Oxygen diffusion in the orthorhombic FeNbO4 material: a computational study†
Received
5th September 2022
, Accepted 17th January 2023
First published on 18th January 2023
Abstract
ABO4-type materials have shown significant potential for applications as luminescence and photocatalytic materials, and the orthorhombic FeNbO4 (o-FeNbO4) material has also shown excellent promise in catalytic electrodes, unlike other common ABO4 materials. However, little computational work has been carried out on the o-FeNbO4 structure, potentially because it is disordered and thus not straightforward to simulate. In this work, we first confirmed the accuracy of the force field parameters obtained from previous studies through optimizations carried out using the GULP code. Next, we found that one ordered configuration of the stoichiometric o-FeNbO4 structure dominates when analysing the probabilities of cation disorder in three supercells (2 × 2 × 1, 2 × 1 × 2, and 1 × 2 × 2). We then studied the bulk properties of this selected o-FeNbO4 through DFT calculations, including the lattice parameters, the mechanical properties and the electronic structures, where no remarkable differences were observed compared to the monoclinic FeNbO4 structure. Finally, because oxygen mobility is key to the successful application of o-FeNbO4 as an electrode material, we have simulated the diffusion pathways of oxygen through both the stoichiometric and non-stoichiometric structures, where the results show that the existence of oxygen vacancies enhances diffusion and the distribution of the Fe and Nb inside the lattice affects the energy barriers and could therefore impact the oxygen diffusion.
1. Introduction
ABO4-type oxides, e.g. FeWO4, FeVO4 or LaTaO4, have shown significant promise as functional oxides in many applications, for example in super-capacitors and solar cell electrodes.1–6 Generally, ABO4 materials can be classified into two types on the basis of the valence of element B, i.e. +5 or +6. ABO4 oxides with B = W6+ or Mo6+ have shown remarkable efficacy when used as super-capacitor and luminescence materials.5,7,8 For example, it has been reported that green photoluminescence emission of BaWO4 polymer nanohybrids was observed when the tetrahedral WO4 structure was changed to the octahedral WO6 structure,7 and an intense blue photoluminescence emission could also occur in BaWO4 powders due to the existence of defects around oxygen.7,9 Other ABO4-type materials (B = V5+, Nb5+ and Ta5+) normally show catalytic behaviour to some degree, as well as having potential as luminescence materials.3,6,10 For example, LaTaO4 doped with Eu is an interesting material for energy applications, as it catalyses aqueous methanol to generate hydrogen, whereas the BiVO4/FeVO4 heterojunction also displays similar photocatalytic performance when used to generate hydrogen through the splitting of water molecules.2,10
FeNbO4, as a typical ABO4 oxide, has shown good electrical and catalytic performance and thus has been developed for many years in applications such as gas sensing devices and solar energy conversion devices.11–15 So far, three main FeNbO4 structures have been observed during phase transitions, i.e. the monoclinic wolframite-type structure (m-FeNbO4, space group P2/c), the orthorhombic α-PbO2-type structure (o-FeNbO4, space group Pbcn) and the rutile-type structure (r-FeNbO4, space group P42/mnm). At temperatures below 1050 °C, m-FeNbO4 is the most stable polymorph, where stable [FeO6] and [NbO6] octahedra are formed through coordinating one Fe3+ or Nb5+ with six oxygen ions.16–18 Moreover, the Fe3+ and Nb5+ cations are distributed in an ordered fashion along zig-zag [FeO6] and [NbO6] octahedral chains.16 Above 1050 °C, which is the temperature at which the cation distribution becomes disordered, a phase transition occurs from the monoclinic to orthorhombic structure which is completed near 1100 °C. The resulting material shows a typical α-PbO2 structure, which remains stable in the temperature range from 1100–1380 °C. The second phase transition to generate the rutile-type structure is observed above 1380 °C and it remains stable until 1450 °C.16,19 In recent years, both m- and o-FeNbO4 materials have been explored for energy storage and conversion applications.20–22 Interestingly, both m- and o-FeNbO4 have shown potential as a replacement material for the Ni metal in the cathodes of solid oxide electrolysis cells (SOEC)21,22 and the anodes of solid oxide fuel cells (SOFC),20 where the splitting of water vapour takes place, although o-FeNbO4 is more suitable in electrodes than m-FeNbO4. It has been reported that, when m-FeNbO4 is used as anode in SOFC, there is a clear weight loss during its operation,20 caused by the ordering within the m-FeNbO4 system, where Fe3+ and Nb5+ layers are distributed alternately through the structure. Fe3+ is unstable, especially when exposed to a hydrogen atmosphere when it is easy for the Fe3+ to be reduced to the Fe2+, at the same time generating a water molecule, which causes the structure to collapse.20 This does not occur in the disordered o-FeNbO4 system and as such o-FeNbO4 may be the more promising material in energy conversion applications.
In previous work,23 we have employed calculations based on the density functional theory (DFT) to study the bulk and surface properties of m-FeNbO4. Due to the disorder of the cations in the o-FeNbO4 phase, it is more difficult to create a computational model to simulate its behaviours and properties, which is presumably the reason why few computational studies have been reported on the orthorhombic phase. In the present study, we have employed the SOD code24 to construct all the inequivalent cation configurations within the o-FeNbO4 structure to identify the most stable structures. In addition, we have investigated the existence of oxygen vacancies, which according to experimental results improve the electronic conductivity when introduced into the structure in small concentrations.20–22,25 There are few experimental reports on the diffusion of oxygen within the material, and in this work, we have also employed DFT calculations to investigate oxygen diffusion pathways, which are important in providing the oxygen required for the reaction with hydrogen, when the materials is used in catalytic electrodes.20–22
2. Computational methods
2.1 Force field parameters
The General Utility Lattice Program (GULP) code26,27 was employed to carry out calculations on a number of bulk properties. GULP is based on the Born model of ionic solids, which defines all atoms as charged spherical ions and describes their interactions through short-range repulsive and van der Waals forces and long-range Coulombic interactions.28–30 The short-range interactions in this work are described through Buckingham potentials and we have applied the core–shell model for the polarizable oxygen ions, where the total charge of the oxygen anions is split between a massive core and a massless shell. The interatomic potential (IP) parameters to describe the interactions between the Fe–O, Nb–O and O–O pairs are applicable to the simulation of both the monoclinic and orthorhombic FeNbO4 phases. The Fe–OS, Nb–OS and OS–OS potentials used in this work are those reported by Woodley and Bush31,32 and are listed in Table 1. In addition, we have studied the effect of oxygen vacancies on the distribution of the cations in the orthorhombic phase, and to compensate the charges in the oxygen-vacant structures, we have assumed that the surplus electrons generated by the oxygen vacancy remain on the neighbouring iron ions, whose charge is reduced from Fe3+ to Fe2+.
Table 1 Interatomic potential parameters used for the FeNbO4
| Pairs |
Buckingham potential parameters |
|
A (eV) |
ρ (Å) |
C (eV Å6) |
| Fe3+–OS−2.513 |
3219.34 |
0.2641 |
0 |
| Fe2+–OS−2.513 |
2763.94 |
0.2641 |
0 |
| Nb5+–OS−2.513 |
3023.18 |
0.3 |
0 |
| OS−2.513–OS−2.513 |
25.41 |
0.6937 |
32.32 |
| Ion |
Core–shell model parameters |
|
K
cs (eV Å−2) |
q
Oc (e) |
q
Os (e) |
| OC+0.513–OS−2.513 |
20.53 |
+0.513 |
−2.513 |
2.2 SOD calculations for the orthorhombic FeNbO4
We have employed the Site Occupancy Disorder (SOD) code24 to generate all of the Fe and Nb cation configurations within the orthorhombic FeNbO4 structure. We have built 2 × 2 × 1, 2 × 1 × 2 and 1 × 2 × 2 supercell models to obtain the energetically optimum mixed structures. All inequivalent configurations within the 2 × 2 × 1, 2 × 1 × 2 and 1 × 2 × 2 models were transferred into the GULP code to calculate the free energies, which were then used in SOD to carry out the analysis of the probability distribution of the inequivalent configurations. We have also carried out the same process on non-stoichiometric 2 × 2 × 1 models, to determine the dominant phases when oxygen vacancies are present in the structure.
2.3 DFT calculations
In this work, all the DFT calculations were carried out using the Vienna Ab initio Simulations Package, VASP (version 5.4.4),33–36 where we obtained the bulk properties of the preferred o-FeNbO4 model selected from the probabilities calculations above. The ion-electron interactions and the exchange–correlation interactions are described through the projector-augmented wave method (PAW)37 and the generalized gradient approximation (GGA)38 with the Perdew–Burke–Ernzerhof (PBE) density functional, respectively. Spin-polarization was employed in all calculations and we have treated the following as valence electrons: Fe (3p63d74s1), Nb (4p65s14d44s2), O (2s22p4). In addition, to improve the description of the electronic structures, we have used the on-site Coulombic interaction (DFT+U)39 for the Fe 3d electrons with the Ueff = U − J value of 4.3 eV. Following test calculations, the kinetic energy cut-off for the plane wave basis was set at 500 eV and the Henkelman algorithm was used to calculate the Bader charges.39 In addition, 3 × 3 × 3 gamma centered Monkhorst Pack grids were used for the simulation of the 2 × 2 × 2 bulk. We have considered the spin distribution of Fe atoms in the orthorhombic phase as a quasi-random one and this phase could be generated through the Alloy Theoretic Automated Toolkit (ATAT), which was employed in the special quasi-random structures (SQS) method.40–43
In the final part of the paper, we have simulated the diffusion of oxygen through the o-FeNbO4 lattice, where we have first placed an extra oxygen atom into an interstitial site inside the lattice and moved this atom in different directions. In order to obtain a good understanding of the diffusion pathways, we have employed the static lattice method, which has been used by Saadoune, Baykov and others,44–46 to determine the possible pathways. In this method, the diffusing atoms are fixed along the pathway, with the other atoms allowed to optimise fully, thereby avoiding the formation of stable structures which are not part of the diffusion pathways. Compared to the climbing image nudged elastic band (CI-NEB) method, which aims to find the saddle point in chemical reactions, the energy of transition states obtained through the static lattice method may be underestimated but it is a good description, and comparison, of different possible diffusion pathways.
3. Results and discussion
3.1 The GULP results
Elucidating the most probable cation configurations in the orthorhombic phase through the SOD and GULP codes requires suitable IP parameters. To this end, we have first employed the listed force field parameters in the GULP code to optimize the ordered monoclinic phase, and by evaluating whether the computational results are close to the experimental data, we could demonstrate the capability of the IP parameters to describe the interatomic interactions and next use them to simulate the orthorhombic phase.
The lattice parameters of the monoclinic FeNbO4 (m-FeNbO4) structure after optimization are listed in Table 2 and compared with a number of experimental results. The four structures obtained from experiments have been plotted in Fig. S1, S2 (ESI†), and by comparing the structure of experiments 3 and 4, we found that exchanging the Fe position for Nb does not affect the octahedral arrangement and the structural properties. The difference between the structures of experiments 1, 2 and 3, 4 is that experiments 1 and 2 show P2/a symmetry, while the structures in experiments 3 and 4 have P2/c symmetry, in which the parameters a and c are swapped compared to experiments 1 and 2. The computational results from the DFT calculations, discussed in detail in our previous work,23 are generally more accurate than those obtained from GULP, where all lattice parameters except for the β angle vary slightly from the DFT data. However, the error is within an acceptable margin and the force field parameters in Table 1 are accurate enough to employ in the rest of the work.
