
A density functional theory study of a water gas shift reaction on Ag(111): potassium effect†
Yan-Xin
Wang
,
Hai-Li
Zhang
,
Hai-Shun
Wu
and
Jian-Feng
Jia
 *
*
Key Laboratory of Magnetic Molecules & Magnetic Information Materials Ministry of Education, The School of Chemical and Material Science, Shanxi Normal University, Taiyuan, 030000, China. E-mail: jiajf@dns.sxnu.edu.cn
First published on 12th December 2022
Abstract
Density functional theory (DFT) calculations are executed to investigate the effect of a potassium (K) promoter on the activity of the water gas shift reaction (WGSR) over an Ag(111) surface. It is found that the WGSR proceeds mainly through the OH(O)-assisted carboxy pathway in which H2O dehydrogenation is the rate-controlling step on both Ag(111) and K/Ag(111) surfaces. Energetic span model analysis shows that K addition can enhance the activity of the WGSR by reducing the apparent activation energy of the whole reaction since it can promote H2O dissociation and stabilize the adsorption of the reactants (CO and H2O). Importantly, the K adatom can stabilize the binding of all oxygenates by direct K–O bonding and the stabilizing effect of K on OH adsorption of the transition state (TS) plays a leading role in promoting H2O dissociation. Moreover, the K–O distance and K coverage are two key factors affecting H2O activation, that is, the shorter the K–O distance (2–3 Å) the more the K coverage (25%) contributes to the stronger promotion effect. For various metals catalyzing the WGSR, K promotes H2O dissociation on inert metals like Ag, Au and Cu better than those on reactive metals (Pd and Ni) since the more inert metal surfaces would weaken the K and O binding and accordingly strengthen the interaction between them, resulting in a higher promotion effect.
1. Introduction
The water–gas-shift reaction (WGSR), that is, CO + H2O → CO2 + H2, is regarded as one of the most vital means to produce hydrogen and to remove CO in industrial production.1 The generally accepted mechanisms for the WGSR include redox, carboxyl, and formate mechanisms, and which mechanism dominates relies on the kinds of metal catalysts as well as their surface configurations, experimental conditions and so on.2,3 For example, the previous DFT calculations show that the WGSR proceeds mainly via the carboxyl pathway on Cu(111)4–8 and Pt(111) surfaces,9,10 while the redox mechanism is identified to be the most dominant one on the Au(111) surface.2,3 Catapan et al. reported that the carboxyl pathway plays a pivotal role on both Ni(111) and Ni(211) surfaces, and the formate route is equally significant on Ni(211).11 All these studies achieve consensus that H2O dehydrogenation is the rate-controlling step in terms of different mechanisms of different metal surfaces.As is known to all of us introducing small amounts of additives to heterogeneous catalysts is thought to be an efficient method to change the catalytic performance through the promotion or inhibition effect.12 Alkali additives, such as potassium (K), have drawn great attention since they serve as special electron donors to improve the activity of many reactions including the WGSR and the promotion effect on the WGSR by alkali has been studied widely.13,14 The study of the WGSR catalyzed by LaNiO3 doped with K shows that K addition makes stable K–OH species form at the interface between Ni nanoparticles and the La2O3 support, and promotes the formation of a formate intermediate and further generation of CO2 products.15 The experimental results of Sanz et al. indicate that K/Cu/TiO2 shows better catalytic performance for the WGSR than the Cu/TiO2 catalyst because the presence of a K promoter greatly enhances the activity of water dissociation, which is the rate-controlling step for K/Cu/TiO2. Additionally, this study also shows that the rate of H2 production gradually increases with the increase of K coverage which reaches the maximum of 0.15 ML and the productivity of H2 begins to decrease when the coverage is higher.16 Ma et al. reported that with increasing K loading on the 100Fe/5.1Si/2Cu/xK (x = 1.25 or 3) catalyst, the WGS rate constant is accordingly increased during the Fischer–Tropsch synthesis (FTS).17 These experimental studies emphasize the significance of alkali addition for the WGSR as well as its coverage. The promotion effect of alkali on the WGSR is also theoretically studied over Cu, Pt and Ni surfaces, in which the interaction between alkali and O in oxygenates contributes to its promotion effect.18–20 Additionally, the K(Na)–O distance is found to play an important role in determining the K effect since the degree to which K(Na) promotes CO dissociation over the Rh(111) surface depends on the distance between K(Na) and molecular CO and the shorter the distance, the stronger the promotion effect.21,22 However, the fundamental reason why alkali coverage and closeness between alkali and oxygenate matters has not been disclosed so far. It has been reported that noble metal-based catalysts are good catalysts for the WGSR due to their high activity, selectivity, and stability.23,24 However, the study of alkali effect on the Ag metal surface at the atomic level is scarce despite the certain catalytic capacity of silver to the WGSR.25–28
In this work, the pathways of the WGSR are investigated by the DFT method on a clean Ag(111) surface and the corresponding K-promoted surface (K/Ag(111)) to explore the effect of K additive on the mechanism and activity of the WGSR on Ag catalysts. In the light of energetic and electronic effect analysis, the origin of the K effect is revealed and factors like the K–O distance and K coverage affecting the K promotion effect are also clarified.
