
Excess-electron capture and energy transfer to bulk water for aqueous DNA nucleotide†
Yan
Zhang
 *a,
Xuanning
Chen
a,
Shuhui
Yin
a,
Yinhua
Ma
a and
Songqiu
Yang
b
*a,
Xuanning
Chen
a,
Shuhui
Yin
a,
Yinhua
Ma
a and
Songqiu
Yang
b
aSchool of Science, Dalian Maritime University, Linghai Road 1, Dalian 116026, China. E-mail: yan_zhang@dlmu.edu.cn
bState Key Laboratory of Molecular Reaction Dynamics, Dalian Institute of Chemical Physics, Chinese Academy of Science, Zhongshan Road 457, Dalian 116023, China
First published on 2nd December 2022
Abstract
We performed QM/MM simulations to investigate excess-electron attachment to four aqueous DNA nucleotide anions (dRT−). The negative QM/MM vertical electron affinities (−0.86 to −0.59 eV) reveal that aqueous dRT− anions improbably capture the excess electron near 0 eV. Comparing with the calculations in the gas phase and without the background charges, it can be found that first-shell water molecules have a larger contribution to the promotion of the ability of the excess-electron capture and the bulk-water polarization has a small effect on vertical electron affinities. The phosphate group hampers the attachment of the very low-energy excess electron to aqueous dRT−. The large adiabatic electron affinities (1.45–1.96 eV) and vertical detachment energies (1.92–2.44 eV) reveal that stable dRT2− dianions could be formed after dRT− anions catch the higher-energy excess electron (>0.59 eV). We computed the energy changes in the dRT2− structural relaxations. The QM-region conformational changes cause small energy alterations (−0.28 to 0.35 eV). The QM/MM energy decreases are 2.31–2.73 eV which mainly come from QM computations (3.49–4.00 eV) embedded in the background charges. The analysis of excess-electron distributions indicates that the polarization of bulk water and structural relaxations of dianions induce the excess-electron redistributions in the QM region and produce large QM-energy decreases. The MM energy changes are −1.27 to −1.11 eV for four aqueous dianions. The negative values demonstrate that the energy of the MM region would increase in dRT2− structural relaxations. In contrast with the values of the polarized QM computations, about 30% of the energy released by the QM region is transferred to bulk water in the MM region. The large energy dissipation probably suppresses DNA damage by the low-energy electron.
1. Introduction
The radiation of living cells by high-energy rays can produce quantities of second electrons (SEs) due to the presence of abundant water molecules.1,2 The SEs can damage DNA to induce cell death and genetic mutations.3 Various computations and experiments revealed that very low-energy SEs (<∼3 eV) can damage dry DNA and its components in the gas phase through the mechanism of dissociative electron attachment.4 The excess electron could be attached to an antibonding orbital to break the covalent bond.5–7An aqueous environment has an evident effect on the nucleobases and nucleoside damage by low-energy electrons. The experiments revealed that the presence of a few water molecules would suppress the dissociative channel of the nucleobases.8 The first-shell water molecules play an important role for the stabilization of the uracil anion.9 They can affect the dissociative channels of thymine and remarkably increase the dissociative barrier of the N–H bond.10 The polarization of bulk-water molecules causes anionic potential energy surfaces (PESs) of aqueous nucleobases and nucleosides to become very steep and could influence their excess-electron capture and trapping.11–14
Deoxyribonucleotides (dRTs) containing the phosphate group are the component units of DNA and important models to investigate DNA damage by low-energy electrons. The protonated dRT (dRTH) is used in the studies considering a tightly bound counterion. The natural dRT (dRT−) is applied in the investigations in which the counterion is not considered. The experiments and computations reveal that the low-energy electron is able to break the dRTH/dRT− molecule.15–22 The energy barriers of bond cleavage in aqueous solutions are evidently higher than the gas calculations. The computations using different solvation models obtain inconsistent activation energies. The molecular dynamics (MD) simulations reported by McAllister and co-workers indicated that the aqueous environment cannot be easily described by the continuum solvation model.20
The photoelectron spectra measurements implied that the excess electron can attach to gas dAMPH.23 The computations of gas 2′-deoxycytidine-3′-monophosphate anion (3′-dCMP−) and its protonated species (3′-dCMPH) revealed that the excess electron is located on the bases of dRT.24 The vertical electron affinities (VEAs) of gas dRTH/dRT− are much lower than the aqueous values, which were calculated by the continuum solvation models.25–28 The VEAs of DNA dRTH/dRT− computed by the solvation models suggested that the counterion does not affect the electron affinity of dRTs in an aqueous solution.22,26 The aqueous dRTH/dRT− could easily capture the excess electron near 0 eV. In contrast, the VEA of dUMP− in the explicit aqueous solvent is −0.31 eV.29 The result implies that the aqueous dUMP− cannot catch the low-energy (∼0 eV) electron. It seems that the computation using the explicit solvent model could give new insights into the excess-electron capture of dRTs.
