radicals, prefer to participate in reactions via addition.
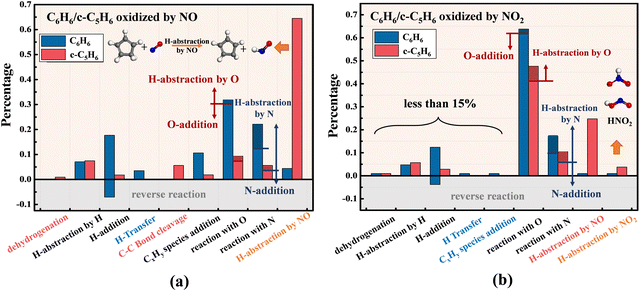 |
| | Fig. 10 Proportion of different first-step reactions of C6H6 (blue columns) and c-C5H6 (red columns) in the presence of (a) NO and (b) NO2 at 2500 K. | |
The presence of NOx greatly increases the number of O radicals in the system, which significantly accelerates PAH oxidation. Therefore, elucidating the reaction pathways of the elementary C6H6/C5H6 + O reaction at high temperature and pressure is of great importance to understanding the reaction mechanisms of hydrocarbon flames as well as the optimization of combustion processes. Fig. 11 shows the reaction pathway for C6H6 + O in the system where C6H6 is oxidized by NO2 since this system has the largest proportion of reactions with O radicals. After connecting to the C atom on C6H6 to form C6H6O (S11), six subsequent reaction pathways are observed as depicted by blue arrows, where bold arrows indicate dominant reaction routes. The H-elimination after introducing O radical into C6H6 is one of the important pathways (C6H6 → S12).45 The H radical then connects to O atoms of S12, forming a hydroxyl group (S12 → phenol), which is eventually abstracted by H radicals to produce H2O and phenyl. The formation of phenol by H-transfer from S11 has also been found with a small probability. The isomerization of S11 via H-transfer between carbon atoms contributes to the ring-opening, as presented by pathway (15). The most important decomposition channel of S11 is to form linear-C6H6O (S15) via C–C β-scission. S15 experiences successive H-transfer, leading to the formation of the –CH2– group in S17, which ultimately decomposes to C5H6 and CO, as shown by pathway (16).
| | | C6H6 + O = C5H6 + CO | (R14) |
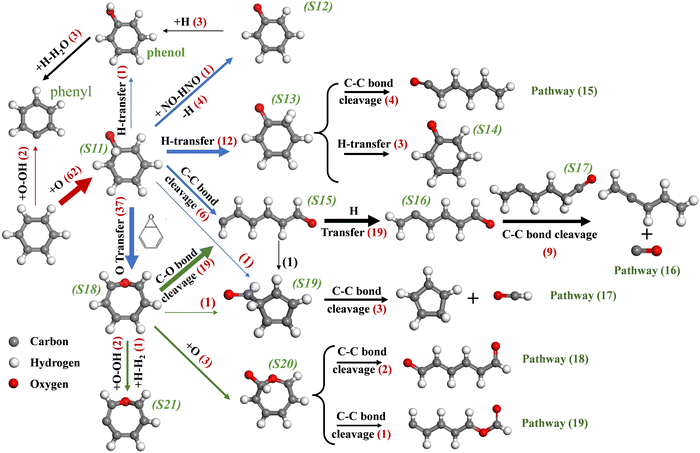 |
| | Fig. 11 Major reaction pathways of C6H6 and O radicals in the system of C6H6 and NO2. | |
The conversion of S11 to a stable species (S18) was found in abundance. The energy of forming S18 is 11.3 kcal mol−1 lower than S11, as calculated in the study of Nguyen et al.46 The bond cleavage of S18 is another key source of S15, followed by pathway (16). The isomerization of S11, S15 and S18 to S19 is found once each. After that, S19 experiences a decomposition reaction to the resonance-stabilized cyclopentadienyl radical and CHO, which agrees with the theoretical results of Hodgson et al.45
| | | C6H6 + O = C5H5 + HCO | (R15) |
NO2 produce large amounts of O radicals at high temperature, which keeps the system at a high concentration of O radicals. Therefore, the continued attack on C6H6O by O radicals is inevitable, which however has rarely been focused on in the literature. For S18, the addition of O radicals on the carbon that connected to the initial oxygen atom is observed. After that, S20 isomerizes to form linear-C6H6O2via two routes, as shown by pathways (18) and (19). At last, the H-abstraction of S18 by O and H radicals to form S21 is found.