2. Calculation methods and models
2.1 Methods
The Vienna ab initio simulation package (VASP)29,30 was used to perform the present DFT calculations. The projector augmented wave (PAW)31,32 method and the Perdew–Burke–Ernzerhof (PBE)33 exchange correlation functional were adopted to describe the valence electron and core interactions. A plane wave basis was carried out with the cut-off energy of 400 eV. The transition states (TSs) were searched using the climbing-image nudged elastic band (CI-NEB) method,34 and further confirmed by the existence of a single imaginary vibrational mode along the reaction coordinate. Charge distributions were estimated using the algorithm developed by Henkelman through Bader's atoms in molecules (AIM) theory.35,36 The binding energy (Ebind) was calculated by the following equation Ebind = Eads/sub − Eads − Esub, in which Eads/sub, Eads and Esub denote the energies of the adsorbate/substrate system, the adsorbate in the gas phase and the substrate, respectively. The activation energies were calculated with Ea = ETS − EIS, where ETS and EIS represent the energies of the transition and initial states, respectively. The reaction energies were calculated with ΔE = EFS − EIS, EFS was the energy of the final state. The charge density difference was evaluated by Δρ = ρads/sub − ρads − ρsub, where ρads/sub represents the total charge density of the adsorbate/substrate system, while ρads and ρsub denote the charge density of the adsorbate molecule and the metal substrate, respectively, fixed at the adsorbed geometry.2.2 Models
To model the Ag(111) surface, a p(3 × 3) supercell with four layers was built, which was separated by 15 Å of a vacuum region. The first two layers as well as the adsorbed molecules were allowed to relax until the forces were less than 0.03 eV Å−1, while the other two layers were fixed. The Brillouin zone sampling was used with the 3 × 3 × 1 Monkhorst–Pack grid.37 Moreover, a p(4 × 4) supercell of the Ag(111) surface with four layers was also constructed for the sake of research and a 2 × 2 × 1 Monkhorst–Pack grid was adopted to sample the Brillouin zone.3. Results
3.1 Adsorption of pertinent intermediates
The optimum adsorption structures of pertinent intermediates involved in the WGSR on Ag(111) and K/Ag(111) surfaces are displayed in Fig. 1. The preferred binding sites and binding energies of such intermediates on both surfaces are summarized in Table 1. From Fig. 1, it can be found that K addition alters the adsorption structures of most intermediates dramatically. The most stable adsorption site for the K adatom on the Ag(111) surface is the face-centered cubic site with a binding energy of −1.96 eV and K tends to go far away from the surface with the distance of K and the nearest Ni atom being 3.44 Å. Once the reaction species adsorb on the K pre-adsorbed surfaces, K atom tends to depart from its preferred face-centered cubic (fcc) site to get close to the O sites in oxygenates. Simultaneously, the geometrical configuration of most adsorbed oxygenates would make readjustments to ensure that the contained O atoms point toward K and remain at a short distance away from K (2.0–3.0 Å). As analyzed above, all the structural changes in adsorbates caused by K addition indicate that a kind of attractive interaction exists between K and O atoms. This result is further confirmed by Bader charge analysis of the single K atom adsorption on the Ag(111) surface in which K is positively charged with +0.80 e, while the vicinal Ag atoms are negatively charged, signifying that the K adatom tends to transmit its valence electrons to the Ag(111) surface. As a result, the electropositive K is apt to attract the much more electronegative O atoms compared to other less electronegative C and H atoms (electronegativity for O, C and H is 3.4, 2.5 and 2.2, respectively). Such electrostatic attraction proves a direct K–O bonding as discussed later. Table 1 shows that the K adatom can stabilize the adsorption of all oxygenates by increasing their binding energies and it is later found that the direct K–O bonding can strengthen their adsorption. However, K addition has little impact on the binding strength of H and H2 on the Ag(111) surface due to the negligible attractive interactions between K and H atoms. | ||