Therefore, we performed the hybrid quantum mechanics/classical mechanics (QM/MM) simulations to investigate the excess-electron capture of four DNA dRT− anions in the aqueous solutions and explore the effect of the explicit water environment. The QM/MM method has been widely utilized in previous studies of various complex molecules.30–32 According to different phosphate-group positions, dRTs have two main forms which are 2′-deoxynucleotide-3′-monophosphate (3′-dRT) and 2′-deoxynucleotide-5′-monophosphate (5′-dRT). In this work, we would study the excess-electron attachment to aqueous DNA 5′-dRT− anions (5′-dAMP−, 5′-dGMP−, 5′-dCMP−, and 5′-dTMP−), which are natural dRTs without the counterion. The dianions would be formed after the excess-electron capture. For convenience, the computational models would be simply denoted by dRT− or dRT2− in the following sections. We calculate the VEA describing the ability of excess-electron capture, adiabatic electron affinity (AEA) which gives the ability of excess-electron trapping, and vertical detachment energy (VDE) exhibiting the dRT2− stability. The excess-electron distributions and change of dRT2− PESs are examined to explore the first-shell and bulk-water effect on the excess-electron capture and trapping.
2. Computational details
The present computational procedure is similar to the previous investigations of aqueous nucleobases and nucleosides.11,12 Each DNA dRT− (dAMP−, dGMP−, dCMP−, or dTMP−) is placed at the center of a water sphere with a 30.0 Å radius. The dRT− structures are shown in Fig. 1. The full systems contain ∼11![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 800 atoms. We first carried out structural optimizations at the MM level and 20 picoseconds (ps) NVT molecular dynamics (MD) simulations to relax the water environment. After aqueous dRT− anions were heated to 300 K, 2000 ps NVT MD simulations were performed to equilibrate the systems. For each model, 40 structures were extracted at 25 ps intervals from MD trajectories of the last 1000 ps. The 40 structures are denoted by S1025–S2000 respectively. They are the initial structures in the following QM/MM computations.
800 atoms. We first carried out structural optimizations at the MM level and 20 picoseconds (ps) NVT molecular dynamics (MD) simulations to relax the water environment. After aqueous dRT− anions were heated to 300 K, 2000 ps NVT MD simulations were performed to equilibrate the systems. For each model, 40 structures were extracted at 25 ps intervals from MD trajectories of the last 1000 ps. The 40 structures are denoted by S1025–S2000 respectively. They are the initial structures in the following QM/MM computations.
 | ||
| Fig. 1 Structures of dAMP−, dGMP−, dCMP−, and dTMP−. | ||
We first optimized the aqueous anionic geometries (dRT−) at the structures of MD sampling. After excess-electron attachment to dRT−, the dianions (dRT2−) were relaxed at the optimized anionic structures. The electrostatic interaction of bulk water would produce a polarized influence on the QM region. Thus, the QM single-point (SP) calculations without background charges (gas-QM) were also executed at the QM/MM optimized structures to analyze the contribution of the bulk water. In the gas-QM calculations, QM atoms consist of dRT and water molecules. The polarized QM calculations (pol-QM) include the background charges on bulk water. The computational formulas of the QM/MM VEA, AEA, and VDE are presented in the recent study.11
All MM MD simulations were performed using CHARMM program.33 TIP3P force field was applied to describe water molecules.34 QM calculations were done by TURBOMOLE 6.4 program and ChemShell 3.5 code was employed to carry out QM/MM calculations.35,36 The electrostatic interactions between QM and MM regions were treated by electrostatic embedding. QM calculations were executed at the B3LYP/6-31+G* level, at which the computations gave reliable electron affinity.37–39 The electrostatic-potential (ESP) charges, which are better to describe charge distribution and through-space charge transfer, were used to explore excess-electron distributions.40–42 The DL-FIND optimizer and high-efficiency double-optimizations-of-buffer-region (DOBR) microiterative scheme were applied in QM/MM structural relaxations of minimal energy.43,44 We averaged the results over 40 snapshots to get the mean values.
3. Results and discussions
