
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Polypropylene carbonate-based electrolytes as model for a different approach towards improved ion transport properties for novel electrolytes†
Anna I.
Gerlitz‡
a,
Diddo
Diddens
 *a,
Mariano
Grünebaum
a,
Andreas
Heuer
ab,
Martin
Winter
a and
Hans-Dieter
Wiemhöfer
*a
*a,
Mariano
Grünebaum
a,
Andreas
Heuer
ab,
Martin
Winter
a and
Hans-Dieter
Wiemhöfer
*a
aHelmholtz Institute Münster, IEK-12, Forschungszentrum Jülich GmbH, Corrensstraße 46, 48149 Münster, Germany. E-mail: d.diddens@fz-juelich.de; hdw@uni-muenster.de
bInstitute of Physical Chemistry, Westfälische Wilhelms-Universität, Corrensstaße 28/30, 48149 Münster, Germany
First published on 18th January 2023
Abstract
Linear poly(alkylene carbonates) such as polyethylene carbonate (PEC) and polypropylene carbonate (PPC) have gained increasing interest due to their remarkable ion transport properties such as high Li+ transference numbers. The cause of these properties is not yet fully understood which makes it challenging to replicate them in other polymer electrolytes. Therefore, it is critical to understand the underlying mechanisms in polycarbonate electrolytes such as PPC. In this work we present insights from impedance spectroscopy, transference number measurements, PFG-NMR, IR and Raman spectroscopy as well as molecular dynamics simulations to address this issue. We find that in addition to plasticization, the lithium ion coordination by the carbonate groups of the polymer is weakened upon gelation, leading to a rapid exhange of the lithium ion solvation shell and consequently a strong increase of the conductivity. Moreover, we study the impact of the anions by employing different conducting salts. Interestingly, while the total conductivity decreases with increasing anion size, the reverse trend can be observed for the lithium ion transference numbers. Via our holistic approach, we demonstrate that this behavior can be attributed to differences in the collective ion dynamics.
Introduction
In the global strive to establish lithium metal batteries as energy storage devices, polymer electrolytes are of great interest. This is due to the fact that polymer electrolytes offer improvements in terms of reduced dendrite formation and leakage of the electrolyte when compared to liquid electrolytes.1 Still, many polymer electrolytes suffer from a low ionic conductivity as compared to liquid electrolytes. Moreover, the partial conductivity of the lithium cations tends to be slower as compared to that of the anions. However, for applications in fast charging and high power devices2,3 electrolytes should not only exhibit a high total conductivity, but also a fast lithium ion transport, corresponding to transference number values close to one. Therefore, the need for polymer electrolytes with enhanced lithium ion transport properties has been in the focus over the last decades.1a,d Polyethylene oxide (PEO)-based electrolytes represent the most studied polymer-based electrolytes and hence are considered as a benchmark.4 This is favored by the ability of PEO to dissolve many different conducting salts at large quantities via the ether functional group.2b,3,4 Low glass transition temperatures Tg are typical for PEO-based systems, thus making them beneficial in terms of processing.Although low Tg values may generally enhance the ionic conductivity, the lithium ion dynamics in PEO melts is comparatively slow, which mainly originates from the coupling to the segmental motion of the polymer chains (Scheme 1),2b,4a which in turn leads to low lithium ion transference numbers around 0.1 (Fig. S1, ESI†).4a,5 In addition to this dynamical coupling, the lithium ions can be transported from one coordination site to another either by intrachain movement (Scheme 1a), i.e. moving from one site to another on the same chain, or by interchain movement (Scheme 1b), i.e. moving between sites on different polymer chains. Most polymer electrolytes such as poly(methyl methacrylate) (PMMA) or poly(vinyl alcohol) (PVA) so far have been observed to roughly mirror this behavior. Alternatively, the polymer may solely function as host material in both dry and gel electrolytes, so that ions are transported via the liquid electrolyte fraction or larger ion clusters, which occurs for example using poly(vinylidene difluoride) (PVdF).2,4a Yet, in such systems the overall flexibility of the polymer segments, often tailored by the liquid content, influences the overall ionic conductivity.6 In PEO, however, ion transport is not only limited by the motion of the polymer segments, but also by its partially crystalline structure below 60 °C, which further decreases the ionic conductivity.
 | ||
| Scheme 1 Lithium ion transport in PEO. Li+ is transported by segmental motion via (a) intrachain movement to a new site at the same polymer chain or via (b) interchain movement to a new site at a different polymer chain. | ||
A particular class of polymer electrolytes which might reduce these drawbacks are polycarbonate-based electrolytes. Polycarbonate electrolytes have gained an increasing interest in the research community within the last years due to their comparatively high ionic conductivities and transference numbers, as well as their sufficient mechanical and electrochemical stability,7 as e.g. demonstrated by Mindemark et al. and Tominaga et al.4a,7b,e,h,8 Prominent examples are based on polyethylene carbonate (PEC) or polypropylene carbonate (PPC), often in combination with lithium bis(trifluoromethylsulfonyl)imide (LiN(SO2CF3)2), LiTFSI or its derivatives. Upon addition of plasticizing agents or large quantities of conducting salts, the lithium ion transference numbers were reported to exceed values of 0.5–0.7 in such systems.7a,e,8a,d,9 Nonetheless, several limitations and drawbacks remain. For instance, studies on polycarbonates such as PEC showed that thermal stability issues arise for certain compositions.8c,10 The conditions for chain degradation of PEC and PPC and the underlying mechanisms were recently studied by Buchheit et al., who concluded that a sterically non-hindered α-carbon in the backbone can readily be attacked by a sufficiently nucleophilic anion if the adjacent carbonate group is polarized by a strong Lewis acid such as Li+.11 This depolymerization is faster for PEC than for PPC due to the additional methyl group in the latter. Furthermore, the rate of chain degradation increases with LiTFSI concentration and with temperature. In contrast, PEC or PPC electrolytes with stronger coordinating anions or less acidic cations are thermally stable.11a Importantly, any degradation of the polymer into smaller oligomer fragments (and finally even EC and PC monomers) would lead to faster dynamics and may thus lead to the misinterpretation of conductivity data. In this study therefore salt concentration, as well as temperature and measurement settings were applied to exclude influences due to depolymerization. Additionally, the investigated conducting salts are all fluorosulfonyl imide based which leads to similar geometrical structures as well as similar charge distribution, which allows one to study the effect of the anion size on the transport properties. Nonetheless, chain degradation cannot explain the improved ion transport properties of thermally stable polycarbonate electrolytes, which is the case at lower temperatures or when using more benign conducting salts. This indicates that apart from potential depolymerization issues, polycarbonate electrolytes based on PEC or PPC show intrinsically high cationic transference numbers. Generally, high cationic transference numbers can be achieved either by slowing down the anion (ultimately yielding single ion conducting polymer electrolytes, SIPE) or by speeding up Li+ (as it is usually achieved by the addition of additives, special structural design, increasing salt content or enhancement of polymer chain movement), or both.
In case of polycarbonates such as PEC or PPC different hypotheses exist on why these phenomena are observed. For example, reports that take contributions from inorganic fillers or other additives into account have been published.7c,d,g,8d,9c Since various PEC and PPC electrolytes with different contents of additives show this behavior, the high transference numbers must be linked to intrinsic properties of the polycarbonates, although the additives themselves are contributing as well. One such intrinsic factor is the observed weaker coordination between the carbonate group and Li+ as compared to the ether groups in PEO.12 This would partly release Li+ from the polymer, leading to less dynamical coupling to it, which corresponds to the second abovementioned mechanism by which Li+ is sped up. Clearly, this partial release of Li+ from the slow polymer will depend on the anion due to the tendency of the salt to form ion pairs or clusters. Another reason could lie in the morphology or structural characteristics of the polymer host, which would reduce anion movement in comparison to PEO. This explanation corresponds to the firstly described scenario above.8a,d A schematic representation of these two main hypotheses is shown in Scheme 2: on the one hand, the Li+ mobility can be increased due to a weakened Li+-polymer coordination, an enhanced Li+ exchange between different coordination sites (including anions) or a vehicular transport by an additive (green arrows). On the other hand, the hindrance of the anion movement due to steric effects or their larger size (compared to Li+) is another possible explanation (blue arrows).
 | ||
| Scheme 2 Possible hypotheses explaining the high transference numbers in polycarbonates such as PPC: enhancement of Li+ mobility or hindrance of anion movement. Fast cation exchange between different sites at polymers and anions are marked by green arrows, which enable enhanced exploration through the polymer matrix. Anion movement through and possible hindrance by the polymer matrix are indicated by dashed blue arrows. | ||
Insight into these underlying mechanisms constitutes a very interesting and relevant topic. In this study we therefore aim to investigate these mechanisms in more detail by a wide scope of experimental and computational techniques. In particular, PPC will be used as polymer matrix, while PEC or PEO are used as reference materials. To determine structure–property relations four different conducting salts are used. While different salts have already been investigated previously,7a our study employs salts with anions which are similar in molecular structure but vary in size. Similar as in our study on polycarbonate degradation,11a we chose LiN(SO2F)2 (LiFSI), LiN(SO2F)(SO2CF3) (LiFTFSI), LiN(SO2CF3)2 (LiTFSI) and LiN(SO2CF2CF3)2 (LiBETI). To investigate the transport properties of the various ions and to answer the question which mechanism is dominating, electrochemical measurements such as impedance spectroscopy and transference number measurements are carried out. Coordination features are observed using IR and Raman spectroscopy. Additionally, molecular dynamics simulations are applied to study the lithium ion coordination and dynamics as well as the cooperative motion of distinct ions.
