
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Excited-state and charge-carrier dynamics in binary conjugated polymer dots towards efficient photocatalytic hydrogen evolution†
Aijie
Liu
*,
Sicong
Wang
,
Hongwei
Song
,
Yawen
Liu
 ,
Lars
Gedda
,
Katarina
Edwards
,
Leif
Hammarström
* and
Haining
Tian
*
,
Lars
Gedda
,
Katarina
Edwards
,
Leif
Hammarström
* and
Haining
Tian
*
Department of Chemistry-Ångström Lab., Box 523, SE 751 20, Uppsala University, Sweden. E-mail: haining.tian@kemi.uu.se; leif.hammarström@kemi.uu.se; aijie.liu@kemi.uu.se
First published on 16th December 2022
Abstract
Aqueous dispersed conjugated polymer dots (Pdots) have shown promising application in photocatalytic hydrogen evolution. To efficiently extract photogenerated charges from type-II heterojunction Pdots for hydrogen evolution, the mechanistic study of photophysical processes is essential for Pdot optimization. Within this work, we use a PFODTBT donor (D) polymer and an ITIC small molecule acceptor (A) as a donor/acceptor (D/A) model system to study their excited states and charge/energy transfer dynamics via steady-state and time-resolved photoluminescence spectroscopy, respectively. Charge-carrier generation and the recombination dynamics of binary Pdots with different D/A ratios were followed using femtosecond transient absorption spectroscopy. A significant spectral relaxation of photoluminescence was observed for individual D Pdots, implying an energetic disorder by nature. However, this was not seen for charge carriers in binary Pdots, probably due to the ultrafast charge generation process at an early time (<200 fs). The results showed slower charge recombination upon increasing the ratio of ITIC in binary Pdots, which further resulted in an enhanced photocatalytic hydrogen evolution, twice that as compared to individual D Pdots. Although binary Pdots prepared via the nanoprecipitation method exhibit a large interfacial area that allows high charge generation efficiencies, it also provides a high possibility for charge recombination and limits the further utilization of free charges. Therefore, for the future design of type-II heterojunction Pdots, suppressing the charge carrier recombination via increasing the crystallinity and proper phase segregation is necessary for enhanced photocatalytic hydrogen evolution.
Introduction
With the increasing global energy demand and environmental crisis, there is an urgent need for the successful utilization of solar energy. Hydrogen has been identified as one of the promising green energy carriers that only produces H2O, which may help to suppress global greenhouse gases and prevent a further air temperature increase. Traditionally, the generation of hydrogen can be achieved via fossil steam methane reforming, which emits CO2. Greener paths are required, such as electrolysis and photoelectrolysis directly from water. One-step photocatalytic water splitting may achieve higher conversion efficiency from solar energy to chemical fuels. With the inspiration from inorganic TiO2 induced charge separation of the photolysis of water,1 Yanagida et al. first reported photocatalytic hydrogen evolution by using conjugated polymer poly(p-phenylene) as a photocatalyst under λ > 290 nm irradiation.2 Compared to inorganic photocatalysts/photosensitizers, conjugated polymers show the advantages of being low cost, having an easy to tune bandgap through molecular design, and high performance. To date, a vast number of conjugated polymers have been synthesized and developed in this field,3–6 including conjugated microporous polymers (CMPs),7 polymeric carbon nitrides (PCNs),8 covalent organic frameworks (COFs),9,10 covalent triazine-based frameworks (CTFs)11 and linear polymers.12,13Linear polymer formed nanoparticles (NPs) of less than 100 nm in size, also called polymer dots (Pdots), dispersed in aqueous solution have been used in wide applications over the last few decades,14 from biomedical to organic photovoltaic (OPV) applications.15,16 In the latter case, it allows the replacement of toxic organic solvents for environmentally-friendly casting.17 Water dispersed Pdots for photocatalytic hydrogen evolution were first introduced in 2016 by our group, with the hydrogen evolution rate being almost five orders of magnitude higher than those of a conventional bulk system.18 Several advantages have made Pdots attract great attention from researchers: (1) easy preparation via the self-assembly of macromolecules or direct growth from monomers;19 (2) tuneable particle sizes and surface hydrophilicity via the modification of preparation methods;20,21 (3) small particle size to reduce the exciton diffusion length since the singlet exciton diffusion length in the most organic semiconductors is limited to be less than 20 nm;22 (4) a high surface area allows sufficient exciton separation and increased catalytic sites and (5) the hydrophilic sponge-structure shows fast proton diffusion through the existing proton channels of the Pdots.20 Owing to the increased interfacial area and lower initial capital cost compared to a film, nanoparticle photocatalysts dispersed in water have been suggested to be the cheapest way to produce hydrogen.23 Therefore, it is the right track to further explore and design Pdots to achieve high performance photocatalytic hydrogen evolution.24
Recently, Kosco et al. introduced binary heterojunction polymer NPs to increase exciton dissociation for enhanced hydrogen evolution, in which the morphology effect was studied in detail.25 A follow-up study indicated that long-lived charges in an organic semiconductor heterojunction are responsible for highly efficient photocatalytic hydrogen evolution.26 Independently, Cooper's group studied a large library of 237 organic binary/ternary nanohybrids and suggested that the key principle from the field of organic photovoltaics can be translated directly into organic polymer NP based photocatalytic reactions. It was also suggested that ternary donor(A)–donor(B)–acceptor compositions outperform binary systems.4 More recently, our group introduced ternary Pdots that exhibit outstanding performance with an external quantum yield of up to 7% at 600 nm, containing PFBT and PFODTBT donor polymers and an ITIC small molecule acceptor as both energy and electron acceptor. Ultrafast energy and charge transfer processes were studied in detail via femtosecond transient absorption spectroscopy. However, it is still unclear as to how the ternary Pdots outperform the binary system. Therefore, in the current study, we use binary Pdots with a type II heterojunction as a model system containing PFOTDTB as a donor (D) and ITIC as an acceptor (A) to further study the photophysical and photocatalytic properties of Pdots. A large composition range of D/A was investigated, with steady-state (st-st) photoluminescence (PL) analysis indicating that upon increasing the amount of A, there is a transition from two steps of electron transfer (ET) along with energy transfer (EnT) processes into one step ET from the excited donor (D*) to A. Moreover, high charge generation yield was observed across the D/A composition range. A significant redshift along with time was observed in D Pdots via time resolved PL analysis, indicating energetic disorder in the D Pdots; however, this was not seen in A dots and binary Pdots. In addition, no characteristic peak broadening or shift were observed in femtosecond transient absorption spectroscopy (TAS) in the study range of 350–750 nm, indicating that charge carrier features may not be affected by the energetic disorder of D. With careful study of the charge carrier recombination dynamics, a prolonged lifetime of the charge carrier was observed upon increasing the amount of A in binary Pdots. Increased crystallinity and charge mobility of A phases with an increasing in A contributed to this slower charge recombination, promoting charge extraction from Pdots and further resulting in higher photocatalytic hydrogen evolution. Despite the high yield charge generation (>80%), the optimal photocatalytic hydrogen evolution of binary Pdots was only double as compared to those individual D Pdots, half the performance of the ternary Pdots in our previous work.27 D/A binary Pdots show much better intermixed phases compared to ternary Pdots, as evidenced from X-ray powder diffraction (XRD) results, with no obvious crystal phases observed for the binary Pdots. Fine phase intermixing helps to accelerate charge generation; however, it also gives more chance for charge recombination. Therefore, suppressing charge recombination via increasing the crystallinity and proper phase segregation are suggested for the future design of type II heterojunction Pdots for enhanced photocatalytic hydrogen evolution.