In the system of c-C5H6 + NO2, about 47% of the c-C5H6 molecules experience O-addition reactions as the initial step. Fig. 12 shows the reaction pathways of c-C5H6 and O radicals. As indicated by the red arrows, O radicals attack carbon (b) or carbon (c) of c-C5H6 to generate C5H6O. O-addition on carbon (b) is more important. Both S22 and S23 are found to form S24. Pathway (20) shows the ring-opening of c-C5H6O (S22) via C–C β-scission. As shown by the blue arrows, both S23 and S24 can proceed with bond cleavage, forming a linear species including S26 and S31. S26 is the main product of ring-opening, which experience H-transfer to S27 with a –CH2– group. In addition, after the H-transfer of S23, S25 undergoes bond cleavage to form S27, which is also an important pathway for generating S27. After that, S27 decomposes into C4H6 and CO.
| | | C5H6 + O = C4H6 + CO | (R16) |
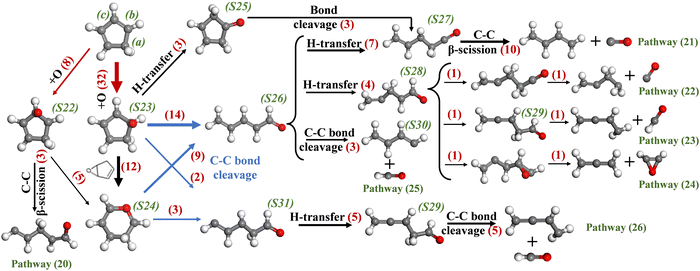 |
| | Fig. 12 Major reaction pathways of c-C5H6 and O radicals in the system of c-C5H6 and NO2. | |
The decomposition route via S23 → S26 → S27 → C4H6 + CO is important for C5H6O consumption, which is in great agreement with the theoretical results by Robinson.47 For the oxidation of C6H6 and C5H6 by O radicals, the key reactions that produce CO are the transfer of the hydrogen from the carbon connected with oxygen to the adjacent carbon, forming –CH2–C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O. After that, CO is removed from the linear C6H6O or C5H6O. In addition, –CH2– formed by successive H-transfer drives the decomposition reaction, as shown by pathways (22), (23) and (24), which produce CO, CHO and C2H2O, respectively. S31 is another isomer of linear-C5H6O after ring-opening, which experiences H-transfer and decomposes to C4H5 and CHO.
O. After that, CO is removed from the linear C6H6O or C5H6O. In addition, –CH2– formed by successive H-transfer drives the decomposition reaction, as shown by pathways (22), (23) and (24), which produce CO, CHO and C2H2O, respectively. S31 is another isomer of linear-C5H6O after ring-opening, which experiences H-transfer and decomposes to C4H5 and CHO.
| | | C5H6 + O = C4H5 + CHO | (R17) |
Among them, (H)CO has an inhibitory effect on soot formation, as it ties up carbon atoms, limiting their involvement in the soot formation processes.
As seen from the decomposition of C6H6O and C5H6O, the formation of –CH2– plays an important role in the fragmentation of linear species. The C–C bond between –CH2– and the carbon attached to the oxygen is easily broken. Notably, linear-C5H6O decomposes more readily than linear-C6H6O, which is probably because the high H/C ratio is more likely to form the –CH2–.