Table 2 Experimental and computational results for the monoclinic FeNbO4 lattice cell. (The experimental data of 1 and 2 show P2/a symmetry, while the data of experiments 3 and 4 show P2/c symmetry; we have swapped the a and c parameters of experiments 1 and 2 to make it consistent with the others)
|
|
Experiment 147 |
Experiment 218 |
Experiment 348 |
Experiment 419 |
DFT23 |
GULP |
|
a
|
5.008 |
4.992 |
4.646 |
4.650 |
4.687 |
4.697 |
|
b
|
5.632 |
5.607 |
5.615 |
5.616 |
5.691 |
5.573 |
|
c
|
4.656 |
4.637 |
4.996 |
4.998 |
5.038 |
5.102 |
|
β
|
90.27 |
90.06 |
89.85 |
90.15 |
90.44 |
87.31 |
| Fe (y) |
0.6742 |
0.6687 |
0.6680 |
0.1713 |
0.1657 |
0.1528 |
| Nb (y) |
0.1807 |
0.1824 |
0.1811 |
0.6799 |
0.6813 |
0.6720 |
| O1 (x) |
0.9402 |
0.919 |
0.2270 |
0.2251 |
0.2311 |
0.2234 |
| O1 (y) |
0.138 |
0.116 |
0.1106 |
0.1187 |
0.1172 |
0.1021 |
| O1 (z) |
0.774 |
0.767 |
0.9227 |
0.9113 |
0.9161 |
0.9134 |
| O2 (x) |
0.425 |
0.418 |
0.2704 |
0.2981 |
0.2754 |
0.2684 |
| O2 (y) |
0.384 |
0.384 |
0.3812 |
0.3827 |
0.3853 |
0.3920 |
| O2 (z) |
0.725 |
0.738 |
0.4177 |
0.437 |
0.4228 |
0.4384 |
To further validate the IP parameters, we have optimised both the monoclinic and orthorhombic FeNbO4 structures. The relaxed lattice energies and structural parameters are listed in Table 3. They show that the lattice energy of m-FeNbO4 is lower than the o-FeNbO4, which is consistent with experiment in that m-FeNbO4 should be more stable than o-FeNbO4.
Table 3 The lattice parameters and lattice energy of m-FeNbO4 and o-FeNbO4 from GULP simulations
|
|
a (Å) |
b (Å) |
c (Å) |
β (°) |
Lattice energy |
|
m-FeNbO4 |
4.697 |
5.573 |
5.102 |
87.31 |
−481.795 |
|
o-FeNbO4 |
4.679 |
5.526 |
5.096 |
90.00 |
−481.687 |
3.2 The dominant phase of the stoichiometric orthorhombic FeNbO4
In the SOD code, the number of inequivalent cation configurations in a 2 × 2 × 2 supercell of the disordered o-FeNbO4 material would reach 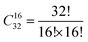 , which is an unrealistic number of configurations to compute. Thus, to still do a comprehensive study of the ordering in the structure, we have generated 2 × 2 × 1, 2 × 1 × 2, and 1 × 2 × 2 supercells. From Table 4, we note that in each of these supercells the total number of inequivalent configurations is
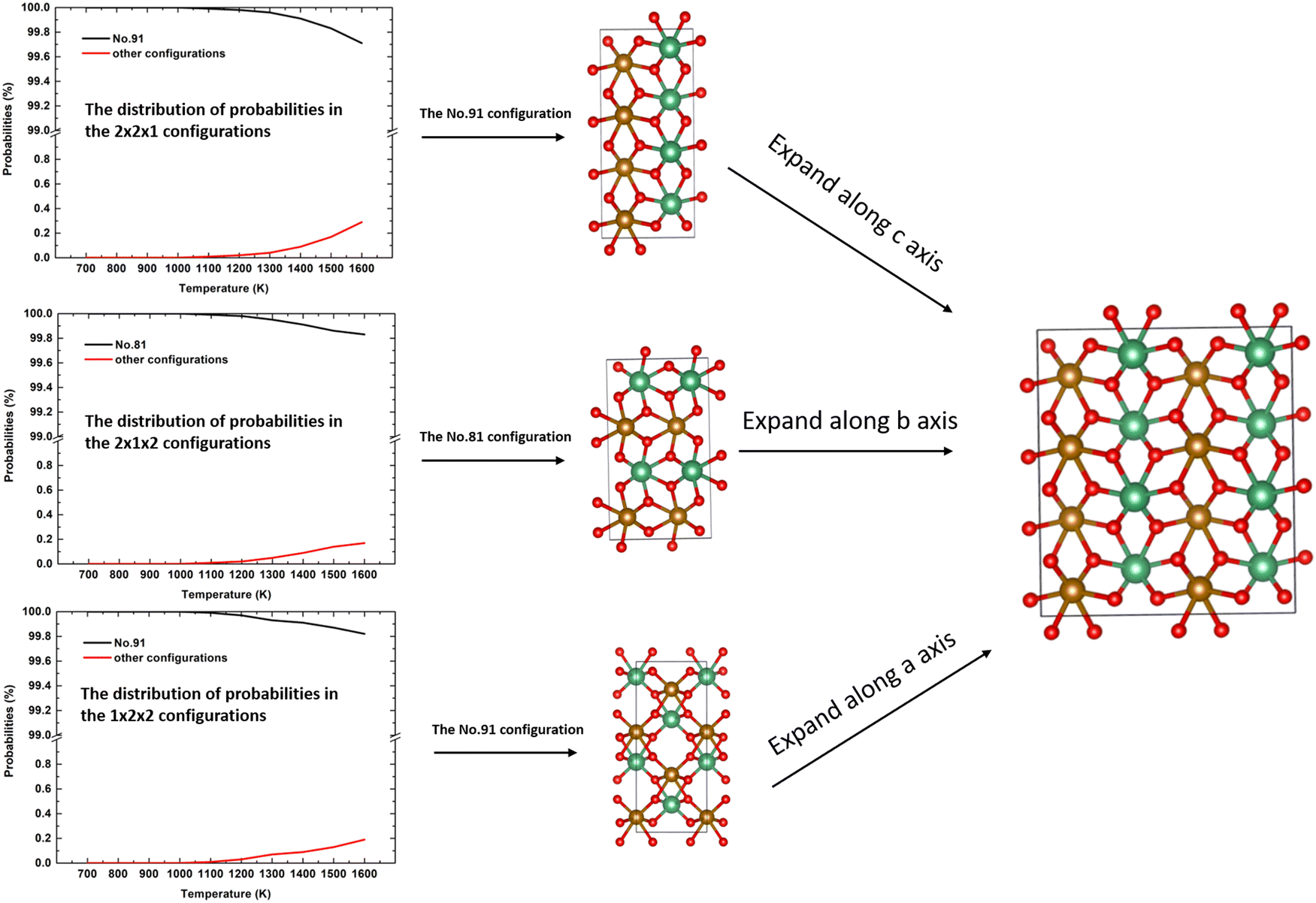
, which is an unrealistic number of configurations to compute. Thus, to still do a comprehensive study of the ordering in the structure, we have generated 2 × 2 × 1, 2 × 1 × 2, and 1 × 2 × 2 supercells. From Table 4, we note that in each of these supercells the total number of inequivalent configurations is 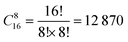 , which number is reduced after consideration of the symmetry, to 482, 448, and 474 for the three supercells, respectively. The results of the calculations of the probability distributions are shown in Fig. 1 from which it is clear that from 700 K to 1600 K, there is one inequivalent configuration (labelled No. 91, No. 81 and No. 91) in each cell whose probability of occurrence exceeds 99.5%. If we expand each cell to a 2 × 2 × 2 supercell, we see that all 3 cells obtained the same dominant and ordered phase as the most probably configuration. As such, from our calculations, it appears that the most stable phase is an ordered structure rather than the disordered material observed experimentally. For the sake of completeness, we have also calculated the contributions of the vibrational entropy to the free energy of both structures, which are listed in Table S2 (ESI†). However, it is clear that the difference in entropy of the two phases is so small that we can ignore its contribution to the structural stabilities.
, which number is reduced after consideration of the symmetry, to 482, 448, and 474 for the three supercells, respectively. The results of the calculations of the probability distributions are shown in Fig. 1 from which it is clear that from 700 K to 1600 K, there is one inequivalent configuration (labelled No. 91, No. 81 and No. 91) in each cell whose probability of occurrence exceeds 99.5%. If we expand each cell to a 2 × 2 × 2 supercell, we see that all 3 cells obtained the same dominant and ordered phase as the most probably configuration. As such, from our calculations, it appears that the most stable phase is an ordered structure rather than the disordered material observed experimentally. For the sake of completeness, we have also calculated the contributions of the vibrational entropy to the free energy of both structures, which are listed in Table S2 (ESI†). However, it is clear that the difference in entropy of the two phases is so small that we can ignore its contribution to the structural stabilities.
Table 4 Inequivalent configurations for the orthorhombic FeNbO4
| Supercell |
Cell numbers (111) |
Composition |
Total configurations (N) |
Inequivalent configurations (M) |
| 2 × 2 × 1 |
4 |
Fe8Nb8O32 |
12870 |
482 |
| 2 × 1 × 2 |
4 |
Fe8Nb8O32 |
12870 |
448 |
| 1 × 2 × 2 |
4 |
Fe8Nb8O32 |
12870 |
474 |
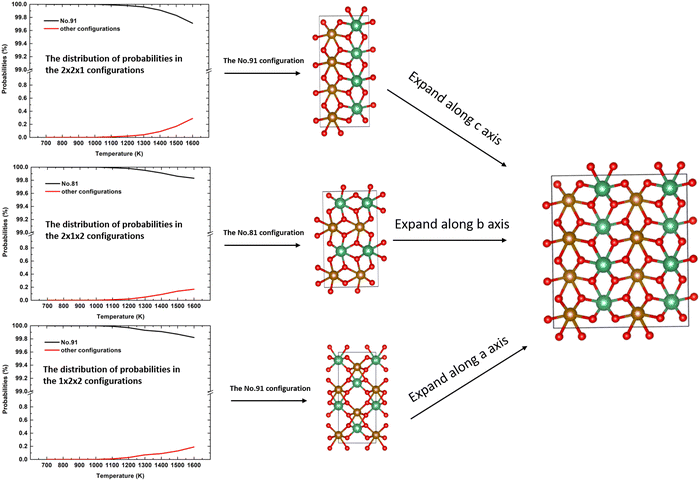 |
| | Fig. 1 The distributions of the probabilities in the 2 × 2 × 1, 2 × 1 × 2, and 1 × 2 × 2 supercells and the related most stable configurations; red is oxygen anion, brown is the Fe cation and green is the Nb cation. | |
3.3 The lattice parameters, mechanical properties and electronic structures
Before optimising the structure, we have first confirmed the magnetic distribution inside the lattice (Fig. S3, ESI†) according to our former study and the work by Lakshminarasimhan and co-workers,23,49 where the orthorhombic structure shows antiferromagnetic behaviour with small areas of spin glass. We next carried out DFT calculations to obtain a range of properties of the selected o-FeNbO4, shown in Fig. 2 and Table 5. As a comparison, we have added the same data for the m-FeNbO4 phase which show no remarkable differences in Bader charges or bond distances between the o-FeNbO4 and m-FeNbO4 structures. Interestingly, in the o-FeNbO4 structure, the magnetic moment of Fe could not maintain the same value as in the m-FeNbO4 structure, indicating that in a spin glass structure, the spin symmetry can be broken. In summary, both the results of the calculation of the probabilities and the electronic structure have demonstrated that the phase transition from the monoclinic to the orthorhombic structure does not impact the bulk properties.
