
Structural insights into self-assembly of a slow-evolving and mechanically robust supramolecular gel via time-resolved small-angle neutron scattering†
Marzieh
Mirzamani‡
 a,
Arnab
Dawn‡
a,
Christopher J.
Garvey
b,
Lilin
He
c,
Hilmar
Koerner
d and
Harshita
Kumari
*a
a,
Arnab
Dawn‡
a,
Christopher J.
Garvey
b,
Lilin
He
c,
Hilmar
Koerner
d and
Harshita
Kumari
*a
aJames L. Winkle College of Pharmacy, University of Cincinnati, 231 Albert Sabin Way, Cincinnati, OH 45267-0004, USA. E-mail: kumariha@ucmail.uc.edu
bHeinz Maier-Leibnitz Zentrum (MLZ), Technische Universität München, Lichtenbergstraße 1, Garching 85748, Germany
cNeutron Scattering Division, Oak Ridge National Laboratory, 1 Bethel Valley Rd., Oak Ridge, TN 37831, USA
dMaterials & Manufacturing Directorate, Air Force Research Laboratory, WPAFB, Ohio 45433, USA
First published on 10th November 2022
Abstract
The supramolecular assembly process is a widespread phenomenon found in both synthetically engineered and naturally occurring systems, such as colloids, liquid crystals and micelles. However, a basic understanding of the evolution of self-assembly processes over time remains elusive, primarily owing to the fast kinetics involved in these processes and the complex nature of the various non-covalent interactions operating simultaneously. With the help of a slow-evolving supramolecular gel derived from a urea-based gelator, we aim to capture the different stages of the self-assembly process commencing from nucleation. In particular, we are able to study the self-assembly in real time using time-resolved small-angle neutron scattering (SANS) at length scales ranging from approximately 30 Å to 250 Å. Systems with and without sonication are compared simultaneously, to follow the different kinetic paths involved in these two cases. Time-dependent NMR, morphological and rheological studies act complementarily to the SANS data at sub-micron and bulk length scales. A hollow columnar formation comprising of gelator monomers arranged radially along the long axis of the fiber and solvent in the core is detected at the very early stage of the self-assembly process. While sonication promotes uniform growth of fibers and fiber entanglement, the absence of such a stimulus helps extensive bundle formation at a later stage and at the microscopic domain, making the gel system mechanically robust. The results of the present work provide a thorough understanding of the self-assembly process and reveal a path for fine-tuning such growth processes for applications such as the cosmetics industry, 3D printing ink development and paint industry.
Introduction
Supramolecular gelation is a fascinating representative self-assembly process that closely mimics important natural and biological events.1–4 The supramolecular nature of such materials imparts the system with reversibility and adaptivity. The individual or collective contributions of various non-covalent interactions, such as hydrogen bonding, π–π stacking, metal–ligand coordination, host–guest interactions, and van der Waals interactions are at the focal point of the structural evolution during an assembly process. Supramolecular gels have been studied extensively in the last few decades, mostly by exploiting functional outputs for technological and medicinal applications.5–21 An excellent example involves enabling the use of an epoxy resin with thermo-activated organic thixotropes, where low-molecular weight diamide-based derivatives of hydroxystearic acid are shown to be thermally activated to facilitate direct writing of epoxy.22 Another example entails utilizing scanning electron and X-ray microscopy to spatially control the cross-linking in a liquid polymer hydrogel solution on the nano- and micrometer scales, facilitating applications in tissue engineering and drug delivery.23 Further examples can be found in carbohydrate-based low-molecular weight gelators (LMWGs) being applied to environmental remediation, 3D printing, ion sensing, catalysis, and drug delivery,24 as well as exploring the molecular structures and rheological properties of LMWGs that make them suitable for use in 3D printing,25 such as a gel with a UV-sensitive moiety permitting rapid curing with a robust structure26 and an easily soluble gel that can used as a sacrificial ink.27 By comparison, purely structural investigations of supramolecular gel materials during the assembly are rather limited.28–33 The lack of convincing structural data during the formation of the gel/gel kinetics has multiple implications, especially controlling the hierarchical self-assembly process of materials in aviation (3D printing). The two most crucial factors for structural investigations are the experimental time scale and the sensitivity of the measurements to different structural aspects during structural evolution. Therefore, the characterization of supramolecular gels is often considered far more challenging than the characterization of solution-phase materials.34 Techniques like liquid state NMR, which are normally very sensitive to the local environment of a freely tumbling molecule in solution, are dominated by signal broadening caused by shorter T2 relaxation times. Electron microscopy, on the other hand, has issues associated with the artifacts caused by the solvent drying method. Similarly, rheological studies, which measure the bulk behavior of a system, cannot directly follow the primary assembly process at the local scale of gel formation.Typically, a stronger gel is considered superior to a weaker gel, and rapid gel formation makes the situation even more complex. Slowing down gel formation might provide an expanded time window for following the self-assembly process. However, control cannot be achieved over gel formation pathways involving sequential and/or continuous intermolecular assembly processes in space aided by various supramolecular interactions. Understanding the self-assembly process will give us the tools to control the process for the variety of applications discussed above. Classically, supramolecular gel formation involves multiple stages of assembly processes. Initially, when gelator molecules are homogenized (usually by a heat-triggered process) in a gel-forming solvent, nucleation occurs with time and/or temperature (by cooling down). In this process, monomeric molecules interact with each other non-covalently to arrange in specific directions, which can be considered as a primary assembly. Because of the molecular/chemical design of gelator molecules, in most cases, the primary assembly propagates in one dimension to form fibers, which are the structural backbone of supramolecular gel formation. However, simple fiber formation is not sufficient to hold the solvent molecules and attain the viscoelastic behavior of a gel. These characteristics can be achieved when the primary assemblies (typically fibers) undergo cross-linking in a supramolecular fashion (in the case of a supramolecular gel) to form a three-dimensional network structure in space. This entire event can occur within a minute or can take several hours, depending on the efficiency of the intermolecular interactions. To obtain insight into the gel formation event, the following issues need to be addressed: (i) whether single or multiple molecular stacks (fibers) are involved in the primary assembly process; (ii) how the fiber dimensions change during the secondary interaction (cross-linking); and (iii) whether formation of the primary and secondary assemblies occurs simultaneously or sequentially. Although there have been attempts to address some of these issues using various approaches,28,30,33,35 the absence of meaningful time-resolved data has limited the effort considerably.