The previous simulations of aqueous nucleobases and nucleosides revealed that the ribose ring could enhance the ability of excess-electron capture.11,12 We compared the averaged QM/MM VEAs and AEAs of aqueous adenine over 5, 10, 20, 30, and 40 snapshots in the investigations. The results of 20 and 30 snapshots are close to those of 40 snapshots. In the present simulations, 40 snapshots are employed to compute the mean values for reducing the standard errors (SE). We would investigate the excess-electron attachment to aqueous dRT− anions and explore the energy redistributions in the explicit solutions. The results of QM/MM simulations could be sensitive to QM-region size. Thus, the convergence of the QM region should be first tested in the study. We calculated the ESP charge on dRT to determine the QM-region size because the electron density on the converged QM region would be stable.30QM-region convergence
We first optimized 40 anionic structures (dRT−) of each system using the minimal QM regions (dRT atoms). At the optimized structures, a series of QM/MM SP calculations of dRT− anion and dRT2− dianion with different QM-region sizes, which range from 1.6 to 4.2 Å, were carried out to compute the ESP charge on dRT. The QM region of RQM being 1.6 Å only contains dRT and those of larger sizes consist of dRT and neighboring water molecules. We averaged the dRT− and dRT2− ESP charges on dRT over 40 snapshots. Fig. 2 displays the mean ESP charges for different QM-region sizes. Observing the curves in the figure, it can be found that the change of the charges on dRT would be very small at larger QM regions (e.g. >3.2 Å). The feature suggests that we can get the converged results as the QM-region size (RQM) is larger than 3.2 Å. In present QM/MM simulations, QM regions, which RQM is equal to 3.4 Å, are used in the simulations of four systems considering the computational cost and accuracy. After the determination of the QM-region sizes, we need to assign all QM atoms for each snapshot. The QM regions include ∼135 atoms (see Table S1, ESI†), and are composed of dRT and more than 30 water molecules around dRT.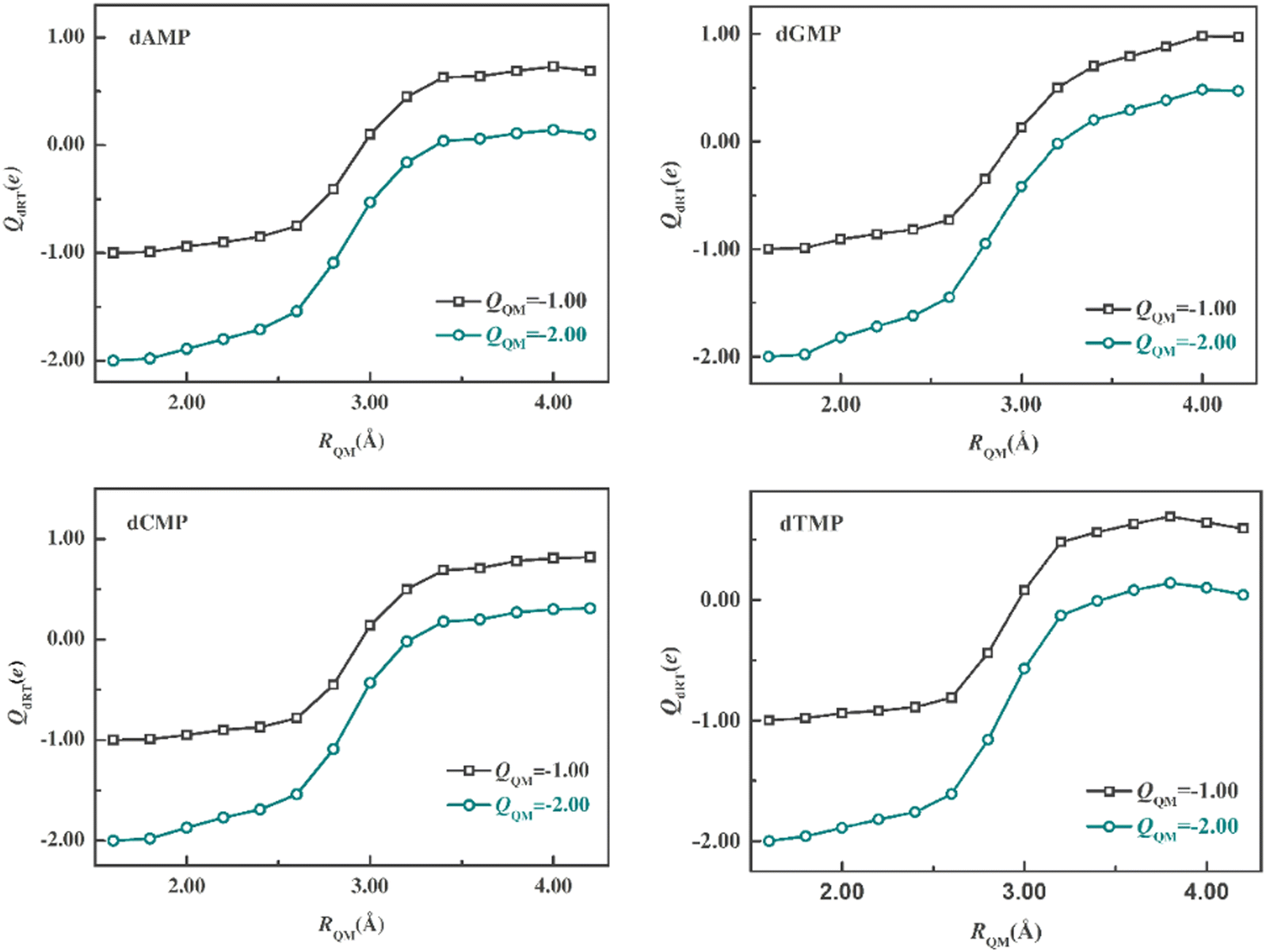 | ||
| Fig. 2 Mean ESP charges of anion and dianion on dRT as the function of QM-region size (RQM), which is the minimal distance between water O and each atom of dRT. | ||
Excess-electron capture of aqueous dRT−
The VEA describes the capability of excess-electron capture. It is the energy difference between the anion and dianion at the optimized anionic structure. We calculated the QM/MM and gas-QM VEAs at QM/MM relaxed dRT− structures. Table 1 lists the VEA values of different computations and corresponding SEs. The negative VEAs imply that the dRT2− energy after excess-electron capture is higher than dRT− anion. The present VEA of gas dCMP− (5′-dCMP−) is −2.42 eV, which is consistent with 3′-dCMP− value (−2.13 eV).13 All VEAs of four dRT− (dAMP−, dGMP−, dCMP−, and dTMP−) in the gas phase are very small (≤−2.00 eV).For comparison, the table also lists VEAs of dRT− in the gas phase and aqueous deoxyribonucleosides
| dAMP− → dAMP2− | dGMP− → dGMP2− | dCMP− → dCMP2− | dTMP− → dTMP2− | |
|---|---|---|---|---|
| a VEA = E(dRT− at optimized dRT− structure) − E(dRT2− at optimized dRT− structure); unit in eV. b QM/MM VEAs of aqueous deoxyribonucleosides from ref. 12. | ||||
| VEAgas | −2.92 | −2.00 | −2.42 | −2.96 |
| VEAgas-QM | −1.11 ± 0.05 | −0.91 ± 0.05 | −1.08 ± 0.05 | −1.02 ± 0.05 |
| VEAQM/MM | −0.79 ± 0.06 | −0.86 ± 0.05 | −0.77 ± 0.05 | −0.59 ± 0.06 |
| VEA(dRN)b | −0.12 | −0.29 | −0.09 | 0.04 |
The mean gas-QM VEAs in Table 1 are −1.11 to −0.91 eV, which are larger than the values in the gas phase by at least 1.00 eV. The results indicate that the water molecules in the first shell have a prominent effect on the VEA and improve the abilities of excess-electron capture for dRT−. The QM/MM values of dAMP−, dCMP−, and dTMP− are higher than gas-QM ones by ∼0.30 eV. It reveals that the polarization of bulk water also enhances the performance of excess-electron capture for the three dRTs. In contrast, the dGMP− VEAs of the QM/MM and gas-QM calculations are very close and the difference is only 0.05 eV. The polarization of bulk water obviously has a negligible contribution to its excess-electron capture. Moreover, the dRT− QM/MM VEA values are lower than those of deoxyribonucleosides (dRN) by ∼0.60 eV. The phosphate group seems to obstruct the capture of the excess electron.
