
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceStructural and dynamical insights into SilE silver binding from combined analytical probes†
Yoan
Monneau
 a,
Cyrielle
Arrault
a,
Coraline
Duroux
a,
Marie
Martin
a,
Fabien
Chirot
b,
Luke
Mac Aleese
b,
Marion
Girod
a,
Clothilde
Comby-Zerbino
b,
Agnès
Hagège
a,
Olivier
Walker
a and
Maggy
Hologne
*a
a,
Cyrielle
Arrault
a,
Coraline
Duroux
a,
Marie
Martin
a,
Fabien
Chirot
b,
Luke
Mac Aleese
b,
Marion
Girod
a,
Clothilde
Comby-Zerbino
b,
Agnès
Hagège
a,
Olivier
Walker
a and
Maggy
Hologne
*a
aUniversité de Lyon, CNRS, UCB Lyon1, Institut des Sciences Analytiques, UMR5280, 5 rue de la Doua, Villeurbanne 69100, France. E-mail: maggy.hologne@univ-lyon1.fr
bUniv Lyon 1, Université Claude Bernard Lyon 1, CNRS, Institut Lumière Matière, UMR5306, Cité Lyonnaise de l’Environnement et de l’Analyse, 5 rue de la Doua, Villeurbanne 69100, France
First published on 16th December 2022
Abstract
Silver has been used for its antimicrobial properties to fight infection for thousands of years. Unfortunately, some Gram-negative bacteria have developed silver resistance causing the death of patients in a burn unit. The genes responsible for silver resistance have been designated as the sil operon. Among the proteins of the sil operon, SilE has been shown to play a key role in bacterial silver resistance. Based on the limited information available, it has been depicted as an intrinsically disordered protein that folds into helices upon silver ion binding. Herein, this work demonstrates that SilE is composed of 4 clearly identified helical segments in the presence of several silver ions. The combination of analytical and biophysical techniques (NMR spectroscopy, CD, SAXS, HRMS, CE-ICP-MS, and IM-MS) reveals that SilE harbors four strong silver binding sites among the eight sites available. We have also further evidenced that SilE does not adopt a globular structure but rather samples a large conformational space from elongated to more compact structures. This particular structural organization facilitates silver binding through much higher accessibility of the involved His and Met residues. These valuable results will advance our current understanding of the role of SilE in the silver efflux pump complex mechanism and will help in the future rational design of inhibitors to fight bacterial silver resistance.
Introduction
Silver-based antiseptics have a wide range of applications in health care due to their high availability, broad spectrum, and safety for humans.1–3 However, silver-resistant Gram-negative bacteria were reported and the resistance mainly relies on the presence of a plasmid-encoded specific silver resistance sil operon4 which is homologous to the copper resistance cus operon. Both operons include the tripartite efflux pump CBA that acts as a proton antiport, an RS system that senses the metal present in the periplasm and enhances the operon gene expression accordingly, and a metal chaperon F that routes metal ions to the efflux pump. The SilE protein does not have any counterpart in the Cus operon but has a homolog in E. coli, PcoE, that displays 48% identity in sequence with SilE. A previous work has reported that the binding of PcoE with Cu+ and Ag+ induces its dimerization and partial structural folding while it has been shown that PcoE can bind up to 6 equivalents of Ag+ before precipitation.5 Gene deletion studies show a complete loss of bacterial silver resistance capacity when the SilE gene is deleted along with the Cus operon.6 SilE has its own promoter and is early expressed while bacteria are exposed to silver.4 Considering that new silver-based drugs are under development to fight multi-resistant bacteria,1,7,8 the appropriate design of such new drugs relies on our understanding of the molecular mechanism underlying silver resistance. Thus, providing insights into silver-bound SilE structure and dynamics will improve our knowledge of the role of this essential sil operon component concerning the efflux pump machinery.In a previous work of Asiani et al., SilE was described as an intrinsically disordered protein (IDP) that acts as a “molecular sponge” dedicated to the early sequestration of silver ions.9 Nevertheless, these previous studies concerning SilE and PcoE lack a clear identification of different silver binding sites at the atomic level and no experimental findings regarding a possible tertiary structure have been reported. Recently, we have improved our knowledge in the field by deriving NMR (nuclear magnetic resonance) structures of SilE mimicking peptides and shown that the sequences encompassing two successive motifs HxxM or MxxH fold into helices in the presence of silver ions.10 In the same order of idea, we have determined low micromolar dissociation constants from the NMR titration analysis.
From the vast landscape of biophysical techniques dedicated to the study of IDPs, NMR spectroscopy is particularly well suited especially in the case of complexes where disorder is at least partly retained.11 Structure and dynamics can be explored at the atomic level even for low-affinity complexes or low-populated states. Although NMR spectroscopy is powerful on its own, the efforts toward our detailed understanding of the conversion between fully IDP and partially disordered complexes introduce significant technical challenges making an integrative structural biology approach necessary. For instance, the stoichiometry for multisite ligand binding is often difficult to assess by NMR spectroscopy. Moreover, spectral quality can significantly decrease due to dramatic line broadening when interconversion rates are at the time regime of chemical shifts.12
In the present work, we have advanced our knowledge regarding the structural organization of SilE compared to previous studies. Therefore, we propose an original combination of analytical techniques to characterize the structure and dynamics of silver binding to SilE, including nuclear magnetic resonance (NMR) spectroscopy and circular dichroism (CD) for local structure and dynamics characterization; small-angle X-Ray scattering (SAXS) and ion-mobility-mass-spectrometry (IM-MS) for global shape analysis; liquid chromatography high-resolution mass-spectrometry (LC-HRMS) and capillary-electrophoresis inductively coupled-plasma mass-spectrometry (CE-ICP-MS) for assessing the stoichiometry of the metal–protein complexes.
In this regard, we will show how these complementary techniques allow the characterization of the SilE binding sites at the atomic level. An assessment of the stoichiometry of the complexes formed but also the structural modifications that SilE undergoes when complexed with Ag+ will be provided. In a final step, we will use a combination of AlphaFold structural prediction, SAXS, and NMR spin relaxation measurements to propose for the first time an insightful structure of the silver-bound SilE protein. Overall, this ensemble of results will allow an in depth overview of the role of SilE in the general context of the silver efflux pump machinery.