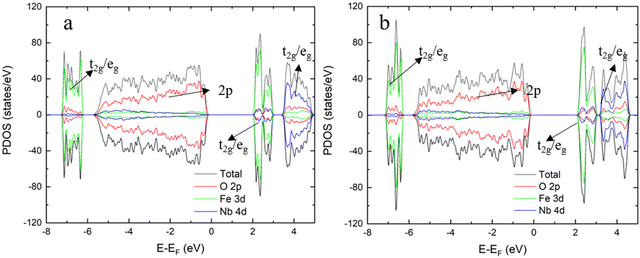 |
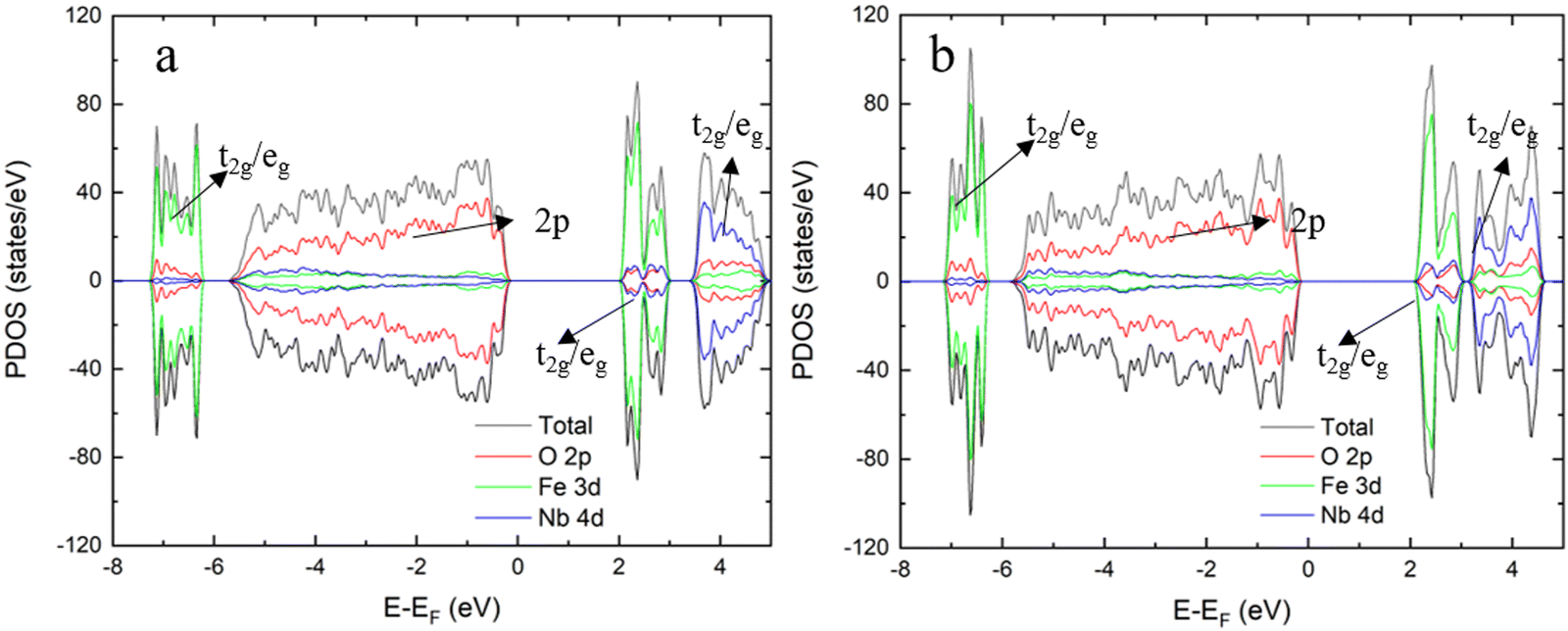
| | Fig. 2 The PDOS for the monoclinic FeNbO4 (a) and the orthorhombic FeNbO4 (b). | |
Table 5 The Bader charge, magnetic moment and bond distance of the m/o-FeNbO4 lattices. Negative atomic magnetic moments indicate antiparallel alignment
|
|
o-FeNbO4 |
m-FeNbO423 |
|
q
Fe (e) |
+1.88 |
+1.89 |
|
q
Nb (e) |
+2.72 |
+2.72 |
|
q
O (e) |
−1.16/−1.31 |
−1.16/−1.30 |
|
μ
Fe (μB) |
(±)4.17–(±)4.25 |
4.27 |
|
μ
Nb (μB) |
0 |
0 |
|
μ
O (μB) |
0 |
0 |
| Fe–O distance (Å) |
2.052 |
2.045 |
| Nb–O distance (Å) |
2.022 |
2.017 |
We have also calculated the mechanical properties of the ordered o-FeNbO4, including the elastic constants (Cij), bulk modulus (B), shear modulus (G) and Young's modulus (E), again comparing with the m-FeNbO4 material from our previous work.23 We have employed VASP and the finite difference technique to obtain the values of the elastic constants Cij, where the elastic tensor was obtained by performing finite distortions in the direction of each Cartesian coordinate of the lattice and deriving Cij through the stress–strain method as:
| | 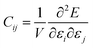 | (1) |
where
E is the total energy of the stressed cell,
V is the equilibrium volume and
ε is the component of the applied strain. Having calculated the values of
Cij, we obtain the values of
B,
G, and
E through the following equations:
| | 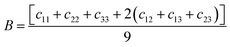 | (2) |
| | 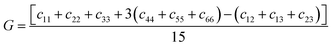 | (3) |
| | 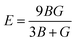 | (4) |
In contrast to the monoclinic phase, there are 9 independent elastic constants (C11, C22, C33, C44, C55, C66, C12, C13, and C23) for the orthorhombic structure, all listed in Table 6. We found that the C11 of m-FeNbO4 is smaller than that of o-FeNbO4, while the converse is true for the C33, indicating that along its a direction the m-FeNbO4 phase is softer, while along the c direction it is harder than the o-FeNbO4 phase. Next, we employed eqn (2)–(4) to calculate the values of the bulk modulus (B), the shear modulus (G) and the Young's modulus (E) and found that all moduli of the m-FeNbO4 phase are a little larger than that of the o-FeNbO4 phase, indicating the ability of the o-FeNbO4 phase to resist compression and elastic shear strain but its tensile stiffness is not as good as that of the m-FeNbO4 phase.
Table 6 The elastic constants and the mechanical properties of the m/o-FeNbO4
| Elastic constant (GPa) |
m-FeNbO423 |
o-FeNbO4 |
|
C
11
|
274.1 |
304.1 |
|
C
22
|
269.8 |
271.4 |
|
C
33
|
318.8 |
253.9 |
|
C
44
|
74.5 |
27.6 |
|
C
55
|
102.1 |
24.7 |
|
C
66
|
57.1 |
72.6 |
|
C
12
|
144.1 |
141.3 |
|
C
13
|
148.5 |
110.7 |
|
C
23
|
119.7 |
140.7 |
|
C
15
|
−6.8 |
— |
|
C
25
|
−11.7 |
— |
|
C
35
|
−22.9 |
— |
|
C
46
|
10.2 |
— |
|
B
|
187.5 |
179.4 |
|
G
|
76.8 |
54.1 |
|
E
|
202.7 |
147.5 |
Finally, we have compared the electronic structures of the m- and o-FeNbO4, whose DOS plots are shown in Fig. 2. Generally, there is a very similar DOS distribution for the m/o-FeNbO4 phases, except for a slight difference in the conduction band, where the integrated orbital is split into two small parts in the DOS of o-FeNbO4 at around 4.0 eV. Both phases show typical semiconductor characteristic with a band gap between the conduction band maximum (CBM) and the valence band minimum (VBM), where the CBM is mainly comprised of the Fe 3d orbitals. Furthermore, it is believed that in the m-FeNbO4 phase, the electron conduction mainly occurs in the FeO6 layers along its a direction while after mixing the Fe and Nb, it is likely that the electron could be conducted through the 3D net inside.
3.4 Oxygen diffusion in the stoichiometric and non-stoichiometric structures
3.4.1 Oxygen diffusion through the stoichiometric o-FeNbO4 structure.
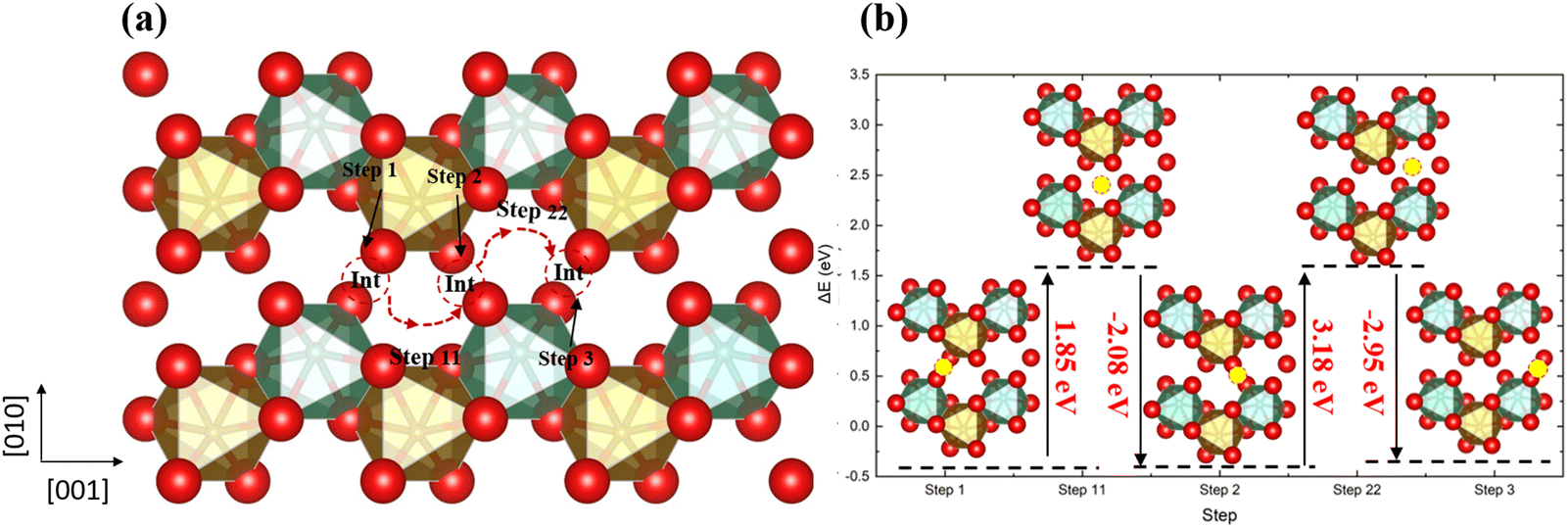
Next, we have introduced an extra interstitial oxygen atom into the 2 × 2 × 2 supercell, whose lattice is large enough that it does not affect the adjacent supercells, and used DFT to calculate the O diffusion pathways. In the stoichiometric o-FeNbO4 structure shown in Fig. 3, the lowest energy location for the insertion of an extra oxygen into the lattice is the interstitial site shown in Fig. S4 (ESI†). The formation energy of the oxygen interstitial atom is relatively high at 2.2 eV and we would therefore not expect this defect to occur widely, but we have investigated this defect here for the sake of completeness. Once an interstitial oxygen atom has been inserted in the supercell, this oxygen atom could move to the adjacent interstitial site through pathways in the 3D networks. We have considered a number of plausible diffusion pathways, along the [100], [010], and [001] directions. The diffusion pathway along the [001] direction has been plotted in Fig. 3, whereas the other pathways are shown in our ESI† (Fig. S5 and S6). Along the [001] direction, shown in Fig. 3, the diffusion starts from the interstitial site, and then moves towards to the gap site, where an energy barrier (up to 3.18 eV) needs to be overcome to diffuse to the adjacent interstitial site. This behaviour is not unique to the [001] direction and can be seen in other pathways in Fig. S5 and S6 (ESI†).