The excess electron could be more easily attached to aqueous dTMP− because its QM/MM VEA value is maximal (−0.59 eV). The negative QM/MM VEAs suggest that four aqueous dRT− anions difficultly catch the SEs near 0 eV. The SEs with the higher energy (e.g. >0.59 eV) could be easily attached to four aqueous dRT− anions and then induce their damage. The phosphate group hampers the excess-electron capture. Hence, the QM/MM VEAs of aqueous dRT− could be maximal as the contribution of the phosphate group is the smallest. The maximal QM/MM VEAs of dAMP−, dGMP−, dCMP−, and dTMP− are −0.16, −0.22, 0.01, and 0.09 eV respectively (see Fig. S1, ESI†). The values approach those of corresponding dRNs without the phosphate group.12 Several bigger dRT− VEAs are near 0 eV among 40 snapshots. The result implies that the aqueous dRT− anions have a low chance to capture an excess electron near 0 eV.
The excess electron could be delocalized over the full QM region including dRT and first-shell water molecules after vertical attachment to aqueous dRT−. We calculated the ESP spin densities on dRT and its components for dRT2− after excess-electron vertical attachment. Table 2 presents the mean spin densities of gas-QM and pol-QM computations. The spin densities on dRT are <0.25 in the gas-QM calculations without background charges. It implies that the main excess electrons are delocalized over water molecules around dRT in the gas-QM computations. The pol-QM spin densities reveal that 47–65% of the excess electron are localized on dRT and most of them are on nucleobases of dRT. Ribose and phosphate groups catch a little excess electron (<10%). The feature is supported by the singly occupied molecular orbital (SOMO) of four aqueous dRT2− dianions for representative snapshots at optimized anionic structures (see Fig. S2, ESI†). The previous studies revealed that the energy of the phosphate-centered radical anion is much higher than that of the base-centered radical one.4,22 The base-centered radical anion is more stable. The present results, which excess electron is localized on the dRT nucleobases, are consistent with the reporting computations.
| dAMP2− | dGMP2− | dCMP2− | dTMP2− | |
|---|---|---|---|---|
| D dRT(gas-QM) | 0.13 ± 0.02 | 0.12 ± 0.02 | 0.15 ± 0.02 | 0.25 ± 0.03 |
| D dRT(pol-QM) | 0.62 ± 0.02 | 0.47 ± 0.02 | 0.57 ± 0.02 | 0.65 ± 0.02 |
| D base(pol-QM) | 0.56 ± 0.03 | 0.43 ± 0.02 | 0.48 ± 0.02 | 0.55 ± 0.02 |
| D ribose(pol-QM) | 0.04 ± 0.01 | 0.02 ± 0.01 | 0.07 ± 0.01 | 0.09 ± 0.01 |
Comparing the pol-QM and gas-QM spin densities and VEAs, it can be found that the bulk-water polarization could drive the excess-electron transfer from water molecules to dRT and improve the ability of excess-electron capture. We calculated the difference of ESP charges on dRT and its components between dRT2− and dRT− to examine the excess-electron distributions. A similar result could also be given by the mean excess-electron distributions of the pol-QM and gas-QM SP calculations in the vertical attachment (see Table S2, ESI†).