Results and discussion
The SilE sequence (Fig. S1, ESI†) used in this work was defined according to the wild-type sequence of the Salmonella Typhimurium SilE (UniProt accession number Q9Z4N3). The first twenty residues of the signal peptide that routes SilE to the periplasm have been removed. Therefore, the SilE21–143 construct has been successfully expressed and isotopically labeled with E. coli cells. As indicated in blue in the sequence, SilE is composed of 6 HxxM, 2 MxxH, and one HxM motifs. As previously described by Chabert et al.,10,13 silver ions can bind His and Met residues and association binding constants have been calculated for each motif.Silver-bound SilE complexes exist at different stoichiometries
As indicated above, the SilE binding sites are widely distributed over its amino acid sequence from M56 to G143, with a minor site around the position M40. Since the global stoichiometry of the silver-bound SilE complex needs to be determined, mass spectrometry is particularly well suited for this analysis. Solutions of SilE containing 0 to 10 equivalents of Ag+ relative to SilE were analyzed using positive electrospray ionization high-resolution mass spectrometry (ESI-HRMS). The same stock solution of silver nitrate (oxidation state I) has been used throughout all titration studies presented hereafter and the accuracy of the Ag+ solution concentration was assessed before any assay using a calibration curve and a silver ion selective electrode.The SilE protein was observed with a charge state distribution from 6+ to 21+ protonated adducts (Fig. S2A, ESI† SilE in the free state) for HR-MS and IM-MS experiments. With an increase in the concentration of Ag+ solution, protonated [SilE:nAg+] complex ions were also detected. As an example, Fig. S2B (ESI†) shows the ESI-MS spectrum of SilE with 5 equivalents of Ag+ solution, where SilE protonated ions and the peaks corresponding to [SilE + nAg + (z − n)H]z+ complexes with n = 1 to 5 were detected (see the inset in Fig. S2B, ESI†). Some hybrid adducts with Ag+ and 1 to 3 Na+ ions were also observed in the MS spectra. To simplify and better observe the different stoichiometries of [SilE:nAg+] complexes, all the recorded ESI-MS spectra were deconvoluted. The deconvoluted spectra, averaged over the end of the elution band in order to avoid chemical artefacts, are shown in Fig. 1(A) for the different molar equivalents of Ag+ in solution. The number of Ag+ bound to one molecule of SilE increases with the concentration of Ag+ present in the solution. A high abundance of 2 to 6 Ag+ per SilE molecule was observed, while complexes with 7 or 8 Ag+ were only observed at low abundances (even with a higher concentration of Ag+). However, higher concentrations of Ag+ did not allow for the observation of more Ag+ complexes, suggesting the saturation of the SilE complex sites. More quantitatively, the relative abundance of the different complexes [SilE-nAg+] were determined by integration of the deconvoluted spectra over the full elution band and are plotted in Fig. 1(B) as a function of molar equivalents Ag+ in solution. The free SilE protein is the most intense species detected for 1 to 6 equivalents Ag+ in the solution (Fig. 1(A)). However, starting from 4 equivalents of Ag+, the distribution shifts significantly towards the higher stoichiometries. Remarkably, only little 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2/1:3 and 1:4 complexes seem to be formed before 3, 4, and 5 equivalents of Ag+ are respectively added to the solution (while 1:5 and 1:6 already appear at 5 and 6 equivalents). After 4 molar equivalents of Ag+ in the solution, the abundances of the 1:1 complex start decreasing while the 1:2 and 1:3 complexes soon reach a plateau and the 1:4 complex progressively takes over. Up to 10 equivalents of Ag+, the abundance of the 1:4 complex remains maximal, pointing towards 4 strong complexation sites.
:2/1:3 and 1:4 complexes seem to be formed before 3, 4, and 5 equivalents of Ag+ are respectively added to the solution (while 1:5 and 1:6 already appear at 5 and 6 equivalents). After 4 molar equivalents of Ag+ in the solution, the abundances of the 1:1 complex start decreasing while the 1:2 and 1:3 complexes soon reach a plateau and the 1:4 complex progressively takes over. Up to 10 equivalents of Ag+, the abundance of the 1:4 complex remains maximal, pointing towards 4 strong complexation sites.
 | ||
| Fig. 1 Stoichiometry of the Ag-SilE complex. (A) Deconvoluted spectra, over the end of the elution band (in order to avoid chemical artefacts), of the 5 μM solutions of SilE and in the presence of 1 to 10 molar equivalents of silver ions. The red lines represent the masses of different stoichiometry [SilE:nAg+] complexes. (B) Relative abundances or branching ratios of different [SilE:nAg+] complexes as a function of molar equivalents Ag+ in solution. (C) Electropherograms obtained in CE-ICP-MS for 5 μM SilE and increasing Ag+ concentrations in the sample for the (1) [SilE:nAg] complex and (2) sorbed Ag+ released by HNO3. Electropherograms are shifted in the 107Ag signal intensity for the sake of clarity, and the background signal is around 50 counts for all. (D) Peak areas measured (C) for Ag species as a function of Ag+ equivalents added in the sample for the [SilE:nAg] complex (black) and sorbed Ag+ released by HNO3 (blue). (E) Electropherograms obtained in CE-ICP-MS for 5 μM SilE and increasing Ag+ concentrations in the electrolyte determined using CE-ICP/MS analyses. Electropherograms are shifted in the 107Ag signal intensity for the sake of clarity. (F) Peak areas measured (E) for the [SilE:nAg] complex as a function of Ag+ concentration in the electrolyte. | ||
To further confirm the observed stoichiometry in ESI-HRMS, the samples were analyzed using CE-ICP-MS. From 1 to 4 equivalents of Ag+ in solution, the quantity of Ag+ bound to SilE linearly increases (Fig. 1(C) and (D)). The nearly quantitative formation of the complex indicates a high affinity of SilE for Ag+. However, for higher concentrations of Ag+, no increase of the silver bound to SilE was detected but only free Ag+ (Fig. 1(D)). This result indicates that either we have reached the saturation of SilE binding sites or the additional labile sites exist. In the latter case, the potential sites could not be detected by using this CE-ICP-MS strategy. Indeed, the CE separation relies on the electric field applied along the capillary and the analyte must join the anode to be analyzed while silver ions are positive. We hypothesized that labile sites are prompted to dissociate while the migration front is quickly depleted from silver. To challenge the potential presence of such labile sites, the same experiments have been performed but with increasing electrolyte Ag+ concentrations (1–3 μM). The results are shown in Fig. 1(E) and (F). Increasing the electrolyte Ag+ concentrations led to an increase of the SilE-bound Ag+ peak areas until a concentration close to 1 μM. For the higher Ag+ electrolyte concentrations, the [SilE:nAg+] peak area remains constant, which reflects the saturation of the protein. The quantification of the signal at the plateau was performed by external calibration and revealed the complexation of 7–8 Ag+ per protein under these non-dissociating conditions. These results agree with those of our ESI-HRMS studies and further corroborate the maximum number of Ag+ that can specifically bind to SilE.
As a consequence, it can be assumed that SilE contains 4 kinetically inert complexation sites, which can be seen by directly injecting the complex, and 3–4 more labile sites for Ag+, which require an equilibrium state all along the separation to be detected.