Results and discussion
In this study, poly[2,7-(9,9-dioctylfluorene)-alt-4,7-bis(thiophen-2-yl)benzo-2,1,3-thiadiazole] (PFODTBT) was used as a donor and the small molecule ITIC was used as an acceptor, and the amphiphilic polymer PS-PEG-COOH was used as a surfactant to stabilize the Pdots in water, with the involved chemical structures shown in Fig. 1a. PFODTBT (D) and ITIC (A) were chosen to form blended binary (D/A) Pdots due to their excellent spectral overlap (Fig. 1b), which improves energy transfer (EnT) efficiency.28 From the energy levels shown in Fig. 1c, the binary D/A system can be seen to have a staggered gap, type II heterojunction, where both electron transfer (ET) from the excited D (D*) to acceptor A and hole transfer (HT) from the excited acceptor (A*) to donor D drive charge separation at the donor/acceptor interfaces for further applications. | ||
| Fig. 1 (a) Chemical structures of PFODTBT, ITIC and amphiphilic polymer PS-PEG-COOH used in this work; (b) steady state absorption (solid lines) and photoluminescence spectra (dashed lines) and (c) energy diagrams of the D and A. | ||
Pdots were prepared via a nanoprecipitation method according to the literature.29 Binary Pdots with a series of blend ratios (20 wt%, 39 wt%, 51 wt%, 62 wt%, 82 wt%, and 95 wt% of acceptor) were prepared. The resulting aqueous Pdots were colloidally stable for over a year at room temperature. The diameter of the binary Pdots was around 100 nm, as determined by dynamic light scattering (DLS, Fig. S1, ESI,† mean intensity), larger than those of the individual donor Pdots and small molecule acceptor dots, which have average diameters of 65 nm and 8 nm, respectively. Cryo-TEM results show that particles' shapes tend to transform from regular to irregular, with acceptor ratios of 20 wt% A (Fig. 2a) and 62 wt% A (Fig. 2b), respectively. For these binary Pdots, a slight increase in the crystallinity was observed when the A concentration was larger than 62 wt%, as shown in the XRD data in Fig. S2 (ESI†). However, the crystallinity of the acceptor in these binary Pdots is much smaller when compared to ternary Pdots that reported,27 and the low-detectable crystal domain of the acceptor presented in this work may indicate much better intermixed D/A phases in the binary Pdots.
 | ||
| Fig. 2 Cryo-TEM images of D/A binary Pdots with mass ratios of A of (a) 20 wt% and (b) 62 wt%. | ||
Steady-state absorption and photoluminescence measurements
The st-st absorption spectra of binary Pdots with a series of blend ratios are shown in Fig. 3a. The st-st PL spectra under excitations of 550 nm and 680 nm are shown in Fig. S3b and c (ESI†). Under excitation at 550 nm, mainly D was excited, although A could also be excited under this excitation wavelength since A exhibits slight absorption at around 550 nm. Under excitation at 680 nm, only A can be excited. According to Fig. S3b (ESI†), the PL intensity of D* was strongly quenched. This donor quenching is probably due to the parallel pathways of EnT and ET from the D* to A, according to the energy levels, as well as the spectral overlap between PL of the *D and the absorption of A (Fig. 1b and c).27 On the other hand, HT is the only pathway for the acceptor quenching. Nevertheless, in order to identify the photophysical pathways in detail, fluorescence excitation spectra were studied under the emission wavelength of 800 nm. Strong overlap between the excitation and absorption spectra for binary Pdots with compositions from 20 wt% to 51 wt% of A was observed (Fig. 3b), providing evidence for efficient EnT from the D* to A. A corresponding spectral overlap was not observed in the mixtures of individual D Pdots and A dots, due to large distance for the EnT process (Fig. S4, ESI†). However, we cannot exclude the ET process completely in the binary dots, considering the large energy offset of 0.35 eV. Therefore, within this concentration range, EnT and ET processes coexist from the D* to A, as we observed the PL intensity of D was decreased with the increasing the amount of A (Fig. S3b and c, ESI†). While for binary Pdots with higher amount of A (≥62 wt%), only the characteristic feature of A was observed in the excitation spectra (Fig. 3b), indicating that EnT from *D is inefficient and instead ET from the D* to A occurs. This ET process from the D* to A completely outcompetes the EnT process for a higher composition of A, which has also been observed in heterojunction thin films.30,31 Increasing the amount of A greatly enhances the interfaces and shortens the distance between D and A, which promotes the ET process after the immediate excitation of D,32 since the charge transfer time is much shorter than for a single EnT step.33 While the pathway of HT from A* to D should exist in all composition ratios, under the excitation of 680 nm, we observed that the PL intensity of A was decreased when the composition of A was lower than 51 wt% (Fig. S3b and c, ESI†), possibly indicating an enhanced HT from A* to D upon increasing the amount of A. Further increasing the amount of A (≥62 wt%) resulted in the increased PL intensity of A, due to limited charge separation efficiencies and solvation effects (discussed in the section on Excited-state dynamics). Therefore, we can conclude at this point that under a relatively low concentration of A (≤51 wt%), donor quenching can be a result of both EnT and ET pathways; while under a high concentration of A (≥62 wt%), the ET pathway is the only quenching process of the donor. Moreover, HT is the only process for acceptor quenching across the entire D/A composition range, as shown in Fig. 3c. | ||