Excess-electron trapping of aqueous dRT2−
After vertical attachment, the structures of dRT2− dianions could be relaxed to reach stable configurations to bind the excess electron. Table 3 gives the mean QM/MM AEAs and VDEs. The two physical quantities describe the performance of the trapping excess electron for stable dianions. The mean QM/MM AEAs and VDEs of four aqueous dRT− anions are all larger than 1.45 eV. The large positive values demonstrate that the stable dRT2− dianions can be formed after excess-electron capture. The QM/MM AEAs and VDEs of pyrimidine anions (dCMP− and dTMP−) are larger than those of purine (dAMP− and dGMP−) by about 0.5 eV. The result indicates that the aqueous pyrimidine dRT− has a higher ability of excess-electron trapping. Moreover, the dRT− QM/MM AEAs and VDEs are smaller than dRN ones by around 0.4 and 0.6 eV respectively. It seems that the phosphate group reduces the stability of the dianions and weakens the ability of excess-electron trapping.| dAMP− → dAMP2− | dGMP− → dGMP2− | dCMP− → dCMP2− | dTMP− → dTMP2− | |
|---|---|---|---|---|
| a AEA = E(dRT− at optimized dRT− structure) − E(dRT2− at optimized dRT2− structure); VDE = E(dRT− at optimized dRT2− structure) − E(dRT2− at optimized dRT2− structure); unit in eV. b From ref. 12. | ||||
| AEAQM/MM | 1.57 ± 0.11 | 1.45 ± 0.14 | 1.96 ± 0.10 | 1.95 ± 0.12 |
| AEAmax/min | 3.06/0.17 | 3.87/−1.28 | 3.52/0.54 | 3.91/0.39 |
| AEA(dRN)b | 1.98 | 2.21 | 2.74 | 2.37 |
| VDEQM/MM | 1.96 ± 0.07 | 1.92 ± 0.11 | 2.32 ± 0.07 | 2.44 ± 0.06 |
| VDEmax/min | 2.90/1.06 | 4.46/0.26 | 3.11/0.58 | 3.38/1.55 |
| VDE(dRN)b | 2.44 | 2.65 | 3.09 | 3.00 |
The QM/MM AEA at each snapshot is shown in Fig. 3A. All AEA values of dAMP−, dCMP−, and dTMP− are positive. The minimal AEAs are near 0 eV for dAMP−, dCMP−, and dTMP− (see Table 3). In contrast, the dGMP− AEA values at three snapshots are negative and the minimal AEA is −1.28 eV. As can be seen in Fig. 3B, all QM/MM VDEs of four dianions are positive. The dGMP2− minimal VDE is 0.26 eV. We compare the QM/MM AEA and VDEs of 40 snapshots. It can be found that dAMP−, dCMP−, and dTMP− anions can easily trap the excess electron for 40 snapshots. The excess electron is difficultly attached to aqueous dGMP− anion at several special structures due to small AEA and VDEs.
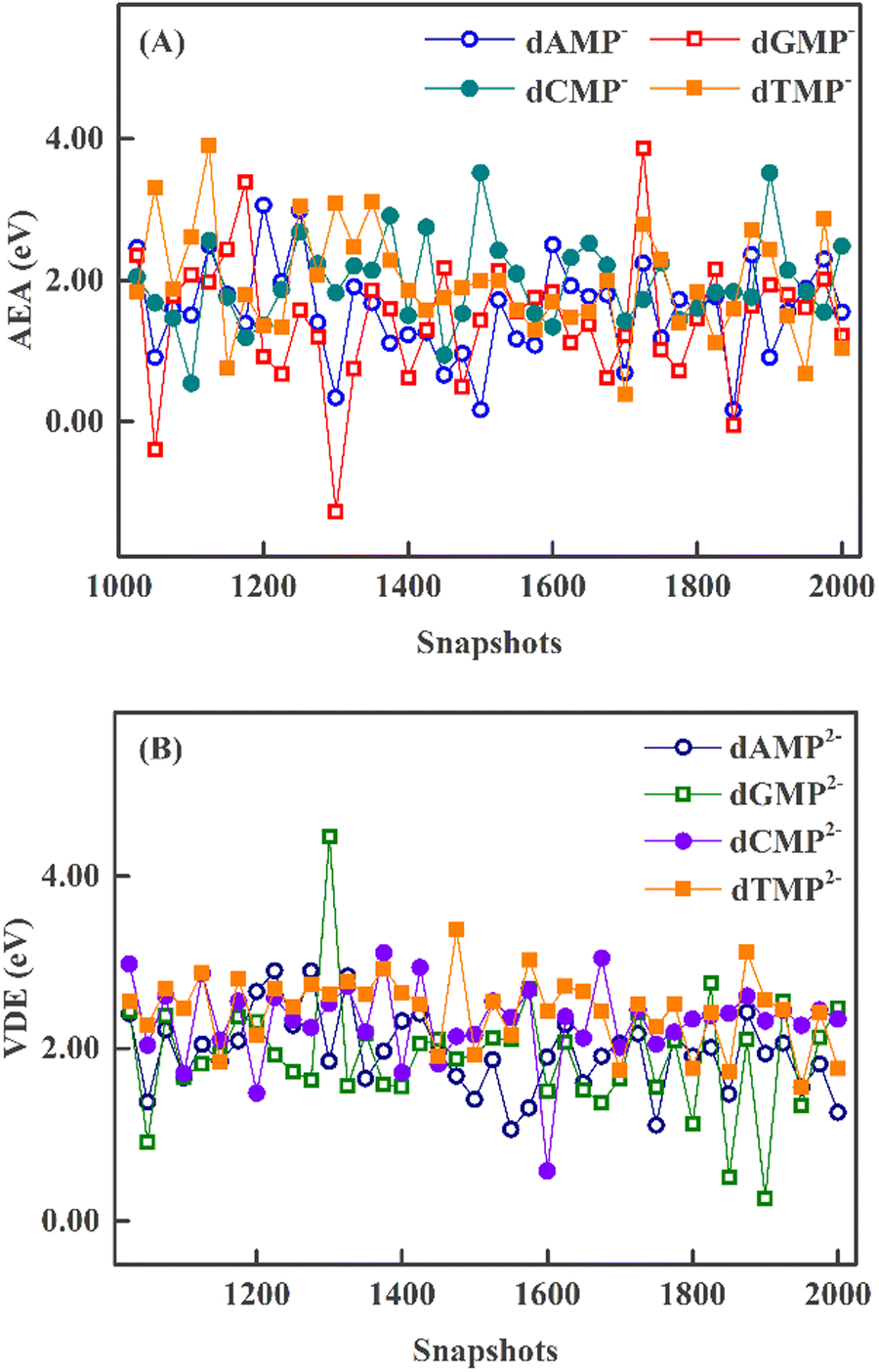 | ||
| Fig. 3 QM/MM AEAs (A) and VDEs (B) for 40 snapshots. | ||
Table 4 lists the averaged dRT2− spin densities on dRT and its components for pol-QM SP calculations at optimized structures of the dianions. The values suggest that most excess electrons (∼85%) are localized on the bases of dRT after the structural relaxations of dianions, and there are ∼10% of excess electrons on ribose and phosphate groups. The excess electron is evidently trapped in the nucleobases of dRT. We also calculated the differences of dRT2− ESP charges at optimized dRT2− and dRT− structures (Table S3, ESI†). The distributions of excess-electron charges exhibit a similar feature which the nucleobases bind the excess electron.