In the present work, we aimed to explore some of the least studied aspects of supramolecular gel formation with the help of time-lapse experiments, primarily by using the SANS technique. SANS is a powerful tool for elucidating structural information in substances at a mesoscopic scale ranging from 1 to 200 nm. In addition to the size, SANS can also provide accurate information about the shape of an assembly structure.36–41 Advantageously, SANS experiments can be performed in the presence of a solvent, allowing the solute–solvent interplay to be taken into account. Some other advantages of SANS (especially over small-angle X-ray scattering (SAXS)) are the scope of isotope labeling and the nondestructive nature of neutrons. As this approach requires a relatively long gelation time to allow for the collection of time-resolved data, we attempted to design a supramolecular gel system in which the balanced intermolecular interactions can result in slow formation of primary and/or secondary assemblies, offering ample time to capture the nucleation process.
Here, our molecular design of 1 consists of three key elements: a hydrogen-bond forming urea motif, a stacking-interaction-forming aromatic motif, and a highly solubilizing tert-butyloxycarbonyl (BOC) group. Bisurea-based gelators are well-known for their tendency to form highly efficient one-dimensional assembly structures via cooperative urea tape formations at two points.42–46 The presence of a single urea group, on the other hand, is expected to alter the assembly structure. Here, the presence of the BOC group plays a vital role by partially weakening the cooperative participation of intermolecular hydrogen bonding and stacking interactions. Furthermore, the BOC group provides the necessary van der Waals interactions to initiate network formation via higher order assembly. Therefore, this molecular design balancing the interaction parameters is expected to offer an ideal platform for time-resolved studies of the assembly process. Indeed, 1 formed a transparent gel in chloroform (gel formation was evaluated using a stable-to-inversion method) and the gelation time could be extended to more than 10 h using a 1% (w/v) gelator concentration. However, sonicating the system during the initial stage shortened the gelation time considerably. Using SANS, the gel samples (sonicated and nonsonicated) were tested in low-q and high-q regions over a period of 8 h, to capture the nucleation event spanning different time scales. There are reports on the use of SANS to study supramolecular gel systems,47–55 including ours.56–58 However, to the best of our knowledge, this is the first report of a time-resolved SANS study on the nucleation behavior of a supramolecular gel system. Using SANS with complementary spectroscopy, rheology, and morphological investigations, the present study offers an important fundamental understanding of the kinetics and propagation of the self-assembly processes involved in supramolecular gelation.
Experimental section
Synthesis of 1
1-(N-Boc-aminomethyl)-4-(aminomethyl)benzene (1 g, 4.2 mmol) was refluxed with benzyl isocyanate (0.6 g, 4.6 mmol) in 100 ml of anhydrous chloroform under a nitrogen atmosphere. The solvent was removed in vacuo and the crude product was purified by column chromatography [silica gel, CHCl3/MeOH = 15![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 (v/v)] to give a yellowish white powder (1.2 g, 77% yield). 1H NMR (400 MHz, DMSO-d6): δ 1.39 (9H, s, –C(CH3)3), 4.12–4.24 (6H, m, ArCH2–NH–), 6.4 (2H, br, –CONHCH2Ar), 7.2–7.35 (9H, m, ArH). MS (ESI): m/z calcd 369.2, found 392.2 [M + Na]+.
:1 (v/v)] to give a yellowish white powder (1.2 g, 77% yield). 1H NMR (400 MHz, DMSO-d6): δ 1.39 (9H, s, –C(CH3)3), 4.12–4.24 (6H, m, ArCH2–NH–), 6.4 (2H, br, –CONHCH2Ar), 7.2–7.35 (9H, m, ArH). MS (ESI): m/z calcd 369.2, found 392.2 [M + Na]+.
NMR spectroscopy
NMR spectra of 1H were acquired on a Bruker AVANCE NEO-400 spectrometer using a single 30° pulse of 14 μs duration. Each spectrum was the result of 16 FIDs with a relaxation delay of 1 s between pulses. The time-dependent signal was then Fourier transformed into the frequency domain.Rheological studies
Rheological studies were performed using a Discovery Hybrid Rheometer DHR-1 obtained from TA Instruments. Gel samples of 1 were prepared in chloroform at room temperature and aged for 24 h before transferring to the rheometer plate, which was kept at 10 °C. A 40-mm steel cone plate geometry with 55 μm truncation was used for all experiments. Stress sweep experiments were performed at a constant frequency of 1 Hz, and frequency sweep experiments were performed at a constant strain of 0.5%.SEM
The prepared gel samples of 1 were cast onto a silicon wafer, dried in vacuo for 2 days and then coated with platinum. SEM images of the samples were obtained using a SCIOS SEM/FIB instrument.SANS
The gel sample was prepared in CDCl3 at a concentration of 2% (w/v) 1 to reduce the amount of incoherent scattering and to improve the contrast. The sample was heated using a heat gun until the gelator was completely dissolved. The hot solution was then injected into a pre-heated Helma cell of 2 mm path length using a heated needle and syringe to reduce the chance of gel formation during sample transfer. Once the cell holders were capped, one sample was sonicated for approximately 1 minute, while the other was left unsonicated. To best obtain an understanding of how the gel structure developed over a period of 8 hours just after homogenization, separate samples were prepared and measured for each instrument configuration. The SANS measurements were performed at 22 °C using the GP-SANS instrument (CG2) in the High Flux Isotope Reactor facility at Oak Ridge National Laboratory (Oak Ridge, TN, USA).59 Two instrument configurations were used to yield an overall q range of 0.001 Å−1 < q < 0.23 Å−1, where q = (4π/λ)sin(θ/2) (q = scattering vector; λ = neutron wavelength; θ = scattering angle). The high-q configuration (0.0081 Å−1 < q < 0.23 Å−1) used a neutron wavelength of λ = 4.75 Å and a sample-to-detector distance (SDD) of 6 m. The low q configuration (0.001 Å−1 < q < 0.019 Å−1) used a neutron wavelength of λ = 12 Å and an SDD of 19.1 m. The wavelength distributions in both configurations were Δλ/λ = 13%. Beginning 3 minutes after the samples were homogenized, a series of 5 minute scans were collected over a period of 8 hours at high-q and low-q. The raw counts on the area detector were converted to I(q) in Mantid60 by first correcting for the dark current, empty cell scattering, and the sensitivity of the individual detector pixels, and then circular averaging the 2D data to obtain 1D data. The reduced data were set to absolute scale by using porous silica as a secondary standard. The high- and low-q data were merged in Igor Pro using macros developed by NIST.61 The data were then fitted in SasView 5.0.362 with a smeared fractal core–shell cylinder model in order to account for the instrumental resolution.