Methods
The tested systems are based either on polyethylene carbonate (PEC) or polypropylene carbonate (PPC) with 1 mol kg−1 of conducting salt, respectively. The used lithium conducting salts were lithium bis(fluorosulfonyl)imide (LiN(SO2F)2, LiFSI), lithium(fluorosulfonyl)(trifluoromethylsulfonyl)imide (LiN(SO2F)(SO2CF3), LiFTFSI), lithium bis(trifluoromethylsulfonyl)imide (LiN(SO2CF3)2, LiTFSI) and lithium bis(pentafluoroethanesulfonyl)imide (LiN(SO2CF2CF3)2, LiBETI). For preparation of corresponding gel electrolytes, a weight fraction of 33 wt% of PC was added to the dry electrolyte samples for each salt.Experimentally, the resulting PEC and PPC electrolytes have been investigated, in dry and gelled state, respectively. Reference data from the benchmark system PEO-LiTFSI were kindly provided by A. Buchheit and used for comparison where appropriate.
Membrane preparation
Polymer electrolytes were prepared using PEC (QPAC25, empower materials, USA, M = 50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000–200000 g mol−1) and PPC (QPAC40, Empower Materials, USA, M = 100000–300000 g mol−1) together with the lithium conducting salts LiFSI (TCI Chemicals, >98%), LiFTFSI (Provisco CS, 98.8%), LiTFSI (Sigma Aldrich, >99.95%) and LiBETI (Iolitec, >99%). Additionally, PC (Merck, SelectiLyte, >99.9%) was used as gelation agent. Prior to membrane fabrication the polymers have been purified to remove residual particles from synthesis.13 For this, they have been dissolved in Chloroform (VWR, 99%) and filtered through a silica-filled column. Afterwards they have been precipitated in methanol (VWR, >99.8%) and dried under reduced pressure (3 × 10−6 mbar, 25 °C, 5–6 weeks) to yield polymers with less than 200 ppm residual water content. The lithium conducting salts LiFSI, LiFTFSI, LiTFSI and LiBETI were dried before use (LiFSI, LiTFSI and LiBETI at 120 °C and LiFTFSI at 80 °C) for 3 days under reduced pressure.
000–200000 g mol−1) and PPC (QPAC40, Empower Materials, USA, M = 100000–300000 g mol−1) together with the lithium conducting salts LiFSI (TCI Chemicals, >98%), LiFTFSI (Provisco CS, 98.8%), LiTFSI (Sigma Aldrich, >99.95%) and LiBETI (Iolitec, >99%). Additionally, PC (Merck, SelectiLyte, >99.9%) was used as gelation agent. Prior to membrane fabrication the polymers have been purified to remove residual particles from synthesis.13 For this, they have been dissolved in Chloroform (VWR, 99%) and filtered through a silica-filled column. Afterwards they have been precipitated in methanol (VWR, >99.8%) and dried under reduced pressure (3 × 10−6 mbar, 25 °C, 5–6 weeks) to yield polymers with less than 200 ppm residual water content. The lithium conducting salts LiFSI, LiFTFSI, LiTFSI and LiBETI were dried before use (LiFSI, LiTFSI and LiBETI at 120 °C and LiFTFSI at 80 °C) for 3 days under reduced pressure.
The preparation and processing of the polymer electrolyte membranes as well as all preparations for measurements were done in a dry room with a dew point lower than −40 °C.
To obtain the polymer electrolyte, 1 g of the dried polymer was pressed into a membrane using a hot-press (60 °C, 200 bar, 3 min). Then the according amount of conducting salt to yield 1 mol kg−1 was added stepwise to the polymer upon repetitive folding and hot-pressing. Sealed in a pouch bag, the mixtures were stored overnight in a climate chamber (50 °C for LiFSI and LiFTFSI and 70 °C for LiTFSI and LiBETI). This was repeated until the conducting salt was dissolved and homogeneous polymer films were obtained. The ratio of carbonate units to lithium ions in the resulting PEC-based electrolyte was 11:1 and in the PPC electrolytes 10:1.
Upon preparation of the gel electrolytes a weight fraction of 33% of PC was added (i.e. polymer/solvent = 2:1) to the prepared dry polymer electrolyte. PC was chosen as gelation agent for both, PEC and PPC, since it is the monomeric form of PPC, structurally similar to the PEC monomer EC, which offers the gelation of both systems with the same agent while maintaining structural similarity to the polymer systems without introducing many different components.
Electrochemical impedance spectroscopy (EIS)
To determine ionic conductivities in the electrolytes, EIS was used. The sample was placed in a container and contacted with coaxial stainless steel electrodes (inner electrode A = 0.785 mm2, outer ring-shaped electrode A > 10 mm2).14 The measurements were conducted in a temperature range of 0 °C to 80 °C, either going from 0 °C to 60 °C or performing a loop going back to 0 °C and up to 60 °C again. The temperatures were adjusted by a climate cabinet. Frequencies from 1 Hz to 1 MHz with an amplitude of 40 mV have been applied using a PGSTAT302N® potentiostat (Deutsche Metrohm). The obtained data was analyzed with Zview® 3.20 (Scribner Inc.) and Origin 2019 software (OriginLab).Transference number measurements
The transference number of lithium tLi+ was determined via the Bruce-Evans method15 using EIS and polarization of the sample in symmetrical Li|Li cells. A circular piece of the respective electrolyte membrane (A = 132.7 mm2) was cut and placed between the two lithium contacts (A = 113 mm2) with the polymer membrane being slightly bigger than the lithium contacts to prevent short circuiting of the cell. In a temperature range from 30 °C to 80 °C the data was collected using a PGSTAT302N® potentiostat (Deutsche Metrohm) with which polarization of the sample was achieved and the resulting current recorded. The data analysis was carried out analogously to the EIS measurements to obtain the impedance before and after the polarization experiment. For plotting and calculating the results Origin 2019 was used.IR spectroscopy
To gain structural information on the coordinational behavior of the polymer, infrared spectroscopy was carried out. Small amounts of the pure polymer and conducting salts as well as the prepared electrolytes were examined using an attenuated total reflection (ATR) unit in a Bruker Vertex 70 with a resolution of 2 cm−1 with a MIR source and a LaDTGS detector. An amount of 5–10 mg of the respective sample was placed on the ATR crystal and the chamber flushed with Ar for 5 min before each measurement was started and before background measurement. Spectra were recorded in a range of 400 cm−1 to 5000 cm−1 with 125 scans. The measurement software used was OPUS and for further data analysis Origin 2019 and Excel 2016 software was used.Raman spectroscopy
Using Raman-spectroscopy the dissociation of the conducting salt was estimated. For this pure polymer samples, conducting salts and solvent as well as the dry and gel electrolyte formulations were investigated using a Vertex 70 (Bruker) for liquid samples and a LabRAM HR Evolution (Horiba) spectrometer for solid samples. Dry and gel electrolyte samples were put in an air tight sample holder (Nanophoton). A laser wavelength of 633 nm, a grating with 1800 lines mm−1 and a 50× objective for the microscope were used. Spectra were recorded with 20 integrations with 10 s integration time in a spectral range from 15–3800 cm−1. LabSpec software and OPUS software were used to perform the measurements and Origin 2019 and Excel 2016 to plot and analyze the acquired data.PFG-NMR
Additional PFG-NMR experiments were carried out to determine the order of magnitude of the molecular movement. A Bruker AVANCE (III) 400 MHz NMR spectrometer was used to record signals of lithium, hydrogen, and fluorine. The measurements were performed at 40 °C and with a sample mass of 10–50 mg filled in a NMR tube. Data analysis was performed using Origin 2019.MD simulations
All simulations have been performed with the GROMACS-2019.3 package.16 The molecular interactions were described by the OPLS-AA force field17 for PPC and PC with charges from Borges Silva et al. specifically parametrized for carbonates18 as well as the CL&P force field19 for the lithium salts (LiFSI, LiFTFSI, LiTFSI and LiBETI). To account for polarization effects, all partial charges of the ions have been scaled by a factor of 0.8 as reported elsewhere in the literature.20The electrolytes in the dry state (i.e. without PC) consisted of 20 ethyl-terminated PPC chains with 30 monomers each and 54 ion pairs of the respective lithium salt, resulting in a ratio of PPC monomers to lithium ions of 11:1 which closely matches the experimental value. The gel electrolytes additionally contained 300 PC molecules in agreement with the experimental mass fraction of PPC to PC of 2:1.