| dAMP2− | dGMP2− | dCMP2− | dTMP2− | |
|---|---|---|---|---|
| D dRT | 0.94 ± 0.01 | 0.87 ± 0.04 | 0.94 ± 0.02 | 0.97 ± 0.01 |
| D base | 0.91 ± 0.02 | 0.84 ± 0.04 | 0.85 ± 0.02 | 0.85 ± 0.01 |
| D ribose | 0.03 ± 0.01 | 0.03 ± 0.01 | 0.09 ± 0.01 | 0.12 ± 0.01 |
Energy transfer to bulk water
The shape of dRT2− PESs is very important to determine the pathways of DNA damage by low-energy SEs. We calculated the energy differences of aqueous dRT2− at optimized anionic and dRT2− structures to explore the energy changes in the dRT2− structural relaxation after excess-electron attachment to aqueous dRT− anions.Table 5 lists mean energy changes in the dRT2− structural relaxations. The decreases of QM/MM energy in the dRT2− structural relaxations are 2.31–2.73 eV for four dRT2− dianions. The energy changes of pol-QM and gas-QM calculations are 3.49–4.00 and −0.28 to 0.35 eV, respectively. The comparison of the QM/MM and pol-QM results reveals that the released energies of full systems mainly come from the relaxation of the QM region which contains dRT and water molecules in the first shell. The values of gas-QM computations are near 0 eV and much smaller than pol-QM simulations. It suggests that the conformational changes of QM-region atoms cause a very small energy alteration. The big QM energy decreases should be produced by the polarization of bulk water which induces the redistribution of electron density. At optimized anionic and dRT2− structures, the dRT2− energy differences between gas-QM and pol-QM calculations are ∼37.0 and ∼40.0 eV respectively (Table S4, ESI†). The bulk-water polarization causes remarkably decreases of QM energy and evidently stabilizes the dRT2− dianions.
| dAMP2− | dGMP2− | dCMP2− | dTMP2− | |
|---|---|---|---|---|
| a ΔE2− = E(dRT2− at optimized dRT− structure) − E(dRT2− at optimized dRT2− structure). | ||||
| ΔE2−(QM/MM) | 2.36 ± 0.11 | 2.31 ± 0.14 | 2.73 ± 0.12 | 2.54 ± 0.13 |
| ΔE2−(pol-QM) | 3.49 ± 0.13 | 3.50 ± 0.13 | 4.00 ± 0.20 | 3.65 ± 0.12 |
| ΔE2−(gas-QM) | −0.02 ± 0.05 | −0.28 ± 0.09 | 0.19 ± 0.05 | 0.35 ± 0.07 |
| ΔE2−(MM) | −1.14 ± 0.12 | −1.20 ± 0.14 | −1.27 ± 0.17 | −1.11 ± 0.11 |
The most interesting thing presented in Table 5 is that MM results are negative (−1.27 to −1.11 eV). The negative values demonstrate that the energy of the MM region containing bulk-water molecules would increase in dRT2− structural relaxations. It means that the energy could be transferred from the QM region to the MM region and dissipated into bulk-water molecules. Moreover, it had been reported that the energy could also be transferred to first-shell water molecules.10 The energy dissipation probably suppresses DNA damage by low-energy SEs. Because of the negative MM energy changes, the pol-QM values are higher than QM/MM results. The changes of the MM energy approach for four dRT2− dianions. It implies that the four dianions have similar capabilities of energy dissipation into bulk water. We compare the values of the pol-QM and MM changes. It can be found that the energy movement is very large. About 30% of the energy released by the QM region is transferred to bulk water for four dianions.
We compare the dRT2− pol-QM spin densities on dRTs, bases, and sugar rings in Tables 2 and 4, which were computed at the optimized structures of anions and dianions. The spin densities on dRT and nucleobases at the relaxed dRT2− configurations both are larger than ones at the optimized dRT− structures by about 0.35. It suggests that ∼35% of excess electrons on QM-region water molecules move to the dRT bases in the dRT2− relaxations. Fig. S3 (ESI†) shows dRT2− SOMOs of pol-QM calculations for the representative snapshots at the optimized dRT− and dRT2− structures. The results indicate that the SOMOs on ribose and phosphate have a little change in the dRT2− relaxations. The energy transport to bulk water is accompanied by the redistribution of excess electrons in the QM region. The excess-electron redistributions in dRT2− relaxations alter the electrostatic interaction among the atoms in the QM region and between QM and MM regions, and drive the dramatic decrease of pol-QM energy. The conformations of the MM region would alter in the dRT2− relaxations and it induces the increase of MM-region energies. The released energy of the QM region would be transferred to bulk water in the MM region. According to the energy wastage, it could be presumed that the polarization of bulk water would lessen DNA damage by low-energy excess electrons.