| Fig. 1 Optimized structures of all pertinent intermediates on Ag(111) and K/Ag(111) surfaces. The red, gray, white and purple balls represent O, C, H and K atoms, respectively. | ||
| Ag(111) | K/Ag(111) | |||||
|---|---|---|---|---|---|---|
| Site | BE/eV | d O–Ag/C–Ag | Site | BE/eV | d O–Ag/C–Ag | |
| H2O | Top | −0.07 | 2.68 | Pointing to K | −0.57 | 3.27 |
| CO | Top | −0.16 | 2.17 | Top | −0.22 | 2.16 |
| OH | fcc | −2.62 | 2.29 | Bridge | −3.21 | 2.43 |
| H | fcc | −2.07 | 1.92 | fcc | −2.09 | 1.94 |
| O | fcc | −3.41 | 2.15 | fcc | −4.34 | 2.22 |
| CO2 | Physisorption | −0.01 | 3.60 | Pointing to K | −0.24 | 3.74 |
| cis-COOH | Top | −1.11 | 2.17 | Top | −1.93 | 2.22 |
| trans-COOH | Top–top | −1.02 | 2.19 | Top | −1.88 | 2.21 |
| HCO | fcc | −0.75 | 2.18 | Top | −1.29 | 2.18 |
| HCOO | Top–top | −2.26 | 2.28 | Top–top | −3.11 | 2.60 |
| H2 | Physisorption | −0.02 | 3.22 | Physisorption | −0.02 | 3.23 |
3.2 WGSR pathway on Ag(111) and K/Ag(111) surfaces
The previously proposed pathways for the WGSR on Ag(111) and K/Ag(111) surfaces containing redox, carboxyl, and formate pathways are taken into account in the present study. No matter which pathway plays the leading role, the primary reaction step is H2O dissociation to produce OH, O and H species, which then oxidize CO through these three pathways to generate the final CO2. The calculated energy barriers, reaction energies and the lengths of forming or breaking bonds at TSs for all elementary steps involved in the three pathways on both surfaces are summarized in Table 2. The TS structures regarding these elementary steps are presented in Fig. 2. The properties of each step involved in H2O and OH activation as well as the three pathways are elucidated next to search for the optimal pathway for the WGSR on both surfaces.| Elementary steps | Ag(111) | K/Ag(111) | |||||
|---|---|---|---|---|---|---|---|
| E a/eV | ΔE/eV | d/Å | E a/eV | ΔE/eV | d/Å | ||
| R1 | H2O → OH + H | 1.77 | 1.06 | 1.65 | 1.41 | 0.92 | 1.77 |
| R2 | OH → O + H | 2.41 | 1.82 | 1.88 | 2.09 | 1.50 | 1.76 |
| R3 | OH + OH → H2O + O | 0.26 | 0.14 | — | 0.34 | 0.15 | — |
| R4 | H + H → H2 | 1.21 | −0.40 | 1.56 | 1.03 | −0.37 | 1.48 |
| R5 | CO + OH → cis-COOH | 0.07 | −0.35 | 2.05 | 0.06 | −0.56 | 1.91 |
| R6 | cis-COOH → trans-COOH | 0.50 | 0.00 | — | 0.52 | −0.04 | — |
| R7 | COOH → CO2 + H | 1.27 | −0.68 | 1.32 | 1.14 | −0.13 | 1.53 |
| R8 | COOH + O → CO2 + OH | — | — | — | — | — | — |
| R9 | COOH + OH → CO2 + H2O | — | — | — | — | — | — |
| R10 | CO + O → CO2 | 0.03 | −2.72 | 2.19 | 0.03 | −2.06 | 2.53 |
| R11 | CO + H → CHO | 0.63 | 0.27 | 1.46 | 0.53 | −0.18 | 1.84 |
| R12 | CHO + O → HCOO | 0.02 | −3.29 | 2.75 | 0.48 | −2.72 | 2.28 |
| R13 | HCOO → CO2 + H | 1.93 | 0.16 | 1.01 | 2.21 | 0.78 | — |
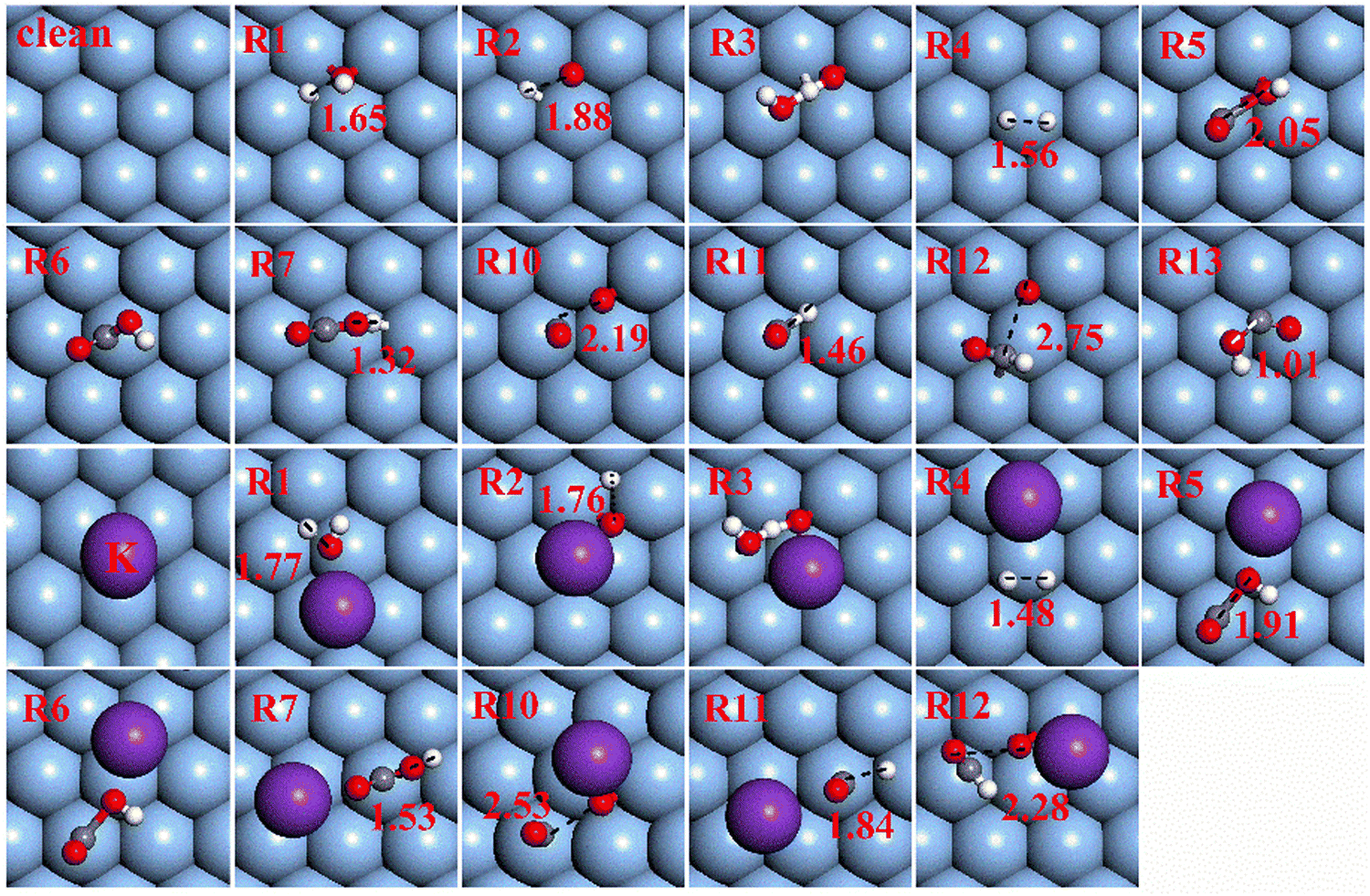 | ||
| Fig. 2 Transition state (TS) structures of elementary steps on Ag(111) and K/Ag(111). The red, gray, white and purple balls represent O, C, H and K atoms, respectively. | ||
When K is introduced to the Ag(111) surface, the initial state (IS), transition state (TS) and final state (FS) geometrical structures involved in H2O and OH activation alter dramatically due to the attractive interaction between K and O. Such an effect of K on O atoms causes the H2O plane to stay far away from the Ag(111) plane with the O atom orienting toward K and two H atoms pointing to the Ag surface. Additionally, K addition makes the adsorption site of OH at both TS and FS structures change from the fcc site of the clean surface to the top site by its O atom pointing to K. The large stabilizing effect of K on OH or O of TS and FS renders H2O and OH dehydrogenation kinetically and thermodynamically favored with both energy barriers decreasing by about 0.30 eV and the endothermy reducing by 0.14 and 0.32 eV, respectively. For OH disproportionation, the K adatom prefers to interact with dual O atoms in IS, TS and FS structures and the kinetics of this reaction is slightly suppressed by K with the activation barrier increasing to 0.34 eV from 0.26 eV on a clean surface, while its endothermy (about 0.14 eV) is barely affected by K addition.