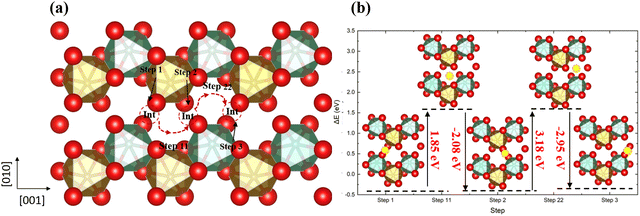 |
| | Fig. 3 The diffusion pathway of the interstitial oxygen atom along the [001] direction in the ordered stoichiometric structure (a), and the variation of the total energy (b); red is oxygen, brown is Fe, green is Nb and the yellow ball in (b) is the interstitial oxygen atom. | |
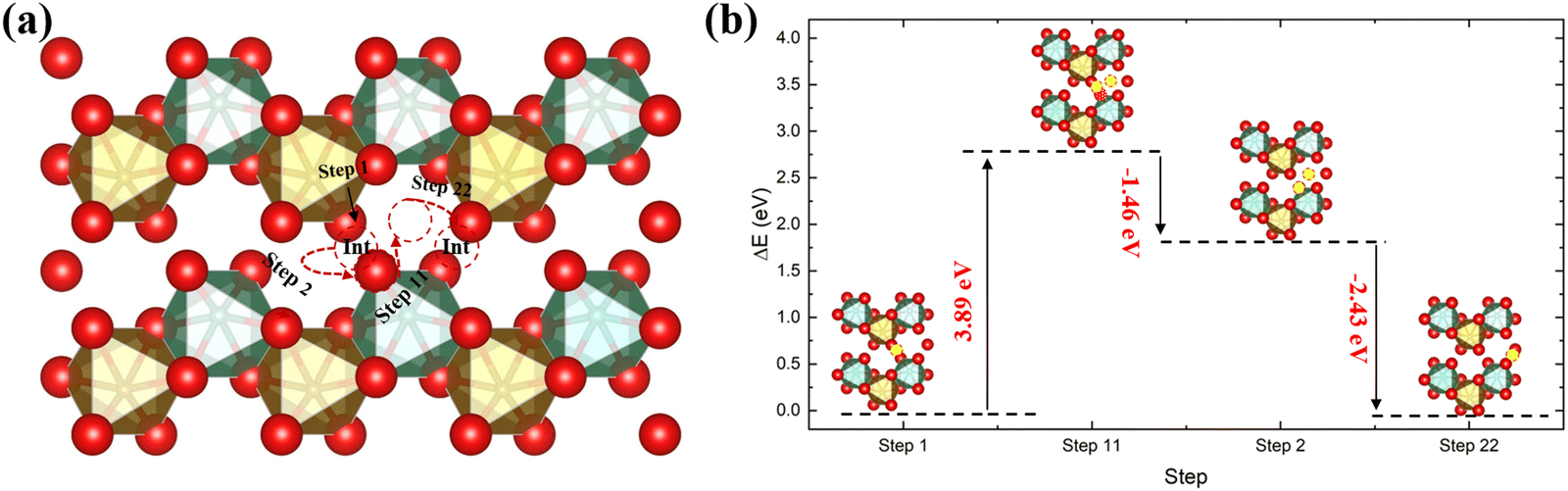
In addition to the direct diffusion pathway, we have also studied the interstitialcy mechanism along the [001] direction, which is the dominant diffusion pathway in many metal oxides.50,51 The result is plotted in Fig. 4, showing an initial energy barrier of 3.89 eV, i.e. considerably larger than in the direct diffusion pathway. This therefore suggests that in FeNbO4 materials the direct diffusion pathway should dominate, owing to the energetic cost of moving a lattice oxygen to the interstitial site compared to it moving through a gap site.
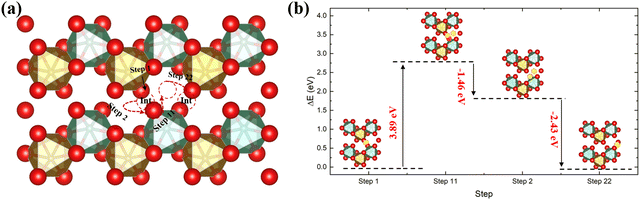 |
| | Fig. 4 The diffusion pathway of the interstitial mechanism along the [001] direction in the ordered stoichiometric structure (a), and the variation of the total energy (b); red is oxygen, brown is Fe, green is Nb and the yellow ball in (b) is the interstitial oxygen atom. | |
3.4.2 Diffusion of oxygen through oxygen-deficient FeNbO4.
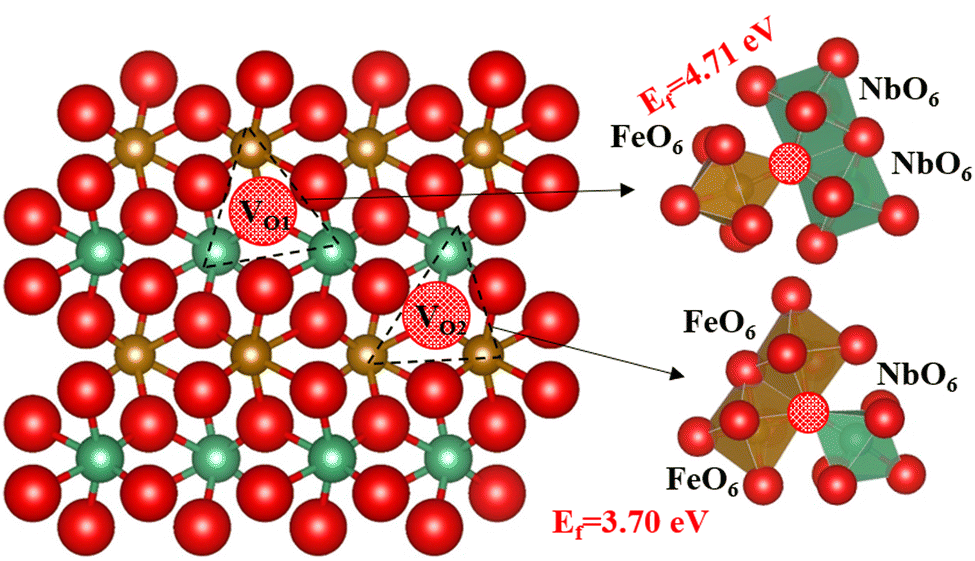
Next, we have introduced an oxygen vacancy into the o-FeNbO4 phase to study the oxygen diffusion in the presence of oxygen vacancies, as can be expected to occur in experimental electrode materials, also using DFT. There are two sites for the oxygen vacancy in the simulation cell of the ordered material, i.e. labelled VO1 which is surrounded by one FeO6 and two NbO6 octahedral, and labelled VO2 which is surrounded by two FeO6 and one NbO6 octahedral, shown in Fig. 5. Before studying the diffusion of the oxygen in this non-stoichiometric phase, we have calculated the formation energies of both vacancy types, also shown in Fig. 5, according to the equation: 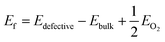 , where Edefective and Ebulk are the total energies of the non-stoichiometric material and the stoichiometric bulk material, respectively, and EO2 refers to the total energy of the oxygen molecule in the triplet ground state. The lattice parameters and lattice energies of the optimized structures are listed in Table S1 (ESI†), which show that the lattice with VO1 type vacancies is the most distorted structure in terms of volume and lattice energy. Overall, VO2 is the preferred oxygen vacancy site in the lattice due to its lower formation energy, although at 3.7 eV it is still a very high value, in agreement with experimental records which show that most oxygen vacancies are generated in the preparation process at temperatures of 1100 °C.16–20 We have therefore introduced the VO2 in the 2 × 2 × 2 supercells to investigate the oxygen diffusion pathways, where we noted that the formation energy of the oxygen vacancies does not change as the supercell is increased in size, which indicates that the reduction is a localised process, also shown by the short-range lattice distortion around the vacancy sites.
, where Edefective and Ebulk are the total energies of the non-stoichiometric material and the stoichiometric bulk material, respectively, and EO2 refers to the total energy of the oxygen molecule in the triplet ground state. The lattice parameters and lattice energies of the optimized structures are listed in Table S1 (ESI†), which show that the lattice with VO1 type vacancies is the most distorted structure in terms of volume and lattice energy. Overall, VO2 is the preferred oxygen vacancy site in the lattice due to its lower formation energy, although at 3.7 eV it is still a very high value, in agreement with experimental records which show that most oxygen vacancies are generated in the preparation process at temperatures of 1100 °C.16–20 We have therefore introduced the VO2 in the 2 × 2 × 2 supercells to investigate the oxygen diffusion pathways, where we noted that the formation energy of the oxygen vacancies does not change as the supercell is increased in size, which indicates that the reduction is a localised process, also shown by the short-range lattice distortion around the vacancy sites.