The initial polymer structures were created with RDKit21 based on SMILES strings, and the corresponding number of polymer chains, PC molecules and ion pairs were inserted into a cubic simulation box without overlapping via the Packmol tool.22 Subsequently, the systems were relaxed at a temperature of T = 700 K over 10 ns before quenching to 450 K during 10 ns. The velocity-rescale thermostat23 and Berendsen barostat24 with a high pressure of p = 1000 bar have been used at this stage to compress the initial structure and yield a dense polymer melt/gel. The elementary integration time step was 1 fs. Afterwards, the systems were equilibrated at T = 450 K and p = 1 bar for 110 ns using a time step of 2 fs. Subsequently, the systems were cooled down to 400 K over a period of 10 ns, at which they were equilibrated for another 110 ns before collecting production runs with a length of 1 μs. During all equilibration and production runs, the following settings were used: temperature and pressure have been maintained by a Nosé–Hoover thermostat25 and a Parrinello–Rahman barostat,26 respectively. The electrostatic interactions were computed by the particle-mesh Ewald method27 using a cut-off radius of 1.6 nm and a grid spacing of 0.1 nm with sixth-order interpolation. Lennard-Jones interactions were truncated at 1.6 nm. All bonds involving hydrogen atoms were constrained by the LINCS algorithm.28 Periodic boundary conditions were applied in all three dimensions. The final systems had sizes of about 4.6 nm for the dry electrolytes and 5.2 nm for the gelled systems.
Results and discussion
Measurement of ion transport properties
The ionic conductivities of the PPC and PEC-based electrolytes were determined in the dry as well as in the gelled state. In Fig. 1 the Arrhenius-plots of the PPC gel electrolytes containing the four different conducting salts are shown. The corresponding Arrhenius-plots of the PEC gel electrolytes as well as the respective dry electrolytes can be found in Fig. S4 in the ESI.† In both polycarbonate matrices the ionic conductivity increases when going from conducting salts with larger to smaller anions, namely BETI− (green symbols) to FSI− (black symbols). | ||
| Fig. 1 Arrhenius-plots of the ionic conductivities in PPC gel electrolytes (1 mol kg−1 salt and 33 wt% PC). The ionic conductivity of systems with small anion sizes is larger than those of systems with larger anions. | ||
The ionic conductivity σ in the PPC gel system was found to range from 1.05 × 10−7 S cm−1 in case of PPC-LiBETI gel to 8.60 × 10−5 S cm−1 in case of PPC-LiFSI gel at elevated temperatures (60 °C). In PEC-gel electrolytes the ionic conductivity was found to be higher compared to the PPC-based system with, for example, the PEC-LiBETI gel possessing a conductivity of 1.60 × 10−5 S cm−1 at 60 °C. The benchmark electrolyte system PEO-LiTFSI displays an ionic conductivity of 8.70 × 10−4 S cm−1 at 60 °C (compare Fig. S2 in the ESI†). When comparing the ionic conductivities of PPC gel electrolytes to the state-of-the-art PEO system, the polycarbonate-based electrolytes show lower values.
Keeping in mind the above-mentioned chain depolymerization, which is occurring mainly in PEC-based electrolytes, we only present measurement data where no degradation was proven to occur, either verified by NMR measurements or by not exceeding known temperature limitations. Because of this, not all data of PEC and PEC gel electrolytes can be shown and hence is only given for comparison if no interference was happening. The observed decrease of σ in Fig. 1 when going from LiFSI to LiBETI also shows that the chain depolymerization is not interfering, because the rate of the depolymerization reaction increases the weaker the coordination by the anion is. Therefore, if the depolymerization would interfere, exactly the opposite trend in σ would be observed. Furthermore, the less pronounced degradation of PPC-based as compared to PEC-based electrolytes can be seen in Fig. 2, where the ionic conductivity of PPC-LiFSI with 33 wt% PC is shown. Within the time to measure σ in the PPC gel systems, ramping from 0 °C up to 80 °C, back down to 0 °C and up to 80 °C again, which corresponds to 15 h at elevated temperatures, it does not vary significantly. Reversible heating and cooling can be performed without severe degradation, which would affect the conductivity measurements. Due to the additional methyl group within the backbone, the degradation is too slow to have a negative impact on the performed experiments.
 | ||
| Fig. 2 Arrhenius-plots of the ionic conductivities in the PPC-LiFSI gel electrolyte (1 mol kg−1 salt and 33 wt% PC). Measurements were performed in a range from 0 °C to 80 °C ramping up, down and up again. | ||
The other PPC-gel electrolytes have also been monitored during repeated heating and cooling and show similar results (Fig. S3, ESI†). In some instances minor deviations between the first and the following cycles can be observed. This could be explained by structural realignment of the polymer chains.
Considering that ion pairing should be stronger for smaller anions and therefore less dissociation should occur, one would naively expect the ionic conductivity to be lower in case of salts with smaller anions. However, the observed behavior seems to indicate that salts with larger anions tend to move slower, resulting in a decreased conductivity. Although this complies with the hypothesis that a morphological hindrance hampers the motion of the anions depending on their respective sizes, the anion species could also affect the Li+-anion pairing and hence shift the equilibrium of the Li+-polymer coordination.
As expected, the ionic conductivity is lower in the dry electrolytes than in the gelled ones (see Fig. 3). The difference between the gelled and dry samples is a few decades in conductivity. The dry electrolytes show conductivities around 1 × 10−7 S cm−1 (blue and black symbols) whereas gelled systems yield up to 4 × 10−5 S cm−1 (red and green symbols). This is in good agreement with the computational results (see below), where the dynamics of the different species in the dry and gelled PPC electrolytes were monitored and an increase in their dynamics by two orders of magnitude at high temperatures due to gelation was observed. This indicates that the addition of PC leads to a plasticizing effect.
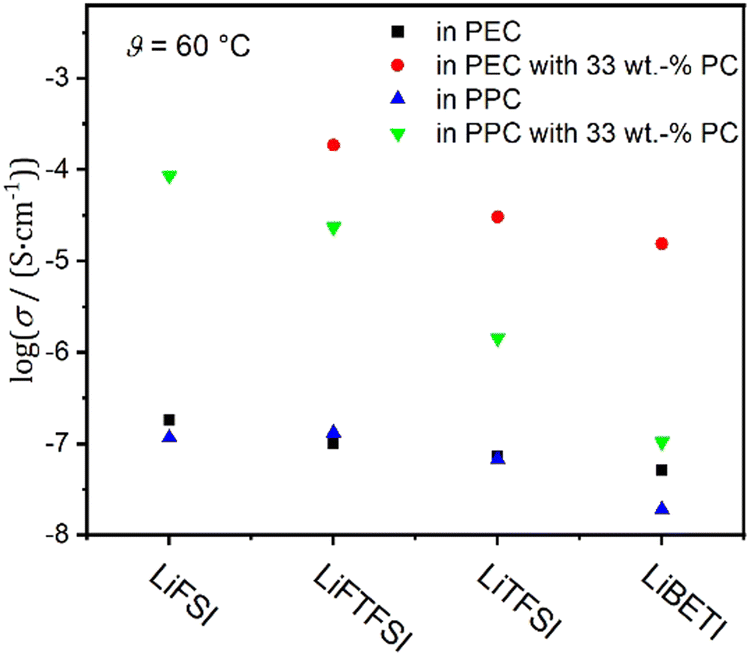 | ||
| Fig. 3 Comparison of ionic conductivities in dry and gelled electrolytes at 60 °C. Values are given in the logarithm of the ionic conductivity. | ||
While differences between dry and gelled electrolytes are expected and well documented3,4a,29 the observed increase of up to three decades is unusually large. When comparing the ionic conductivities of different conducting salts at the same temperature (Fig. 3) a correlation between σ and the anion size is again observed in the gel systems (green and red symbols). This can hardly be described by enhanced chain movement mechanism only as often observed with polymer electrolytes.30 The dissociation of the conducting salt typically governs the transport properties, which usually also depends on the anion31,32 and hence could explain the observations from Fig. 3. However, the PPC-based electrolytes tend to show lower ionic conductivities than the PEC-based, which suggests that the flexibility of the polymer backbone also affects the conductivity, especially in the limit of long chains. Therefore, a size-dependent slowdown of the anions could equally cause the trend in Fig. 3.