SilE is mainly disordered in the free state
Using 3D standard NMR experiments, the backbone assignment of SilE has been performed (HN, Hα, Cα, Cβ, and N). The 2D 1H–15N HSQC NMR spectrum including amino acid numbering is shown in Fig. 2(A).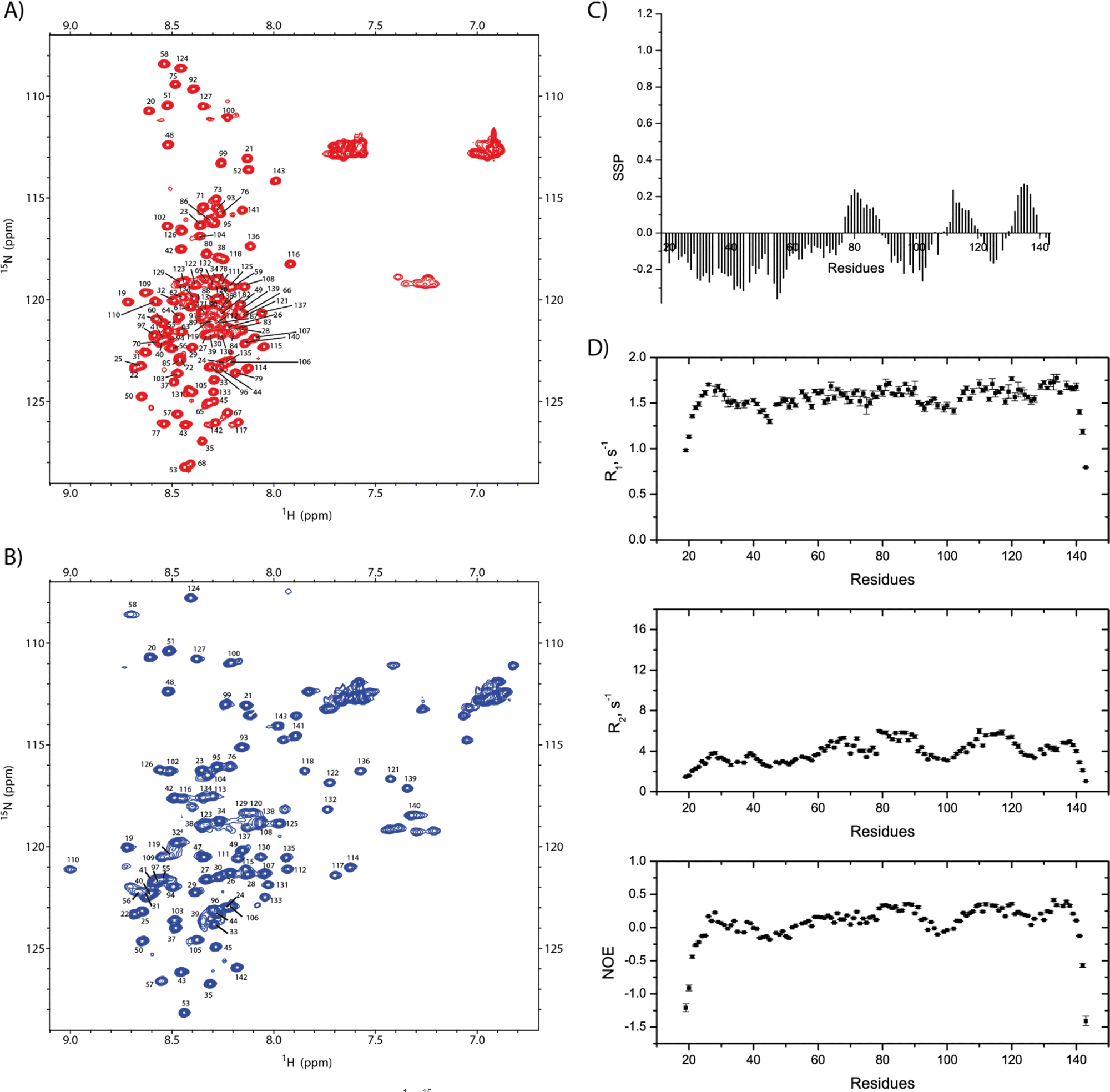 | ||
| Fig. 2 Backbone NMR assignments and dynamics of SilE. 2D 1H–15N HSQC NMR spectra of SilE at 140 μM (A) in the free state and (B) in the presence of 6 equivalents of silver ions. (C) Secondary structure propensity calculated from the NMR backbone assignments of the free SilE. The positive values indicate the α-helical structure propensity. (D) NMR relaxation data 15N-R1, and -R2 and hetNOEs for the free SilE. | ||
Resonances are dispersed in a narrow spectral range from 8 to 8.7 ppm, which is the hallmark of an intrinsically disordered protein.14 The observed chemical shifts result from the average of a conformational ensemble the protein may adopt if the exchange time scale is much faster than the chemical shift time scale. Consequently, the propensity to form different secondary structures can be assessed from the analysis of the backbone chemical shifts.15
For the amino acid regions 76–88, 112–117, and 132–139, we observe a propensity for the transient α-helix structure between 10% and 27% while the rest of the SilE sequence appeared in a fully extended conformation (Fig. 2(C)). To support these findings, we have set up NMR spin relaxation experiments that are closely related to the global and local motion of the protein. For the region that exhibits a propensity for the α-helix, the mean value of R2 reaches 5.1 s−1 while it is 3.8 s−1 for the rest of the sequence (excluding N- and C-ter). Additionally, the average hetNOEs show values of 0.30 and 0.1 for the same regions, respectively. The latter one is a typical value for IDP.16 This effect is less pronounced in R1 experiments due to its higher sensitivity for local motion compared to global motion. The HetNOEs are mainly dependent on the local N–H bond rigidity or flexibility, while R2 is also affected by the global motion. The N- and C-termini present highly negative hetNOE values indicating high flexibility. This corroborates that more rigid conformations are sampled by the regions 76–88, 112–117, and 132–139 compared to the purely intrinsically disordered behavior of the rest of the protein (Fig. 2(D)).
SilE has several silver binding sites
To investigate the silver binding areas, we have performed NMR titration experiments, recording a series of 1H–15N HSQC spectra on 15N-SilE by gradually increasing the Ag+ solution concentration (Fig. S3, ESI†). Along the titration experiments, we can make different observations that correspond to different exchange regimes. First and foremost, a set of 40 peaks experienced a slow exchange rate. Second, 33 other peaks exhibit a very strong decrease of their signal intensity so that they completely disappeared from the different spectra under saturating conditions. At this level, we can rule out a paramagnetic effect due to Ag+ binding. Indeed, Ag+ has an electron configuration of 5s0 4d10 so that it does not show any unpaired electron and paramagnetic properties.17 Consequently, signal disappearance may be ascribed to the intermediate exchange regime or oligomerization.18 This statement is also corroborated by the very strong increase of the transverse relaxation rate in this region (see below) and is likely due to the significant contribution of Rex to the global R2. Therefore, we can rule out any oligomerization effect in the present case. The expected number of peaks was never recovered even with the addition of a large excess of Ag+ (up to 50 eq.). As can be seen in Fig. S4 (ESI†), any attempt to recover these peaks was also unsuccessful either by modifying magnetic field (600 and 900 MHz), temperature (from 283 to 323 K), pH (5.2 to 7.8), ionic strength (20 to 300 mM NaF) or pressure (high pressure NMR experiments up to 2250 bars).Therefore, we have assigned the chemical shifts of SilE under silver saturating conditions (HN, Hα, Cα, Cβ, and N). All visible 1H–15N correlations correspond to residues 19–58, 76, and 93–143 (Fig. 2(B)). Unfortunately, residues 59 to 92 are then out of reach for studying binding, structures, and dynamics by NMR. It is noteworthy that both spectra in the free and silver-bound states are in stark contrast compared to the ones previously published where neither assignment nor structural information have been derived.9
The NMR chemical shift perturbations (CSP) upon the addition of 6 equivalents of Ag+ were calculated for each amino acid (Fig. 3). From S104 to A142, free and bound states are visible due to the slow exchange regime. The proportion of the bound state can be estimated from the NMR signal ratio between both states (Fig. S5, ESI†). For 1 equivalent of Ag+, 35% of the SilE region encompassing residues S104 to A142 is in its bound conformation and reaches 91% for 6 equivalents of Ag+. No significant changes occur for 6 to 9 equivalents of Ag+.