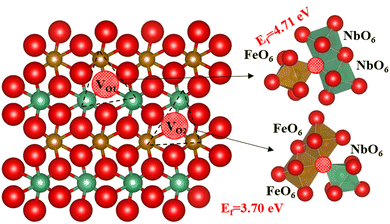 |
| | Fig. 5 The types of oxygen vacancies in the ordered o-FeNbO4; oxygen is red, Fe is brown, Nb is green and the red ball with black pattern represents the oxygen vacancy. | |
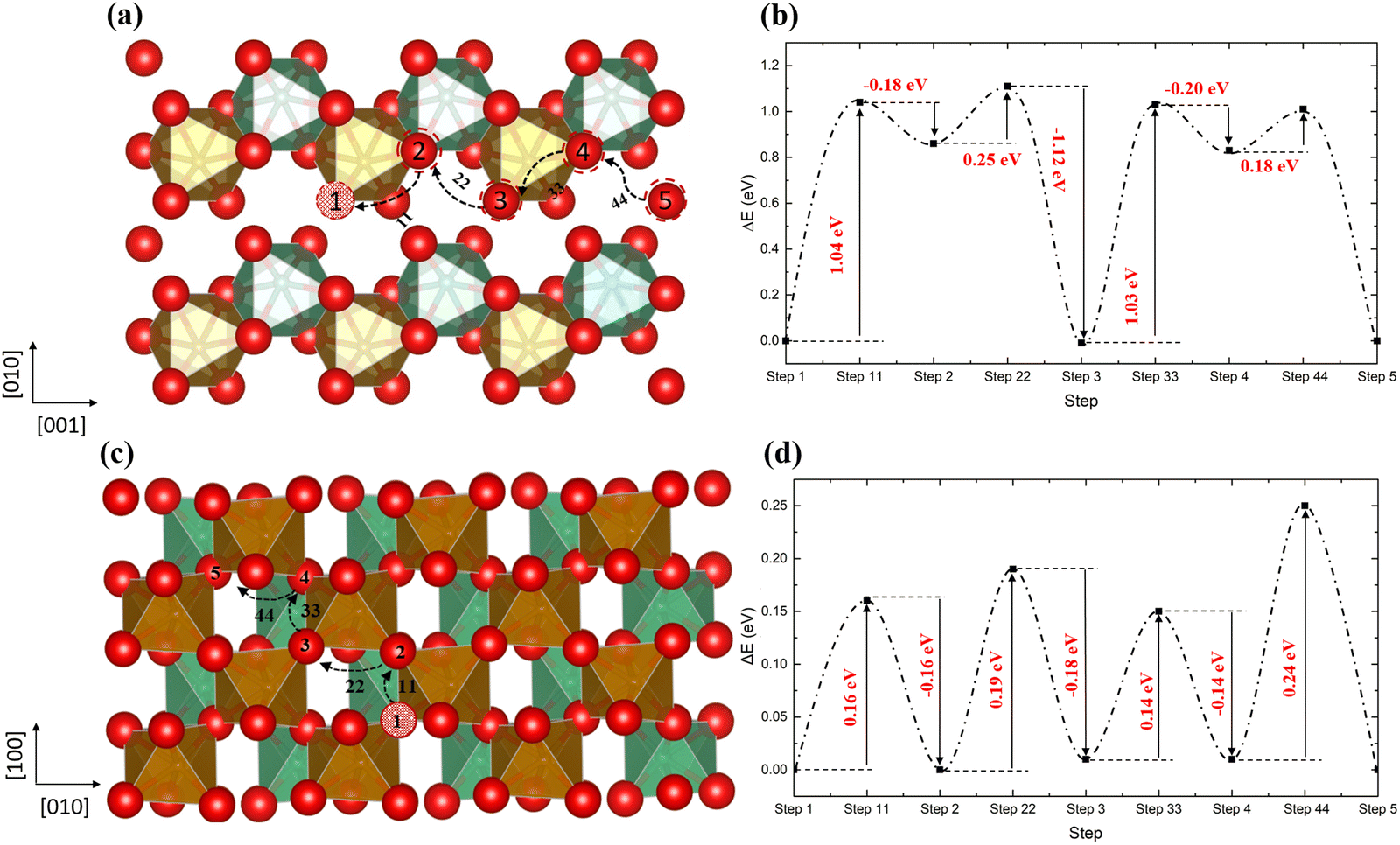
Usually, the oxygen atom in the oxygen vacancy-containing oxide materials prefers to diffuse through a hopping mechanism, similar to the process reported in BaFeO3, SrCoO3, and CeO2 materials.52–55Fig. 6 shows two possible pathways for this type of diffusion, where in Fig. 6a, the oxygen atom hopping occurs along the [001] direction with the nearest neighbouring oxygen hopping into the original and newly generated vacancy sites. The energy barriers are shown in Fig. 6b, where the vacancy sites 2 and 4 (type VO1) are less stable than the other three (1, 3 and 5 which are type VO2 vacancies). Thus, from step 1 to 2, there is an energy barrier of ∼1.04 eV, similar to the barrier from step 2 to 3 (∼1.03 eV). Finally, when the vacancy is transferred to site 5 (VO2) from 4 (VO1), the structure is stabilised again.
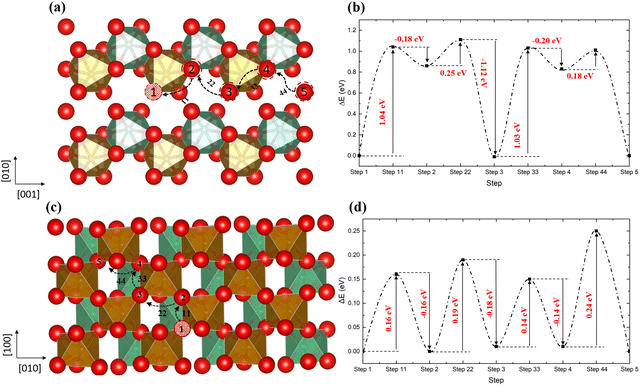 |
| | Fig. 6 The diffusion pathways of the oxygen atom along the [001] direction (a) and the [110] direction (c) in the ordered non-stoichiometric structures with an oxygen vacancy and their corresponding energy barriers (b and d); red is oxygen, brown is Fe, green is Nb and the red ball with black pattern represents the oxygen vacancy. | |
Along the [110] direction (Fig. 6c), which can be seen as a combination of the diffusion pathways along [010] and [100], all the energy barriers have similar values (∼0.2 eV), since all vacancies along this pathway are type VO2 vacancies. Therefore, although the oxygen could diffuse through the vacancy sites in the [100] way, the [110] pathway is energetically favourable. As expected, the presence of oxygen vacancies benefits oxygen diffusion through the lattice. In addition, we have noticed that for the oxygen atom, diffusing in the [110] direction is much easier than the [001] direction, since in the ordered structure all the vacancies along the [110] belong to the VO2 type.
3.4.3 The effect of cation disorder on the oxygen diffusion.
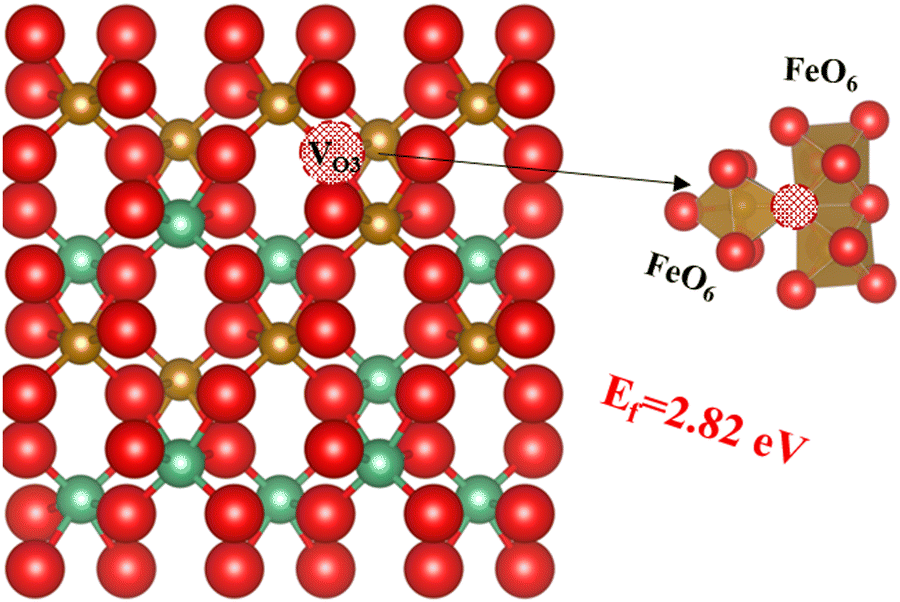
Since experimentally the orthorhombic phase is found to be disordered, we can also expect configurations with different Nb and Fe cation distributions to exist within the material. It has been reported that oxygen vacancies are found in the orthorhombic FeNbO4 structure, which will increase cation mobility and the likelihood of cation disorder in the lattice.25,47,56 We have demonstrated that the formation energy of oxygen vacancies depends on the oxygen site, and our earlier results suggest that if an oxygen were to be surrounded by three FeO6 octahedra, the formation energy of the oxygen vacancy will be reduced even further. To test this hypothesis using our DFT calculations, we have created a 2 × 2 × 1 supercell with this type of oxygen site and created the oxygen vacancy VO3, as shown in Fig. 7. As hypothesised, the oxygen formation energy at 2.82 eV is indeed lower than that for the former two oxygen vacancy types (3.70 eV and 4.71 eV for VO2 and VO1 respectively). Next, we have investigated this oxygen vacancy-containing configuration with SOD and GULP to simulate the probabilities of its inequivalent configurations. Note that for these simulations, we assigned the two electrons left behind by the O atoms to the two Fe cations closer to the O vacancy, which oxidation number was reduced to +2.
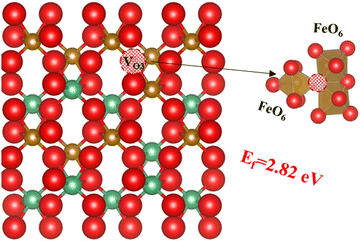 |
| | Fig. 7 The third type of oxygen vacancy; oxygen is red, Fe is brown, Nb is green and the red ball with black pattern represents the oxygen vacancy. | |
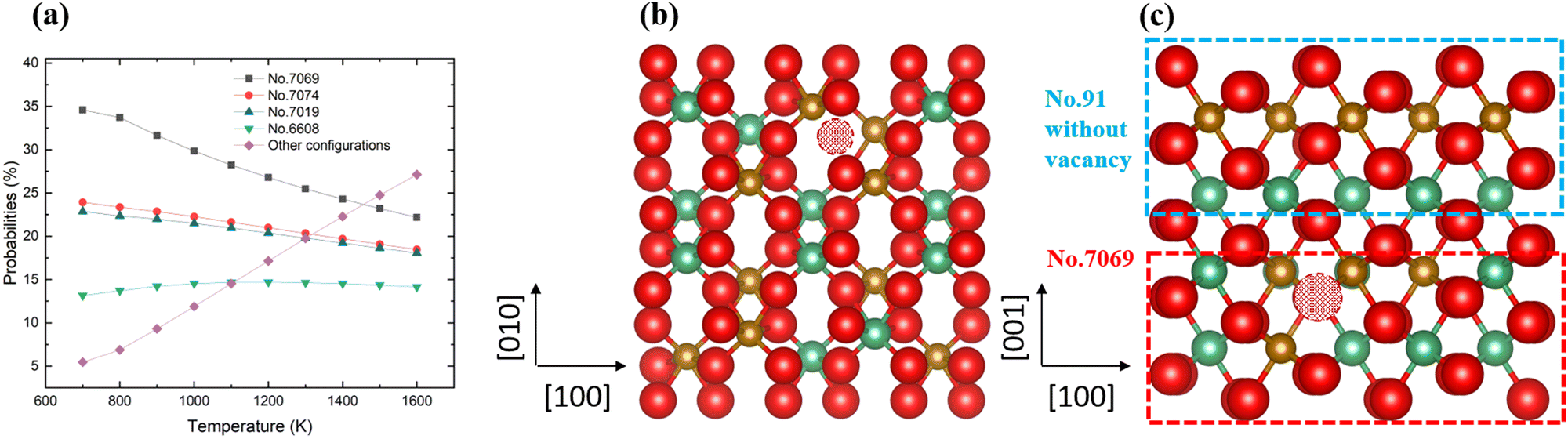
The probabilities of the non-stoichiometric structures containing oxygen vacancies are plotted in Fig. 8, where we observe four main structures from 700 K to 1600 K, in addition to the dominant structure, No. 7069, which is shown in Fig. 8b. These configurations are all different from the dominant ordered phase, discussed earlier, and our findings therefore suggest that when oxygen vacancies are present, cation disorder is more likely to occur, which could be the origin of the disordered materials found in experiment.