Results and discussion
Gel samples studied
Gelator 1 gave transparent gels in chloroform. Gel formation (as evaluated by the stable-to-inversion method) was slow and took over 10 h for a concentration of 1% (w/v). To influence the kinetics of the gel formation process, we adopted two different approaches for preparing the gel samples. In the first approach, we homogenized 1 in the solvent at a high temperature and then left the sample undisturbed at room temperature (RT, 20 °C) for nucleation and subsequent gel formation. In the second approach, after 30 min of mixing, we sonicated the homogenized system (sol) containing 1 for 1 min and then left the sample undisturbed at RT for nucleation and subsequent gelation. Interestingly, the sonicated system formed a gel in 3 h for a concentration of 1% (w/v). Sonication-induced gelation can be approximated as a rapid crystallization process that facilitates the formation of intermolecular interactions by suppressing and/or destroying any intramolecular interactions.63 The presence of such an external stimulus is expected to influence first, the gelation kinetics, and second, the overall assembly behavior of 1. Therefore, using the various characterization methods employed in this study, systems prepared with and without sonication were tested in parallel.NMR, rheology, and morphology
1H NMR is highly sensitive towards the molecular conformation, molecular mobility, translational diffusion and intermolecular interactions, offering qualitative valuable short range information about molecular assembly particularly in the early stages of assembly. The perspective offered here by a high resolution 1H liquid NMR spectrometer and probe is on the monomers and smaller oligomers since efficient T2 relaxation64 precludes efficient detection of immobile protons due to probe deadtime effects. This effect has been used previously to provide kinetic details of starch depolymerization;65,66 however in this discussion, we restrict ourselves to qualitative observations around the behavior of monomers and small aggregates.Solvents like DMSO, which are strong hydrogen-bond accepters, inhibit molecular assembly by disfavoring the formation of intermolecular hydrogen bonds; thus, gelator molecules frequently exist as monomers in such solvent systems. Initially, we assessed various proton signals of 1 in the well-dispersed monomeric state in DMSO-d6 (ESI,† Fig. S1). As expected, distinct and well-resolved signals were observed for all the protons, including the urea protons, signifying the absence of a molecular assembly. Next, to assess the assembly properties of 1, we performed a series of single pulse 1H NMR studies in a gel-forming solvent (CDCl3) as the function of time and sonication (Fig. 1). During the initial stage of gel formation, the NMR spectra associated with the sonicated and nonsonicated systems are very similar. Three signals appear in the range of 4.7–5.2 ppm, these signals are ascribed to the two urea protons and the proton attached to nitrogen near the BOC group in 1. With time, these signals broaden and become less distinct. The broadening is most distinct in the other resonances in the sonicated sample. This is consistently less of the oligomeric form in the gel formed from the sonicated solution. This differential behavior over DMSO-d6 could be the result of either the increased presence of free molecules vs. more assembled molecules, or a conformational restriction of 1 gel-forming in CDCl3. Either of these scenarios signifies a molecular assembly. Because of anticipated very fast T2 relaxation in a dense molecular assembly, it is reasonable to consider that the observed signals are mostly coming from oligomeric species. However, continuous signal broadening over time could still be related to higher order assembly resulting in denser packing. Two additional physical effects that could lead to this effect include more dipole interaction and chemical shift anisotropy. Surprisingly, the urea hydrogens exhibit an upfield shift, signifying a shielding event. It is possible that the nearby benzene ring adopts a conformation that brings it in close proximity to the urea hydrogen, inducing a shielding effect. This effect outweighs the conventional downfield shift caused by hydrogen-bonding interactions.67 A comparison of the sonicated and nonsonicated systems reveals two important findings. First, the kinetics of assembly formation differ, as evidenced by the changes in the signals associated with different protons attached to nitrogen with time.
 | ||
| Fig. 1 Structure of 1 (top) and 1H NMR spectra of 1 in CDCl3 at various time intervals after homogenization: (a) nonsonicated and (b) sonicated (red and blue arrows indicate the signals associated with the protons linked to N and aromatic hydrogens, respectively). | ||