| Fig. 3 Binary Pdots with various amounts of A: (a) normalized UV-vis absorption spectra; (b) excitation spectra under the emission of 800 nm; (c) photophysical pathways within the binary Pdots. | ||
Excited-state dynamics
Time-resolved (TR) PL spectroscopy, at 550 nm excitation and employing streak camera detection, was used to study the excited state dynamics of individual Pdots as well as binary Pdots. Here, binary Pdots with 51 wt% of A were chosen for this study because of their large spectral overlap between the absorption and excitation spectra, and because they give the highest charge separation efficiencies (Fig. S3b and c, ESI†). Photon absorption efficiency at 550 nm for the donor in the binary Pdots (51 wt% A) is 75%, according to Fig. S3a (ESI†). Fig. 4a shows a PL plot of the D Pdots, a gradual redshift was observed over time, from 650 nm at 60 ps to 690 nm at 750 ps (Fig. 4b), this relaxation of excitons in the density of states has also been observed by another group34 and is due to the disorder of polymer packing in the Pdots. However, this time-dependent redshift of PL was not observed in either A dots or binary Pdots, as shown in Fig. S5 (ESI†) and Fig. 4c, respectively. The TR-PL spectra of D/A Pdots also shows PL peaks centred at around 750 nm, consistent with the st-st PL studies (Fig. S3, ESI†).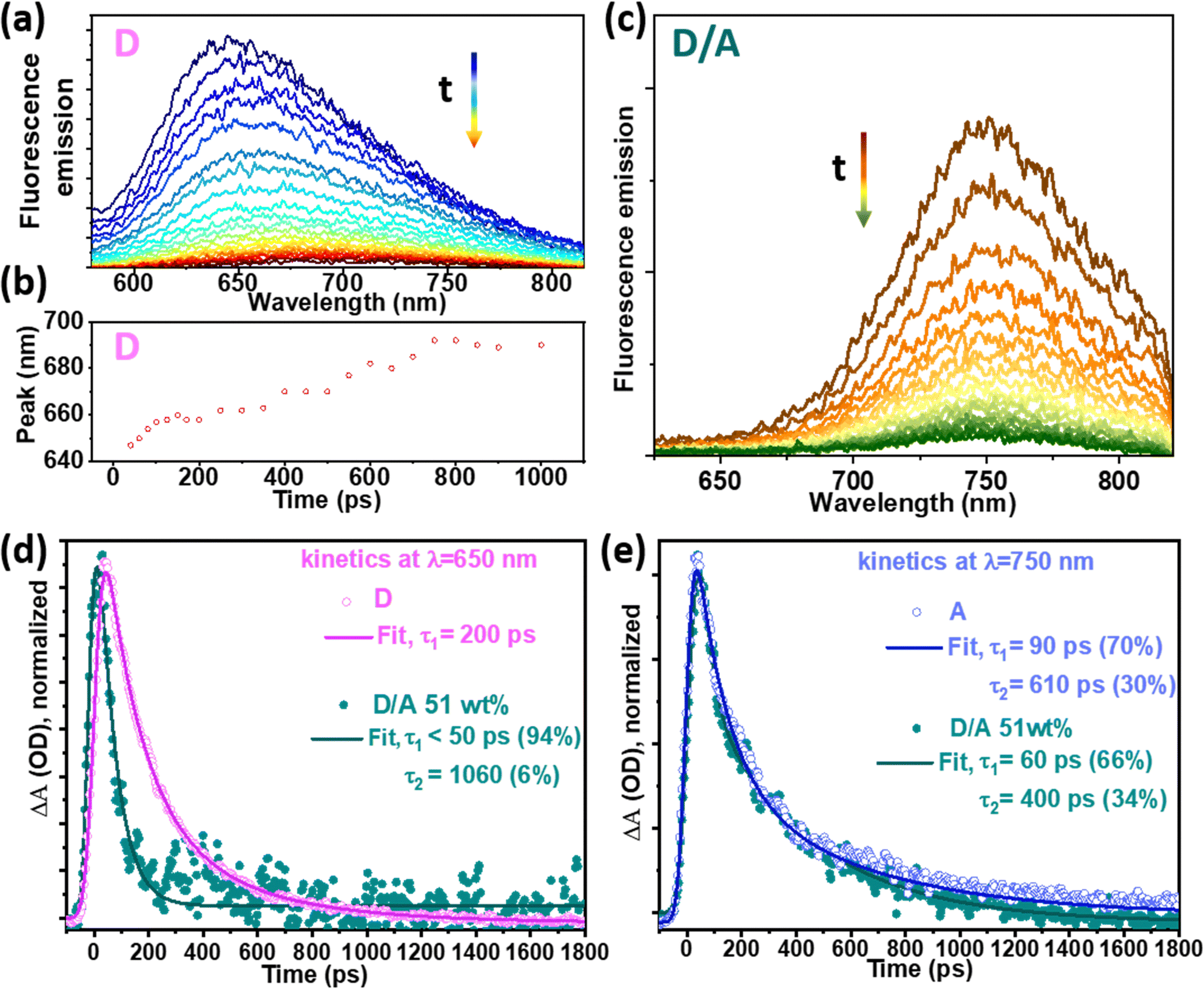 | ||
Fig. 4 Time-resolved photoluminescence spectra under excitation at 550 nm: (a) D; (b) A; (c) D/A binary with A 51 wt%; (d) kinetic traces at 650 nm of D and D/A and (e) kinetic traces at 750 nm of A and D/A binary Pdots. The residual plots of the fittings in (d) and (e) are shown in Fig. S6 (ESI†). Note: the comparison of the averaged lifetimes and the st-st intensities are mathematically correct, since the st-st intensities are proportional to the area under the corresponding time resolved traces, since the integral of 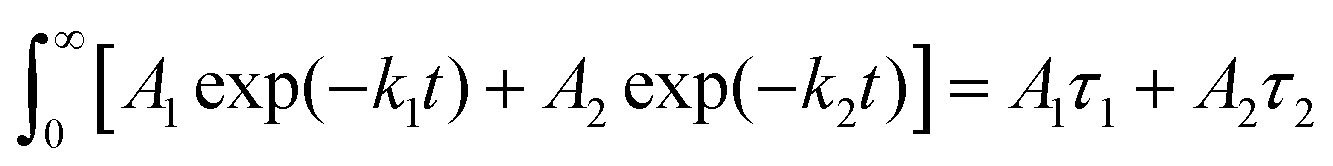 . . | ||
The PL kinetic traces of all the samples were fitted using a biexponential decay that was deconvoluted with the instrument response function (IRF, 50 ps). The amplitude-weighted average lifetime (τa) of D was 200 ps for individual D Pdots and was quenched to 60 ps in binary Pdots (Fig. 4d), resulting in 70% quenching yield. The PL kinetic traces of A were fitted at 750 nm, and a similar risetime of A in binary Pdots and bare A dots were observed, both within the IRF (τ < 50 ps), indicating a fast energy transfer process in the binary dots, consistent with our previous report.27 The averaged decay lifetime of A dots obtained from a streak camera was τa = 220 ps, consistent with the time-correlated single photon counting results (Fig. S7, ESI†). Notably, the lifetime was not reduced much for A in the binary Pdots as compared to the bare A dots, with averaged lifetimes of τa = 190 ps and τa = 220 ps, respectively (Fig. 4e). In fact, in contrast to the large quenching that was observed in the st-st PL intensity, a slight reduction in the PL lifetime, or even a prolonged lifetime of A, was observed in binary Pdots with a high composition of A, e.g., 229 ps for 82 wt% A (Fig. S8, ESI†). At first glance, the lifetime of A* in the D/A Pdots might seem unquenched compared to the A dots, according to Fig. 4e, with a possible explanation for this phenomenon being as follows. The D matrix may act as a “good solvent” for A and reduced the aggregation of A, thus, the aggregation induced self-quenching was suppressed and resulted in a prolonged PL lifetime of A in the D/A Pdots as compared to neat A dots. What is worth noting that the PL lifetime of A is much longer in a good solvent, for example, it gives a PL lifetime of 1 ns when in THF solution (Fig. S7, ESI†). This PL lifetime is almost 5 times longer than A dots in water. A prolonged PL lifetime of A was also observed in McNeil's work with acceptors blended in conjugated polymer nanoparticles.28 In fact, the PL lifetime of the A in the D/A system was even longer than A in the good solvent of THF.28 Therefore, in our system, there was a high possibility that the intrinsic PL lifetime of A* in D/A Pdots was much longer than those in the A dots. After the excitation of D, there was EnT from D* to A (for D/A, ≤51 wt%), while charge separation in the form of HT from A* to D (and/or ET from D* to A) resulted in quenching of A*, and finally resulted in a similar PL lifetime of A* in D/A as compared to A dots. This also explained the observation of a slightly prolonged lifetime of A* in the D/A Pdots with a composition of 82 wt% as compared to 51 wt%, since the acceptor quenching efficiency of D/A 82 wt% is lower than of D/A 51 wt% (Fig. S3c, ESI†).