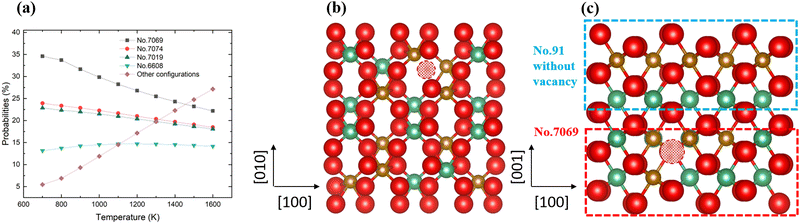 |
| | Fig. 8 The distributions of the probabilities in the defect 2 × 2 × 1 supercell (a); the most stable No. 7069 configuration (b) and the 2 × 2 × 2 supercell used in this work (c). | |
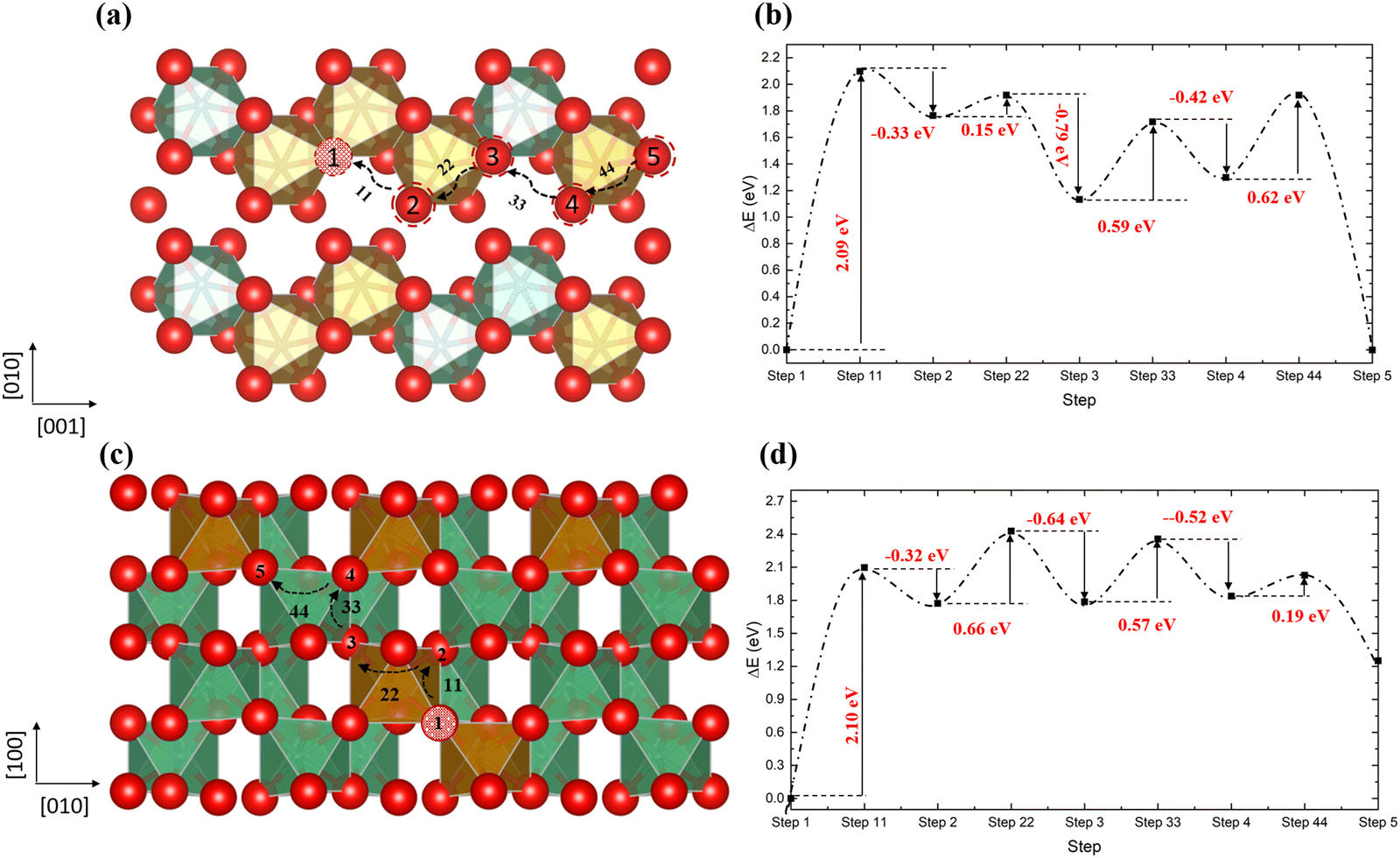
In order to gain insight into the impact of the cation distribution on the oxygen diffusion, we have employed the 2 × 2 × 2 supercell in Fig. 8c, with different cation distributions along the diffusion pathways. In order to make the calculations tractable, we have selected pathways where the oxygen diffuses through a vacancy hopping mechanism. The same two pathways as identified in the ordered structure are plotted in Fig. 9. Along the [001] direction, the sequence of vacancy types is: VO3(step1)–VO1(step2)–VO2(step3)–VO2(step4)–VO3(step5), in contrast to the ordered structured where VO1 and VO2 alternate. From step 1 to step 2, the energy barrier to be surmounted is 2.09 eV to create VO1, where the total energy of the system is almost at its highest, before the vacancy hops to the more stable VO2 type, only having to overcome a very small further barrier of 0.15 eV. Next on the pathway, another VO2 is created, for which an energy barrier of 0.59 eV has to be overcome, followed by a similar barrier of 0.62 eV to finally form another VO3. Similar results are found in the [110] direction, where in the first two steps, the vacancy moves from VO3 to VO1 with a barrier of 2.10 eV, but releasing 0.66 eV to form a VO1 site. When we compare these results with the diffusion in the ordered structure in Fig. 6, we note that in the disordered structure the initial energy barrier for the oxygen to move into the VO3 site at about 2.10 eV is much higher than in the ordered structure, owing to the stability of the VO3 vacancy, and oxygen diffusion through the disordered structure will therefore be slower.
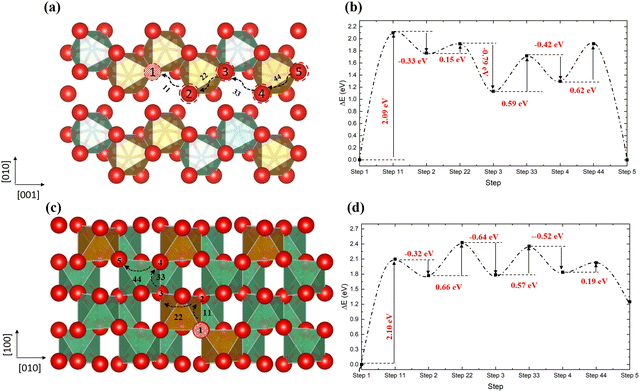 |
| | Fig. 9 The diffusion pathways of the oxygen atom along the [001] direction (a) and the [110] direction (c) in the disordered non-stoichiometric structures with an oxygen vacancy and their corresponding energy barriers (b and d); red is oxygen, brown is Fe, green is Nb and the red ball with black pattern represents the oxygen vacancy. | |
4. Conclusion
In this work, we have created 2 × 2 × 1, 1 × 2 × 2 and 2 × 1 × 2 supercells of the o-FeNbO4 material to confirm the dominant cation configuration(s) within the o-FeNbO4 structures, and we have studied the related bulk properties of the dominant o-FeNbO4 phase by DFT calculations. For the stoichiometric material, the results all confirmed a single dominant, ordered configuration with a probability of occurrence that exceeds 99.5%, which was selected for more in-depth study. The bulk properties of this selected o-FeNbO4 phase showed that there is no marked difference from the monoclinic FeNbO4 phase, which was studied in previous work. We have also investigated the probabilities of cation disorder in the non-stoichiometric material containing an oxygen vacancy, where we identified a number of potential cation configurations. We therefore suggest that oxygen vacancies, which are present in experimental materials, may encourage the cation disorder in the orthorhombic FeNbO4 phase observed experimentally.
We have simulated oxygen diffusion pathways in the ordered stoichiometric material and in the non-stoichiometric ordered and disordered structures. In the stoichiometric material, the diffusion of an interstitial oxygen atom could occur only by surmounting an energy barrier of around 3 eV, whereas in the non-stoichiometric ordered structure, it is much easier for the oxygen to diffuse in a vacancy hopping mechanism along the [110] direction (energy barrier up to ∼0.24 eV) than along the [001] direction (up to ∼1.04 eV), since in the [110] direction all the diffusion sites belong to the VO2 vacancy type, while in the [001] direction, some sites are of the energetically less favourable VO1 type. In addition, we found that the distribution of the cations in the structure could impact the energy barriers along the pathways. In particular, during the vacancy hopping mechanism, vacancies are more stable in sites which are coordinated by fewer Nb–O bonds and more Fe–O bonds. When a disordered structure contains the low-energy VO3 vacancy sites, the initial energy barrier for vacancy hopping is ∼2.10 eV, and oxygen diffusion through non-stoichiometric disordered structures should therefore be slower.
We consider that this work has contributed detailed atomic-level insight into the orthorhombic FeNbO4 material, where our results suggest that intrinsic oxygen vacancies promote oxygen diffusion through the vacancy hopping mechanism, but cation disorder may slow oxygen diffusion by allowing more stable vacancy sites to be formed. These are therefore two parameters that could be the focus in experimental syntheses to improve the efficacy of o-FeNbO4 as a cathode material in solid oxide fuel cells.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
Xingyu Wang acknowledges the China Scholarship Council (CSC) [No. 201906460008] and the University of Leeds for a PhD scholarship. This work has used the computational facilities of the Advanced Research Computing at Cardiff (ARCCA) Division, Cardiff University and HPC Wales, and the high performance computing facilities (ARC4) provided by the University of Leeds. We also acknowledge computing resources on the UK's national supercomputing service ARCHER2 facility (http://www.archer2.ac.uk) via our membership of the UK's HEC Materials Chemistry Consortium, which is funded by EPSRC (EP/L000202).
References
- E. S. Kim, C. J. Jeon, P. G. Clem and A. Feteira, Effects of Crystal Structure on the Microwave Dielectric Properties of ABO4 (A = ni,Mg,Zn and B = mo,W) Ceramics, J. Am. Ceram. Soc., 2012, 95, 2934–2938 CrossRef CAS.
- A. H. Krumpel, P. Boutinaud, E. van der Kolk and P. Dorenbos, Charge Transfer Transitions in the Transition Metal Oxides ABO4:Ln3+ and ABO4:Ln3+ (a = La, Gd, Y, Lu, Sc; B = V, Nb, Ta; Ln = Lanthanide), J. Lumin., 2010, 130, 1357–1365 CrossRef CAS.