When K is added to this co-adsorption system, K adatom prefers to interact with the adsorbed O atom and the C–O distance would elongate to 2.53 Å at the TS. The oxidation of CO is kinetically as easy as on a clean surface with an energy barrier of 0.03 eV and thermodynamically less favored with an exothermicity of 2.06 eV.
3.2.4.1 CO oxidation by OH to form COOH. In addition to being oxidized by O, CO can also be oxidized by OH to generate surface carboxyl species (COOH). The most stable co-adsorption sites for CO and OH are similar to those for CO and O, that is, CO binds to the top site and OH occupies the fcc site. This C–O bond formation process is also facile with a reaction barrier of 0.07 eV, but is thermodynamically less favored with an exothermicity of 0.35 eV, much less than that of 2.72 eV for CO oxidation by O. We notice that the activation barrier of CO oxidation by OH on the Ag(111) surface is much lower than that on Cu(111) (0.54 eV), Pt(111) (0.31 eV) and Ni(111) (0.96 eV) and CO oxidation by O also needs higher energy barriers of 0.48, 0.91 and 0.97 eV on Cu, Pt and Ni surfaces, respectively.18–20 The reason can be attributed to that compared to other metals, more inert metals like Ag would weaken CO and OH (O) binding with the surface and accordingly strengthen the ability to combine with each other.
When K is introduced into the co-adsorption system for CO and OH, OH would shift from the preferred fcc site to the bridge one with the O–H axis parallel to the surface so that its O can better interact with K with an O–K distance of 2.45 Å, while CO still binds to the top site with O inclining toward K and the distance between them equals to 2.90 Å. K addition rarely affects the kinetics of this oxidation step with an activation barrier of 0.06 eV, but enhances the thermodynamics by decreasing the reaction energy to −0.56 eV from the original −0.35 eV.
3.2.4.2 Transformation of cis-COOH to trans-COOH. The produced COOH species is featured by its H atom pointing upwards relative to the surface, named cis-COOH, in which the atomic H is unable to fall to the surface. As a result, the cis-COOH would convert to another form with the H atom pointing to the surface (namely, trans-COOH) before further dehydrogenation. The transformation from cis-COOH to trans-COOH is thermoneutral with an energy barrier of 0.50 eV.
The translation from cis-COOH to trans-COOH is also considered on the K/Ag(111) surface, which is thermally neutral (−0.04 eV) with an energy barrier of 0.52 eV, the same reactivity as on the corresponding pure surface.
3.2.4.3 Trans-COOH dehydrogenation. One possible way for CO2 generation is through trans-COOH direct dehydrogenation with an exothermicity of 0.68 eV, which is thermodynamically advantageous. Nevertheless, this step is still hard to occur due to so large activation barrier (1.27 eV). K addition can enhance the kinetics of trans-COOH dehydrogenation by decreasing the activation barrier to 1.14 eV from the original 1.27 eV, but restrain the thermodynamics with a lower exothermicity of 0.13 eV (compared to 0.68 eV on a clean surface). In this way, trans-COOH direct dehydrogenation is still a tough process under the effect of K.
Besides direct dehydrogenation, COOH dehydrogenation assisted by OH or O species supplies an alternative way to yield CO2. Whether on the pure or on the K-adsorbed Ag(111) surface, once OH or O species adsorbs in the position near COOH, COOH species would automatically transfer its H atom to adjacent OH or O, leading to CO2 and H2O (OH) formation. Hence, assisted by OH and O species, abstraction of H from trans-COOH turns into a spontaneous process which needs no reaction barrier on both surfaces. Consequently, OH(O)-assisted COOH dehydrogenation rather than its direct dehydrogenation dominates the carboxyl pathway.