Charge generation dynamics
Photoinduced charge generation and recombination dynamics were studied by using femtosecond transient absorption spectroscopy (TAS) measurements. According to the type II heterojunction in the D/A binary Pdots, charge generation can be achieved via both ET and HT processes. The TAS of individual D Pdots and A dots were studied in our previous work.27 In the current work, for binary Pdots with 62 wt% of A, immediately after the excitation at 550 nm, the ground state bleaching (GSB) of D at 402 nm and 575 nm, and the GSB of A at 625 nm and 675 nm were observed, as shown in Fig. S9 (ESI†). At this early time, an additional positive absorption between 440 nm to 510 nm with a risetime below the IRF (<200 fs) was observed, and the weak net positive absorption at 475 nm was assigned to the reduced A (A˙−) species,27 since it is in good agreement with the sum of the difference spectra for the oxidized donor and reduced acceptor. The two species of the charge separated state resulted in a weak net positive absorption at around 475 nm due to A˙− (Fig. S9, ESI†), at which the excited A* dots gave no negligible signal (Fig. S10c and d, ESI†) and the excited *D Pdots showed an isosbestic point (Fig. S13, ESI†). The results indicate that the one-step charge transfer process (ET or HT) between D and A is ultrafast in the binary Pdots. Similar results were also observed in binary Pdots with A of 39 wt% in our earlier study,27 where both ultrafast ET and HT within IRF (<200 fs) were observed, contributing to fine intermixed D/A phases, the delocalization of hot excitons and possibly an intermolecular (or domain) electric field.30,35,36 It is worth noting that the energetic disorder of the charge carriers was not observed in our binary Pdots, as shown in Fig. S9 (ESI†), as well as in our previous results,27 where there was no obvious peak shift in the GSB of the donor, acceptor and A˙− species. This may indicate that ultrafast charge generation in binary Pdots was completed on a very early timescale (<200 fs), so that the generated charge carriers were not affected by the energetic disorder of the pristine donor polymer. This phenomenon is different from that of the bulk blended thin film,34 in which the energetic disorder of the pristine donor polymer leads to density of states broadening of the charge carriers, resulting in charge trapping in solar cells. Although exciton delocalization contributes to fast charge carrier generation in D/A systems, and it has been generally accepted for both NPs and bulk thin films,38,39 we suggest that charge generation could be enhanced in Pdots for one or more of the following reasons: (1) the solid-water interface with higher dielectric constant could promote exciton dissociation;20,40 (2) D/A phases are much better intermixed in binary Pdots that are prepared via a nanoprecipitation method,37–41 as also supported by our XRD results, as no obvious crystal phases were observed; (3) the exciton diffusion volume is limited for Pdots, resulting in increased possibilities for exciton dissociation at the D/A interface considering that the typical exciton diffusion length for PFODTBT type Pdots is around 10 nm.42 However, the last two reasons could also give more chances for charge carrier recombination. Therefore, it is necessary to understand the nature of charge carrier recombination dynamics in D/A blended Pdots to extract free charges efficiently for further utilization.Charge carrier recombination dynamics
Charge carrier recombination dynamics were studied by following the decay kinetics of A˙− species at 475 nm. Similar decay kinetics of A˙− species were observed under the excitation of D (λex = 550 nm) and A (λex = 710 nm) with a pump intensity of 80 μW, as shown in Fig. S11 (ESI†), which means that charge recombination kinetics may be independent of the excitation nature of the donor or acceptor, considering that the charge generation process was completed within the first 200 fs. Fig. 5a shows the decay kinetics of A˙− in D/A with an A of 39 wt%, where excitation intensity-dependent decay kinetics were observed as the charge carrier recombination is concentration-dependent, therefore higher concentration accelerated the charge pair and nongeminate recombination under the higher excitation intensity. As shown in Fig. 5a, under a low pump intensity of 80 μW, the kinetic traces can be fitted by a single exponential decay with a lifetime of 431 ps. A slight increase in pump intensity to 140 μW resulted in a large portion of fast decay with a lifetime of 3.4 ps (43.9%). In the Pdots, the charge carriers have much less diffusion space compared to those within a bulk thin film, therefore a slight increase in the pump intensity increased the number of charge carriers per diffusion volume significantly, and also led to a large D/A interface in Pdots, with both factors contributing towards accelerating charge recombination.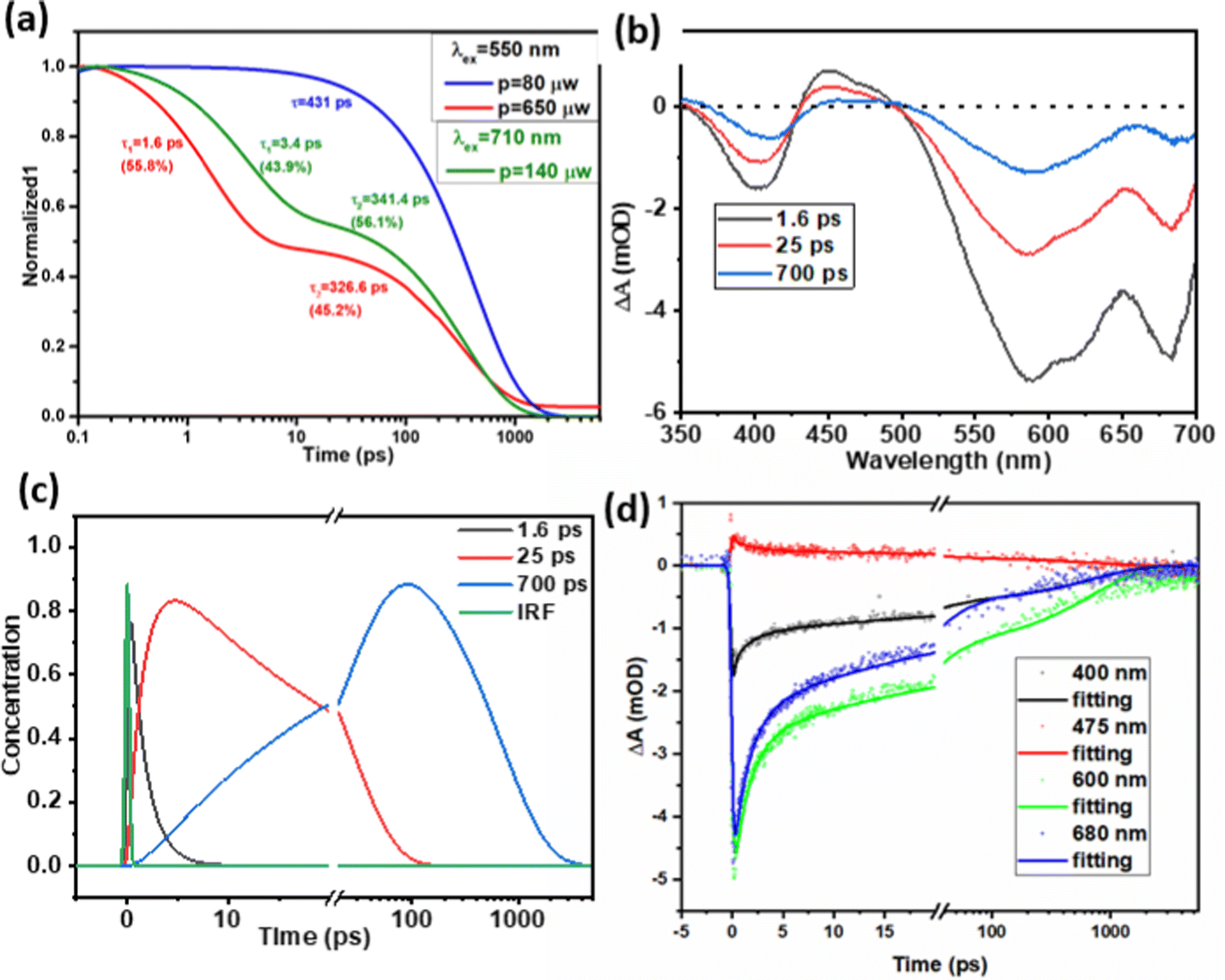 | ||
| Fig. 5 (a) Normalized TAS decay kinetics at 475 nm for D/A binary Pdots with 39 wt% A under an excitation wavelength of 550 nm, pump intensity of 80 μW and 650 μW, excitation wavelength at 710 nm with an intensity of 140 μW, where the raw data are presented in the ESI,† Fig. S10; (b) and (c) evolution associated difference spectra (EADSs) and concentrations of transient species from a global fit of TA data for binary Pdots with 62 wt% A under pumping at 550 nm, an intensity of 140 μW (note, to have a proper signal for analysis, 140 μW was chosen here to compare). (d) Selected kinetic traces at 400, 475, 600 and 680 nm for binary Pdots with 62 wt% A under pumping at 550 nm and an intensity of 140 μW, which show a good quality of fit. | ||
With a further increase in the pump intensity from 140 μW to 650 μW, an increase in amplitude for the first fitting component was observed: 3.4 ps (43.9%) for 140 μW and 1.6 ps (55.8%) for 650 μW. Although the signal at 475 nm is within the excited state absorption (ESA) region of D*, we believe we can exclude D*–D* annihilation by comparison with experiments for neat D Pdots. Thus, we monitored the kinetics at 475 nm under an excitation laser power of 572 μW, as shown in Fig. S13 (ESI†). Since the 475 nm signal is very close to the isosbestic point (at 477 nm) of D* and D, no significant signal or kinetics were observed under this low absorption. This is consistent with our previous work where the sum of ESA and GSB of D at 475 nm was zero under a low excitation power (80 μW).20 Furthermore, we can also exclude the possibility of the A*–A* annihilation of A* by comparing with experiments for neat A dots. As shown in Fig. S10c and d (ESI†), there is only very weak positive absorption in the wavelength range of 400–500 nm under an excitation of 710 nm and laser power of 160 μW. Moreover, the fast component for the D/A Pdots at higher pump intensity (3.4 ps at 140 μW, 1.6 ps at 540 μW) shows strong features of donor GSB recovery (see the bottom global fitting analysis data in Fig. 5b), which is inconsistent with A*–A* annihilation. With the above investigation, we can exclude the possibilities that the fast decay component at 475 nm for binary Pdots under high excitation intensity (≥140 μW) is due to the biexciton annihilation of D* or A*, instead, this is assigned to faster non-geminate recombination of separated charges that is more likely due to low phase segregation and limited charge diffusion space in the Pdots compared to bulk thin films.