The differential effect of sonication on gel formation prompted us to study the rheology of the associated systems. To minimize solvent loss during the rheological studies, experiments were performed using chloroform gels of 1 (2%, w/v) at 10 °C. While all the tested gel systems of 1 exhibit extremely high mechanical strength, the stress sweep experiment reveals that the nonsonicated system has a higher storage modulus and yield stress than the sonicated system (Fig. 2a). Therefore, the sonication stimuli can significantly affect the higher order assembly such as entanglement and bundling of fibers. A practically frequency-independent modulus with G′ ≫ G′′, on the other hand, signifies a stable assembly structure over the examined time scale for both the sonicated and nonsonicated systems (Fig. 2b).
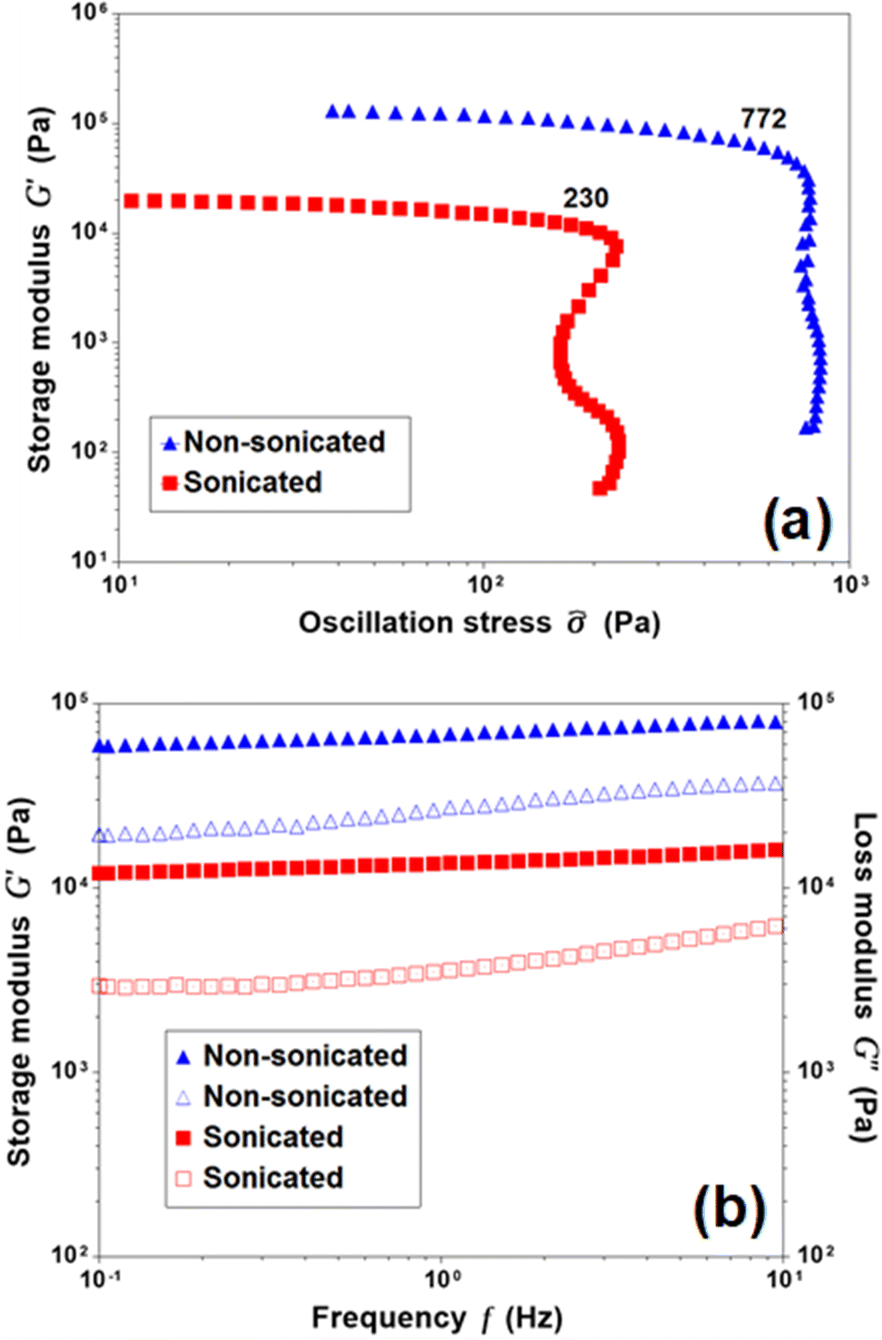 | ||
| Fig. 2 Rheology of sonicated and nonsonicated gels of 1 in CDCl3 (2%, w/v) at 10 °C: (a) stress sweep experiments at a constant frequency of 1 Hz (yield stress values are indicated), and (b) frequency sweep experiments at a constant strain of 0.5% (filled and empty symbols correspond to G′ and G′′, respectively). | ||
To gain insight into the morphology of the above systems, scanning electron microscopy (SEM) was performed on the gel samples of 1 at the initial and final stages of gel formation. In general, the morphology of the gel systems was fibrous with some finer differentiations observed as functions of time and sonication. At the initial stage, the fibrous structures of the sonicated and nonsonicated gels are practically the same with similar fiber thicknesses (Fig. 3a and b). However, after complete gel formation (over 24 h), there are significant differences in the fiber structures (Fig. 3c and d). First, the fiber dimensions show remarkable uniformity in the sonicated system, whereas the nonsonicated system exhibits a wide distribution of fiber dimensions. Second, bundling of fibers is very prominent in the absence of sonication. In the sonicated system, the fibers also become thicker, but to a lesser extent and without losing the uniformity in distribution. This preferential fiber bundling might explain the high mechanical strength of the nonsonicated system in the rheological study (Fig. 2).
 | ||
| Fig. 3 SEM images of nonsonicated and sonicated gels of 1 in CDCl3 prepared at different time intervals after homogenizing: (a) nonsonicated gel after 30 min, (b) sonicated gel after 30 min, (c) nonsonicated gel after 24 h, and (d) sonicated gel after 24 h. The scale bars represent 3 μm. | ||
Time-resolved SANS study
SANS measurements were performed on sonicated and nonsonicated gels formed in CDCl3. SANS provides structural perspectives over a broad range of length-scales in reciprocal space, characterized by the scattering vector q, where q = (4π/λ)sin(θ) and λ is the wavelength of the scattered radiation and 2θ is the scattering angle. SANS measurements were performed at two instrument configurations where the low q region corresponds to a longer length scale and the high q corresponds to a shorter length scale. The low-q and high-q regions for both gels were each measured over a period of 8 h using a series of 5 min acquisitions. Since a fresh gel sample was made before each measurement to ensure that little to no gel structure had formed before the first SANS scan, we assume a matching time course for each gel measurement allowing us to follow how the entire gel structure formed after homogenization. The high-q scans spanned a q-range of 0.0081–0.23 Å−1 (corresponding to a length scale of ∼30–700 Å), while the low-q scans covered a q-range of 0.0010–0.019 Å−1 (length scale of ∼350–3500 Å); in combined form, the full-q ranges from 0.001 to 0.23 Å−1.
The SANS curve overlays for the non-sonicated gel and the sonicated gel and their fits using a fractal core–shell cylinder model are shown in Fig. 4, and fits to the full q-range are shown in Fig. S5 of the ESI.† Other models were attempted, such as fractal flexible cylinder, lamellar models, and empirical models, but these were not able to sufficiently fit the combination of the low-q slope and intensity and Bessel oscillations with two maxima at q = 0.05 Å−1 and q = 0.1 Å−1. A noticeable difference can be observed in the maximum at q = 0.05 Å−1 for the nonsonicated and sonicated gels: it is broader in the nonsonicated gel, suggesting a more polydispersed structure; additionally, the scattering intensity of the maximum is more intense for the nonsonicated gel, indicating that there is a greater amount of the repeating structure contributing to the maximum. The position of this maximum remains consistent for the entire duration of the experiment and is the same for both the non-sonicated and sonicated gels, indicating that the repeating structure contributing to this maximum does not change significantly over time.