The N radical is one of the key products of the high-temperature reaction of NOx. Fig. 13 shows the reaction pathway of C6H6 with N radicals in the system of C6H6 + NO since C6H6 has the highest proportion of initial reactions with N radicals compared to other systems. S32 is formed after the N-addition of C6H6. The study of Balucani et al.48 shows that the N-addition of C6H6 proceeds through the formation of a van der Waals complex (T1), which is barrierless. Then, T1 isomerizes to form S32 through the nitrogen bridging of two carbon atoms of the ring, with a low barrier of 0.7 kcal mol−1.49 The rapid reaction leads to difficulty in the observation of T1. S32 can evolve in two different ways. Firstly, it can undergo C–C bond breaking to form the seven-membered c-S33, where a nitrogen atom is inserted into the C–C bond of C6H6. Secondly, the hydrogen of S32 transfers from carbon to the nitrogen atom, then anilino radical (S34) is produced by the cleavage of the HC–NH bond. Notably, the hydrogen on the carbon attached to the nitrogen tends to transfer to the nitrogen to form –CNH, which is also observed after the N-addition reaction of c-C5H6, which will be discussed later. The route of C6H6 → S32 → S34 is the most important pathway for N-addition reactions of C6H6, and S34 is the most stable isomer of C6H6N.48 S34 species isomerize to S35, which subsequently lose an NH2 group, and the following reaction is proposed:
| | | C6H6 + N = C6H4 + NH2 | (R18) |
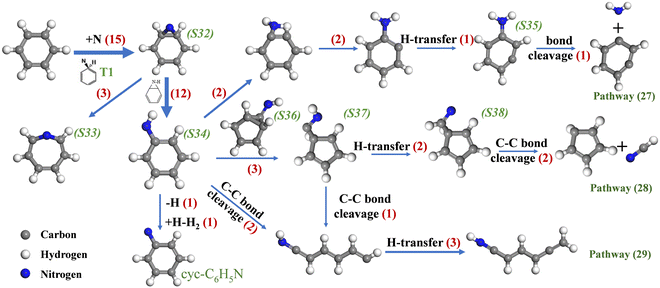 |
| | Fig. 13 Major reaction pathways of C6H6 and N radicals in the system of C6H6 and NO. | |
Pathway (28) shows the main decomposition reaction of C6H6N (S34):
| | | C6H6 + N = C5H5 + HCN | (R19) |
S34 isomerizes to S36 via C–C ring-closure and then S37 is produced from the bicyclic intermediate (S36) through C–C cleavage. After that, S37 experiences H-transfer to form S38 and then decomposes to c-C5H5 and HCN, which have been proven to be reasonable with a low energy barrier height.49 Overall, O and N radicals can react with C6H6 and form the resonance-stabilized cyclopentadienyl radical. Notably, the ring-opening reaction of C6H6 assisted by N radicals has not been found at low temperatures. In the present work, pathway (29) shows the route of C6H6N isomerization to linear-C6H6N in high-temperature systems for the first time. In addition, H-elimination of S34 forming cyc-C6H5N is observed in the system.
The reaction of N radicals with c-C5H6 has received less attention in previous studies, which however has important implications for assisting in developing comprehensive kinetic models. The pathways in the system of c-C5H6 + NO and c-C5H6 + NO2 are investigated as indicated by blue and black, respectively, as shown in Fig. 14. The red arrows indicate that the reaction is found in both systems. Attacking carbon (b) and carbon (c) of c-C5H6 are two main routes for N-addition of c-C5H6, which generate S39 and S42, respectively. After that, S39 produces S40 and S43 sequentially via successive H-transfer and finally experiences the cleavage of the C–C bond as shown by pathway (31). In addition, S41 forms via C–N ring-closure of S40 in the system of c-C5H6 and NO, which finally breaks the C–C bond to a linear-C5H6N. Three reaction routes are observed for S42. Among them, C–C β-scission of S42, shown by pathway (32), is observed in both the C5H6 +NO and C5H6 +NO2 systems. In the system of c-C5H6 and NO2, the hydrogen atom of S42 shifts from carbon to the nitrogen atom to form the –NH group of S43, which subsequently experiences ring-opening as mentioned above. The third reaction route of S42 is found in the system of c-C5H6 and NO. As shown by pathway (33), the isomer (S44) is formed via C–N bond fusion of S42, which then isomerizes to linear-C5H6N via C–C bond cleavage. In summary, both C6H6 and c-C5H6 take the ring-opening and fragmentation with the assistance of N radicals.