For binary Pdots with a higher concentration of A (62 wt%), to figure out the charge recombination lifetime of binary Pdots with higher concentration of A (62 wt%), a corresponding sequential model for global fitting was performed, as shown in Fig. 5. Well-fitted evolution associated difference spectra (EADSs) and concentrations of the transient species and three-time constants (1.6, 25 and 700 ps) were obtained. The slower charge recombination process with a lifetime of 700 ps is more than twice that of D/A 39 wt% (341 ps) under an excitation power of 140 μW, as shown in Fig. 5. This slower charge recombination at a higher concentration of A has also been observed in bulk blended thin films,25 contributed to by the greater extent of aggregation of A, which allows more effective charge migration, therefore reducing the possibility of charge carrier recombination.43,44 The residual plots for kinetic traces at 400, 475, 600 and 680 nm from global fitting analysis are shown in Fig. S14 (ESI†).
Photocatalytic hydrogen evolution
After observing the photophysical dynamics in these binary Pdots, we aimed to verify the application of these binary Pdots in photocatalytic hydrogen evolution. A series of binary Pdots containing varying amounts of A was tested with 6 wt% Pt as a cocatalyst. The yield of hydrogen evolution after 24 h is presented in Fig. 6. Upon increasing the amount of A, the photocatalytic activity of binary Pdots increased initially. It was noted that there was almost no difference in the XRD results for Pdots with 20 wt%, 39 wt%, and 52 wt% of A, while a slight increase in the HER along with the increasing amount of A is probably because there is more ITIC exposed on the surface of the Pdots that thus benefits the electron transfer from the ITIC to the Pt cocatalyst. The highest photocatalytic activity was obtained for binary Pdots with A of 60 wt% to 80 wt%. Increasing the concentration of A increased the charge mobility and slowed the charge-carrier recombination, which further led to a higher probability of extracting electrons from the binary Pdots to the Pt cocatalyst, resulting in better performance in photocatalytic hydrogen evolution. Further increasing the amount of A to 95 wt% resulted in relatively reduced hydrogen evolution, probably due to the lowered absorption of D. The external quantum efficiency (EQE) measurement of the binary Pdots with 62 wt% of A indicates that it gives EQEs within a wavelength range from 420 nm to 700 nm, which matches well with the absorption spectrum, as shown in Fig. S15 (ESI†). The maximum EQE obtained was 2.1% at 650 nm. However, the optimal photocatalytic activity of the binary Pdots in the current work exhibits only half of the performance of the ternary Pdots in our previous work,27 in which PFBT was used as an additional donor and formed a type I heterojunction with PFODTBT (here, D) as well as with ITIC (here, A). In addition to the lack of photon absorption around the 450 nm region compared to ternary Pdots, the major difference for the binary Pdots in this work is the lack of crystallinity in the acceptor phase and better intermixed D/A phases, which may lead to the higher possibility of charge recombination and lower photocatalytic hydrogen evolution. | ||
| Fig. 6 Amount of H2 evolved for the binary Pdots over 24 h as a function of the mass ratio of A. Reaction conditions: 60 μg with 6 wt% of Pt in 2 mL of a 0.2 M ascorbic acid solution, pH 4, in a 9 mL air-tight vial. The data for the HER versus time are presented in Fig. S16 (ESI†). | ||
Conclusions
In conclusion, we studied the excited-state, photoinduced charge carrier generation, and recombination dynamics in detail by using PFODTBT/ITIC (D/A) binary Pdots as a model system. With increasing the amount of A in the binary PFODTBT-ITIC Pdots, photophysical process transition from multi-steps of energy transfer together with electron transfer to one step of electron transfer from excited D to A was identified by steady-state photoluminescence and excitation spectra. Time-resolved photoluminescence and absorption spectra results indicate that ultrafast energy transfer and charge generation occurred on a sub-picosecond timescale for the studied D/A mass ratios, and that charge generation dynamics may not be affected by the energetic disorder of the donor polymer. A slower charge carrier recombination was observed with increasing ITIC mass ratio, contributing to higher crystallinity and a comparably better phase segregation. This may further contribute to higher charge extraction efficiencies from the Pdots to the co-catalyst Pt NPs, resulting in enhanced photocatalytic hydrogen evolution. From this study, as also suggested in the organic photovoltaics field, suppressing charge recombination was found to allow sufficient charge extraction. For type II heterojunction Pdots, increasing the crystallinity of the heterojunction components, and proper phase segregation between the donor and acceptor, are essential factors to prolong the charge carrier lifetime, therefore enhancing photocatalytic hydrogen evolution.Experimental
Materials
The semiconducting polymer PFODTBT polymer (Mw 50-80 kDa) was purchased from Solaris Chem. The n-type nonfullerene acceptor 3,9-bis(2-methylene-(3-(1,1-dicyanomethylene)-indanone))-5,5,11,11-tetrakis(4-hexylphenyl)-dithieno[2,3-d:2′,3′-d′]-s-indaceno[1,2-b:5,6-b′] dithiophene (ITIC) was purchased from Ossila, UK. The copolymer, polystyrene grafted with ethylene oxide and carboxyl groups (PS-PEG-COOH, backbone chain Mw 8500, graft chain Mw 4600, total chain Mw 36![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 500), was purchased from Polymer Source Inc., Canada. All other chemical reagents were purchased from Sigma Aldrich and used as received unless indicated otherwise.