 | ||
| Fig. 4 SANS curves of (a) the nonsonicated gel and (b) the sonicated gel of 1 in deuterated chloroform at a concentration of 2 w/v% showing the fits to the data over the full q-range using a fractal core–shell cylinder model (solid black lines). All curves are offset by powers of 3 for visual clarity. | ||
The data were fitted with a smeared fractal core–shell cylinder model. In other words, this model consists of the fractal structure factor proposed by Teixeira68 with the form factor of the core–shell cylinder model69 replacing that of a solid sphere. The general equation describing the 1D scattering intensity consisting of a structure factor and a form factor is68
| I(q) = P(q)S(q) + background |
where Df is the fractal dimension, ξ is the correlation length, and rt is the total radius of the core–shell cylinder (i.e. core + shell thickness). For randomly oriented cylinders, the form factor is

where
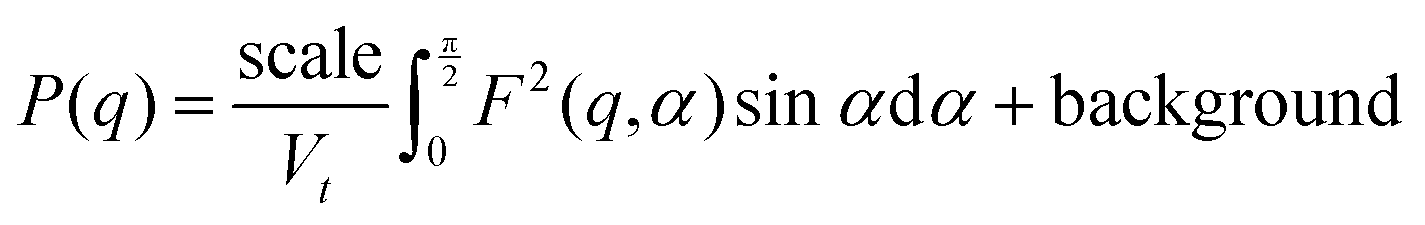
and
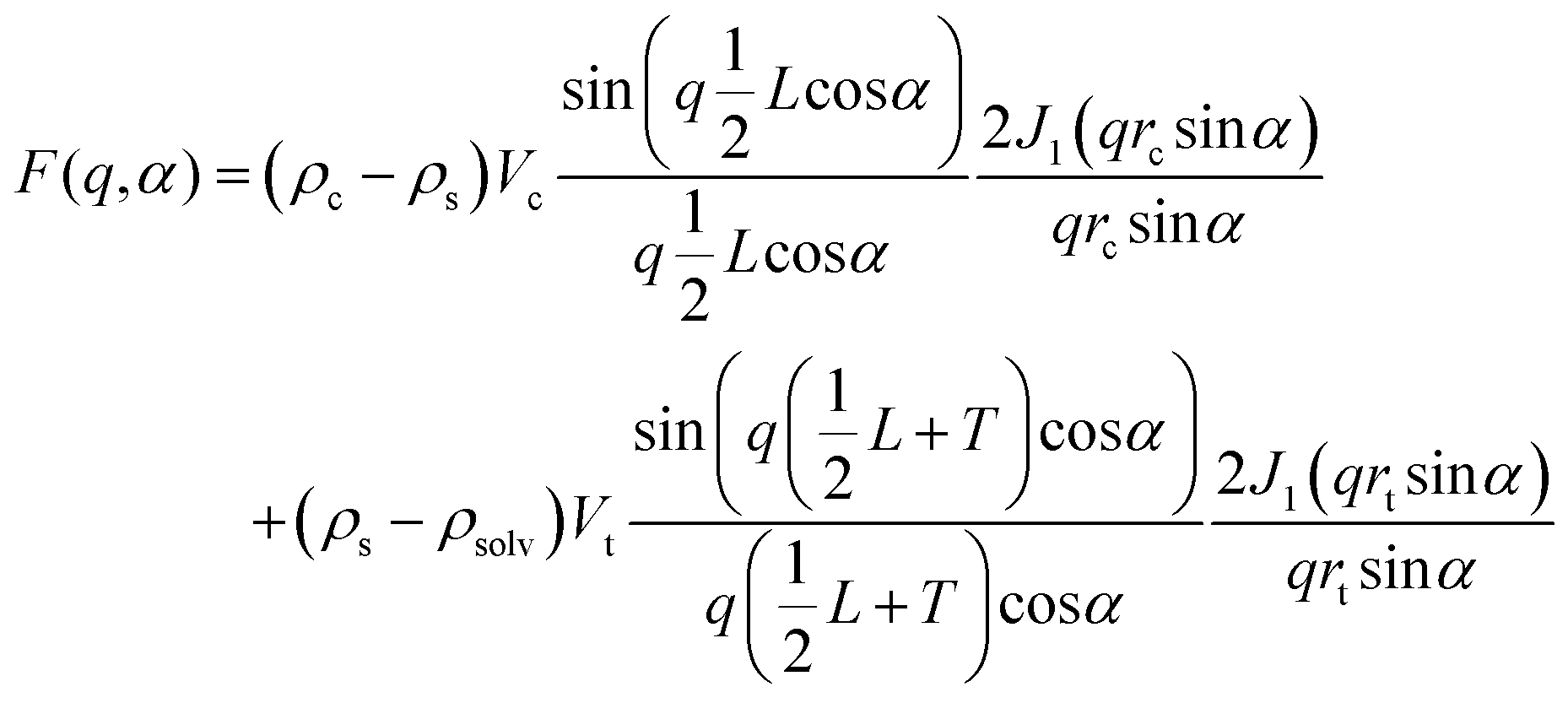
| Vt = πrt2(L + 2T) |
![[q with combining right harpoon above (vector)]](https://www.rsc.org/images/entities/i_char_0071_20d1.gif) , Vt is the total volume of the core–shell cylinder, Vc is the volume of the core, rc is the radius of the core, L is the length of the core, T is the shell thickness, J1 is the first-order Bessel function, and ρc, ρs, and ρsolv are the scattering length densities of the core, the shell, and the solvent respectively.
, Vt is the total volume of the core–shell cylinder, Vc is the volume of the core, rc is the radius of the core, L is the length of the core, T is the shell thickness, J1 is the first-order Bessel function, and ρc, ρs, and ρsolv are the scattering length densities of the core, the shell, and the solvent respectively.
The parameters—i.e. the volume fraction, core radius, shell thickness, polydispersity of the shell thickness, and fiber length—are plotted as a function of time for both types of gels as shown in Fig. 5. A number of differences between the two gels can be seen in the volume fraction (Fig. 5a): (1) the volume fraction for the sonicated gel increased over time, while the volume fraction for the nonsonicated gel decreased slightly over time, (2) the sonicated gel equilibrated approximately 50 min after homogenization, whereas the nonsonicated gel needed 100 min to equilibrate, and (3) the volume fraction for the nonsonicated gel was about 50% greater than that of the sonicated gel (0.015 for the nonsonicated gel vs. 0.01 for the sonicated gel). Both were less than the calculated volume fraction determined from the concentration of the sample (2 wt/v%) and the molecular volume calculated with MULCh,70 indicating that a portion of the gelator in each system remained dissolved in its monomeric state. Furthermore, this means that more of the gelator in the nonsonicated gel (approximately 64%) participated in the formation of the gel structure compared to the sonicated gel (approximately 42%), and thus the nonsonicated gel system has more structures than the sonicated gel system. The thickness of the shell (Fig. 5b) increased over time for both gels before equilibrating after 50 min for the sonicated gel and 100 min for the nonsonicated gel, with the shell of the sonicated gel fiber being approximately 25% thicker than that of the nonsonicated gel (50 Å vs. 40 Å, respectively). The shell of the sonicated gel fibers was also very monodisperse compared to that of the nonsonicated gel fiber (0.05 vs. 0.225), with similar equilibration times, as shown in Fig. 5c. Despite the time needed to equilibrate, however, the shell thicknesses did not increase substantially, indicating that the shell structure was mostly formed immediately after homogenization. The core radius values shown in Fig. 5d show the same equilibration times as the volume fraction and shell thickness equilibration times for both gels, except that the core radius values decrease slightly over time, while the volume fraction and shell thickness values increase over time. When considered with the changes in the shell thickness over time, it further indicates that the full core–shell geometry is formed soon after homogenization. Additionally, the core radius of the nonsonicated gel is about 5 Å larger than that of the sonicated gel. The overall radius of the sonicated gel fiber (core radius + shell thickness) is still larger than that of the nonsonicated gel fiber, with an overall radius of 92 Å vs. 87 Å, respectively. Interestingly, the overall diameter of these fibers as well as the shell thickness are much larger than the length of 1 if it were hypothetically fully extended (ESI,† Fig. S3). Although the number of gelator molecules surrounding the core cannot be determined, the gelators may arrange themselves via π–π stacking and H-bonding to form the core–shell cylindrical structure as shown in Fig. 6. Based on the shell thickness values, it appears that the gelator arrangement is affected by the use of sonication, such that the gelators in the nonsonicated gel arrange themselves end-to-end to achieve the 40 Å shell thickness, while the gelators in the sonicated gel arrange themselves in an offset, antiparallel double layer to reach a shell thickness of 50 Å. These arrangements also explain the core–shell geometry, as the lipophilic tert-butyl ends face toward the solvent, thus allowing the hydrophilic urea groups to remain inside the fiber away from the solvent. Finally, the fiber lengths (Fig. 5e) for both the nonsonicated and sonicated gels surprisingly took about 100 min to equilibrate, with the sonicated gel fibers being approximately 50% longer than the nonsonicated gel fibers (1650 Å vs. 1100 Å, respectively). The fractal dimension as a function of time is shown in the ESI† (Fig. S4), which indicates the different network structures formed in the two gels.