In the next step we therefore investigate whether only the anions or entire cation–anion pairs/clusters are hindered in their motion. If the morphology impedes dissociated anions only, no effect on the Li+ dynamics should be observed. In conventional polymer electrolytes, the lithium transport is rather small due to its strong coupling to the polymer. For instance, transference numbers around 0.1–0.2 are typically reported in standard PEO-based electrolytes.33,34 In agreement with this, we observe a value of 0.11 for our PEO-LiTFSI reference system (see Fig. S1 in the ESI†). It should be mentioned that the determination of the lithium transference number was not feasible for every sample at each temperature in this study due to marginal conductivity values or stability issues towards lithium. However, reproducible results were obtained with the stable PPC electrolytes in dry and gelled state. The values of the transference numbers in PPC-gel electrolytes are given in Table 1.
| Electrolyte | T/°C | t Li+ | σ total/S cm−1 | σ Li+/S cm−1 |
|---|---|---|---|---|
| PPC-LiFSI gel | 50 | 0.30 | 4.77 × 10−5 | 1.43 × 10−5 |
| PPC-LiFTFSI gel | 50 | 0.44 | 1.36 × 10−5 | 5.96 × 10−6 |
| PPC-LiTFSI gel | 50 | 0.61 | 5.58 × 10−7 | 3.40 × 10−7 |
| PPC-LiBETI gel | 50 | 0.65 | 2.0 × 10−8 | 1.30 × 10−8 |
The determined transference numbers vary from 0.30 in case of PPC-LiFSI with 33 wt% PC to values of 0.65 in case of PPC LiBETI and were around 0.4 to 0.5 for most of the investigated samples (Table S1, ESI†). These values are quite high, which is in agreement with previous studies.7h,8a Often such high values are only obtained by multi-step synthesized polymer electrolytes of which many exhibit dissolution issues, although other polycarbonate-based and polymers containing carbonyl groups, such as polycaprolactone (PCL) or poly(trimethylene carbonate) (PTMC), were found to reach values of 0.5–0.8 as well.4a,12,35
It can be seen that the transference number increases with increasing anion size, starting from 0.30 in the case of PPC-LiFSI gel to 0.46 in the case of PPC-LiFTFSI gel and up to 0.65 in case of PPC-LiBETI gel (compare Fig. S5 in the ESI†). When comparing this behavior to the previously described ionic conductivity measurements the correlation of movement and anion size is reversed. If the lithium ions were strongly attached to the polymer chains and only transported by segmental motion, the transference number should not be linked this closely to the anion size but rather to the polymer dynamics, which seems not to be the case. The opposite trends in ionic conductivity and transference number, however, make sense if either (a) larger ions are hindered more strongly as compared to smaller ones (and hence become slower as compared to Li+) or (b) if the cooperative motion of Li+ and anions changes with the anion size due to different interionic interactions. Besides the anion-size dependence, we also find that gelation leads to slightly lower tLi+ as compared to analogous dry polymer electrolytes, the latter ranging from 0.37 up to 0.86 (Table S1, ESI†). Notably, both in dry and gelled PPC the correlation between anion size and increase in transference number is observed, although one would naively expect a weaker morphological anion hindrance for the latter due to the less dense packing of polymer chains.
Next, we used PFG-NMR measurements to investigate the diffusion coefficients of 7Li and 19F in the PPC electrolytes. In the dry PPC electrolytes containing the different fluorosulfonyl imides LiFSI, LiFTFSI, LiTFSI and LiBETI the diffusion coefficients derived from the 7Li and 19F signals were in the order of 10−12 m2 s−1. In the gelled PPC electrolytes an increase of one order of magnitude of the diffusion coefficients was observed, resulting in values of around 10−11–10−10 m2 s−1 for the cation and the four different anions (compare Fig. S16 and S17, ESI†). These results are in the same range as the MD data with 2 × 10−13 m2 s−1 in case of 7Li and 19F diffusion coefficients in the dry PPC electrolytes and 3 × 10−11 m2 s−1 in the gel PPC electrolytes (although the MD values for the dry electrolytes might be slightly overestimated when compared to the gel; see detailed discussion below). Obviously, due to the gelation and the resulting overall increase in the dynamics, the dissolved conducting salts can move faster, in agreement with the observed conductivity increase (Fig. 1 and 3; for additional information regarding the NMR experiments, see ESI†).
Vibrational spectroscopy
As the dynamical information contained in σ, tLi+ and the diffusion coefficients alone cannot distinguish between the two abovementioned hypotheses (ion correlations vs. anion hindrance, both depending on the anion species), IR and Raman spectroscopy were performed. Via IR measurements it was examined whether the coordination takes place at the carbonyl oxygen atoms (denoted as “![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O” in the following) or the alkoxy (or ethereal) oxygen atoms (denoted as “–O–”) of PPC or PC.
O” in the following) or the alkoxy (or ethereal) oxygen atoms (denoted as “–O–”) of PPC or PC.
To assign the observed signals at the respective wavenumbers to coordinating or non-coordinating oxygen atoms, spectra were recorded for the single compounds as well as their binary and ternary mixtures. In Fig. 4 the signal of the O oxygen can be observed for the different compositions. A difference in position of the adsorption band is observed whether the O signal is originating from PC (∼1790 cm−1) or PPC (∼1740 cm−1). In the region of the –O– signal, 1280–1230 cm−1, however, no change is observed, suggesting that the Li+ coordination mainly takes place via the O group and not via the –O– unit (see MD part for further discussion). For the O band of PC (dark blue curve) and of PPC (bright green curve) a splitting into two signals can be observed when a lithium salt is dissolved, which means a coordinating and non-coordinating behavior of O is distinguishable. Therefore, bands can be attributed to coordinating (∼1713 cm−1 in PPC-LiFSI, bright green curve) and non-coordinating O (∼1740 cm−1 in PPC-LiFSI, bright green curve) groups from PPC and coordinating (∼1765 cm−1 in LiFSI in PC, dark blue curve) and non-coordinating O groups (∼1784 cm−1 in LiFSI in PC, dark blue curve) from PC. There are slight shifts of the bands observable depending on whether only one compound or a combination of solvent, polymer, conducting salt and PC are present. Thus, the relative ratio between coordinating and non-coordinating O groups can be determined by fitting the bands of the single and combined bands via a Gaussian function and using the parameters obtained to fit the complex PPC gel electrolyte system. This procedure was analogous for all investigated electrolytes (Fig. S6–S8 in the ESI†). The resulting percentage of coordination in the different PPC gel electrolytes in comparison to the coordination in the dry PPC electrolytes is given in Fig. 5.
 | ||
| Fig. 4 Comparison of obtained IR spectra for PPC-LiFSI as gel electrolyte and in the dry state. Non-coordinating and coordinating O groups were identified using single components and binary mixtures of the electrolyte. Note that the obtained spectra for the conducting salts LiFTFSI, LiTFSI and LiBETI in PPC and PPC gel look similar. | ||
 | ||
| Fig. 5 Percentage of O-groups coordinating Li+ in PPC electrolytes determined via IR-spectroscopy. The amount of coordination in dry PPC electrolytes (SPE) is shown in blue bars and in PPC gel electrolytes in grey and orange bars. The latter splits in the contribution of PPC (orange) and PC (grey). | ||
It can be seen that in the gel electrolytes (orange and grey bars) most of the coordination is realized by the O groups of PC. Compared to the dry systems (blue bars) the overall amount of coordination does not differ much, but the proportion of coordination by PPC is clearly decreased, which means upon addition of the gelation agent PC a slightly favored coordination of Li+ by O of PC occurs, which corresponds to less coordination to the polymer and therefore a weaker dependence of the Li+ transport on the polymer dynamics. In total, no significant trend concerning the impact of the anion can be observed from Fig. 5. We will return to this point in the MD section below.
Similarly, whether the lithium ions are associated with anions or may even exist in agglomerates has not yet been identified. Conceivably, the interaction may affect the overall transport mechanism, since similar phenomena were observed in systems containing large amounts of conducting salt that tend to form larger agglomerates, in which the ion transport is rather independent from the surrounding matrix.36 To investigate this issue for the PPC gel electrolytes, Raman spectroscopy was employed. The approach for analyzing the data derived from these measurements was analogous to the IR data. The single compounds were characterized, which enabled us to assign the peaks and to determine their relative ratios in the electrolytes. There was a noticeable shift of the signal in the obtained spectra depending on whether the salt was in an agglomerated state (∼747 cm−1) or dissolved by PC (∼741 cm−1) or PPC (∼743 cm−1) (see Fig. 6). A distinction between different variations of ion pairing and agglomeration, however, was not possible due to the close proximity of the different bands. This also made the fitting difficult in cases where solvated species largely exceeded agglomerated species, which is why the ratios derived from the Raman data rather give a qualitative picture. Note that the obtained spectra for the systems containing the other conducting salts LiFSI, LiFTFSI and LiBETI in PPC and PPC gel look similar (see Fig. S9 and Table S2 in the ESI†).
 | ||
| Fig. 6 Section of the obtained Raman-spectra for the PPC-LiTFSI gel and dry electrolyte for comparison. | ||
The results for the different PPC gel electrolytes are listed in Table 2. The ratios of agglomerated and solvated state are given in percentages. In most gel electrolytes, it seems that agglomeration is very low to non-existent. Only in case of PPC-LiTFSI the agglomeration of 30% is larger than within the other electrolytes, although this outlier should be taken with care as argued above. This means that no larger clusters are formed within the PPC gel electrolytes that could facilitate vehicular transport of Li+. In such systems the coordination is strongly bound to the anions, leading to a cooperative transport with Li+.4,37 Not surprisingly, we observed the same trend as in Table 2 for gel electrolytes containing the chemically similar PEC instead of PPC as matrix (see Table S2, ESI†).
| Electrolyte with 33 wt% PC | Agglomerated state/% | Solvated state/% |
|---|---|---|
| PPCLiFSI | 1 | 99 |
| PPC-LiFTFSI | 6 | 94 |
| PPC-LiTFSI | 30 | 70 |
| PPC-LiBETI | 3 | 97 |
Moreover, the agglomeration in the dry PPC electrolytes does not vary significantly from the gel PPC electrolytes (Table S2 in ESI†). Although gelation could in principle further reduce agglomeration due to dilution and the presence of additional Li+ coordination sites, the fraction of agglomerated ions is already low in the dry state. Therefore, such minute differences cannot be resolved due to the aforementioned uncertainties when fitting the Raman peaks.