500), was purchased from Polymer Source Inc., Canada. All other chemical reagents were purchased from Sigma Aldrich and used as received unless indicated otherwise.
Preparation of Pdots
Pdots in aqueous solutions were prepared using a modified method according to the literature.6,27 In brief, PFODTBT was dissolved in tetrahydrofuran (THF) at a concentration of 0.1 mg mL−1. ITIC and PS-PEG-COOH at a concentration of 1.0 mg mL−1 in THF were prepared. 10 mL of PFODTBT, 3 mL of PS-PEG-COOH and various amounts of ITIC solutions were mixed and then sonicated for 2–5 min. The resulting mixture was then added to 20 mL of deionized water and left at room temperature in a fume-hood overnight to remove THF completely. Pure PFODTBT Pdots and ITIC dots were prepared using the same procedure as that of the binary Pdots. All samples were filtered through a 0.45 μm syringe filter before further use. The exact composition of the obtained binary Pdots was determined as follows, 100 μL of sample was freeze-dried using liquid nitrogen and dissolved in a certain amount of THF, then the solution was measured by UV-vis to determine the final weight of the Pdots in solution.Photocatalytic hydrogen generation
The concentration of all the Pdot solutions for photocatalysis were adjusted to 40 μg mL−1 (without accounting for PS-PEG-COOH). Photocatalytic hydrogen evolution was performed in 9 mL gastight vials. A 1.5 mL amount of Pdots (60 μg) with various ITIC weight ratios was first mixed with 6 wt% of Pt precursor and purged with argon (Ar) for 20 min; then, 0.5 mL of pre-purged ascorbic acid aqueous solution (0.8 M, pH 4 adjusted by 2 M KOH) was added into the above solution. The mixture was purged with Ar for a further 30 min in order to completely remove oxygen. An LED PAR38 lamp (17 W, 5000 K, Zenaro Lighting GmbH, λ > 420 nm) was used as the light source. The light intensity illuminated on the active area of the sample was 50 mW cm−2, which was measured using a pyranometer (CM11, Kipp & Zonen, Delft, Holland). The LED light source basically has a similar intensity to standard 1 sun conditions between 420 and 750 nm. The generated hydrogen was quantified by an HPR-20 benchtop gas analysis system (HIDEN analytical) using Ar as the carrier gas.External quantum efficiency
External quantum efficiency (EQE) was tested by using 2 mL ternary Pdots with 62 wt% ITIC (0.2 M ascorbic acid, pH 4, 6 wt% of Pt) in a 9 mL airtight vial. The solution was illuminated using a 300 W Xe lamp (AULTT CEL-HXF300/CEL-HXUV300) as a light source equipped with an AM1.5 filter and different band pass filters (CEAULIGHT, 450, 500, 550, 600, 650, 700 and 765 nm) were used to select a particular wavelength. The hydrogen was measured by an HPR-20 benchtop gas analysis system (HIDEN Analytical) using Ar as a carrier gas. The EQE was calculated using the following equations:
where np represents the moles of incident photons, I is the radiant power, λ is the light wavelength, t is the irradiation time (excluding the induction time), h is Planck's constant, NA is the Avogadro constant, and c is the speed of light.

Cryo-transmission electron microscopy (Cryo-TEM)
Samples were analyzed by cryo-TEM as described earlier.45 Samples were equilibrated at 25 °C under a high relative humidity within a climate chamber. A small drop of each sample was deposited on a carbon-sputtered copper grid covered with a perforated polymer film. Excess liquid was thereafter removed by blotting with filter paper, leaving a thin film of the solution on the grid. The sample was vitrified in liquid ethane and transferred to a microscope, continuously kept below −160 °C and protected against atmospheric conditions. Analyses were performed with a Zeiss Libra 120 transmission electron microscope (Carl Zeiss AG, Oberkochen, Germany) operated at 80 kV in zero-loss bright-field mode. Digital images were recorded under low-dose conditions with a BioVision Pro-SM Slow Scan CCD camera (Proscan Elektronische Systeme GmbH, Scheuring, Germany).Dynamic light scattering (DLS) measurements
The hydrodynamic diameter of the samples was measured using a Zetasizer Nano-S from Malvern Instruments Nordic AB. Average data were obtained from at least five runs of measurements.Steady-state absorption and photoluminescence measurements
Steady-state UV-vis measurements were analyzed by using a Varian Cary 5000 spectrometer. Steady-state photoluminescence spectra were analyzed by using a Fluorolog 3-222 emission spectrophotometer (Horiba Jobin-Yvon) together with the FluorEssence software.Time-resolved photoluminescence measurements (streak camera)
The pumping light was generated by a high-power femtosecond laser with a wavelength of 1030 nm (Jasper 10, Fluence). After wavelength tuning from an optical parametric amplifier (Harmony, Fluence), an output laser with wavelengths of 550 nm was generated. The excitation laser beam was focused on the samples at an angle of 45° by a convex mirror. After photoexcitation, the photoluminescence emission was collected at a right angle from the pump light and diffracted by a Bruker SPEC 2501S spectrograph, collimated and then focused by two lenses with a 50 mm focal length. A long-pass filter with 550 nm was used before the detection system. The PL emission was collected and sent to a 100 μm width input slit of a spectrograph with a grating of 50 lines mm−1. Then, the PL emission arrived at a streak camera (C5680 + M5675, Hamamatsu) with a temporal instrument response function (IRF) of 50 ps. Finally, the signal was received by a digital camera (C4742-95, Hamamatsu). The background signal and camera sensitivity were corrected after measuring and data processing. The data was processed via SurfaceXplore and fitted to a trial mono-exponential decay model or biexponential decay model by convolution a Gaussian instrument response function.Time-correlated single photon counting (TCSPC) measurements were carried out in a time window of 50 ns by using a pulsed diode laser source (Edinburgh Instruments EPL470, λex = 470 nm).