 | ||
| Fig. 5 The volume fraction (a), shell thickness (b), polydispersity of the shell (c), the core radius (d), and cylinder length (e) values over time after homogenization as determined from the SANS modeling (error bars are too small to be distinguished from the symbols). The blue diamonds show the results for the non-sonicated gel, whereas the orange squares show the results for the sonicated gel. | ||
 | ||
| Fig. 6 Fiber cross-section across the long axis (top row) and side view (bottom row) showing the gelator arrangement inside the shell of the nonsonicated (a) and sonicated (b) fibers. | ||
Despite the difference in macroscopic gelation times for sonicated and nonsonicated gels (3 h vs. 12 h), each system equilibrates after about 50 min and 100 min, respectively. However, the nonsonicated gel shows more noticeable changes occurring over that somewhat longer period of time compared to the sonicated gel, showing that the stimulus affected the interactions between the gelator molecules and thereby influenced the final structure of the fully formed gel. The differential behaviors in bulk property, on the other hand, originate mainly from the secondary assembly comprising of fiber bundling, fiber entanglement etc.
Structural analysis
From the time-resolved experiments supported by the NMR, morphology, and rheology data, a few conclusions can be drawn unambiguously: (i) the overall assembly patterns including the hierarchy are quite similar for the gels in the presence and absence of sonication; (ii) the structural evolution differs mostly in first 100 min for sonicated and nonsonicated gels; (iii) the structural evolution with time is more consistent for the sonicated system; (iv) polydispersity in ordered structures is more prominent in the nonsonicated system; and (v) higher order structures (bundling and entanglement) are more abundant in the nonsonicated gel. Thus, there are two key areas of interest: the structural evolution of each type of system, and their comparative natures in the presence and absence of the stimulus. It should be noted that the time-lapse SANS study specifically addresses the local growth processes occurring on the short length scale (high q region), as the resolution is not high enough to draw conclusions from the processes occurring on longer length scales. In contrast, morphology and rheology can take long-range interactions into account. 1H NMR, on the other hand, can capture both domains but with limited finer detail. As the primary assembly structure of a supramolecular gel relies on the chemical structure of the gelator and the solvent system, it is expected that both the sonicated and nonsonicated systems will follow similar nucleation paths. Indeed, there is no striking difference between the two systems in terms of the assembly pattern (in this case, fiber formation). At the primary stage of nucleation, one expects the growth of a one-dimensional assembly of 1via intermolecular hydrogen bonding (urea groups) and stacking interactions (aromatic groups). The overall fiber radius (core radius + shell thickness) obtained from SANS data indicates that the fibers are several folds thicker than hypothetically stretched 1 (ESI,† Fig. S3), which is indicative of a core–shell cylinder geometry of the fiber, where the gelator monomers are arranged in the shell, while the solvent is within the core and surrounding the cylinder (Fig. 7b). This result is quite significant because even though the gel formation is quite slow, primary cluster formation occurs within the first 50 min in the sonicated gel and within the first 100 min in the nonsonicated gel, as evident from SANS data. The presence of a tertiary butyl group at one end of 1 might facilitate van der Waals interactions in a direction perpendicular to the one-dimensional growth axis or enable the core–shell geometry by acting as the lipophilic group. This core–shell fibril arrangement is probably responsible for the extremely high mechanical strength of the gels. A quick and consistent levelling of core radius, shell thickness and fiber length (columnar fibers in the true sense) for the sonicated system indicates a more organized hierarchical event in the presence of the mechanical stimuli (sonication) at the expense of the number of fiber bundles formed. Notably, the intermediate core–shell assembly structure is fairly uniform in shell thickness, as evident from the SANS fitting results (Fig. 5c). This result supports the formation of a hollow columnar structure comprising defined numbers of gelator monomers in the shell oriented perpendicularly to the long axis of the columnar structure, as shown in Fig. 6. At the local scale, we observed quite different growth kinetics for the sonicated and nonsonicated systems; however, the difference between values of a specific parameter (as summarized in Fig. 5) for sonicated and non-sonicated systems is never nullified. Therefore, the effect of the mechanical stimulus becomes more crucial than the time. Clearly, sonication helps disperse the assemblies uniformly from a random aggregated state. Furthermore, sonication helps achieve structural orderliness. Finally, sonication initiates quick core–shell formation at local scale by efficient intermolecular interaction. The same is also reflected in more prominent aromatic stacking in the sonicated gel, as indicated by the severe broadening of the associated aromatic signals in the NMR spectrum (Fig. 1b). However, at the same time, sonication minimizes random interactions among the fibril clusters, thereby eliminating extensive bundling at the microscopic domain as evident from the SEM images of the fully matured gels (Fig. 3c and d). Interestingly, the structurally less ordered nonsonicated gel appeared mechanically more robust (Fig. 2a) because of the formation of higher order bundles especially at the microscopic domain, as visualized by SEM (Fig. 3c). These key findings are illustrated in Fig. 7.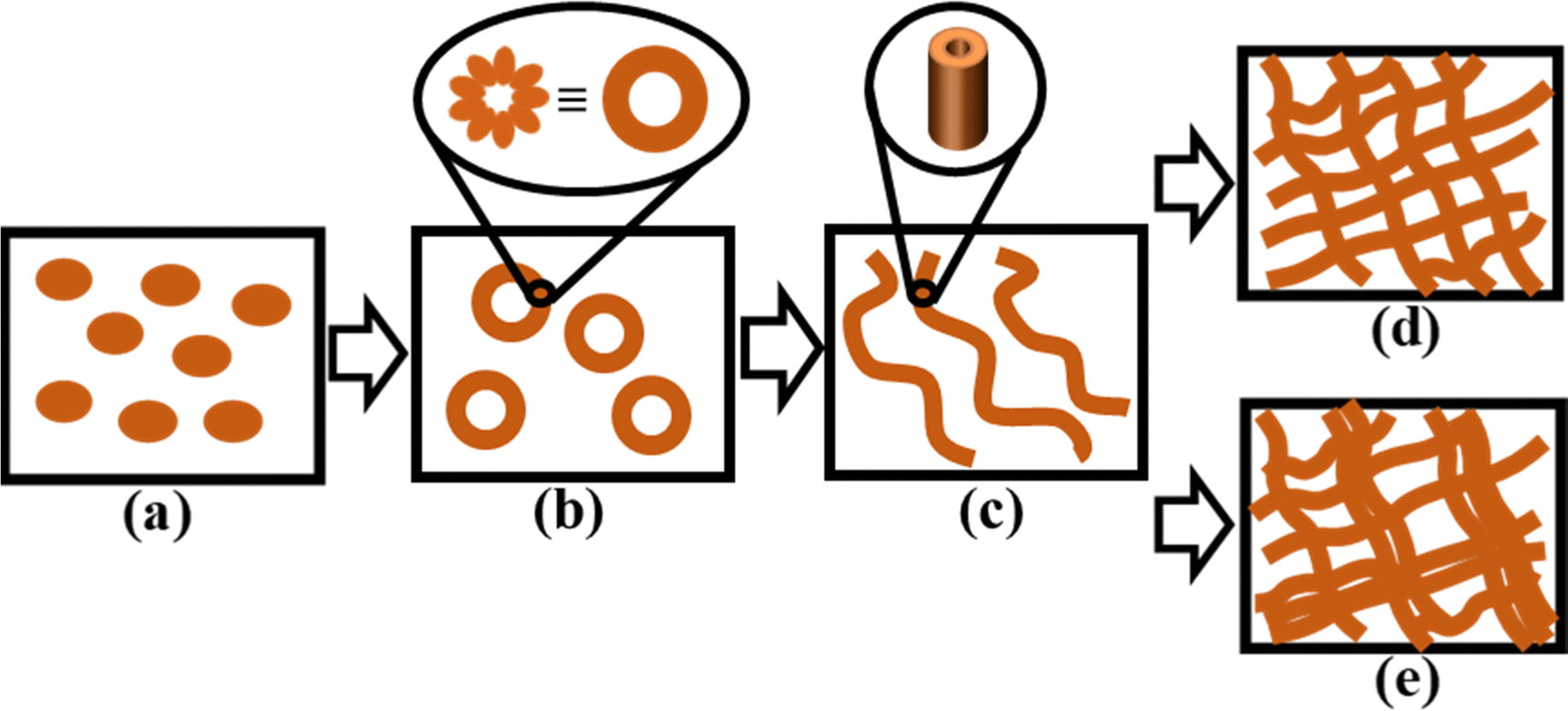 | ||