Molecular dynamics simulations
So far, we collected evidence for both scenarios described in the beginning, i.e. anion-dependent decoupling of Li+ from PPC and size-dependent slowdown of the anions by the polymer matrix. In particular, either mechanism could rationalize the opposing trends for the overall conductivity (Fig. 1 and 3) and the Li+ transference number (Table 1) alone, but also the combination of both effects seems possible. To obtain further insights at the molecular level, we additionally performed MD simulations. In a first step, we studied the lithium ion coordination by polymer, solvent and anions. Fig. S10 in the ESI† shows the radial distribution functions (RDFs) between Li+ on the one hand and carbonyl oxygen atoms (O), alkoxy (ethereal) oxygen atoms (–O–) of PPC and PC, or oxygen atoms of the anions on the other hand. In all cases except the –O– atoms of PC, a significant first coordination peak is observed, in which the O and anion oxygen atoms show the largest peaks similar to the results of other recent MD studies on polycarbonate electrolytes.35,38 The comparatively small peak of the –O– atoms of PC can be attributed to the cyclic nature of the molecule, such that the oxygen atoms embedded in the ring only weakly coordinate Li+ due to steric reasons. Contrarily, in linear carbonates the –O– atoms can in principle coordinate in a bidentate fashion, which would result in a more stable coordination of this type for PPC. In fact, for dimethyl carbonate, this binding mode has been revealed by DFT calculations,39 although it is important to stress that for the coordination by PPC, polymer-specific effects may play a role as well. To quantify the Li+ coordination in more detail, all coordinating oxygen atoms in the first coordination shell defined by the RDFs were counted. In particular, O atoms were defined as coordinating if their distance to Li+ was not larger than 0.35 nm, corresponding to the position of the minimum after the first coordination peak in the RDFs (see Fig. S10 in the ESI†). Likewise, –O– atoms of PPC were considered as bound if their distance did not exceed 0.34 nm, whereas for –O– atoms of PC a smaller value of 0.29 nm was used due to the reasons mentioned above. Oxygen atoms of the anions were defined as bound up to a distance of 0.34 nm. The resulting average coordination numbers N are shown in Fig. 7.
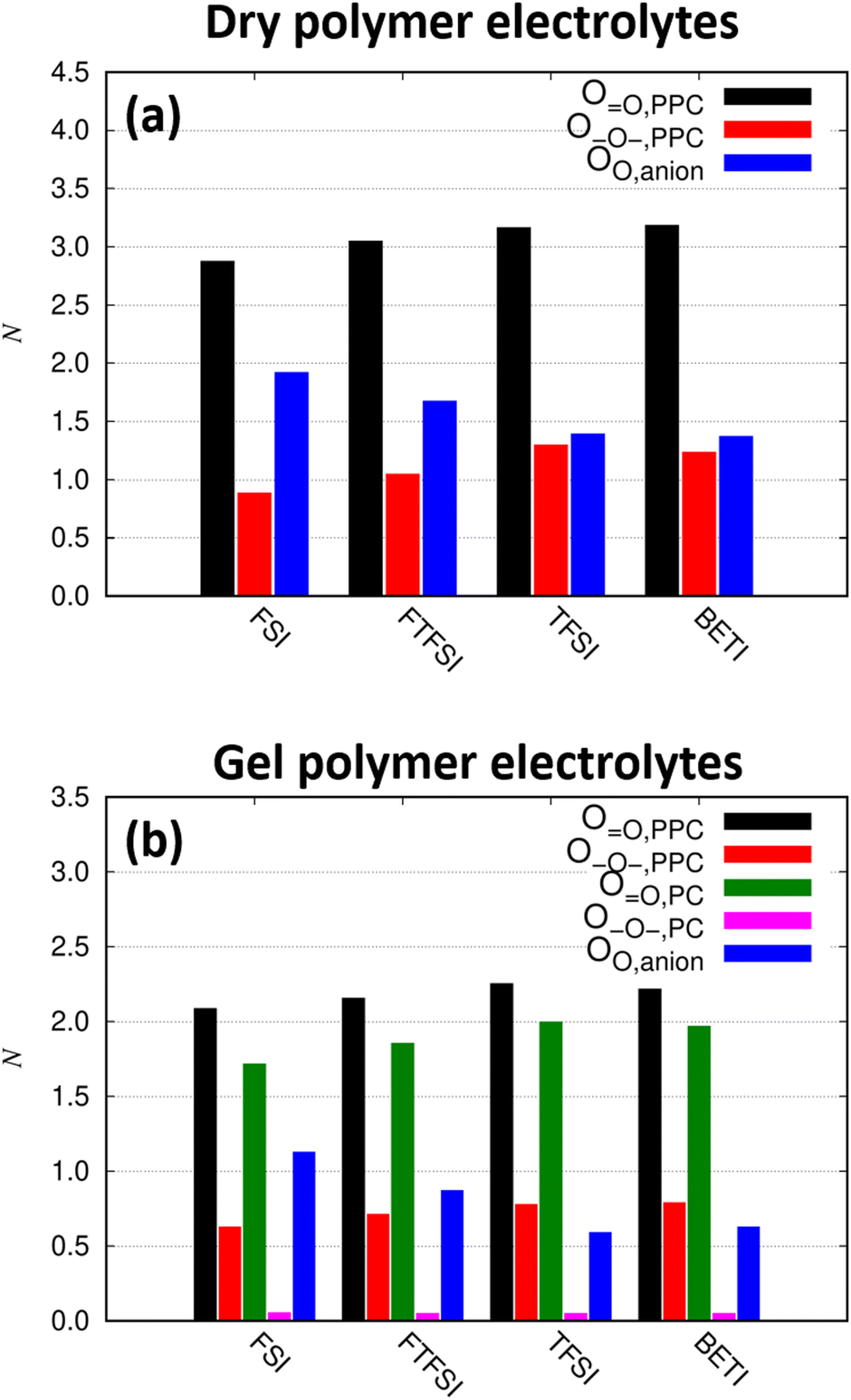 | ||
| Fig. 7 Average coordination numbers for the different types of coordinating oxygen atoms for (a) the dry electrolytes and (b) the gel polymer electrolytes. | ||
For the dry polymer electrolytes, we find from Fig. 7 that Li+ is typically coordinated by 2.8–3.2 O atoms and 0.9–1.3 –O– atoms from the PPC chains as well as 1.4–1.9 oxygen atoms from the anions, resulting in an overall coordination number of 5–6 consistent with previous studies.35,38 This is in good agreement with the results from IR-spectroscopy where it was observed that coordination is rather originating from O than from –O– (compare Fig. 4). The coordination by –O– atoms increases slightly in the order FSI < FTFSI < TFSI < BETI at the expense of the anion coordination, that is, larger anions tend to cluster less in the dry electrolytes. In total, almost two (i.e. 1.8–1.9) PPC chains and slightly more than one anion (i.e. 1.2 on average) coordinate each lithium ion on average (Fig. S11 in the ESI†).
For the gel polymer electrolytes, the lithium ions are coordinated by roughly equal fractions of O atoms of PPC and O atoms of PC (between 1.7–2.2 each), in addition to 0.6–0.8 –O– atoms of PPC and 0.6–1.1 anionic oxygen atoms. This is consistent with the results obtained from IR-spectroscopy where a shared coordination by PPC and PC in the gel electrolytes was observed (compare Fig. 5). Again, larger anions generally exhibit lower coordination numbers. As expected, the coordination of –O– atoms of PC are rare, reflected by small average coordination numbers of only 0.1. Due to the addition of PC, Li+ is on average only coordinated by 1.4 PPC polymer chains as opposed to about 1.8 for the dry case (Fig. S11, ESI†), indicating that the lithium ions are less strongly coupled to the polymer matrix in the gelled systems. Furthermore, only 0.5–0.9 anions coordinate a given lithium ion, suggesting that the additional PC coordination also reduces the degree of ion pairing. The reduced PPC and anion coordination numbers are counterbalanced by about 1.4 coordinating PC molecules (Fig. S11, ESI†). Nonetheless, the vast majority of all lithium ions is still coordinated to at least one PPC chain and only 2–3% of the lithium ions are fully detached from the polymer chains. This rules out a transport mechanism in which the lithium ions are completely decoupled from the polymer chain and also leads to the conclusion that a vehicular transport can be excluded as primary mechanism.