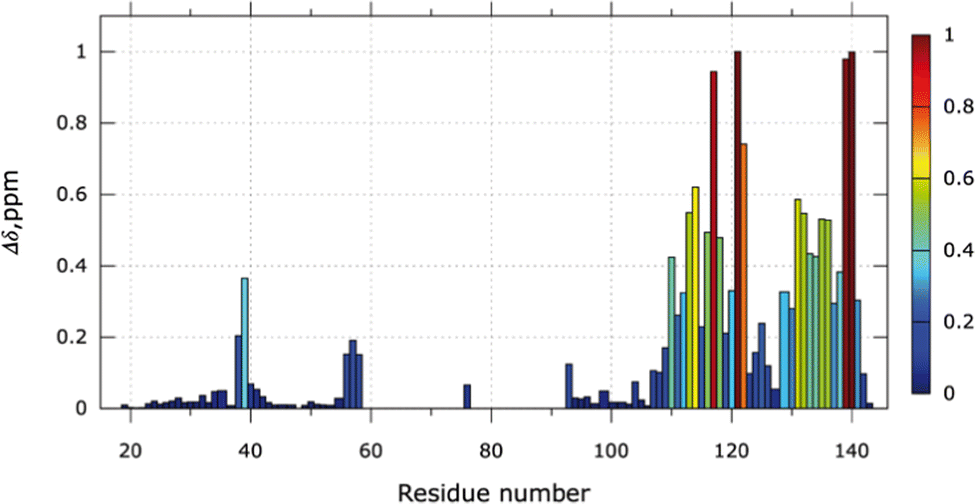 | ||
| Fig. 3 Silver binding sites. CSP observed at the end of the titration for 6 Ag+ equivalent on a 140 μM 15N-SilE (20 mM MES buffer pH 6.8 and 20 mM NaF). Residues 59–92 (except 76) are missing due to the large signal broadening in the 1H–15N HSQC spectra. | ||
Large variations of CSP are observed for residues E110 to N122 and residues H129 to S141. In these two regions, several His (H111, H118, H129, H136) and Met (M121, M132, M139) residues are present and their associated perturbations are in good agreement with the perturbations seen on the SilE-mimicking peptides.10 Additionally, we have detected measurable shifts in the regions H80 to M90 and M59 to M72 before the signal disappearance where 3 HxxM and 1 MxxH motifs are present. The latter one (59MDQH63) harbors visible signals until 4 equivalents of Ag+ while the others disappeared after the addition of 2 equivalents of Ag+ (Fig. S5B, ESI†). It is also noteworthy that the 59MDQH63 motif is involved in a less stable helical formation.9 The motif H38 to M40 also binds silver but displays significant CSPs from 5 to 9 equivalents of Ag+ and their corresponding signals are in a fast exchange regime. Overall, we have identified two major areas of SilE that binds silver ions with different dynamics, one in a slow exchange regime and the second one in an intermediate exchange regime. The third one is the weakest area of binding and has fast exchange regime dynamics. All the identified binding motifs of the entire SilE protein are in perfect agreement with the ones found in previous peptide-mimicking SilE10 and suggest that (i) the individual SilE-mimicking peptides may be used individually to describe the SilE/Ag+ interaction and (ii) each helical binding motif can be treated as independent segments in the SilE protein.
Although the general analysis of CSP is widely used to describe the protein/ligand binding interface, the discrimination between direct binding and a local conformational rearrangement upon ligand binding may be problematic. This is particularly true for IDP proteins which have no defined structure and possibly fold to stabilize their ligands.19
To further confirm this binding site mapping onto the SilE sequence, we have performed a differential digestion analysis monitored by mass spectroscopy. The SilE protein in the free state or in the presence of 6 equivalents of Ag+ was digested and the resulting peptides were analyzed by LC-MS/MS. Among the digested peptides, 3 peptides containing the 59MDQH63, 69HETM72, 108MNEH111, 118HEFM121, and 129HQAM132 motifs were detected as protonated adducts from the digested solution of SilE (Table S1A, ESI†). When digestion was performed on the SilE protein in interaction with Ag+, only the peptides containing the 59MDQH63, 69HETM72, and 108MNEH111 motifs were detected (Table S1B, ESI†). This result could indicate that the strong binding between silver ions and the two motifs, 118HEFM121 and 129HQAM,132 may disturb the digestion process due to a particular structural organization (or would not allow the protonated form of the peptide to be observed). In contrast, the 59MDQH63, 69HETM72, and 108MNEH111 motifs are observed in the protonated peptides under both conditions and do not perturb the digestion process when Ag+ is present.
Silver-bound SilE displays secondary structure folds
A series of CD spectra were recorded for the free SilE and in the presence of 1 to 8 equivalents of silver ions relative to SilE (Fig. 4(A)). The ellipticity significantly decreases at 222 nm and increases at 190 nm upon the addition of up to 4 equivalents of Ag+ in solution. These data demonstrate the formation of α-helices upon the addition of silver. We can also point out the presence of a clear isodichroic point at 206 nm indicating a two-step process between the free-form and the silver-bound form for SilE. From 5 to 8 equivalents of Ag+ in solution, the ellipticity only slightly changes toward more α-helix folds. This structuration is further confirmed by NMR since new peaks appeared out of the 8 to 8.7 ppm area of the 1H–15N HSQC spectra upon the addition of 2 to 9 equivalents of Ag+ in solution, which is a strong indicator of a protein structuration. The secondary structure propensities15 calculated from the backbone chemical shift assignment for each residue of the silver-bound SilE in the presence of 6 equivalents of Ag+ in solution further confirmed the α-helix folding upon silver addition (Fig. 4(B)). The folded area encompasses more residues compared to the free form (segments 110–123 + 128–142 vs. 112–117 + 132–139 without silver) and the α-helix propensity increases from 18% in the free state up to 65% in the bound state.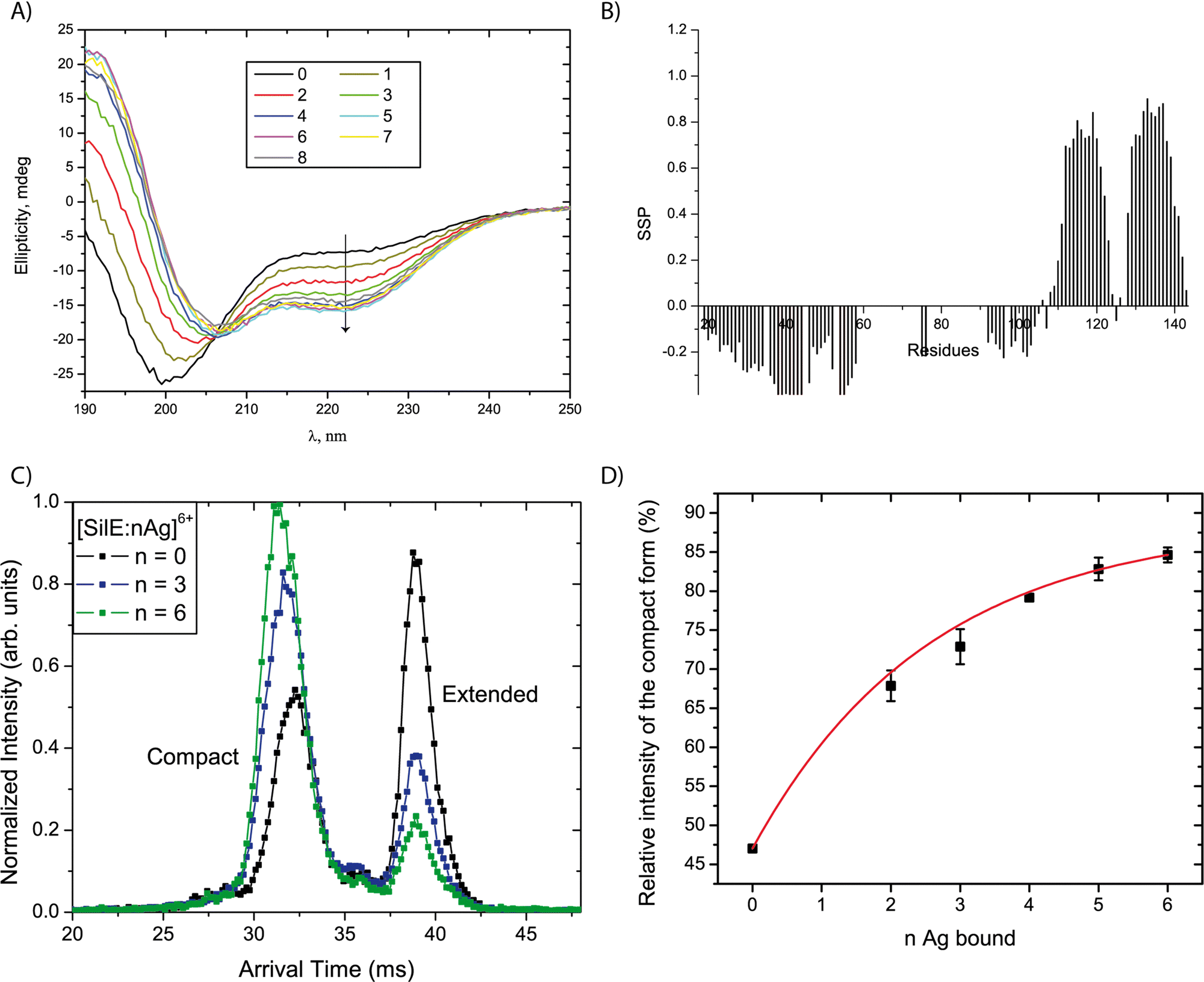 | ||
| Fig. 4 Structuration of SilE upon silver addition. (A) CD curves recorded at 25 °C for the free SilE and in the presence of 1 to 8 equivalents of silver ions ([SilE] = 20 μM in 20 mM MES buffer at pH 6.8 implemented with 20 mM NaF). The decrease of the ellipticity at 222 nm upon the addition of silver ions demonstrate the progressive formation of α-helices. (B) Secondary structure propensity calculated from NMR backbone assignments of SilE + 6 equivalents of Ag+. (C) Extracted arrival time distributions for selected [SilE:nAg]6+ complexes with different stoichiometries emphasizing the relative increase of the compact (low arrival times) species as n increases. (D) Evolution of the relative intensity of the compact form as a function of the number n of Ag bound to SilE6+. Error bars correspond to 95% confidence intervals from repeated measurements with different relative Ag+ concentrations in the sample. The full line represents a fit using an exponential decay function. | ||
To gain insight into the structuration of the SilE part that undergoes strong NMR signal attenuation, we designed a truncated SilE57–95. A series of CD experiments were performed and confirmed the formation of α-helix upon silver binding for this shorter construct (Fig. S6, ESI†) as observed for the full-length SilE. We can also point out the presence of a clear isodichroic point at 206 nm indicating a two-step process between the apo-form and the silver-bound form of SilE57–95.