| Fig. 7 Simplified illustrations of different stages of gel formation: (a) well-dispersed 1 in monomeric form immediately after homogenizing the substrate with the solvent, (b) intermediate molecular core–shell formation, trapping the solvent within the formation, (c) stacks of molecular core–shell formations to form a hollow columnar structure, (d) uniformly distributed cross-linked assembly structure in the sonicated gel, and (e) polydispersity in the randomly distributed cross-linked assembly structure in the nonsonicated gel, which contains higher order (bundled) structures. | ||
The different characterization methods employed here provided independent, valuable, and complementary information. NMR spectroscopy, which works primarily in the molecular domain with the reflection of supramolecular events in cases, followed the formation of assemblies over time without finer differentiation between primary and secondary assemblies. SANS provided unprecedented insight into the local assembly structures and their evolution with time. In addition, SANS revealed the presence of polydispersity in the nonsonicated system. It is found that sonication helps to organize the structure from very early stage periodically and consistently over time. It is worth noticing that the formation of primary assembly is not only sonication-independent but also completes within about 100 min, while macroscopic properties keep changing for about 10 h. Therefore, such stimuli would be more influential towards the bulk behavior of the samples rather than early nucleation. Adopting a monodisperse assembly with the help of sonication by inhibiting extensive bundling could be crucial in the development of functional gels in which structural orderliness could amplify the functional output. In contrast, as revealed by SEM and rheological studies, bundle formation at the later stages of gelation improved the mechanical strength, which is beneficial for many biomedical applications. Therefore, the present study not only offers an unprecedented insight into the time-resolved phenomenon of supramolecular gel formation process but also provides important information for programming the functional applications of a gelator.
Conclusions
In conclusion, the present work uses a slow-evolving supramolecular gel derived from a bis-urea gelator, in the presence and absence of sonication as the stimulus, to capture time-lapsed structural insights at various domains ranging from nanoscopic to microscopic scales. Time-lapse SANS experiments capture the quick formation of primary assembly composed of the core–shell arrangement of monomers within the shell and solvent in the core and then of stacking to form hollow columnar fibers, where the sonicated system is found to be more monodisperse in the shell thickness but less abundant in terms of the overall gel structure. Sonication induces a periodic and consistent growth and hierarchy in the assembly process at the local scale with time, which practically levelled after 50–100 min irrespective of the stimulus. While sonication favors entanglement at the local scale, in the absence of such a stimulus, the gel structure of the nonsonicated system is formed with a majority of the gelator in the system and undergoes extensive and random bundling at the microscopic domain as captured from SANS, NMR, morphological and rheological studies. This could be the reason behind the greater mechanical strength of the nonsonicated system compared to the sonicated system. The information provided by the present work will help develop an in-depth understanding of supramolecular hierarchical assembly processes and their stimuli responsiveness together with the associated structure–function relationships, connecting the molecular level to the macroscopic level. This understanding can then be applied to tune the assembly processes of prospective gelators to produce faster curing or more robust 3D printing inks, controlling 3D printing in aviation applications, more effective scaffolds for tissue engineering, or to identify the best stage of gel assembly for efficient loading of a drug into the gel for drug delivery.Author contributions
MM: data curation, investigation, formal analysis, visualization, writing—original draft, and writing—review and editing; AD: data curation, investigation, formal analysis, visualization, writing—original draft, and Writing—review and editing; CJ: Validation and Writing—review and editing; LH: investigation, validation, and writing—review and editing; HKo: writing—review and editing; and HKu: funding, conceptualization, supervision, project administration, and writing—review and editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The current work was funded through start-up funds provided by CoP, UC (HK). A portion of this research used resources at the High Flux Isotope Reactor, a DOE Office of Science User Facility operated by the Oak Ridge National Laboratory. This work benefited from the use of the SasView application, originally developed under NSF Award DMR-0520547. SasView also contains codes developed with funding from the EU Horizon 2020 programme under the SINE2020 project Grant No 654000.Notes and references
- B. J. G. E. Pieters, M. B. van Eldijk, R. J. M. Nolte and J. Mecinović, Chem. Soc. Rev., 2016, 45, 24–39 CAS.