- J. Li, W. Zhao, Y. Guo, Z. Wei, M. Han, H. He, S. Yang and C. Sun, Facile Synthesis and High Activity of Novel BiVO4/FeVO4 Heterojunction Photocatalyst for Degradation of Metronidazole, Appl. Surf. Sci., 2015, 351, 270–279 CrossRef CAS.
- Y. Tian, Y. Tian, P. Huang, L. Wang, Q. Shi and C. E. Cui, Effect of Yb3+ Concentration on Upconversion Luminescence and Temperature Sensing Behavior in Yb3+/Er3+ Co-Doped YnBO4 Nanoparticles Prepared Via Molten Salt Route, Chem. Eng. J., 2016, 297, 26–34 CrossRef CAS.
- Z. Gu and X. Zhang, Nico2o4@Mnmoo4 Core–Shell Flowers for High Performance Supercapacitors, J. Mater. Chem. A, 2016, 4, 8249–8254 RSC.
- M. Nyman, M. A. Rodriguez, L. E. S. Rohwer, J. E. Martin, M. Waller and F. E. Osterloh, Unique LaTaO4 Polymorph for Multiple Energy Applications, Chem. Mater., 2009, 21, 4731–4737 CrossRef CAS.
- Y. Oaki and H. Imai, Room-Temperature Aqueous Synthesis of Highly Luminescent BaWO4–Polymer Nanohybrids and Their Spontaneous Conversion to Hexagonal WO3 Nanosheets, Adv. Mater., 2006, 18, 1807–1811 CrossRef CAS.
- V. B. Mikhailik, H. Kraus, V. Kapustyanyk, M. Panasyuk, Y. Prots, V. Tsybulskyi and L. Vasylechko, Structure, Luminescence and Scintillation Properties of the MgWO4–MgMoO4 system, J. Phys.: Condens. Matter, 2008, 20, 365219 CrossRef.
- G. Zhou, M. Lü, Z. Xiu, S. Wang, H. Zhang and W. Zou, Polymer Micelle-Assisted Fabrication of Hollow BaWO4 Nanospheres, J. Cryst. Growth, 2005, 276, 116–120 CrossRef CAS.
- J. Deng, J. Jiang, Y. Zhang, X. Lin, C. Du and Y. Xiong, FeVO4 as a Highly Active Heterogeneous Fenton-Like Catalyst Towards the Degradation of Orange Ii, Appl. Catal., B, 2008, 84, 468–473 CrossRef CAS.
- C. Balamurugan, A. R. Maheswari, D. W. Lee and A. Subramania, Selective Ethanol Gas Sensing Behavior of Mesoporous N-Type Semiconducting FeNbO4 Nanopowder Obtained by Niobium–Citrate Process, Curr. Appl. Phys., 2014, 14, 439–446 CrossRef.
- K. I. Gnanasekar, V. Jayaraman, E. Prabhu, T. Gnanasekaran and G. Periaswami, Electrical and Sensor Properties of FeNbO4: A New Sensor Material, Sens. Actuators, B, 1999, 55, 170–174 CrossRef CAS.
- S. Kanti Biswas, T. Gnanasekaran, T. Kumar Ghorai and P. Pramanika, Sensing Properties of Chemically Synthesized Pristine and Pt-Impregnated Nanosized FeNbO4 in Hydrogen, Ammonia, and Lpg, J. Electrochem. Soc., 2008, 155, J26–J31 CrossRef.
- S. H. Ahmed, M. Bakiro and A. Alzamly, Visible-Light-Driven Photocatalytic Formation of Propylene Carbonate Using FeNbO4/Reduced Graphene Oxide Composites, Materialia, 2020, 12, 100781 CrossRef CAS.
- N.-S. Cho, S. Lee, J. H. Noh, G. K. Choi, H. S. Jung, D. W. Kim and K. S. Hong, Visible-Light-Induced Photocatalytic Activity in Fenbo4 Nanoparticles, J. Phys. Chem. C, 2008, 112, 18393–18398 CrossRef.
- R. Theissmann, H. Ehrenberg, H. Weitzel and H. Fuess, Domain Structure and Lattice Strains in FeNbO4, Solid State Sci., 2005, 7, 791–795 CrossRef CAS.
- S. Ananta, R. Brydson and N. W. Thomas, Synthesis, Formation and Characterisation of FeNbO4 Powders, J. Eur. Ceram. Soc., 1999, 19, 489–496 CrossRef CAS.
- V. M. Harder and H. Muller-Buschbau, FeNbO4: Untersuchungen an Einkristallen Rnit Wolf Ramit- Und AiNbO4-Struktur, Z. Anorg. Allg. Chem., 1979, 456, 99–105 CrossRef.
- R. Theissmann, H. Ehrenberg, H. Weitzel and H. Fuess, Nanostructured Cation Distribution in FeNbO4:A Synchrotron Powder Diffraction and Transmission Electron Microscopy Investigation, J. Mater. Sci., 2002, 37, 4431–4436 CrossRef CAS.
- X. Liu, D. Xie, J. T. S. Irvine, J. Ni and C. Ni, An FeNbO4-Based Oxide Anode for a Solid Oxide Fuel Cell (Sofc), Electrochim. Acta, 2020, 335, 135692 CrossRef CAS.
- X. Liu, J. Zhou, D. Xie, J. Ni and C. Ni, FeNbO4-Based Oxide Cathode for Steam Electrolysis, Solid State Ionics, 2020, 345, 115181 CrossRef CAS.
- C. Ni, J. Feng, J. Cui, J. Zhou and J. Ni, Ann-Type Oxide Fe0.5Mg0.25Ti0.25Nb0.9Mo0.1O4-Δ for Both Cathode and Anode of a Solid Oxide Fuel Cell, J. Electrochem. Soc., 2017, 164, F283–F288 CrossRef CAS.
- X. Wang, D. Santos-Carballal and N. H. de Leeuw, Density Functional Theory Study of Monoclinic FeNbO4: Bulk Properties and Water Dissociation at the (010), (011), (110), and (111) Surfaces, J. Phys. Chem. C, 2021, 125, 27566–27577 CrossRef CAS.
- R. Grau-Crespo, S. Hamad, C. R. A. Catlow and N. H. de Leeuw, Symmetry-Adapted Configurational Modelling of Fractional Site Occupancy in Solids, J. Phys.: Condens. Matter, 2017, 19, 256201 CrossRef.
- L. Li, Y. Li, Y. Li, H. Ye, A. Lu, H. Ding, C. Wang, Q. Zhou, J. Shi and X. Ji, Influences of Fe Mn Ratio on the Photocatalytic Performance of Wolframite (FexMn1-XWO4), Chem. Geol., 2021, 575, 120253 CrossRef CAS.
- J. D. Gale, Gulp: A Computer Program for the Symmetry-Adapted Simulation of Solids, J. Chem. Soc., Faraday Trans., 1997, 93, 629–637 RSC.
- J. D. Gale and A. L. Rohl, The General Utility Lattice Program, Mol. Simul., 2003, 29, 291–341 CrossRef CAS.
- M. Born and J. E. Mayer, Zur Gittertheorie Der Ionenkristalle, Phys. B, 1932, 75, 1–18 CAS.
- J. E. Mayer, Dispersion and Polarizability and the van der Waals Potential in the Alkali Halides, J. Chem. Phys., 1933, 1, 270–299 CrossRef CAS.
- M. S. Islam, M. Cherry and C. R. A. Catlow, Oxygen Diffusion in LaMnO3 and LaCoO3 Perovskite-Type Oxides: A Molecular Dynamics Study, J. Solid State Chem., 1996, 124, 230–237 CrossRef CAS.
- T. S. Bush, J. D. Gale, C. R. A. Catlow and P. D. Battle, Self-Consistent Interatomic Potentials for the Simulation of Binary and Ternary Oxides, J. Mater. Chem., 1994, 4, 831–837 RSC.
- S. M. Woodley, P. D. Battle, J. D. Gale and C. R. A. Catlow, The Prediction of Inorganic Crystal Structures Using a Genetic Algorithm and Energy Minimisation, Phys. Chem. Chem. Phys., 1999, 1, 2535–2542 RSC.
- G. Kresse and J. Furthmiiller, Efficiency of Ab-Initio Total Energy Calculations for Metals and Semiconductors Using a Plane-Wave Basis Set, Comput. Mater. Sci., 1996, 6, 15–20 CrossRef CAS.
- G. Kresse and J. Furthmuller, Efficient Iterative Schemes for Ab Initio Total-Energy Calculations Using a Plane-Wave Basis Set, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 11169–11186 CrossRef CAS PubMed.
- G. Kresse and J. Hafner, Ab Initio Molecular Dynamics for Open-Shell Transition Metals, Phys. Rev. B: Condens. Matter Mater. Phys., 1993, 48, 13115–13118 CrossRef CAS PubMed.
- G. Kresse and J. Hafner, Norm-Conserving and Ultrasoft Pseudopotentials for First-Row and Transition Elements, J. Phys.: Condens. Matter, 1994, 6, 8245–8260 CrossRef CAS.
- P. E. Blochl, Projector Augmented-Wave Method, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 50, 17953–17979 CrossRef PubMed.
- J. P. Perdew, K. Burke and M. Ernzerhof, Generalized Gradient Approximation Made Simple, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed.
- S. L. Dudarev, G. A. Botton, S. Y. Savrasov, C. J. Humphreys and A. P. Sutton, Electron-Energy-Loss Spectra and the Structural Stability of Nickel Oxide: An Lsda+u Study, Phys. Rev. B: Condens. Matter Mater. Phys., 1998, 57, 1505–1509 CrossRef CAS.
- K. Jun, J. U. Lee, M. H. Chang and T. Oda, A Comparative Study on Modeling of the Ferromagnetic and Paramagnetic States of Uranium Hydride Using a DFT+U Method, Phys. Chem. Chem. Phys., 2019, 21, 17628–17639 RSC.
- A. van de Walle, Multicomponent Multisublattice Alloys, Nonconfigurational Entropy and Other Additions to the Alloy Theoretic Automated Toolkit, CALPHAD: Comput. Coupling Phase Diagrams Thermochem., 2009, 33, 266–278 CrossRef CAS.
- A. van de Walle, M. Asta and G. Ceder, The Alloy Theoretic Automated Toolkit: A User Guide, CALPHAD: Comput. Coupling Phase Diagrams Thermochem., 2002, 26, 539–553 CrossRef CAS.
- A. van de Walle, P. Tiwary, M. de Jong, D. L. Olmsted, M. Asta, A. Dick, D. Shin, Y. Wang, L. Q. Chen and Z. K. Liu, Efficient Stochastic Generation of Special Quasirandom Structures, CALPHAD: Comput. Coupling Phase Diagrams Thermochem., 2013, 42, 13–18 CrossRef CAS.
- I. Saadoune, J. A. Purton and N. H. de Leeuw, He Incorporation and Diffusion Pathways in Pure and Defective Zircon ZrSiO4: A Density Functional Theory Study, Chem. Geol., 2009, 258, 182–196 CrossRef CAS.
- M. Lontsi-Fomena, A. Villesuzanne, J. P. Doumerc, C. Frayret and M. Pouchard, A Density Functional Theory Study of Oxygen Diffusion in LaAlO3 and SrTiO3, Comput. Mater. Sci., 2008, 44, 53–60 CrossRef CAS.