Next, we investigate the lithium ion dynamics in the PPC gel electrolytes and compare it to the dry electrolyte as a reference. To get a first impression of the motion of the lithium ions, Fig. 8 shows so-called topological maps, i.e. a contact map between a given lithium ion and all potentially coordinating oxygen atoms – consecutively numbered – as a function of time. In each of these plots, a single lithium ion is regarded and all coordinating oxygen atoms at time t are marked by a dot. For the dry PPC-LiTFSI electrolyte, the lithium ion is essentially only coordinated to oxygen atoms of a few PPC chains over the entire simulation period of 1 μs in agreement with earlier findings38 (black points in Fig. 8, note that the dashed horizontal lines indicate different PPC chains within the otherwise consecutively numerated index). Similarly, only coordinations to a small number of different oxygen atoms of a few TFSI anions are observed (blue points). In contrast, for the gel polymer electrolyte, it is found that the Li+ under consideration visits a large fraction of all PPC chains contained in the simulation box, demonstrating the rapid exchange in its coordination sphere. Even faster dynamics can be observed for the brief coordination to PC molecules (red points) or anions. This indicates that due to the addition of PC, the relaxation of the local environment becomes dramatically enhanced, leading to a rapid exchange in the Li+ coordination shell as opposed to the long-lived Li+ coordination in the dry electrolyte. This is matching the experimental observations on ionic conductivity well, where a drastically enhanced ionic motion for the gels as compared to the dry PPC was observed (see Fig. 3).
 | ||
| Fig. 8 Topological maps showing the time dependent evolution of the coordination shell of a given lithium ion in (a) the dry PPC-LiTFSI electrolyte and (b) PPC-LiTFSI gelled with PC. All potentially coordinating oxygen atoms are consecutively numerated, where dashed horizontal lines indicate the different oxygen types (PPC, PC and TFSI) or different PPC chains, respectively. PPC coordinations are shown in black, PC coordinations in red and TFSI coordinations in blue. For PPC and PC, coordinations to O oxygen atoms are displayed in full colours, whereas coordinations to –O– oxygen atoms are shown in lighter colours (note that the latter coordination is less prominent and hence less visible). | ||
Only a minor fraction of all PPC-Li+ contacts exists long enough to allow the lithium ion to diffuse along the PPC backbone by exchanging coordinating oxygen atoms of the same chain. This is in stark contrast to PEO-based electrolytes, in which lithium ions typically reside at a given PEO chain for a substantial amount of time.40 While Li+ can explore the PEO chain it is attached to during this period, its motion remains confined to the same chain, which significantly slows down its overall dynamics.40b,c Nonetheless, a certain degree of Li+ motion along the backbone is observed, which was also reported for other linear polycarbonates.38 Due to the fast exchange in the gelled electrolytes, the Li+ dynamics is less hindered by the slow polymer chains, rationalizing their good transport properties. Together with the experimental findings this shows a coordination of Li+ to the polymer which is strong enough to affect its transport but is weaker than the coordination by PEO, which suggests that a certain amount of decoupling from segmental motion is achieved. Note that for the other lithium salts, the topological maps look similar to Fig. 8. In contrast to PEO electrolytes, this fast Li+ dynamics gives rise to the overall high transference numbers observed within all investigated PPC electrolytes (compare Table 1).
To quantify the exchange of the lithium ion solvation shell in more detail, Fig. 9 shows the average lifetimes of the individual coordination types (see ESI† for details of the calculation). Note that these results are only shown for the gel electrolytes, as the corresponding relaxation times for the dry systems are in the same range as the entire simulation time, such that we expect the statistical uncertainties to be considerable. Consistent with the observations from Fig. 8, it can be seen that both O and –O– oxygen atoms of PPC coordinate a given Li+ for 3–5 ns. Since lithium ions may exchange oxygen atoms from the same PPC chain, the overall residence time at a given PPC chain is somewhat longer, i.e., on the order of 10–20 ns. Nonetheless, this is still significantly shorter than the corresponding residence times observed for PEO electrolytes40b,c,41 – also bearing in mind that these simulation studies were carried out at a higher temperature. Overall, these findings support the hypothesis of a weak Li+ coordination to the polycarbonate backbone, leading to a rapid exchange of coordinating polymer chains. Interestingly, the average Li+ residence time at a given PPC chain slightly increases with the anion size, consistent with the experimental observation that the overall transport becomes slower. As expected from Fig. 8, the lifetimes of the Li+-PC coordination are significantly shorter (1–2 ns) than the corresponding residence times of the PPC monomers (i.e. O and –O– of PPC). Note that in Fig. 9, the PC coordination lifetime has been computed for the entire molecule (i.e. irrespective of the oxygen type) as the lifetime of the –O– coordinations are extremely short. Due to the short Li+-PC coordination lifetime, the PC molecules presumably only travel very short distances together with the lithium ions, and therefore do not act as an ion transporting shuttle in the strict sense, which has been observed for additives bearing oligoether side chains.41b,42 This is also compatible with the fact that only 2–3% of the lithium ions are not connected to any PPC chain. Finally, the anions display coordination lifetimes comparable to those of the PPC monomers (about 5 ns, cf.Fig. 8), and slightly decrease with increasing anion size as a result of the reduced ion pairing.
 | ||
| Fig. 9 Average lithium ion coordination lifetimes for specific oxygen atom types of the PPC chain (carbonyl oxygen atoms “O” and ethereal oxygen atoms “–O–”) in the gel electrolytes with different lithium salts. The average lithium ion residence time at a given PPC chain, PC molecule or salt anion is also shown. | ||
To study the dynamics in more detail, we present the mean squared displacements (MSDs) of the individual components of the dry and gel polymer electrolytes with LiTFSI in Fig. 10. Most notably, when comparing the gel electrolyte (solid curves and filled symbols) with the dry electrolyte (dashed curve and open symbols), the dynamics of all compounds become drastically enhanced by the presence of PC, as reflected by an increase of the MSDs by several orders of magnitude. Therefore, the addition of PC not only strongly reduces the average Li+ residence time at a given PPC chain (Fig. 8 and 9), but also acts as a plasticizer which enhances the polymer motion, similar to ionic liquid additives in PEO electrolytes.41a,43 Interestingly, the MSDs of Li+ and TFSI in the gel electrolyte are almost identical throughout the entire simulated time range. This supports the experimental finding of rather large transference numbers, although it should be stressed that the latter quantity is not only affected by the self-diffusion of both ion species, but also by cross correlation between distinct ions (see below).44 Furthermore, the MSDs of Li+ and TFSI in Fig. 10 are comparable to that of the PPC segments (measured by the MSD of the O atoms) on short time scales (i.e. up to about 0.3–3 ns), indicating that the motion of the ions is coupled to the polymer dynamics in this regime. However, on larger time scales, the ion MSDs become diffusive showing that the ions’ dynamics become decoupled from the polymer chains in agreement with the observations from Fig. 8 and 9 as well as the experimental findings. Note that for the dry electrolyte (dashed curve and open symbols), we do not observe this decoupling on the accessible time scales as the overall dynamics is too slow. For the diffusion coefficients of the ions, we find values in the order of 2 × 10−13 m2 s−1 for the dry electrolytes and 3 × 10−11 m2 s−1 for the gel electrolytes (see Fig. S13 and Table S3 in the ESI†), which is in the same range as the experimental data (Fig. S16 and S17 in the ESI†). It has to be kept in mind, though, that the temperature in the simulations was significantly higher than in the experiments, that is, the simulations predict too slow dynamics. This can be attributed to the absence of polarization in the simulation model, for which it is known that the dynamics is underestimated.45 Nevertheless, the dynamical trends and structural properties19c are generally correctly reproduced. Both the Li+ and anion diffusion coefficients decrease slightly with the anion size (Fig. S13–S15 in the ESI†). Of all components in the gel electrolytes, PC exhibits the fastest motion, which can be rationalized by its low molecular weight in combination with its comparatively short-lived coordination to Li+ (Fig. 8 and 9).
 | ||
| Fig. 10 Mean squared displacements (MSDs) of the individual components in the gel polymer electrolyte (solid curves and filled symbols) and dry polymer electrolyte (dashed curve and open symbols) with LiTFSI. The specifications “seg.” and “c.o.m.” denote the segmental and center-of-mass motion of PPC, respectively. | ||
Although the data in Fig. 9 and 10 is largely consistent with the experimental observations, the precise role of the anion remains elusive so far. For this purpose, we additionally calculated the individual contributions to the overall conductivity. In particular, we separate the total conductivity σ as40a,46
| σ = σS+ + σd++ + σS− + σd−− − 2σ+− |
 | ||
| Fig. 11 (a) Conductivity contributions of cation and anion self-diffusion (σS+ and σS−) as well as the cooperative motion of distinct cations (σd++), distinct anions (σd−−) as well as cations and anions (σ+−). Panel (b) additionally shows the quantities σ+ = σS+ + σd++ − σ+−, σ− = σS− + σd−− − σ+− and σ = σ+ + σ−. | ||
Overall, our findings demonstrate that the drastically enhanced lithium ion transport in gelled PPC electrolytes arises from both a substantially faster relaxation of the lithium ion coordination sphere and globally increased dynamics of the polymer host due to plasticization. Notably, this Li+ dynamics is also affected by the employed anion species, as the different cation–anion interactions gives rise to different dynamical ion correlations. However, a direct solvent- or anion-mediated transport, in which the lithium ions that are solely coordinated by PC or anions and travel larger distances in this way can be ruled out.