Transient absorption spectroscopy (TAS)
Femto-second transient spectroscopy was performed using a 3 kHz 800 nm output of a Ti:sapphire amplifier (1.5 mJ, 45 fs FWHM, Libra, Coherent), which was split into pump and probe. The excitation wavelength at λ = 550 and 710 nm was generated by a femtosecond optical parametric amplifier (TOPAS-PRIME). The excitation was made pseudo-unpolarized by a depolarizer (Thorlabs) to avoid the major effects of rotational depolarization. A fundamental laser (probe, 800 nm) passed through the delay stage (8.5 ns and 1–2 fs step size) and was focused in a CaF2 optical window in order to generate white light. The pump beam profile was assumed to have a Gaussian distribution, with a full width at half-maximum (FWHM) of around 300 μm, and the time resolution, i.e., the instrument response function, was ca. 150–200 fs. The probe spectra were recorded by a custom-made 200–1000 nm silicon diode array (Newport). The femtosecond transient absorption data were analysed by using SurfaceXplore, and Glotaran software based on the fitting package TIMP.46,47 The kinetic traces were fitted using a sum of convoluted exponentials:where

 and IRF represent the width of the instrument response function (full width at half-maximum), t0 is zero time, Ai and τi are amplitude and decay times, respectively, and * is the convolution operator.
and IRF represent the width of the instrument response function (full width at half-maximum), t0 is zero time, Ai and τi are amplitude and decay times, respectively, and * is the convolution operator.
Photocatalytic hydrogen generation
Photocatalytic hydrogen evolution was performed in 9 mL gastight vials. 1.5 mL of Pdots with various ITIC weight ratios was mixed with a specific amount of aqueous potassium hexachloroplatinate solution containing 4 μg of Pt and purged with argon (Ar) for 20 min. Then, 0.5 mL of pre-purged ascorbic acid aqueous solution (0.8 M, pH 4 adjusted by 2 M KOH) was added into the above solution. The mixture was purged with Ar for a further 30 min in order to completely remove the oxygen. An LED PAR38 lamp (17 W, 5000 K, Zenaro Lighting GmbH, λ > 420 nm) was used as the light source. The light intensity illuminated on the active area of the sample was 50 mW cm−2, which was measured by a pyranometer (CM11, Kipp&Zonen, Delft/Holland). The LED light source basically has a similar intensity to standard 1 sun conditions between 420 and 750 nm. The generated hydrogen was quantified by an HPR-20 benchtop gas analysis system (HIDEN Analytical) using Ar as a carrier gas.Powder X-ray diffraction (PXRD)
PXRD patterns were collected at ambient temperature using a Simons D5000 diffractometer (Cu Kα, λ = 0.154 18 nm) at 45 kV and 40 mA, using a step size of 0.02° and a scan speed of 2 s per step in the range of 2–50°. The diffractometer was equipped with parallel beam optics (mirror + mirror) for grazing-incidence XRD measurements.Author contributions
H. T. and A. L. conceived the project. A. L. conducted the experiments and analysed the data. S. W. contributed to the st-st analysis, EQE analysis and TCSPC analysis. H. S. contributed to the global fitting. Y. L. contributed to the XRD analysis. L. G. and K. E. helped with the cryo-TEM analysis and discussion. L. H., H. T. and A. L. discussed the st-st and time resolved spectroscopy data, as well as participating in discussion on manuscript writing. All authors contributed to the discussion and manuscript editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by Olle Engkvist foundation and Wallenberg Academy Fellow program from K&A Wallenberg Foundation. The authors thank Yocefu Hattori (UU) for the help with the femtosecond laser and streak camera introduction and Mariia Pavliuk (UU) for the introduction to XRD.References
- A. Fujishima and K. Honda, Nature, 1972, 238, 37–38 CrossRef CAS PubMed.
- S. Yanagida, A. Kabumoto, K. Mizumoto, C. Pac and K. Yoshino, J. Chem. Soc., Chem. Commun., 1985, 474–475 RSC.
- J. Kosco, M. Sachs, R. Godin, M. Kirkus, L. Francas, M. Bidwell, M. Qureshi, D. Anjum, J. R. Durrant and I. McCulloch, Adv. Energy Mater., 2018, 8, 1802181 CrossRef.
- H. Yang, X. Li, R. S. Sprick and A. I. Cooper, Chem. Commun., 2020, 56, 6790–6793 RSC.
- C. Dai and B. Liu, Energy Environ. Sci., 2020, 13, 24–52 RSC.
- P. B. Pati, G. Damas, L. Tian, D. L. A. Fernandes, L. Zhang, I. B. Pehlivan, T. Edvinsson, C. M. Araujo and H. Tian, Energy Environ. Sci., 2017, 10, 1372–1376 RSC.
- J.-Z. Cheng, L.-L. Liu, G. Liao, Z.-Q. Shen, Z.-R. Tan, Y.-Q. Xing, X.-X. Li, K. Yang, L. Chen and S.-Y. Liu, J. Mater. Chem. A, 2020, 8, 5890–5899 RSC.
- G. Zhang, L. Lin, G. Li, Y. Zhang, A. Savateev, S. Zafeiratos, X. Wang and M. Antonietti, Angew. Chem., Int. Ed., 2018, 57, 9372–9376 CrossRef CAS PubMed.
- Y. Ma, Y. Fu, W. Jiang, Y. Wu, C. Liu, G. Che and Q. Fang, J. Mater. Chem. A, 2022, 10, 10092–10097 RSC.
- W. Zhao, P. Yan, B. Li, M. Bahri, L. Liu, X. Zhou, R. Clowes, N. D. Browning, Y. Wu, J. W. Ward and A. I. Cooper, J. Am. Chem. Soc., 2022, 144, 9902–9909 CrossRef CAS PubMed.
- Z.-A. Lan, M. Wu, Z. Fang, Y. Zhang, X. Chen, G. Zhang and X. Wang, Angew. Chem., Int. Ed., 2022, 61, e202201482 CAS.
- M. V. Pavliuk, S. Wrede, A. Liu, A. Brnovic, S. Wang, M. Axelsson and H. Tian, Chem. Soc. Rev., 2022, 51, 6909–6935 RSC.
- C. Han, S. Xiang, P. Xie, P. Dong, C. Shu, C. Zhang and J.-X. Jiang, Adv. Funct. Mater., 2022, 32, 2109423 CrossRef CAS.
- C. Wu, B. Bull, C. Szymanski, K. Christensen and J. McNeill, ACS Nano, 2008, 2, 2415–2423 CrossRef CAS PubMed.
- S. Kim, C.-K. Lim, J. Na, Y.-D. Lee, K. Kim, K. Choi, J. F. Leary and I. C. Kwon, Chem. Commun., 2010, 46, 1617–1619 RSC.
- X. Pan, A. Sharma, R. Kroon, D. Gedefaw, S. Elmas, Y. Yin, G. G. Andersson, D. A. Lewis and M. R. Andersson, Front. Mater., 2020, 7, 281 CrossRef.
- A. Holmes, E. Deniau, C. Lartigau-Dagron, A. Bousquet, S. Chambon and N. P. Holmes, ACS Nano, 2021, 15, 3927–3959 CrossRef CAS PubMed.