K addition tends to promote the formation of CHO in kinetics and thermodynamics with the energy barrier decreasing by 0.10 eV and reaction energy decreasing to −0.18 eV from the original 0.53 eV. However, whether CHO oxidation or HCOO dissociation reaction, the effect of the K adatom on them is adverse with activation barriers increasing by 0.46 eV for the C–O bond formation and 0.28 eV for C–H bond breaking, while the exothermicity reduces by 0.57 and 0.62 eV, respectively.
The above analysis shows that the formate pathway is the most undesirable pathway on both Ag(111) and K/Ag(111) surfaces due to the difficulty of C–H bond breaking from HCOO to form CO2. Both redox and carboxyl pathways are favorable for the WGSR on both surfaces, since C–O bond formation in both pathways is facile and C–H bond cleavage to form CO2 in OH(O)-assisted carboxyl pathway is also kinetically favored. However, the redox pathway is more sensitive to the availability of O, which derives from OH disproportionation in advance. Thus, the OH-assisted carboxyl pathway rather than the redox pathway is the optimal one for the WGSR on both surfaces. For all elementary reaction steps involved in the OH(O)-assisted carboxyl pathway, H2O dissociation is the most difficult step with the highest energy barrier and thus would be the rate-controlling step. K addition can lower the energy barrier of such rate-controlling step and consequently enhance the activity of the whole the WGSR.
3.3 Energetic span model analysis
Fig. 3 exhibits the free energy profiles on Ag(111) and K/Ag(111) surfaces for the OH-assisted carboxyl pathway since such pathway dominates the WGSR of both surfaces as expounded above. The typical experimental condition of 500 K and 100 kPa is used to calculate the free energies. The entropy contributions for the adsorption of CO and H2O reactants and the desorption of CO2 and H2 products are drawn into the free energy calculations. The translational, rotational, and vibrational entropy contributions have been considered for these species in their gaseous and adsorption states. For the gas-phase species, the entropies are obtained from the NIST Chemistry WebBook.38 The Campbell Sellers (CS) equation, a semi-empirical relationship between the gas phase and surface entropies (seen in eqn (1)), was proposed to accurately evaluate the entropies of weakly bound molecular adsorbates.39 In the present work, the entropies of reactants and products (CO, H2O, CO2 and H2) are computed using the CS equation on account of their weak adsorption strength and low frequency modes on both Ag(111) and K/Ag(111) surfaces. The CS equation is as follows:| Sad0(T) = 0.7Sgas0(T) − 3.3R | (1) |
 | ||
| Fig. 3 Free energy profiles of the WGSR via OH-assisted carboxyl mechanism over Au(111) and K/Au(111) at 500 K. | ||
The activity of the rate-controlling step has been determined, but it is not enough to determine the whole reaction activity since the steps around it also work. Instead, the apparent activation energy would be a more suitable indicator to evaluate the activity of the entire cycle and it would be analyzed by the energetic span model proposed by Kozuch et al.40 with regard to the carboxyl pathway of the whole WGSR on both clean and K-promoted surfaces. In this model, the TOF-determining transition state (TDTS) and TOF-determining intermediate (TDI) play a key role to define the energy span (δE), namely, the apparent activation energy of the entire cycle, which is expressed as follows:

For both Ag(111) and K/Ag(111) surfaces, the TDTSs are the TSs for H2O → OH + H with free energies of 2.48 and 1.56 eV, and the TDIs are the coadsorption of CO2 and H2 in the gas phase with free energies of −0.52 eV on both surfaces. Since the TDTSs appear before TDI, the energy spans are calculated to be 2.48 and 1.56 eV, respectively, on Ag(111) and K/Ag(111) surfaces. Apparently, K addition can dramatically decrease the apparent activation energy on the clean Ag(111) surface, due to the much lower position of TDTSs on K/Ag(111) than on clean Ag(111), caused by the dual effects of smaller energy barrier of H2O dissociation and stronger binding strength of reactants with the help of K.
4. Discussion
4.1 Origin of the K affects the activity of the WGSR
As analyzed above, K addition can enhance the activity of the WGSR by reducing the apparent activation energy of the WGSR circle through decreasing the energy barrier of H2O dissociation (the rate-controlling step) and stabilizing the adsorption of reactants (CO and H2O). To further explore the origin of the K promoting H2O dissociation, the energetic effect analysis adopting the energetic decomposition method proposed by Hammer41 will be indispensable.| Ea = Ebond(XY) – EISXY + ETSX + ETSY + ETSint | (2) |
By eqn (2), the decomposition results for the energy barrier of H2O dissociation have been acquired and illustrated in Fig. 4. It can be found that K additive would alter the magnitude of each term to varying degrees. On the one hand, the K adatom tends to stabilize H2O binding with the Ag(111) surface, which is not conducive to dissociation of the H2O molecule, but on the other hand, K prefers to slightly weaken the interaction between OH and H fragments at the TS and meanwhile strengthen their adsorption, especially OH adsorption on the Ag(111) by greatly increasing OH binding energy, contributing to H2O cleavage. Given the above, the stability effect of K on OH adsorption of TS plays a leading role in promoting H2O dissociation. Actually, the K adatom would stabilize almost all the oxygenates adsorbed on the Ag(111) surface including the reactants (CO, H2O) by increasing their binding energies (see Table 1). It is amazingly found that the interaction between the oxygenated adsorbates and Ag atoms become weakened, as evidenced by the elongated O–Ag and C–Ag bonds (see Table 1). Besides interacting with surface Ag atoms, these oxygenates also tend to interact with K since their O atoms and K adatom get close to each other, as shown in Fig. 1 and 2 for the co-adsorbed structures of adsorbates and K. Thus it is supposed that the attractive interaction between oxygenates and the K adatom render their adsorption more stable than their counterparts on pure surfaces. Yet the origin of such interactions between them is vague and remains to be disclosed further through electronic effect analysis.