Conclusion
In this study, we presented a combined experimental and numerical analysis of the ion coordination and dynamics in dry and gelled polypropylene carbonate-based electrolytes using different conducting salts. Gelation with 33 wt% PC causes a plasticizing effect and leads to a significant increase in the ionic conductivity by about two orders of magnitude. Interestingly, we observe experimentally that although the total ionic conductivity decreases with increasing anion size (in particular FSI, FTFSI, TFSI and BETI), the lithium ion transference number shows the reverse trend, i.e. it increases with increasing anion size. In principle, two mechanisms could be responsible for this observation: first, an anion-dependent decoupling of the lithium ions from the slow polymer chain or second, a significant size-dependent slowdown of the anions by the polymer matrix. Notably, this behavior does not depend on gelation.We demonstrate both experimentally and numerically that the addition of PC weakens the interaction between lithium ions and the polymer, and gives rise to a strongly enhanced exchange of the lithium ion coordination shell as compared to the dry state, which rationalizes the remarkable transport properties of the gel polymer electrolytes. This interplay is also affected by the anion species, as the molecular dynamics simulations revealed that the slightly different cation–anion interaction results in differences in the overall collective motion of the ions, thus rationalizing the anion-size dependence of both conductivities and transference numbers. However, due to the rapid relaxation of the lithium ion coordination shell, we did not observe a solvent- or anion-mediated shuttle mechanism, in which lithium ions move cooperatively over larger distances with the solvent or the anions. For further improvement, the use of chelating solvents or additives – such as oligoethers – could open up a promising research avenue. In this way, combining the advantages of various polymer and additive classes as well as conducting salts offers a new perspective to consider when designing new polymer electrolytes.
Author contributions
AIG and DD wrote the major part of the manuscript. AIG developed the preparation of the salt-in-polymer electrolytes and carried out the analytical, thermal and electrochemical characterizations and analysis. PN performed the PFG-NMR measurements and analysis. DD performed all the theoretical calculations and modelling and contributed to the comparative analysis of ion interactions with the polymers. HDW and MG originated and advised the research. AIG, DD and MG discussed and interpreted the results together with AH, HDW and MW. HDW, MG, AH and MW joined in on discussions and advised the research. All authors contributed to writing the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors would like to acknowledge the German Federal Ministry for Education and Research (BMBF) for funding within the project FestBatt (13XP0175A & 03XP0174B) and BenchBatt (03XP0047B). The authors also acknowledge Anirban Sharma for helpful discussions and Pinchas Nürnberg (University of Münster) for all performed calculations and NMR-measurements.Notes and references
- (a) B. Dunn, H. Kamath and J.-M. Tarascon, Science, 2011, 334, 928–935 CrossRef CAS PubMed; (b) M. Winter, B. Barnett and K. Xu, Chem. Rev., 2018, 118, 11433–11456 CrossRef CAS PubMed; (c) T. Placke, R. Kloepsch, S. Dühnen and M. Winter, J. Solid State Electrochem., 2017, 21, 1939–1964 CrossRef CAS; (d) R. Schmuch, R. Wagner, G. Hörpel, T. Placke and M. Winter, Nat. Energy, 2018, 3, 267–278 CrossRef CAS; (e) L. I. J. R. Nair, G. Brunklaus and M. Winter, Electrochem. Soc. Interface, 2019, 28, 55–61 CrossRef; (f) M. H. Ryou, Y. M. Lee, Y. Lee, M. Winter and P. Bieker, Adv. Funct. Mater., 2015, 25, 834–841 CrossRef CAS; (g) G. Bieker, M. Winter and P. Bieker, Phys. Chem. Chem. Phys., 2015, 17, 8670–8679 RSC; (h) M. M. Hiller, M. Joost, H. J. Gores, S. Passerini and H. D. Wiemhöfer, Electrochim. Acta, 2013, 114, 21–29 CrossRef CAS.
- (a) S. B. Aziz, T. J. Woo, M. Kadir and H. M. Ahmed, J. Sci.: Adv. Mater. Dev., 2018, 3, 1–17 Search PubMed; (b) D. Zhou, D. Shanmukaraj, A. Tkacheva, M. Armand and G. Wang, Chem, 2019, 5, 2326–2352 CrossRef CAS.
- K. Xu, Chem. Rev., 2004, 104, 4303–4417 CrossRef CAS PubMed.
- (a) J. Mindemark, M. J. Lacey, T. Bowden and D. Brandell, Prog. Polym. Sci., 2018, 81, 114–143 CrossRef CAS; (b) J. Song, Y. Wang and C. Wan, J. Power Sources, 1999, 77, 183–197 CrossRef CAS.
- (a) W. H. Meyer, Adv. Mater., 1998, 10, 439–448 CrossRef CAS PubMed; (b) S. Yusuf, S. Yusof, M. Kufian and L. Teo, Mater. Today Proc., 2019, 17, 446–458 CrossRef CAS.
- Y. Wu, Y. Li, Y. Wang, Q. Liu, Q. Chen and M. Chen, J. Energy Chem., 2022, 64, 62–84 CrossRef CAS.
- (a) Y. Tominaga, K. Yamazaki and V. Nanthana, J. Electrochem. Soc., 2015, 162, A3133–A3136 CrossRef CAS; (b) K. Kimura, H. Matsumoto, J. Hassoun, S. Panero, B. Scrosati and Y. Tominaga, Electrochim. Acta, 2015, 175, 134–140 CrossRef CAS; (c) H. Huang, F. Ding, H. Zhong, H. Li, W. Zhang, X. Liu and Q. Xu, J. Mater. Chem. A, 2018, 6, 9539–9549 RSC; (d) Z.-J. He and L.-Z. Fan, Rare Metals, 2018, 37, 488–496 CrossRef CAS; (e) T. Okumura and S. Nishimura, Solid State Ionics, 2014, 267, 68–73 CrossRef CAS; (f) Z. Li, R. Mogensen, J. Mindemark, T. Bowden, D. Brandell and Y. Tominaga, Macromol. Rapid Commun., 2018, 39, 1800146 CrossRef PubMed; (g) Z. Li, H. Matsumoto and Y. Tominaga, Polym. Adv. Technol., 2018, 29, 820–824 CrossRef CAS; (h) Y. Tominaga, V. Nanthana and D. Tohyama, Polym. J., 2012, 44, 1155–1158 CrossRef CAS.
- (a) Y. Tominaga, Polym. J., 2017, 49, 291–299 CrossRef CAS; (b) K. Kimura, J. Motomatsu and Y. Tominaga, J. Polym. Sci., Part B: Polym. Phys., 2016, 54, 2442–2447 CrossRef CAS; (c) K. Kimura, M. Yajima and Y. Tominaga, Electrochem. Commun., 2016, 66, 46–48 CrossRef CAS; (d) Y. Tominaga and K. Yamazaki, Chem. Commun., 2014, 50, 4448–4450 RSC; (e) J. Mindemark, L. Imholt and D. Brandell, Electrochim. Acta, 2015, 175, 247–253 CrossRef CAS; (f) J. Mindemark, B. Sun and D. Brandell, Polym. Chem., 2015, 6, 4766–4774 RSC; (g) J. Mindemark, R. Mogensen, M. J. Smith, M. M. Silva and D. Brandell, Electrochem. Commun., 2017, 77, 58–61 CrossRef CAS; (h) B. Sun, J. Mindemark, K. Edström and D. Brandell, Solid State Ionics, 2014, 262, 738–742 CrossRef CAS; (i) L. Yue, J. Ma, J. Zhang, J. Zhao, S. Dong, Z. Liu, G. Cui and L. Chen, Energy Storage Mater., 2016, 5, 139–164 CrossRef.
- (a) Y. Tominaga and K. Yamazaki, Chem. Commun., 2014, 50, 4448–4450 RSC; (b) Y. Tominaga, Polym. J., 2017, 49, 291–299 CrossRef CAS; (c) K. Kimura, J. Hassoun, S. Panero, B. Scrosati and Y. Tominaga, Ionics, 2015, 21, 895–900 CrossRef CAS; (d) T. Okumura and S. Nishimura, Kobunshi Ronbunshu, 2017, 74, 139–143 CrossRef CAS.
- B. Commarieu, A. Paolella, S. Collin-Martin, C. Gagnon, A. Vijh, A. Guerfi, K. Zaghib and J. Power, Sources, 2019, 436, 226852 CrossRef CAS.