4. Conclusions
The excess-electron capture and trapping are closely related to the mechanism of DNA damage by low-energy SEs. In the study, QM/MM simulations were performed to investigate excess-electron attachment to four aqueous DNA nucleotide anions (dAMP−, dGMP−, dCMP−, and dTMP−). Their VEAs, AEAs, and VDEs are −0.86 to −0.59, 1.45–1.96, 1.92–2.44 eV respectively. The negative VEA values indicate that <0.59 eV SEs could be difficultly attached to dRT− anions in the water environment. The large AEAs and VDEs suggest that stable dRT2− dianions could be formed after the excess electron with slightly higher energy is attached to the dRT− anions.We also computed the gas-QM VEAs without background charges. The results reveal that water molecules in the first shell prominently raise the ability of dRT− excess-electron capture and bulk-water polarization has smaller contributions. Compared with dRN VEAs, the phosphate group reduces VEAs by ∼0.6 eV to hamper the attachment of very low-energy excess electrons. More than 85% of excess electrons delocalize over first-shell water molecules around dRT without bulk-water polarization. When the QM region is polarized by bulk water, ∼50% of excess electrons are localized on dRT. The most excess electrons on dRT are captured by nucleobases. The polarization of bulk water drives excess-electron movement from first-shell water molecules to dRT bases.
The energy decreases in the relaxations of four aqueous dRT2− dianions are 2.31–2.73 eV for QM/MM computations and 3.49–4.00 eV for pol-QM calculations. The gas-QM and MM energy changes are −0.28 to 0.35 and −1.27 to −1.11 eV respectively. The analyses of excess-electron distributions in the optimizations of aqueous dRT2− indicate that structural relaxations cause QM-region excess-electron redistributions to release a large amount of energy. The gas-QM values near 0 eV suggest that the QM-region conformational change has little contribution to energy alterations. The negative changes of MM energies reveal that ∼30% of the energy released by the QM region would be transferred to bulk water in the MM region. The energy dissipation into bulk water probably suppresses DNA damage by the excess-electron attachment.
Author contributions
Yan Zhang: conceptualization, investigation, methodology, writing – original draft. Xuanning Chen: investigation, visualization. Shuhui Yin: writing – review & editing. Yinhua Ma: investigation. Songqiu Yang: resource.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (22173014 and 21873100), and an open fund of the state key laboratory of molecular reaction dynamics in DICP, CAS.References
- S. M. Pimblott and J. A. LaVerne, Radiat. Phys. Chem., 2007, 76, 1244–1247 CrossRef CAS.
- E. Alizadeh and L. Sanche, Chem. Rev., 2012, 112, 5578–5602 CrossRef CAS PubMed.
- E. Alizadeh, T. M. Orlando and L. Sanche, Annu. Rev. Phys. Chem., 2015, 66, 379–398 CrossRef CAS PubMed.
- J. Gu, J. Leszczynski and H. F. Schaefer III, Chem. Rev., 2012, 112, 5603–5640 CrossRef CAS PubMed.
- J. Simons, Acc. Chem. Res., 2006, 39, 772–779 CrossRef CAS.
- J. Zhao, M. Wang, A. Fu, H. Yang and Y. Bu, ChemPhysChem, 2015, 16, 2348–2356 CrossRef CAS PubMed.
- J. Narayanan S J, D. Tripathi and A. K. Dutta, J. Phys. Chem. Lett., 2021, 12, 10380–10387 CrossRef CAS PubMed.
- J. Kočišek, A. Pysanenko, M. Fárník and J. Fedor, J. Phys. Chem. Lett., 2016, 7, 3401–3405 CrossRef PubMed.
- C. S. Anstöter, M. DelloStritto, M. L. Klein and S. Matsika, J. Phys. Chem. A, 2021, 125, 6995–7003 CrossRef PubMed.
- M. McAllister, N. Kazemigazestane, L. T. Henry, B. Gu, I. Fabrikant, G. A. Tribello and J. Kohanoff, J. Phys. Chem. B, 2019, 123, 1537–1544 CrossRef CAS PubMed.
- Y. Zhang, P. Xie, S. Yang and K. Han, J. Phys. Chem. B, 2019, 123, 1237–1247 CrossRef CAS PubMed.
- Y. Zhang, J. Wang and S. Yang, Phys. Chem. Chem. Phys., 2019, 21, 8925–8932 RSC.
- M. Mukherjee, D. Tripathi and A. K. Duttaa, J. Chem. Phys., 2020, 153, 044305 CrossRef CAS PubMed.
- M. Mukherjee, D. Tripathi, M. Brehm, C. Riplinger and A. K. Dutta, J. Chem. Theory Comput., 2021, 17, 105–116 CrossRef CAS PubMed.
- J. Gu, J. Wang and J. Leszczynski, J. Am. Chem. Soc., 2006, 128, 9322–9323 CrossRef CAS PubMed.
- P. Schyman and A. Laaksonen, J. Am. Chem. Soc., 2008, 130, 12254–12255 CrossRef CAS PubMed.
- P. Schyman, A. Laaksonen and H. W. Hugosson, Chem. Phys. Lett., 2008, 462, 289–294 CrossRef CAS.
- M. Smyth and J. Kohanoff, J. Am. Chem. Soc., 2012, 134, 9122–9125 CrossRef CAS PubMed.
- J. Kopyra, Phys. Chem. Chem. Phys., 2012, 14, 8287–8289 RSC.
- M. McAllister, M. Smyth, B. Gu, G. A. Tribello and J. Kohanoff, J. Phys. Chem. Lett., 2015, 6, 3091–3097 CrossRef CAS PubMed.