 | ||
| Fig. 4 The decomposition results for the energy barriers of H2O dissociation on Ag(111) and K/Ag(111) surfaces. | ||
 | ||
| Fig. 5 Calculated PDOS of K 3p states and O 2p states for TS of H2O dissociation and K2O bulk. | ||
4.2 The factors affecting the K promotion effect
 | ||
| Fig. 6 The energy barriers and reaction energies of H2O dissociation versus K coverage. | ||
4.3 Comparison of the K promotion effect on Au, Ag, Cu, Ni and Pt surfaces
Promotion effects of K on H2O dissociation over different metal surfaces (such as Au(111), Cu(111), Ni(111) and Pt(111)) have been reported previously.2,3,18–20 Comparing these results with that over the Ag(111) surface, it is found that K addition reduces energy barriers on inert metal (Au, Ag and Cu) surfaces to a much larger extent (by 0.29–0.31 eV) than those on reactive metals like Ni and Pt (by 0.05–0.07 eV) and thus we deduce that the strength of K promotion effects may be associated with the activities of catalytic metal surfaces. To confirm this, we quantitatively plot the relationship between K-induced barrier reduction on various metal surfaces and the d-band center of the surfaces, as displayed in Fig. 7. The d-band center can be calculated by the formula: , where ρd represents the density of states projected onto metal atom's d-band and E is the energy of the d-band. Fig. 7(a) shows energy barrier differences for different metals linearly relate to the corresponding d-band center and the closer the d-band center to the Femi energy level, the lower the dissociation barrier difference, that is, the weaker the promotion effect of K. According to the rule proposed by Nørskov et al., the higher or more positive the d-band, the more active the metal surface.42 Consequently, K has a stronger promotion effect over more inert metals. As discussed in the last section, the direct K–O bonding forming at the TSs is the major cause of the K promotion effect on H2O dissociation, hence the stronger K–O bonding (i.e., the shorter the K–O bond) results in a stronger promotion effect, which is further confirmed by the fact that the lengths of K–O bonds on different metal surfaces are found well correlated with the K promotion effect (see Fig. 7(b)). The strengths of K–O bonds at the TSs for H2O dissociation on Ag(111) and Ni(111) surfaces are measured by the projected crystal orbital Hamilton population (pCOHP) method developed by the Dronskowski group43 and the related results are presented in Fig. S4 (ESI†) which shows that the K–O bonds on Ag(111) is much stronger than those on the Ni(111) surface. The length of the K–O bond on the Ag(111) surface is 2.44 Å, shorter than 2.60 Å on the Ni(111) and the result reconfirms that the shorter K–O bond means a stronger K–O bonding. Then why does stronger K–O bonding appear over the more inert metals? The reason may be that the more inert metal surfaces would weaken K and O binding and accordingly strengthen the interaction between them.
, where ρd represents the density of states projected onto metal atom's d-band and E is the energy of the d-band. Fig. 7(a) shows energy barrier differences for different metals linearly relate to the corresponding d-band center and the closer the d-band center to the Femi energy level, the lower the dissociation barrier difference, that is, the weaker the promotion effect of K. According to the rule proposed by Nørskov et al., the higher or more positive the d-band, the more active the metal surface.42 Consequently, K has a stronger promotion effect over more inert metals. As discussed in the last section, the direct K–O bonding forming at the TSs is the major cause of the K promotion effect on H2O dissociation, hence the stronger K–O bonding (i.e., the shorter the K–O bond) results in a stronger promotion effect, which is further confirmed by the fact that the lengths of K–O bonds on different metal surfaces are found well correlated with the K promotion effect (see Fig. 7(b)). The strengths of K–O bonds at the TSs for H2O dissociation on Ag(111) and Ni(111) surfaces are measured by the projected crystal orbital Hamilton population (pCOHP) method developed by the Dronskowski group43 and the related results are presented in Fig. S4 (ESI†) which shows that the K–O bonds on Ag(111) is much stronger than those on the Ni(111) surface. The length of the K–O bond on the Ag(111) surface is 2.44 Å, shorter than 2.60 Å on the Ni(111) and the result reconfirms that the shorter K–O bond means a stronger K–O bonding. Then why does stronger K–O bonding appear over the more inert metals? The reason may be that the more inert metal surfaces would weaken K and O binding and accordingly strengthen the interaction between them.
 | ||
| Fig. 7 Plot of energy barrier difference for water dissociation on clean and K-doped metal surfaces versus (a) the d-band center and (b) the length of K–O bond. | ||
5. Conclusion
The effect of the K promoter on the activity of the WGSR over the Ag(111) surface has been probed by DFT calculations systematically. The conclusions are summarized as follows:(1) The K adatom can stabilize the adsorption of all oxygenates by increasing their binding energies and the direct K–O bonding would strengthen their adsorption. However, K addition has little impact on the binding strength of H and H2 on the Ag(111) surface since negligible attractive interaction exits between K and H atoms.