- V. I. Baykov, P. A. Korzhavyi and B. Johansson, Diffusion of Interstitial Mn in the Dilute Magnetic Semiconductor (Ga,Mn)As: The Effect of a Charge State, Phys. Rev. Lett., 2008, 101, 177204 CrossRef CAS PubMed.
- E. Schmidbauer, Electrical Resistivity, Thermopower, and Fe Mössbauer Study of FeNbO4, J. Solid State Chem., 1997, 134, 253–264 CrossRef CAS.
- H. Ehrenberg, G. Wltschek, R. Theissman, H. Weitzel, H. Fuess and F. Trouw, The Magnetic Structure of Fenbo4, J. Magn. Magn. Mater., 2000, 218, 261–265 CrossRef CAS.
- N. Lakshminarasimhan, A. K. N. Kumar, S. S. Chandrasekaran and P. Murugan, Structure-Magnetic Property Relations in FeNbO4 Polymorphs: A Spin Glass Perspective, Prog. Solid State Chem., 2019, 54, 20–30 CrossRef CAS.
- J. Xu, J. Wang, A. Rakhmatullin, S. Ory, A. J. Fernández-Carrión, H. Yi, X. Kuang and M. Allix, Interstitial Oxide Ion Migration Mechanism in Aluminate Melilite La1+XCa1–XAl3O7+0.5x Ceramics Synthesized by Glass Crystallization, ACS Appl. Energy Mater., 2019, 2, 2878–2888 CrossRef CAS.
- M. Youssef and B. Yildiz, Predicting Self-Diffusion in Metal Oxides from First Principles: The Case of Oxygen in Tetragonal ZrO2, Phys. Rev. B: Condens. Matter Mater. Phys., 2014, 89, 024105 CrossRef.
- C. Chen, Z. M. Baiyee and F. Ciucci, Unraveling the Effect of La a-Site Substitution on Oxygen Ion Diffusion and Oxygen Catalysis in Perovskite BaFeO3 by Data-Mining Molecular Dynamics and Density Functional Theory, Phys. Chem. Chem. Phys., 2015, 17, 24011–24019 RSC.
- J. Koettgen, T. Zacherle, S. Grieshammer and M. Martin, Ab Initio Calculation of the Attempt Frequency of Oxygen Diffusion in Pure and Samarium Doped Ceria, Phys. Chem. Chem. Phys., 2017, 19, 9957–9973 RSC.
- A. Kushima, D. Parfitt, A. Chroneos, B. Yildiz, J. A. Kilner and R. W. Grimes, Interstitialcy Diffusion of Oxygen in Tetragonal La2CoO4+Δ, Phys. Chem. Chem. Phys., 2011, 13, 2242–2249 RSC.
- C. Mitra, T. Meyer, H. N. Lee and F. A. Reboredo, Oxygen Diffusion Pathways in Brownmillerite SrCoO2.5: Influence of Structure and Chemical Potential, J. Chem. Phys., 2014, 141, 084710 CrossRef PubMed.
- B. Balzer and H. Langbei, Solid Solutions in the FeNbO4-TiO2 System, Cryst. Res. Technol., 1996, 31, 93–98 CrossRef CAS.
|
| This journal is © the Owner Societies 2023 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  ,
David
Santos-Carballal
,
David
Santos-Carballal
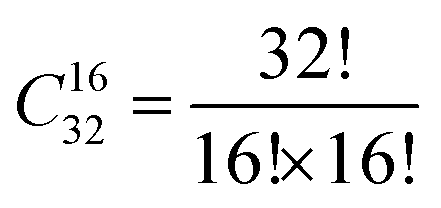 , which is an unrealistic number of configurations to compute. Thus, to still do a comprehensive study of the ordering in the structure, we have generated 2 × 2 × 1, 2 × 1 × 2, and 1 × 2 × 2 supercells. From Table 4, we note that in each of these supercells the total number of inequivalent configurations is
, which is an unrealistic number of configurations to compute. Thus, to still do a comprehensive study of the ordering in the structure, we have generated 2 × 2 × 1, 2 × 1 × 2, and 1 × 2 × 2 supercells. From Table 4, we note that in each of these supercells the total number of inequivalent configurations is 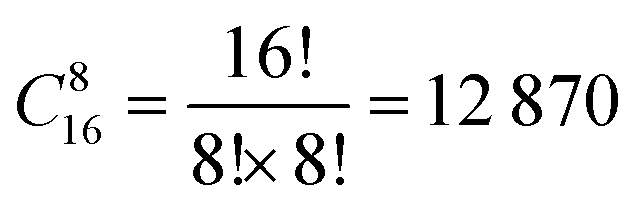 , which number is reduced after consideration of the symmetry, to 482, 448, and 474 for the three supercells, respectively. The results of the calculations of the probability distributions are shown in Fig. 1 from which it is clear that from 700 K to 1600 K, there is one inequivalent configuration (labelled No. 91, No. 81 and No. 91) in each cell whose probability of occurrence exceeds 99.5%. If we expand each cell to a 2 × 2 × 2 supercell, we see that all 3 cells obtained the same dominant and ordered phase as the most probably configuration. As such, from our calculations, it appears that the most stable phase is an ordered structure rather than the disordered material observed experimentally. For the sake of completeness, we have also calculated the contributions of the vibrational entropy to the free energy of both structures, which are listed in Table S2 (ESI†). However, it is clear that the difference in entropy of the two phases is so small that we can ignore its contribution to the structural stabilities.
, which number is reduced after consideration of the symmetry, to 482, 448, and 474 for the three supercells, respectively. The results of the calculations of the probability distributions are shown in Fig. 1 from which it is clear that from 700 K to 1600 K, there is one inequivalent configuration (labelled No. 91, No. 81 and No. 91) in each cell whose probability of occurrence exceeds 99.5%. If we expand each cell to a 2 × 2 × 2 supercell, we see that all 3 cells obtained the same dominant and ordered phase as the most probably configuration. As such, from our calculations, it appears that the most stable phase is an ordered structure rather than the disordered material observed experimentally. For the sake of completeness, we have also calculated the contributions of the vibrational entropy to the free energy of both structures, which are listed in Table S2 (ESI†). However, it is clear that the difference in entropy of the two phases is so small that we can ignore its contribution to the structural stabilities.
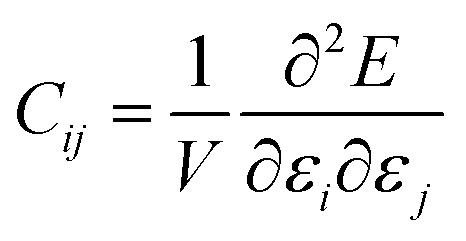
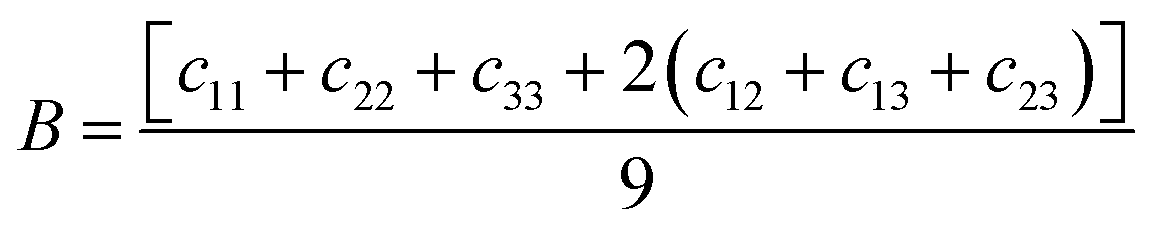
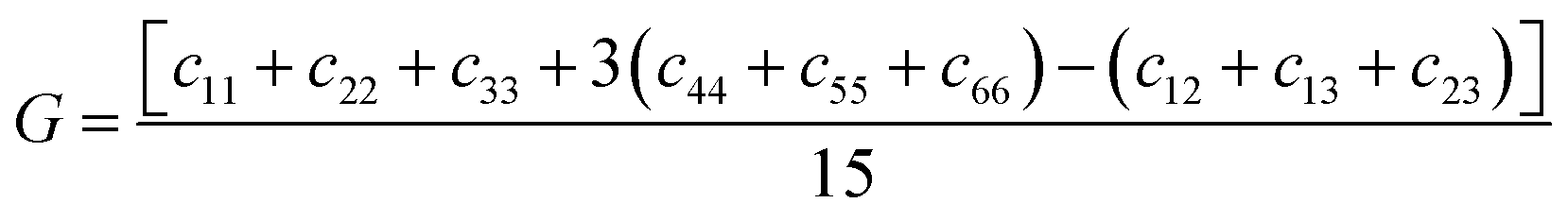
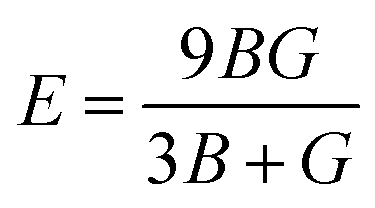
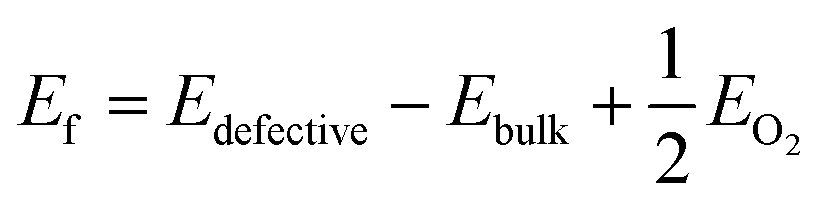 , where Edefective and Ebulk are the total energies of the non-stoichiometric material and the stoichiometric bulk material, respectively, and EO2 refers to the total energy of the oxygen molecule in the triplet ground state. The lattice parameters and lattice energies of the optimized structures are listed in Table S1 (ESI†), which show that the lattice with VO1 type vacancies is the most distorted structure in terms of volume and lattice energy. Overall, VO2 is the preferred oxygen vacancy site in the lattice due to its lower formation energy, although at 3.7 eV it is still a very high value, in agreement with experimental records which show that most oxygen vacancies are generated in the preparation process at temperatures of 1100 °C.16–20 We have therefore introduced the VO2 in the 2 × 2 × 2 supercells to investigate the oxygen diffusion pathways, where we noted that the formation energy of the oxygen vacancies does not change as the supercell is increased in size, which indicates that the reduction is a localised process, also shown by the short-range lattice distortion around the vacancy sites.
, where Edefective and Ebulk are the total energies of the non-stoichiometric material and the stoichiometric bulk material, respectively, and EO2 refers to the total energy of the oxygen molecule in the triplet ground state. The lattice parameters and lattice energies of the optimized structures are listed in Table S1 (ESI†), which show that the lattice with VO1 type vacancies is the most distorted structure in terms of volume and lattice energy. Overall, VO2 is the preferred oxygen vacancy site in the lattice due to its lower formation energy, although at 3.7 eV it is still a very high value, in agreement with experimental records which show that most oxygen vacancies are generated in the preparation process at temperatures of 1100 °C.16–20 We have therefore introduced the VO2 in the 2 × 2 × 2 supercells to investigate the oxygen diffusion pathways, where we noted that the formation energy of the oxygen vacancies does not change as the supercell is increased in size, which indicates that the reduction is a localised process, also shown by the short-range lattice distortion around the vacancy sites.