To gain further insight into the structural changes and their diversity as a function of stoichiometry of the complexes, ion mobility coupled to mass spectrometry (IM-MS) spectra were recorded using solutions with 0 to 12 equivalents of silver ions relative to the SilE protein. Two factors lead to the sorting of the [SilE:nAg+] complexes in IM-MS (with n the number of bound Ag+): the mass of the complexes and their arrival times. The latter can be linked to the overall shape of the complexes. We detect species from n = 0 to 6, which is consistent with the HRMS data, considering that low-abundant complexes with n = 7 and 8 have not been detected due to the lower sensitivity of the IM-MS instrument compared to that of the HRMS instrument. The species with n = 1 was not detected with sufficient intensity, whatever the protein:silver ratio in the solution. The arrival time distributions (ATDs) were extracted for the different complexes detected. A selection of ATDs for different stoichiometries is shown in Fig. 4(C). All ATDs are undoubtedly bimodal indicating the coexistence of two ion populations with different mobilities. We interpreted these two populations as two conformational families since no evidence for isobaric SilE oligomers with higher charge states was found in the mass spectra. As generally observed for proteins, each peak is broader than expected from ion diffusion,20 which indicates that each family is a collection of different conformers with a close structure.21 The overall shape of the ATD would be compatible with a relatively slow interconversion between the two families during the course of the separation due to the plateau between the two peaks.22 However, no sign of this process was observed when selecting either of the population in a tandem-IMS scheme.23 Consequently, the two observed peaks can be attributed to the existence of two distinct conformational families, either pre-existing in solution or more likely resulting from structural reorganization upon desolvation. In the following, we focus on the relative intensity of the main two populations. The ATDs in Fig. 4(C) clearly indicate that the relative intensity of the two forms depends on the number of Ag bound in the complex. The peak at lower arrival times corresponds to species with a relatively more compact structure leading to a DTCCSHe value of 1385 Å2 and the second peak corresponds to an extended form characterized by a DTCCSHe value of 1693 Å2. Both forms are nevertheless detected for the bare SilE, with a predominance of the extended form. As the number of bound Ag+ increases, the relative population of the compact form increases to become predominant, as seen in Fig. 4(D). The data present the average ratio from ATDs for the same ionic species originating from solutions with different relative stoichiometries in Ag+. Importantly, the relative intensity of the two forms does not seem to be affected by the relative concentration of Ag+ in the solution, but only depends on the stoichiometry of the complex. Consequently, the observed evolution is not driven by a change in the conformational preferences of SilE due to a change in its environment, but rather reflects a structural change induced by Ag+ binding. As already mentioned, the gas phase structures of the complexes probably differ from their original solution structures. However, the observed ATDs can be understood as an indirect mapping of the conformational space sampled by the complexes in solution.24,25 Following this line, each gas phase conformer corresponds to the end point of a distinct portion of the solution-phase conformational space, after conformational relaxation. The observed changes can then be attributed to a modification in the solution-phase conformational space of SilE upon silver binding. In this context, we tentatively interpret the relative increase of the population of the most compact conformer for complexes with an increased silver content as the global compaction of the complexes in solution.
This interpretation is supported by the small angle scattering data (SAXS) of free SilE and SilE + 6 equivalents of Ag+ in solution (Fig. S7, ESI†).26 Through the Guinier plot analysis, we derived a radius of gyration Rg = 37.7 ± 0.7 Å for free SilE and Rg = 32.9 ± 0.2 Å for SilE + 6 equivalents Ag+. Therefore, we can conclude the compactness of the structure in the presence of silver as observed in IM-MS experiments. At this level, it is important to clarify the meaning of compactness according to IM-MS and SAXS techniques. The compactness is due to the formation of α-helices and is different from a globular structure which refers to a tertiary structure.
Interestingly, HRMS enables the observation of an inverse relationship between the number of silver ions observed on SilE and the charge state under which it is detected. This is independent of the concentration of Ag+ in solution, and is illustrated in Fig. S2B (ESI†) in the case of 5 equivalents of Ag+: at high charge states (18+, 19+), the dominant forms are SilE:Ag+ complexes 1:0 and 1.1, i.e. relatively depleted in silver. In contrast, at low charge states (9+, 10+, 11+), the dominant complexes are 1:3 and 1:4. This indicates that SilE bound to many silver ions is less prone to bearing many charges. A general trend in MS is that disordered species can unfold and extend to accommodate more charges during electrospray ionization and tend to be observed with larger and higher charge state distributions. Along this line, the HRMS observation suggests that silver-bound SilE is generally less disordered than silver-free SilE, which is consistent with the NMR and CD data. This is also consistent with the IM-MS and SAXS conclusion on the preference for more compact structures for silver binding SilE. Overall, a strong indication is that silver binding yields important structural adjustments in SilE.