(2) For both Ag(111) and K/Ag(111) surfaces, the WGSR proceeds mainly through the OH-assisted carboxy pathway and H2O dehydrogenation is found to be the rate-controlling step on both surfaces. K addition can enhance the activity of the WGSR by reducing the apparent activation energy of the whole reaction since it can promote H2O dissociation and stabilize adsorption of the reactants (CO and H2O). It is the ionic K–O bonding, the predominant interaction between K and O, which stabilizes the adsorption of oxygenates (including reactants of CO and H2O) and the stabilization effect on OH adsorption of TS contributes to promoting H2O dissociation.
(3) The K–O distance is a key factor to affect H2O activation, that is, when the K–O distance is long, the electrostatic interaction exists between them and the reduction of the dissociation barrier is small, while if the K–O distance is short, the direct K–O bonding dominates, which results in the barrier reduction to a large extent. Another factor of K coverage also has influence on its promotion effect and the dissociated OH and H are much more stabilized by K in its higher coverage, resulting in a remarkable decrease of the reaction energy and barrier of H2O dissociation when K coverage is high (25%).
(4) For various metals catalyzing the WGSR, K promotes H2O dissociation on inert metals like Ag, Au and Cu further than those on reactive metals (Pd and Ni) since the more inert metal surfaces would weaken K and O binding and accordingly strengthen the interaction between them, resulting in a higher promotion effect.
Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
This work is financially supported by the Applied Basic Research Project of Shanxi Province (No. 20210302124212) and the 1331 Engineering of Shanxi Province.References
- D. C. Grenoble, M. M. Estadt and D. F. Ollis, The chemistry and catalysis of the water gas shift reaction: 1. The kinetics over supported metal catalysts, J. Catal., 1981, 67, 90–102 CrossRef.
- C. H. Lin, C. L. Chen and J. H. Wang, Mechanistic studies of water-gas-shift reaction on transition metals, J. Phys. Chem. C, 2011, 115, 18582–18588 CrossRef.
- S. C. Huang, C. H. Lin and J. H. Wang, Trends of water gas shift reaction on close-packed transition metal surfaces, J. Phys. Chem. C, 2010, 114, 9826–9834 CrossRef.
- A. A. Gokhale, J. A. Dumesic and M. Mavrikakis, On the mechanism of low-temperature water gas shift reaction on copper, J. Am. Chem. Soc., 2008, 130, 1402–1414 CrossRef.
- Q. L. Tang, Z. X. Chen and X. A. He, Theoretical study of the water gas shift reaction mechanism on Cu (111) model system, Surf. Sci., 2009, 603, 2138–2144 CrossRef.
- R. J. Madon, D. Braden, S. Kandoi, P. Nagel, M. Mavrikakis and J. A. Dumesic, Microkinetic analysis and mechanism of the water gas shift reaction over copper catalysts, J. Catal., 2011, 281, 1–11 CrossRef.
- Z. Y. Li, N. Li, N. Wang, B. Zhou, P. Yin, B. Y. Song, J. Yu and Y. S. Yang, Mechanism investigations on water gas shift reaction over Cu (111), Cu (100), and Cu (211) surfaces, ACS Omega, 2022, 7, 3514–3521 CrossRef CAS PubMed.
- H. Prats, L. Álvarez, F. Illas and R. Sayós, Kinetic monte carlo simulations of the water gas shift reaction on Cu (111) from density functional theory based calculations, J. Catal., 2016, 333, 217–226 CrossRef.
- L. C. Grabow, A. A. Gokhale, S. T. Evans, J. A. Dumesic and M. Mavrikakis, Mechanism of the water gas shift reaction on Pt: First principles, experiments, and microkinetic Modeling, J. Phys. Chem. C, 2008, 112, 4608–4617 CrossRef.
- J. P. Clay, J. P. Greeley, F. H. Ribeiro, W. N. Delgass and W. F. Schneider, DFT comparison of intrinsic WGS kinetics over Pd and Pt, J. Catal., 2014, 320, 106–117 CrossRef.
- R. C. Catapan, A. A. M. Oliveira, Y. Chen and D. G. Vlachos, DFT study of the water-gas shift reaction and coke formation on Ni(111) and Ni(211) surfaces, J. Phys. Chem. C, 2012, 116, 20281–20291 CrossRef.
- G. Ertl, H. Kniizinger, J. Weitkamp, S. Professor and K. S. Suslick, Handbook of heterogeneous catalysis, VCH, Weinheim, 1997, pp. 2449–2479 Search PubMed.
- H. P. Bonzel, Alkali-metal-affected adsorption of molecules on metal surfaces, Surf. Sci. Rep., 1988, 8, 43–125 CrossRef.
- R. D. Diehl and R. McGrath, Structural studies of alkali metal adsorption and coadsorption on metal surfaces, Surf. Sci. Rep., 1996, 23, 43–171 CrossRef CAS.
- T. Maneerung, M. K. Hidajat and S. Kawi, K-doped LaNiO3 perovskite for high-temperature water-gas shift of reformate gas: Role of potassium on suppressing methanation, Int. J. Hydrogen Energy, 2017, 42, 9840–9857 CrossRef CAS.
- J. A. Rodriguez, E. R. Remesal, P. J. Ramírez, I. Orozco, Z. Liu, J. Graciani, S. D. Senanayake and J. F. Sanz, Water-gas shift reaction on K/Cu(111) and Cu/K/TiO2(110) surfaces: Alkali promotion of water dissociation and production of H2, ACS Catal., 2019, 9, 10751–10760 CrossRef CAS.
- W. Ma, G. Jacobs, U. M. Graham and B. H. Davis, Fischer-tropsch synthesis: Effect of K loading on the water-gas shift reaction and liquid hydrocarbon formation rate over precipitated iron catalysts, Top. Catal., 2014, 57, 561–571 CrossRef CAS.