- (a) A. Buchheit, M. Grünebaum, B. Teßmer, M. Winter and H.-D. Wiemhöfer, J. Phys. Chem. C, 2021, 4371–4378 CrossRef CAS; (b) A. Buchheit, M. Grünebaum, A. I. Gerlitz, B. Tessmer and H.-D. Wiemhöfer, in Electrochemical Conference on Energy and the Environment (ECEE 2019): Bioelectrochemistry and Energy Storage (July 21–26, 2019), ECS, 2019.
- M. P. Rosenwinkel, R. Andersson, J. Mindemark and M. Schönhoff, J. Phys. Chem. C, 2020, 124, 23588–23596 CrossRef CAS.
- D. Mankovsky, D. Lepage, M. Lachal, L. Caradant, D. Aymé-Perrot and M. Dollé, Chem. Commun., 2020, 56, 10167–10170 RSC.
- M. Grünebaum, H.-D. Wiemhöfer and M. M. Hiller, DE Pat., 102013004204A1, 2013 Search PubMed.
- P. G. Bruce, J. Evans and C. A. Vincent, Solid State Ionics, 1988, 28, 918–922 CrossRef.
- M. Abraham, T. Murtola and R. Schulz, SoftwareX, 2015, 1, 19–25 CrossRef.
- W. L. Jorgensen, D. S. Maxwell and J. Tirado-Rives, J. Am. Chem. Soc., 1996, 118, 11225–11236 CrossRef CAS.
- L. B. Silva and L. C. G. Freitas, THEOCHEM, 2007, 806, 23–34 CrossRef CAS.
- (a) J. N. Canongia Lopes, J. Deschamps and A. A. Pádua, J. Phys. Chem. B, 2004, 108, 2038–2047 CrossRef; (b) J. N. Canongia Lopes and A. A. Pádua, J. Phys. Chem. B, 2004, 108, 16893–16898 CrossRef; (c) J. N. C. Lopes and A. A. Pádua, Theor. Chem. Acc., 2012, 131, 1–11 Search PubMed.
- (a) I. Leontyev and A. Stuchebrukhov, J. Chem. Theory Comput., 2010, 6, 3153–3161 CrossRef CAS PubMed; (b) N. Molinari, J. P. Mailoa and B. Kozinsky, Chem. Mater., 2018, 30, 6298–6306 CrossRef CAS; (c) A. Thum, A. Heuer, K. Shimizu and J. N. C. Lopes, Phys. Chem. Chem. Phys., 2020, 22, 525–535 RSC.
- RDKit, in RDKit: Open-Source Cheminformatics Software, 2021 Search PubMed.
- L. Martínez, R. Andrade, E. Birgin and J. Martínez, J. Comput. Chem., 2009, 30, 2157–2164 CrossRef PubMed.
- G. Bussi, D. Donadio and M. Parrinello, J. Chem. Phys., 2007, 126, 014101 CrossRef PubMed.
- H. J. Berendsen, J. V. Postma, W. F. van Gunsteren, A. DiNola and J. R. Haak, J. Chem. Phys., 1984, 81, 3684–3690 CrossRef CAS.
- (a) S. Nosé, Mol. Phys., 1984, 52, 255–268 CrossRef; (b) W. G. Hoover, Phys. Rev. A: At., Mol., Opt. Phys., 1985, 31, 1695–1697 CrossRef PubMed.
- (a) M. Parrinello and A. Rahman, J. Appl. Phys., 1981, 52, 7182–7190 CrossRef CAS; (b) S. Nosé and M. Klein, Mol. Phys., 1983, 50, 1055–1076 CrossRef.
- (a) T. Darden, D. York and L. Pedersen, J. Chem. Phys., 1993, 98, 10089–10092 CrossRef CAS; (b) U. Essmann, L. Perera, M. L. Berkowitz, T. Darden, H. Lee and L. G. Pedersen, J. Chem. Phys., 1995, 103, 8577–8593 CrossRef CAS.
- B. Hess, H. Bekker, H. J. Berendsen and J. G. Fraaije, J. Comput. Chem., 1997, 18, 1463–1472 CrossRef CAS.
- A. M. Stephan and K. Nahm, Polymer, 2006, 47, 5952–5964 CrossRef CAS.
- G. Appetecchi, F. Croce, L. Persi, F. Ronci and B. Scrosati, Electrochim. Acta, 2000, 45, 1481–1490 CrossRef CAS.
- R. Kumar, B. Singh and S. Sekhon, J. Mater. Sci., 2005, 40, 1273–1275 CrossRef CAS.
- Y. Wang, F. Fan, A. L. Agapov, T. Saito, J. Yang, X. Yu, K. Hong, J. Mays and A. P. Sokolov, Polymer, 2014, 55, 4067–4076 CrossRef CAS.
- K. Timachova, H. Watanabe and N. P. Balsara, Macromolecules, 2015, 48, 7882–7888 CrossRef CAS.
- B. Scrosati, F. Croce and L. Persi, J. Electrochem. Soc., 2000, 147, 1718 CrossRef CAS.
- B. Sun, J. Mindemark, E. V. Morozov, L. T. Costa, M. Bergman, P. Johansson, Y. Fang, I. Furó and D. Brandell, Phys. Chem. Chem. Phys., 2016, 18, 9504–9513 RSC.
- (a) D. W. McOwen, D. M. Seo, O. Borodin, J. Vatamanu, P. D. Boyle and W. A. Henderson, Energy Environ. Sci., 2014, 7, 416–426 RSC; (b) A. Subramania, N. K. Sundaram, A. S. Priya and G. V. Kumar, J. Membr. Sci., 2007, 294, 8–15 CrossRef CAS; (c) Z. Gadjourova, Y. G. Andreev, D. P. Tunstall and P. G. Bruce, Nature, 2001, 412, 520–523 CrossRef CAS PubMed; (d) L. Mathies, D. Diddens, D. Dong, D. Bedrov and H. Leipner, Solid State Ionics, 2020, 357, 115497 CrossRef CAS.
- D. T. Hallinan Jr and N. P. Balsara, Annu. Rev. Mater. Res., 2013, 43, 503–525 CrossRef.
- M. Ebadi, T. Eriksson, P. Mandal, L. T. Costa, C. M. Araujo, J. Mindemark and D. Brandell, Macromolecules, 2020, 53, 764–774 CrossRef CAS PubMed.
- O. Borodin, M. Olguin, P. Ganesh, P. R. Kent, J. L. Allen and W. A. Henderson, Phys. Chem. Chem. Phys., 2016, 18, 164–175 RSC.
- (a) F. Müller-Plathe and W. F. van Gunsteren, J. Chem. Phys., 1995, 103, 4745–4756 CrossRef; (b) O. Borodin and G. D. Smith, Macromolecules, 2006, 39, 1620–1629 CrossRef CAS; (c) D. Diddens, A. Heuer and O. Borodin, Macromolecules, 2010, 43, 2028–2036 CrossRef CAS.
- (a) D. Diddens and A. Heuer, ACS Macro Lett., 2013, 2, 322–326 CrossRef CAS PubMed; (b) D. Diddens, E. Paillard and A. Heuer, J. Electrochem. Soc., 2017, 164, E3225–E3231 CrossRef CAS.
- J. Atik, D. Diddens, J. H. Thienenkamp, G. Brunklaus, M. Winter and E. Paillard, Angew. Chem., Int. Ed., 2021, 60, 11919–11927 CrossRef CAS PubMed.
- (a) D. Diddens and A. Heuer, J. Phys. Chem. B, 2014, 118, 1113–1125 CrossRef CAS PubMed; (b) L. T. Costa, B. Sun, F. Jeschull and D. Brandell, J. Chem. Phys., 2015, 143, 024904 CrossRef PubMed.
- F. Wohde, M. Balabajew and B. Roling, J. Electrochem. Soc., 2016, 163, A714–A721 CrossRef CAS.
- (a) T. Yan, C. J. Burnham, M. G. Del Pópolo and G. A. Voth, J. Phys. Chem. B, 2004, 108, 11877–11881 CrossRef CAS; (b) F. Dommert, K. Wendler, R. Berger, L. Delle Site and C. Holm, ChemPhysChem, 2012, 13, 1625–1637 CrossRef CAS PubMed; (c) M. Salanne, Phys. Chem. Chem. Phys., 2015, 17, 14270–14279 RSC; (d) D. Bedrov, J.-P. Piquemal, O. Borodin, A. D. MacKerell Jr, B. Roux and C. Schröder, Chem. Rev., 2019, 119, 7940–7995 CrossRef CAS PubMed.
- (a) D. R. Wheeler and J. Newman, J. Phys. Chem. B, 2004, 108, 18362–18367 CrossRef CAS; (b) K. Oldiges, D. Diddens, M. Ebrahiminia, J. Hooper, I. Cekic-Laskovic, A. Heuer, D. Bedrov, M. Winter and G. Brunklaus, Phys. Chem. Chem. Phys., 2018, 20, 16579–16591 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2cp03756d |
| ‡ Current address: MEET Battery Research Center, Institute of Physical Chemistry, University of Münster, Corrensstaße 46, 48149 Münster, Germany. |
| This journal is © the Owner Societies 2023 |