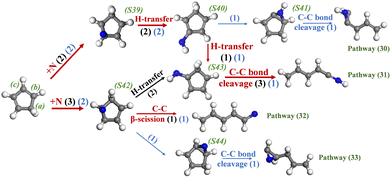 |
| | Fig. 14 Major reaction pathways of c-C5H6 and N radicals in the system of c-C5H6 and NO (blue arrows) and system of c-C5H6 and NO2 (black arrows). The red arrows indicate that the reaction occurs in both systems. | |
4. Conclusion
In this study, ReaxFF MD simulations are performed to investigate both the pyrolysis and oxidation of C6H6 and c-C5H6 at high temperatures and pressures. The pyrolysis of c-C5H6 mainly starts with the dehydrogenation in the methylene (–CH2–) group. However, C6H6 prefers to undergo addition reactions with H radicals and CxHy species. C22H6 and C21H3 are the largest species in the pyrolysis system of C6H6 and c-C5H6. The size growth is accompanied by an increase in mass as well as an increase in the C/H ratio. Among the three oxidizers of O2, NO and NO2, NO2 is the most effective in aromatic elimination, followed by NO and O2. The chain-branching reaction of H + O2 = O + OH leads to the formation of O radicals in systems with O2 as the oxidizer. The presence of NOx effectively inhibits the growth of the species due to the formation of abundant O and N radicals in the high-temperature decomposition of NOx by reactions: NO2 = NO + O, NO = N + O, and N + NO = N2 + O. Note that NO shows a remarkable ability to abstract hydrogen atoms from the –CH2– group of c-C5H6. However, H-abstraction by NO on C6H6 is not dominant. The reaction pathways of O and N radicals with C6H6 and c-C5H6 have been studied. The addition of O radicals to c-C5H6 is more likely to occur on the carbon adjacent to –CH2–. Addition reactions of O and N radicals lead to the ring-opening of C6H6 and c-C5H6, and the formation of –CH2– by H-transfer promotes the decomposition of linear-C6H6O and C5H6O. Anilino radical is the main product of N-addition to C6H6. N radicals are added at both the ortho- and para-carbon sites of –CH2– of c-C5H6. Species containing nitrogen readily form –CNH, and the addition of N radicals assists the ring-opening. Notably, the addition of O and N radicals to C6H6 also assists in their decomposition to the resonance-stabilized cyclopentadienyl radical and HCO/HCN. Overall, novel insights into the key reactive events related to the pyrolysis and oxidation of C6H6 and c-C5H6 are obtained, which could improve comprehensive kinetic mechanisms and soot reduction strategies.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (Grant No. U2233201) and the Ministry of Science and Technology, China (2021YFA 0716200/2022YFB 4003900). Qian Mao acknowledges the research fellowship from Alexander von Humboldt Foundation.
References
- B. Frank, R. Schlögl and D. S. Su, Environ. Sci. Technol., 2013, 47, 3026–3027 CrossRef CAS PubMed.
- J. D. Kubicki, Environ. Sci. Technol., 2006, 40, 2298–2303 CrossRef CAS PubMed.
- A. Drakon, A. Eremin, M. Korshunova and E. Mikheyeva, Combust. Flame, 2021, 232, 111548 CrossRef CAS.
- M. Commodo, K. Kaiser, G. De Falco, P. Minutolo, F. Schulz, A. D'Anna and L. Gross, Combust. Flame, 2019, 205, 154–164 CrossRef CAS.