- L. Wang, R. Fernández-Terán, L. Zhang, D. L. A. Fernandes, L. Tian, H. Chen and H. Tian, Angew. Chem., Int. Ed., 2016, 55, 12306–12310 CrossRef CAS PubMed.
- C. M. Aitchison, R. S. Sprick and A. I. Cooper, J. Mater. Chem. A, 2019, 7, 2490–2496 RSC.
- A. Liu, C.-W. Tai, K. Holá and H. Tian, J. Mater. Chem. A, 2019, 7, 4797–4803 RSC.
- Z. Hu, Z. Wang, X. Zhang, H. Tang, X. Liu, F. Huang and Y. Cao, iScience, 2019, 13, 33–42 CrossRef CAS PubMed.
- Y. Tamai, H. Ohkita, H. Benten and S. Ito, J. Phys. Chem. Lett., 2015, 6, 3417–3428 CrossRef CAS PubMed.
- B. A. Pinaud, J. D. Benck, L. C. Seitz, A. J. Forman, Z. Chen, T. G. Deutsch, B. D. James, K. N. Baum, G. N. Baum, S. Ardo, H. Wang, E. Miller and T. F. Jaramillo, Energy Environ. Sci., 2013, 6, 1983–2002 RSC.
- J. Kosco, S. Gonzalez-Carrero, C. T. Howells, W. Zhang, M. Moser, R. Sheelamanthula, L. Zhao, B. Willner, T. C. Hidalgo, H. Faber, B. Purushothaman, M. Sachs, H. Cha, R. Sougrat, T. D. Anthopoulos, S. Inal, J. R. Durrant and I. McCulloch, Adv. Mater., 2021, 2105007 Search PubMed.
- J. Kosco, M. Bidwell, H. Cha, T. Martin, C. T. Howells, M. Sachs, D. H. Anjum, S. Gonzalez Lopez, L. Zou, A. Wadsworth, W. Zhang, L. Zhang, J. Tellam, R. Sougrat, F. Laquai, D. M. DeLongchamp, J. R. Durrant and I. McCulloch, Nat. Mater., 2020, 19, 559–565 CrossRef CAS PubMed.
- J. Kosco, S. Gonzalez-Carrero, C. T. Howells, T. Fei, Y. Dong, R. Sougrat, G. T. Harrison, Y. Firdaus, R. Sheelamanthula, B. Purushothaman, F. Moruzzi, W. Xu, L. Zhao, A. Basu, S. De Wolf, T. D. Anthopoulos, J. R. Durrant and I. McCulloch, Nat. Energy, 2022, 7, 340–351 CrossRef CAS.
- A. Liu, L. Gedda, M. Axelsson, M. Pavliuk, K. Edwards, L. Hammarström and H. Tian, J. Am. Chem. Soc., 2021, 143, 2875–2885 CrossRef CAS PubMed.
- X. Wang, L. C. Groff and J. D. McNeill, J. Phys. Chem. C, 2014, 118, 25731–25739 CrossRef CAS.
- S. Stainmesse, A.-M. Orecchioni, E. Nakache, F. Puisieux and H. Fessi, Colloid Polym. Sci., 1995, 273, 505–511 CrossRef CAS.
- S. De, T. Kesti, M. Maiti, F. Zhang, O. Inganäs, A. Yartsev, T. Pascher and V. Sundström, Chem. Phys., 2008, 350, 14–22 CrossRef CAS.
- S. De, T. Pascher, M. Maiti, K. G. Jespersen, T. Kesti, F. Zhang, O. Inganäs, A. Yartsev and V. Sundström, J. Am. Chem. Soc., 2007, 129, 8466–8472 CrossRef CAS PubMed.
- S. N. Clafton, D. M. Huang, W. R. Massey and T. W. Kee, J. Phys. Chem. B, 2013, 117, 4626–4633 CrossRef CAS PubMed.
- S. K. Pal, T. Kesti, M. Maiti, F. Zhang, O. Inganäs, S. Hellström, M. R. Andersson, F. Ostwald, F. Langa, T. Österman, T. Pascher, A. Yartsev and V. Sundström, J. Am. Chem. Soc., 2010, 132, 12440–12451 CrossRef CAS PubMed.
- F. Etzold, I. A. Howard, R. Mauer, M. Meister, T.-D. Kim, K.-S. Lee, N. S. Baek and F. Laquai, J. Am. Chem. Soc., 2011, 133, 9469–9479 CrossRef CAS PubMed.
- H. Yao, Y. Cui, D. Qian, C. S. Ponseca, A. Honarfar, Y. Xu, J. Xin, Z. Chen, L. Hong, B. Gao, R. Yu, Y. Zu, W. Ma, P. Chabera, T. Pullerits, A. Yartsev, F. Gao and J. Hou, J. Am. Chem. Soc., 2019, 141, 7743–7750 CrossRef CAS PubMed.
- S. Gélinas, A. Rao, A. Kumar, S. L. Smith, A. W. Chin, J. Clark, T. S. van der Poll, G. C. Bazan and R. H. Friend, Science, 2014, 343, 512 LP–516 CrossRef PubMed.
- K. N. Schwarz, S. B. Farley, T. A. Smith and K. P. Ghiggino, Nanoscale, 2015, 7, 19899–19904 RSC.
- F. Laquai, D. Andrienko, C. Deibel and D. Neher, in Elementary Processes in Organic Photovoltaics, ed. K. Leo, Springer International Publishing; Cham, 2017, pp. 267–291 Search PubMed.
- Y. Song, S. N. Clafton, R. D. Pensack, T. W. Kee and G. D. Scholes, Nat. Commun., 2014, 5, 4933 CrossRef PubMed.
- X. Ma, M. Li, P. Pfeiffer, J. Eisener, C.-D. Ohl and C. Sun, J. Colloid Interface Sci., 2022, 606, 1380–1394 CrossRef CAS PubMed.
- C. Xie, T. Heumüller, W. Gruber, X. Tang, A. Classen, I. Schuldes, M. Bidwell, A. Späth, R. H. Fink, T. Unruh, I. McCulloch, N. Li and C. J. Brabec, Nat. Commun., 2018, 9, 5335 CrossRef CAS PubMed.
- L. C. Groff, X. Wang and J. D. McNeill, J. Phys. Chem. C, 2013, 117, 25748–25755 CrossRef CAS.
- M. Stolterfoht, A. Armin, S. Shoaee, I. Kassal, P. Burn and P. Meredith, Nat. Commun., 2016, 7, 11944 CrossRef CAS PubMed.
- D. T. Scholes, P. Y. Yee, J. R. Lindemuth, H. Kang, J. Onorato, R. Ghosh, C. K. Luscombe, F. C. Spano, S. H. Tolbert and B. J. Schwartz, Adv. Funct. Mater., 2017, 27, 1702654 CrossRef.
- M. Almgren, K. Edwards and G. Karlsson, Colloids Surf., A, 2000, 174, 3–21 CrossRef CAS.
- J. J. Snellenburg, S. Laptenok, R. Seger, K. M. Mullen and I. H. M. van Stokkum, J. Stat. Softw., 2012, 49, 1–22 Search PubMed.
- I. H. M. van Stokkum, D. S. Larsen and R. van Grondelle, Biochim. Biophys. Acta, Bioenerg., 2004, 1657, 82–104 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2cp04204e |
| This journal is © the Owner Societies 2023 |