- Y. X. Wang and G. C. Wang, A systematic theoretical study of water gas shift reaction on Cu(111) and Cu(110): Potassium effect, ACS Catal., 2019, 9, 2261–2274 CrossRef CAS.
- Y. X. Wang and G. C. Wang, A systematic theoretical study of the water gas shift reaction on the Pt/ZrO2 interface and Pt(111) face: Key role of a potassium additive, Catal. Sci. Technol., 2020, 10, 876–892 RSC.
- M. Zhou and B. Liu, DFT investigation on the competition of the water-gas shift reaction versus methanation on clean and potassium-modified nickel (111) surfaces, ChemCatChem, 2015, 7, 3928–3935 CrossRef CAS.
- Z. P. Liu and P. Hu, An insight into alkali promotion: A density functional theory study of CO dissociation on K/Rh (111), J. Am. Chem. Soc., 2001, 123, 12596–12604 CrossRef.
- L. Y. Gan, R. Y. Tian, X. B. Yang and Y. J. Zhao, Theoretical study of the influence of Na on CO adsorption and dissociation on Pd(111): Long-range or short-range interactions between co-adsorbates?, Chem. Phys. Lett., 2011, 511, 33–38 CrossRef.
- M. G. Castano, T. R. Reina, S. Ivanova, M. A. Centeno and J. A. Odriozola, Pt vs. Au in water-gas shift reaction, J. Catal., 2014, 314, 1–9 CrossRef.
- R. J. Lin, H. L. Chen, S. P. Ju, F. Y. Li and H. T. Chen, Quantum-chemical calculations on the mechanism of the water–gas shift reaction on nanosized gold cluster, J. Phys. Chem. C, 2012, 116, 336–342 CrossRef.
- M. Bingham and A. Mills, Catalytic and photocatalytic water gas shift reaction (WGSR) using a continuous flow, gas phase reactor, J. Photochem. Photobiol., A, 2021, 409, 113–133 CrossRef.
- A. Arab, D. Sharafie and M. Fazli, Theoretical study of water-gas shift reaction on the silver nanocluster, J. Phys. Chem. Solids, 2017, 109, 100–108 CrossRef.
- D. Sharafie, A. Arab and M. Fazli, The formate and redox mechanisms of water-gas shift reaction on the surface of Ag: A nanocluster model based on DFT study, Iran. J. Catal., 2019, 9, 213–221 Search PubMed.
- F. Boccuzzi, A. Chiorino, M. Manzoli, D. Andreeva, T. Tabakova, L. Ilieva and V. Iadakiev, Gold, silver and copper catalysts supported on TiO2 for pure hydrogen production, Catal. Today, 2002, 75, 169–175 CrossRef.
- G. Kresse, Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 11169–11187 CrossRef PubMed.
- G. Kresse and P. Furthmuller Effeciency, of ab-initio total energy calculations for metals and semiconductors using a plane-wave basis set, Comp, Mater. Sci., 1996, 6, 15–50 Search PubMed.
- P. E. Blöchl, Projector augmented-wave method, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 50, 17953–17979 CrossRef PubMed.
- G. Kresse and D. Joubert, From ultrasoft pseudopotentials to the projector augmented-wave method, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 59, 1758–1775 CrossRef CAS.
- J. P. Perdew, K. Burke and M. Ernzerhof, Generalized gradient approximation made simple, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed.
- G. Henkelman, B. P. Uberuaga and H. Jónsson, A climbing image nudged elastic band method for finding saddle points and minimum energy paths, J. Chem. Phys., 2000, 113, 9901–9904 CrossRef CAS.
- E. Sanville, S. D. Kenny, R. Smith and G. Henkelman, Improved grid-based algorithm for bader charge allocation, J. Comput. Chem., 2007,(28), 899–908 CrossRef CAS PubMed.
- G. Henkelman, A. Arnaldsson and H. Jonsson, A fast and robust algorithm for bader decomposition of charge density, Comput. Mater. Sci., 2006, 36, 354–360 CrossRef.
- H. J. Monkhorst and J. D. Pack, Special points for Brillouin-zone integrations, Phys. Rev. B: Solid State, 1976, 13, 5188–5192 CrossRef.
- NIST Chemistry WebBook: https://webbook.nist.gov/chemistry/, in NIST Standard Reference Database Number 69, June 2005 Release, ed. W. G. Mallard, National Institute of Standards and Technology, 2005.
- C. T. Campbell and J. R. V. Sellers, The entropies of adsorbed molecules, J. Am. Chem. Soc., 2012, 134, 18109–18115 CrossRef CAS PubMed.
- S. Kozuch and S. Shaik, How to conceptualize catalytic cycles? The energetic span model, Acc. Chem. Res., 2011, 44, 101–110 CrossRef CAS.
- B. Hammer, Adsorption, diffusion, and dissociation of NO, N and O on flat and stepped Ru(0001), Surf. Sci., 2000, 459, 323–348 CrossRef CAS.
- B. Hammer and J. K. Nørskov, Electronic factors determining the reactivity of metal surfaces, Surf. Sci., 1995, 343, 211–220 CrossRef CAS.
- V. L. Deringer, A. L. Tchougreeff and R. Dronskowski, Crystal orbital Hamilton population (COHP) analysis as projected from plane-wave basis sets, J. Phys. Chem. A, 2011, 115, 5461–5466 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2cp03757b |
| This journal is © the Owner Societies 2023 |