Complementary experimental observations finally allow drawing a global multiscale picture of the changes in the SilE structure upon silver binding. At the secondary structure level, the NMR and CD data suggest that α-helices are formed as the relative silver concentration increases. Moreover, SAXS, IM-MS, and HRMS data show that this local structuration is accompanied by global compaction. Finally, the IM-MS and HRMS data provide evidence that this compaction is correlated with the number of silver cations bound to the complex, rather than in the relative silver concentration.
Silver-bound SilE complexes adopt different conformations
To understand how SilE may accommodate up to eight silver ions, we have built a putative model of SilE based on ColabFold,27 running the AlphaFold prediction protocol.28 Indeed, AlphaFold has demonstrated its capability to accurately predict protein structures by using deep learning methods to analyze coevolutionary information. Based on the SilE sequence, we have modeled five structures that adopt a compact or an elongated organization. All the predicted structures display four helical segments that perfectly match the structure of SilE-mimicking peptides (Fig. S8, ESI†).10 However, we can rule out the AlphaFold structures that exhibit the weakest pLDDT for the helical regions (Fig. S9A and C, ESI†). The rank1 structure also suggests that AlphaFold is quite confident about the relative position of α1 with respect to α2 or α3 with respect to α4, due to its low PAE (Fig. S9B, ESI†). The relative positioning of different helices along the SilE sequence is also confirmed by our spin relaxation measurements of the silver-bound SilE (Fig. 5). From residues T21 to T52 and Q94 to L105, R1, R2, and hetNOE parameters exhibit similar or close values compared to the ones seen in the case of SilE in the free state. This result indicates that these parts remain as flexible as they were in the free form. For the predicted helical regions α3 and α4, our data show a slight decrease in the average R1 from the free to the silver-bound state (1.67 to 1.53 s−1, respectively). This value is concomitant with a strong increase of the average R2 and hetNOE in the silver-bound state compared to the free state (from 5.0 to 10.7 s−1 and from 0.32 to 0.63, respectively).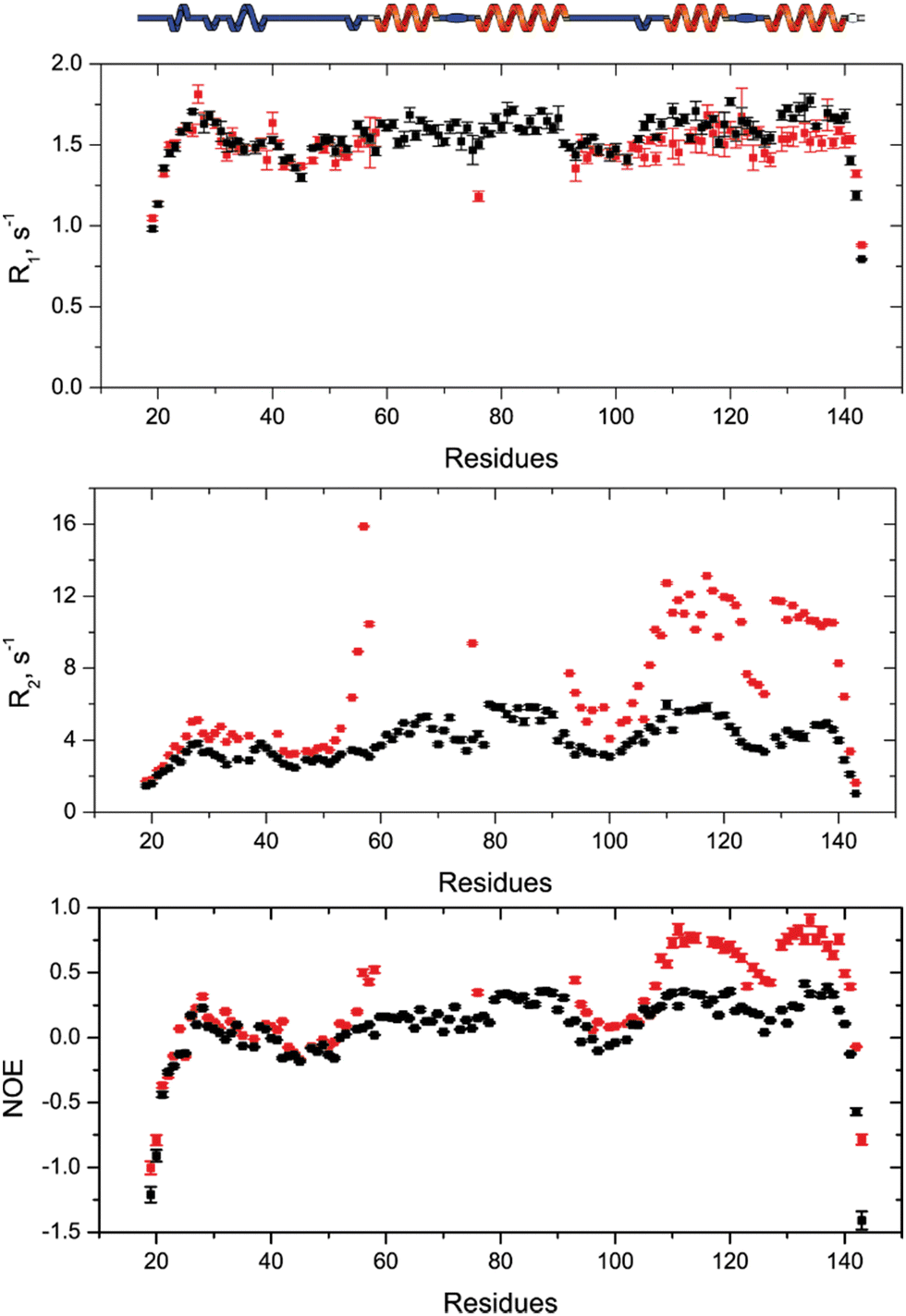 | ||
| Fig. 5 NMR spin relaxation data of the [SilE:nAg] complex. On top, the [SilE:nAg] complex secondary structure deduced from AlphaFold is sketched. NMR spin relaxation data 15N-R1, and -R2 and hetNOE for SilE in the presence of 6 equivalents of Ag+ in solution are displayed in red. For the sake of comparison, the NMR relaxation data of the free SilE have been added in black. | ||
This effect could be explained by a drastic change in the microdynamic parameters with a strong increase of the order parameter S2 along with a decrease in the local motion τloc (Fig. S10, ESI†). The modification of these two parameters undoubtedly reflects the rigidification and formation of helices α3 and α4. Despite the absence of NMR signals in the α1/α2 regions, it is noticeable that there is a tendency for a significant increase of the R2 and hetNOE parameters, further denoting the presence of helical formation and strong conformational exchange. This fact is further supported by the CD spectra recorded for the SilE57–95 construct that concludes to the formation of an α-helix for this region (Fig. S6, ESI†).
The local increase of the hetNOE values that exactly follow the secondary structure rather than a general increase led also us to conclude that silver-bound SilE has no defined tertiary structure. Therefore, the prediction of different helical regions added to our relaxation data is in good agreement with IM-MS and HRMS that conclude in higher compactness of SilE when bound to silver ions. At this level, it is important to highlight that the compactness is due to the silver-induced helical folding of SilE compared to the disordered state of the free SilE.