- M. Frenklach and A. M. Mebel, Phys. Chem. Chem. Phys., 2020, 22, 5314–5331 RSC.
- K. Kashiwa, T. Kitahara, M. Arai and Y. Kobayashi, Fuel, 2018, 230, 185–193 CrossRef CAS.
- C. Saggese, A. Frassoldati, A. Cuoci, T. Faravelli and E. Ranzi, Combust. Flame, 2013, 160, 1168–1190 CrossRef CAS.
- H. Wang, Z. Liu, S. Gong, Y. Liu, L. Wang, X. Zhang and G. Liu, Combust. Flame, 2020, 212, 189–204 CrossRef CAS.
- J. H. Kiefer, R. S. Tranter, H. Wang and A. F. Wagner, Int. J. Chem. Kinet., 2001, 33, 834–845 CrossRef CAS.
- R. D. Kern, Q. Zhang, J. Yao, B. S. Jursic, R. S. Tranter, M. A. Greybill and J. H. Kiefer, Symp. (Int.) Combust., [Proc.], 1998, 27, 143–150 CrossRef.
- M. Baroncelli, Q. Mao, S. Galle, N. Hansen and H. Pitsch, Phys. Chem. Chem. Phys., 2020, 22, 4699–4714 RSC.
- Q. Mao, L. Cai and H. Pitsch, Combust. Flame, 2020, 222, 423–433 CrossRef CAS.
- C. Qiu, A. F. Khalizov and R. Zhang, Environ. Sci. Technol., 2012, 46, 9464–9472 CrossRef CAS PubMed.
- F. Arens, L. Gutzwiller, U. Baltensperger, H. W. Gäggeler and M. Ammann, Environ. Sci. Technol., 2001, 35, 2191–2199 CrossRef CAS PubMed.
- A. K. Agarwal, A. P. Singh and R. K. Maurya, Prog. Energy Combust. Sci., 2017, 61, 1–56 CrossRef.
- I. P. Kandylas, O. A. Haralampous and G. C. Koltsakis, Ind. Eng. Chem. Res., 2002, 41, 5372–5384 CrossRef CAS.
- M. Jeguirim, V. Tschamber, J. F. Brilhac and P. Ehrburger, J. Anal. Appl. Pyrolysis, 2004, 72, 171–181 CrossRef CAS.
- M. Abián, E. Peribáñez, Á. Millera, R. Bilbao and M. U. Alzueta, Combust. Flame, 2014, 161, 280–287 CrossRef.
- A. Setiabudi, M. Makkee and J. A. Moulijn, Appl. Catal., B, 2004, 50, 185–194 CrossRef CAS.
- M. Shrivastava, A. Nguyen, Z. Zheng, H.-W. Wu and H. S. Jung, Environ. Sci. Technol., 2010, 44, 4796–4801 CrossRef CAS PubMed.
- M. Sander, A. Raj, O. Inderwildi, M. Kraft, S. Kureti and H. Bockhorn, Carbon, 2009, 47, 866–875 CrossRef CAS.
- K. L. Wray and J. D. Teare, J. Chem. Phys., 1962, 36, 2582–2596 CrossRef CAS.
- Q. Zhong, Q. Mao, J. Xiao, A. C. T. van Duin and J. P. Mathews, Combust. Flame, 2018, 198, 146–157 CrossRef CAS.
- A. C. Van Duin, S. Dasgupta, F. Lorant and W. A. Goddard, J. Phys. Chem. A, 2001, 105, 9396–9409 CrossRef CAS.
- H. M. Aktulga, J. C. Fogarty, S. A. Pandit and A. Y. Grama, Parallel Comput., 2012, 38, 245–259 CrossRef.
- M. Kowalik, C. Ashraf, B. Damirchi, D. Akbarian, S. Rajabpour and A. C. Van Duin, J. Phys. Chem. B, 2019, 123, 5357–5367 CrossRef CAS PubMed.