- L. M. Cornetta, K. Coutinho, S. Canuto and M. T. do N. Varella, Eur. Phys. J. D, 2016, 70, 176 CrossRef.
- J. Kočišek, B. Sedmidubská, S. Indrajith, M. Fárník and J. Fedor, J. Phys. Chem. B, 2018, 122, 5212–5217 CrossRef PubMed.
- S. T. Stokes, A. Grubisic, X. Li, Y. J. Ko and K. H. Bowen, J. Chem. Phys., 2008, 128, 044314 CrossRef.
- J. Gu, Y. Xie and H. F. Schaefer III, J. Am. Chem. Soc., 2006, 128, 1250–1252 CrossRef CAS PubMed.
- X. Bao, J. Wang, J. Gu and J. Leszczynski, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 5658–5663 CrossRef CAS.
- A. Kumar and M. D. Sevilla, J. Phys. Chem. B, 2007, 111, 5164–5474 Search PubMed.
- H.-Y. Chen, P.-Y. Yang, H.-F. Chen, C.-L. Kao and L.-W. Liao, J. Phys. Chem. B, 2014, 118, 11137–11144 CrossRef CAS PubMed.
- J. Gu, Y. Xie and H. F. Schaefer III, ChemPhysChem, 2006, 7, 1885–1887 CrossRef CAS PubMed.
- Y. Zhang, X. Chen, S. Yin and S. Yang, Chin. J. Chem. Phys., 2022, 35, 375–382 CrossRef CAS.
- H. Lin and D. G. Truhlar, Theor. Chem. Acc., 2007, 117, 185–199 Search PubMed.
- S. Shaik, S. Cohen, Y. Wang, H. Chen, D. Kumar and W. Thiel, Chem. Rev., 2010, 110, 949–1017 CrossRef CAS PubMed.
- L. W. Chung, W. M. C. Sameera, R. Ramozzi, A. J. Page, M. Hatanaka, G. P. Petrova, T. V. Harris, X. Li, Z. Ke, F. Liu, H.-B. Li, L. Ding and K. Morokuma, Chem. Rev., 2015, 115, 5678–5796 CrossRef CAS PubMed.
- B. R. Brooks, C. L. Brooks III, A. D. Mackerell, L. Nilsson, R. J. Petrella, B. Roux, Y. Won, G. Archontis, C. Bartels, S. Boresch, A. Caflisch, L. Caves, Q. Cui, A. R. Dinner, M. Feig, S. Fischer, J. Gao, M. Hodoscek, W. Im, K. Kuczera, T. Lazaridis, J. Ma, V. Ovchinnikov, E. Paci, R. W. Pastor, C. B. Post, J. Z. Pu, M. Schaefer, B. Tidor, R. M. Venable, H. L. Woodcock, X. Wu, W. Yang, D. M. York and M. Karplus, J. Comput. Chem., 2009, 30, 1545–1615 CrossRef CAS PubMed.
- W. L. Jorgensen, J. Chandrasekhar, J. D. Madura, R. W. Impey and M. L. Klein, J. Chem. Phys., 1983, 79, 926–935 CrossRef CAS.
- TURBOMOLE V6.4 2012, a development of University of Karlsruhe and Forschungszentrum Karlsruhe GmbH, 1989–2007, TURBOMOLE GmbH, since 2007; available from https://www.turbomole.com Search PubMed.
- P. Sherwood, A. H. de Vries, M. F. Guest, G. Schreckenbach, C. R. Catlow, S. A. French, A. A. Sokol, S. T. Bromley, W. Thiel, A. J. Turner, S. Billeter, F. Terstegen, S. Thiel, J. Kendrick, S. C. Rogers, J. Casci, M. Watson, F. King, E. Karlsen, M. Sjøvoll, A. Fahmi, A. Schäfer and C. Lennartz, THEOCHEM, 2003, 632, 1–28 CrossRef CAS.
- X. Li, Z. Cai and M. D. Sevilla, J. Phys. Chem. A, 2002, 106, 1596–1603 CrossRef CAS.
- J. Gu, Y. Xie and H. F. Schaefer, J. Chem. Theory Comput., 2014, 10, 609–612 CrossRef CAS PubMed.
- P. Dedíková, P. Neogrády and M. Urban, J. Phys. Chem. A, 2011, 115, 2350–2358 CrossRef PubMed.
- U. C. Singh and P. A. Kollman, J. Comput. Chem., 1984, 5, 129–145 CrossRef CAS.
- F. Martin and H. Zipse, J. Comput. Chem., 2005, 26, 97–105 CrossRef CAS PubMed.
- D. Jacquemin, T. Le Bahers, C. Adamobc and I. Ciofini, Phys. Chem. Chem. Phys., 2012, 14, 5383–5388 RSC.
- J. Kästner, J. M. Carr, T. W. Keal, W. Thiel, A. Wander and P. Sherwood, J. Phys. Chem. A, 2009, 113, 11856–11865 CrossRef PubMed.
- Y. Zhang, P. Xie, X. He and K. Han, J. Chem. Theory Comput., 2016, 12, 4632–4643 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Numbers of atoms in QM region; Excess-electron distributions in vertical electron attachment; Excess-electron distributions in adiabatic electron attachment; Energy differences between gas-QM and pol-QM calculations; VEAs and excess-electron distributions; SOMOs at optimized anionic structures; SOMOs at optimized structures of anions and dianions. See DOI: https://doi.org/10.1039/d2cp03592h |
| This journal is © the Owner Societies 2023 |