Whether SilE forms a globular or elongated structure still presents an unresolved conundrum, but a realistic low-resolution tertiary structure may be inferred from the SAXS data. Indeed, Alphafold predicts the correct secondary structure of SilE with a perfect match of the four helices but is not suitable to establish the highly flexible tertiary structure of SilE in the presence of Ag+.29 Therefore, we have used MultiFoXS30 for computing N-state (N = 1 to 5) models of the [SilE:nAg+] complex combined with a divide and conquer approach. As a starting point, we have used the compact rank 1 [SilE:nAg+] complex structure predicted by Alphafold that provides the lowest pLDDT and we have progressively increased the number of possible flexible regions until reaching the full flexibility of the SilE intervening linkers (Table S2, ESI† for the detailed flexible regions under study).
This strategy allows the sampling of an extensive conformational space accessible to SilE. In the case of the rank1 globular structure, the only allowed flexible part has been defined to be the N-terminus. As can be seen in Fig. S11A (ESI†), MultiFoXS could not find a suitable structural ensemble capable of reproducing the SAXS data and shows a χ score of 37.4 (Table S2, ESI†). Additionally, the associated radius of gyration equals 24.6 ± 2.0 Å in complete disagreement with the one experimentally determined from the SAXS data (32.9 ± 0.2 Å). This important result indicates that the [SilE:nAg+] complex does not adopt a globular structure in the liquid state and that the rank1 structure, predicted by AlphaFold, contradicts our experimental data. To maintain the positioning of α1,2 and α3,4, we have added the possibility of SilE to remain flexible in a region spanning the linker between α2 and α3. The χ score decreases from a one-state model to a three-states model (from 7.3 to 6.3 respectively) while a further increase in the number of conformations to 4 and 5 states did not improve the χ score. It is also noteworthy that the χ score significantly decreases compared to the one obtained for the globular structure and therefore supports the fact that the [SilE:nAg+] complex structure adopts a more elongated conformation in the liquid state (Fig. S11B, ESI†). Thus, we have added another degree of flexibility by allowing the linker between α1,2 or α3,4 region to move freely. While the χ score significantly decreases, we did not observe any differences when flexibility affects the linker between α1 and α2 or α3 and α4 with a χ score of 1.9 for this ensemble of structures in the case of a 3-state model. In both cases, the different ratios that represent the conformational sampling are rather similar (Fig. S11C and D, ESI†). The derived average Rg for the structure that presents a flexible α1,2 intervening linker is 28.9 ± 1.4 Å compared to an average Rg of 31.9 ± 4.0 Å for the structure exhibiting flexibility between the α3,4 linker. Finally, the χ score decreases to 0.9 for a 3-state model when the different linkers of the [SilE:nAg+] complex structure are allowed to move freely (Fig. 6). For the 3-state model, the average Rg of 33.6 ± 3.7 Å (Table S2, ESI†) is in good agreement with the experimental Rg derived from the SAXS data (32.9 ± 0.2 Å). This result allows the accurate mapping of the conformational landscape of the [SilE:nAg+] complex and supports the fact that it could be described by different conformations co-existing in solution, from elongated to intermediate states, as concluded by the IM-MS data analysis. Overall, the combined use of AlphaFold structure prediction with the NMR-derived structures of silver-bound SilE peptides and SAXS data support the fact that (i) the [SilE:nAg+] complex secondary structure exhibits four helical regions and (ii) different structural organizations of SilE co-exist in solution where Met and His residues may be favorably oriented and accessible to bind silver ions.
 | ||
| Fig. 6 SAXS analysis of the [SilE:nAg] complex. On top, the [SilE:nAg] complex secondary structure deduced from Alphafold is sketched. The different flexible segments used in the MultiFoXS calculation are coloured in blue. The SAXS data (blue circle), back-calculated SAXS data (red line), and residuals are represented in the left panel. On the right panel, the representation of the top-scoring N-ensemble of structures and their respective weight that best fit the SAXS data are presented close to each structure and are determined using the MultiFoXS server. Residuals per ith SAXS data point have been calculated according to the following expression: (I(qi)exp − I(qi)calc)/σi. I(qi)exp is the measured scattering intensity, where I(qi)calc is the intensity calculated using the MultiFoXS for scattering vector qi and σi is the experimental error. | ||
Conclusions
Several metals remain essential to life in extant organisms and play a diversity of roles in many different physiological processes. Sometimes, the toxicity of a given element may vary significantly because of speciation, so different chemical species containing the same metal may have a very different impact on living organisms. In the case of silver, Gram-negative bacteria have developed a resistant system based on a specific efflux pump dedicated to the removal of silver ions. Within this system, SilE has been recognized as an essential component for the appropriate function of the efflux pump by sequestering silver ions. From the vast panorama of the known metal binding proteins, SilE displays specific features in the sense that it is disordered in its free state and is prone to bind several silver ions while folding upon binding.In the present article, we have advanced our knowledge regarding the unknown SilE structure, dynamics, and interaction with silver ions. By combining experimental methods and AlphaFold prediction, we have determined up to eight binding sites that localize in four well-defined helical segments. Beyond these identified binding sites, four of them exhibit a strong interaction with silver ions. In particular, we have demonstrated that SilE does not adopt a globular structure upon silver binding but rather samples a large conformational space from elongated to more compact structures. This significant result deviates from the hypothesis established previously9 and leads us to ask this dangling question: “How SilE may accommodate up to eight silver ions and what is the advantage of such a structural organization?”. To answer this crucial question, we must resort to the state of the art and recall that SilE is synthesized only during growth in the presence of silver4 and is mandatory to avoid the disruption of the efflux pump machinery. At this level, we may hypothesize that SilE acts as a regulator that retains silver ions through His and Met residues and avoids metal saturation in the periplasm. Therefore, to be efficient, this process needs high solvent accessibility of these residues. To lend credence to our hypothesis, we have calculated the solvent accessible surface area (SASA) for (i) the globular single structure and (ii) the ensemble of the three structures found in the case of fully flexible SilE (Fig. 6). As seen in Table S3 (ESI†), the total percentage of SASA contrasts between the two cases. For the globular case, the average SASA on the interacting His and Met residues is 37% while it reaches 70% for the ensemble of elongated structures. Consequently, this structural organization facilitates silver binding through much higher accessibility of the involved His and Met residues while the globular structure strongly prevents close contact with silver ions. The most elongated structure is an amazing way to bind silver and allow the bacteria to remain active even at high silver concentrations. To the best of our knowledge, this is the first example of an intrinsically disordered protein that folds into helices to bind several metal ions at the same time.
For a deeper understanding of the cytotoxic mechanism of silver and how its antimicrobial properties are used in medicine, we refer the reader to an extensive review.3 Nevertheless, some Gram-negative bacteria have developed resistance to silver and impair the appropriate functioning of its antimicrobial properties. Thus, we may hypothesize that this resistance process is triggered by a high silver concentration in the cell. According to our aforementioned results, SilE can sequestrate up to eight silver ions. The SilCBA pump is also located in the periplasm and oversees the silver efflux out of the cell. Since we have identified two different binding affinities with silver ions (4 strong and 3–4 labile sites), it allows a fine tune of the silver release to the periplasm before extrusion by the efflux pump. This last hypothesis needs to be confirmed by further studies of the different interacting partners in the whole system.