- Y. Wang, Q. Mao, Z. Wang, K. H. Luo, L. Zhou and H. Wei, Combust. Flame, 2023, 248, 112571 CrossRef CAS.
- M. Feng, X. Z. Jiang and K. H. Luo, Proc. Combust. Inst., 2019, 37, 5473–5480 CrossRef CAS.
- C. Barckholtz, T. A. Barckholtz and C. M. Hadad, J. Am. Chem. Soc., 1999, 121, 491–500 CrossRef CAS.
- R. D. Kern and K. Xie, Prog. Energy Combust. Sci., 1991, 17, 191–210 CrossRef CAS.
- A. Mebel, M.-C. Lin, T. Yu and K. Morokuma, J. Phys. Chem. A, 1997, 101, 3189–3196 CrossRef CAS.
- D. Hou and X. You, Phys. Chem. Chem. Phys., 2017, 19, 30772–30780 RSC.
- F. Ossler, S. E. Canton and J. Larsson, Carbon, 2009, 47, 3498–3507 CrossRef CAS.
- F. Ossler, S. E. Canton, L. R. Wallenberg, A. Engdahl, S. Seifert, J. P. Hessler and R. S. Tranter, Carbon, 2016, 96, 782–798 CrossRef CAS.
- Q. Mao, A. C. Van Duin and K. Luo, Carbon, 2017, 121, 380–388 CrossRef CAS.
- H. Michelsen, Proc. Combust. Inst., 2017, 36, 717–735 CrossRef CAS.
- L. Madden, L. Moskaleva, S. Kristyan and M.-C. Lin, J. Phys. Chem. A, 1997, 101, 6790–6797 CrossRef CAS.
- Y. Guo and J. J. Grabowski, J. Am. Chem. Soc., 1991, 113, 5923–5931 CrossRef CAS.
- V. S. Rao and G. B. Skinner, J. Phys. Chem., 1988, 92, 2442–2448 CrossRef CAS.
- W. Sun, A. Hamadi, S. Abid, N. Chaumeix and A. Comandini, Proc. Combust. Inst., 2021, 38, 891–900 CrossRef CAS.
- K. Roy, C. Horn, P. Frank, V. G. Slutsky and T. Just, Symp. (Int.) Combust., 1998, 27, 329–336 CrossRef.
- J. Rodriguez-Mirasol, A. C. Ooms, J. R. Pels, F. Kapteijn and J. A. Moulijn, Combust. Flame, 1994, 99, 499–507 CrossRef CAS.
- R. Ananthula, T. Yamada and P. H. Taylor, J. Phys. Chem. A, 2006, 110, 3559–3566 CrossRef CAS PubMed.
- Z. F. Xu, C.-H. Hsu and M. C. Lin, J. Chem. Phys., 2005, 122, 234308 CrossRef CAS PubMed.
- D. Hodgson, H.-Y. Zhang, M. R. Nimlos and J. T. McKinnon, J. Phys. Chem. A, 2001, 105, 4316–4327 CrossRef CAS.
- T. L. Nguyen, J. Peeters and L. Vereecken, J. Phys. Chem. A, 2007, 111, 3836–3849 CrossRef CAS PubMed.
- R. K. Robinson and R. P. Lindstedt, Combust. Flame, 2011, 158, 666–686 CrossRef CAS.
-
N. Balucani, L. Pacifici, D. Skouteris, A. Caracciolo, P. Casavecchia and M. Rosi, Computational Science and Its Applications – ICCSA, 2018, pp. 763–772 Search PubMed.
- C.-H. Chin, T. Zhu and J. Z. H. Zhang, Phys. Chem. Chem. Phys., 2021, 23, 12408–12420 RSC.
|
| This journal is © the Owner Societies 2023 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  *b,
Zhanyuan
Wang
a and
Haiqiao
Wei
*a
*b,
Zhanyuan
Wang
a and
Haiqiao
Wei
*a