Author contributions
MH coordinated and managed the overall project, and wrote the manuscript. MH analyzed NMR spin relaxation. YM engineered the SilE plasmid, optimized the production and purification protocols, and performed NMR assignments and titration analysis. CA and MM prepared the SilE samples and performed the CD analysis. AH and CD performed and analyzed the CE-ICP-MS experiments. FC and CCZ performed and analyzed the IM-MS experiments. MG performed the HRMS experiments and in collaboration with LMA analyzed the MS data. OW performed the MultiFoXs analysis by combining the AlphaFold simulation and SAXS data interpretation. YM, FC, LMA, MG, AH, CCZ and OW assisted in writing the manuscript, reviewed the final version, and approved the content and submission.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the CNRS through the MITI interdisciplinary program Metallomix and the French ANR (grant # ANR-17-CE29-0013-01). Financial support from the IR INFRANALYTICS FR2054 for conducting the research is gratefully acknowledged. The 900 MHz and the high-pressure NMR experiments have been performed at IMEC-IS-UCCS Lille and ICSN Gif sur Yvette, respectively.Notes and references
- K. Mijnendonckx, N. Leys, J. Mahillon, S. Silver and R. Van Houdt, BioMetals, 2013, 26, 609–621 CrossRef PubMed.
- E. Terzioğlu, M. Arslan, B. G. Balaban and Z. P. Çakar, World J. Microbiol. Biotechnol., 2022, 38, 158 CrossRef PubMed.
- S. Medici, M. Peana, V. M. Nurchi and M. A. Zoroddu, J. Med. Chem., 2019, 62, 5923–5943 CrossRef PubMed.
- A. Gupta, K. Matsui, J.-F. Lo and S. Silver, Nat. Med., 1999, 5, 183–188 CrossRef CAS PubMed.
- M. Zimmermann, S. R. Udagedara, C. M. Sze, T. M. Ryan, G. J. Howlett, Z. Xiao and A. G. Wedd, J. Inorg. Biochem., 2012, 115, 186–197 CrossRef CAS PubMed.
- C. P. Randall, A. Gupta, N. Jackson, D. Busse and A. J. O'Neill, J. Antimicrob. Chemother., 2015, 70, 1037–1046 CrossRef CAS PubMed.
- N. D. Savić, B. B. Petković, S. Vojnovic, M. Mojicevic, H. Wadepohl, K. Olaifa, E. Marsili, J. Nikodinovic-Runic, M. I. Djuran and B. Đ. Glišić, Dalton Trans., 2020, 49, 10880–10894 RSC.
- F. Paladini and M. Pollini, Materials, 2019, 12, 2540 CrossRef CAS PubMed.
- K. R. Asiani, H. Williams, L. Bird, M. Jenner, M. S. Searle, J. L. Hobman, D. J. Scott and P. Soultanas, Mol. Microbiol., 2016, 101, 731–742 CrossRef CAS PubMed.
- V. Chabert, M. Hologne, O. Seneque, O. Walker and K. M. Fromm, Chem. Commun., 2018, 54, 10419–10422 RSC.
- A. Prestel, K. Bugge, L. Staby, R. Hendus-Altenburger and B. B. Kragelund, in Intrinsically Disordered Proteins, ed. E. Rhoades, 2018, vol. 611, pp.193–226 Search PubMed.
- J. C. Ferreon, M. A. Martinez-Yamout, H. J. Dyson and P. E. Wright, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 13260–13265 CrossRef CAS PubMed.
- V. Chabert, M. Hologne, O. Seneque, A. Crochet, O. Walker and K. M. Fromm, Chem. Commun., 2017, 53, 6105–6108 RSC.
- H. J. Dyson and P. E. Wright, Curr. Opin. Struct. Biol., 2021, 70, 44–52 CrossRef CAS PubMed.
- J. A. Marsh, V. K. Singh, Z. Jia and J. D. Forman-Kay, Protein Sci., 2006, 15, 2795–2804 CrossRef CAS PubMed.
- N. Salvi, A. Abyzov and M. Blackledge, Prog. Nucl. Magn. Reson. Spectrosc., 2017, 102, 43–60 CrossRef PubMed.
- G. Parigi, E. Ravera and C. Luchinat, Curr. Opin. Struct. Biol., 2022, 74, 102386 CrossRef PubMed.
- J. Danielsson, R. Pierattelli, L. Banci and A. Gräslund, FEBS J., 2007, 274, 46–59 CrossRef PubMed.
- A. R. Camacho-Zarco, V. Schnapka, S. Guseva, A. Abyzov, W. Adamski, S. Milles, M. R. Jensen, L. Zidek, N. Salvi and M. Blackledge, Chem. Rev., 2022, 122, 9331–9356 CrossRef PubMed.
- H. E. Revercomb and E. A. Mason, Anal. Chem., 1975, 47, 970–983 CrossRef.
- S. L. Koeniger, S. I. Merenbloom and D. E. Clemmer, J. Phys. Chem. B, 2006, 110, 7017–7021 CrossRef PubMed.
- S. Poyer, C. Comby-Zerbino, C. M. Choi, L. MacAleese, C. Deo, N. Bogliotti, J. Xie, J.-Y. Salpin, P. Dugourd and F. Chirot, Anal. Chem., 2017, 89, 4230–4237 CrossRef CAS PubMed.
- A. Le Fèvre, P. Dugourd and F. Chirot, Anal. Chem., 2021, 93, 4183–4190 CrossRef PubMed.
- D. Stuchfield and P. Barran, Curr. Opin. Chem. Biol., 2018, 42, 177–185 CrossRef CAS PubMed.
- F. Gondelaud, M. Bouakil, A. Le Fèvre, A. E. Miele, F. Chirot, B. Duclos, A. Liwo and S. Ricard-Blum, Matrix Biol. Plus, 2021, 12, 100081 CrossRef CAS PubMed.
- P. V. Konarev, V. V. Volkov, A. V. Sokolova, M. H. J. Koch and D. I. Svergun, J. Appl. Crystallogr., 2003, 36, 1277–1282 CrossRef CAS.
- M. Mirdita, K. Schütze, Y. Moriwaki, L. Heo, S. Ovchinnikov and M. Steinegger, Nat. Methods, 2022, 19, 679–682 CrossRef CAS PubMed.
- J. Jumper, R. Evans, A. Pritzel, T. Green, M. Figurnov, O. Ronneberger, K. Tunyasuvunakool, R. Bates, A. Žídek, A. Potapenko, A. Bridgland, C. Meyer, S. A. A. Kohl, A. J. Ballard, A. Cowie, B. Romera-Paredes, S. Nikolov, R. Jain, J. Adler, T. Back, S. Petersen, D. Reiman, E. Clancy, M. Zielinski, M. Steinegger, M. Pacholska, T. Berghammer, S. Bodenstein, D. Silver, O. Vinyals, A. W. Senior, K. Kavukcuoglu, P. Kohli and D. Hassabis, Nature, 2021, 596, 583–589 CrossRef CAS PubMed.
- K. M. Ruff and R. V. Pappu, J. Mol. Biol., 2021, 433, 167208 CrossRef CAS PubMed.
- D. Schneidman-Duhovny, M. Hammel, J. A. Tainer and A. Sali, Nucleic Acids Res., 2016, 44, W424–W429 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental methods, protein sequence, NMR data, MS data, CD data, MultiFoXs analysis, and the Alphafold results. See DOI: https://doi.org/10.1039/d2cp04206a |
| This journal is © the Owner Societies 2023 |