- W. Du, X. Hu, W. Wei and G. Liang, Bioconjugate Chem., 2018, 29, 826–837 CrossRef CAS PubMed.
- Y. Bai, Q. Luo and J. Liu, Chem. Soc. Rev., 2016, 45, 2756–2767 RSC.
- F. Chiti and C. M. Dobson, Annu. Rev. Biochem., 2017, 86, 27–68 CrossRef CAS PubMed.
- P. Terech and R. G. Weiss, Chem. Rev., 1997, 97, 3133–3159 CrossRef CAS PubMed.
- N. M. Sangeetha and U. Maitra, Chem. Soc. Rev., 2005, 34, 821–836 RSC.
- M.-O. M. Piepenbrock, G. O. Lloyd, N. Clarke and J. W. Steed, Chem. Rev., 2010, 110, 1960–2004 CAS.
- A. Dawn, T. Shiraki, S. Haraguchi, S. Tamaru and S. Shinkai, Chem. – Asian J., 2011, 6, 266–282 CrossRef CAS PubMed.
- L. E. Buerkle and S. J. Rowan, Chem. Soc. Rev., 2012, 41, 6089–6102 RSC.
- S. S. Babu, V. K. Praveen and A. Ajayaghosh, Chem. Rev., 2014, 114, 1973–2129 CrossRef CAS PubMed.
- Y. Feng, Y. M. He and Q. H. Fan, Chem. – Asian J., 2014, 9, 1724–1750 CrossRef CAS PubMed.
- L. Zhang, X. Wang, T. Wang and M. Liu, Small, 2015, 11, 1025–1038 CrossRef CAS PubMed.
- B. O. Okesola and D. K. Smith, Chem. Soc. Rev., 2016, 45, 4226–4251 RSC.
- C. D. Jones and J. W. Steed, Chem. Soc. Rev., 2016, 45, 6546–6596 RSC.
- S. Bhattacharya and S. K. Samanta, Chem. Rev., 2016, 116, 11967–12028 CrossRef CAS PubMed.
- W. Fang, Y. Zhang, J. Wu, C. Liu, H. Zhu and T. Tu, Chem. – Asian J., 2018, 13, 712–729 CrossRef CAS PubMed.
- A. Dawn, Int. J. Mol. Sci., 2019, 20, 781 CAS.
- F. Fages and K. Araki, Low molecular mass gelators: design, self-assembly, function, Springer, Berlin, New York, 2005 Search PubMed.
- R. G. Weiss and P. Terech, Molecular gels: materials with self-assembled fibrillar networks, Springer, Dordrecht, 2006 Search PubMed.
- B. Escuder and J. F. Miravet, Functional molecular gels, Royal Society of Chemistry, Great Britain, 2013 Search PubMed.
- A. Dawn, B. Roy and S. Shinkai, in Chemoresponsive Materials: stimulation by chemical and biological signals, ed. H.-J. Schneider, Royal Society of Chemistry, 2015, p. xxvi, 529 pages Search PubMed.
- R. McKenzie and H. Koerner, Addit. Manuf., 2020, 31, 100905 CAS.
- T. Gupta, E. Strelcov, G. Holland, J. Schumacher, Y. Yang, M. B. Esch, V. Aksyuk, P. Zeller, M. Amati, L. Gregoratti and A. Kolmakov, ACS Nano, 2020, 14, 12982–12992 CrossRef CAS PubMed.
- J. Morris, J. Bietsch, K. Bashaw and G. Wang, Gels, 2021, 7, 24 CrossRef CAS PubMed.
- M. C. Nolan, A. M. Fuentes Caparrós, B. Dietrich, M. Barrow, E. R. Cross, M. Bleuel, S. M. King and D. J. Adams, Soft Matter, 2017, 13, 8426–8432 RSC.
- Z. Zhou, M. Samperi, L. Santu, G. Dizon, S. Aboarkaba, D. Limón, C. Tuck, L. Pérez-García, D. J. Irvine, D. B. Amabilino and R. Wildman, Mater. Des., 2021, 206, 109792 CrossRef CAS.
- A. Chalard, M. Mauduit, S. Souleille, P. Joseph, L. Malaquin and J. Fitremann, Addit. Manuf., 2020, 33, 101162 CAS.
- A. Dawn and H. Kumari, Chem. – Eur. J., 2018, 24, 762–776 CrossRef CAS PubMed.
- A. Dawn, T. Shiraki, H. Ichikawa, A. Takada, Y. Takahashi, Y. Tsuchiya, T. N. Lien le and S. Shinkai, J. Am. Chem. Soc., 2012, 134, 2161–2171 CrossRef CAS PubMed.
- W. Weng, J. B. Beck, A. M. Jamieson and S. J. Rowan, J. Am. Chem. Soc., 2006, 128, 11663–11672 CrossRef CAS PubMed.
- Y. Zhang and R. G. Weiss, J. Colloid Interface Sci., 2017, 486, 359–371 CrossRef CAS PubMed.
- V. Ajay Mallia and R. G. Weiss, Soft Matter, 2016, 12, 3665–3676 RSC.
- M. Yan, S. K. P. Velu, M. Maréchal, G. Royal, J. Galvez and P. Terech, Soft Matter, 2013, 9, 4428–4436 RSC.
- G. Yu, X. Yan, C. Han and F. Huang, Chem. Soc. Rev., 2013, 42, 6697–6722 RSC.
- N. Nonappa and E. Kolehmainen, Soft Matter, 2016, 12, 6015–6026 RSC.
- M. J. Hollamby, K. Aratsu, B. R. Pauw, S. E. Rogers, A. J. Smith, M. Yamauchi, X. Lin and S. Yagai, Angew. Chem., Int. Ed., 2016, 55, 9890–9893 CrossRef CAS PubMed.
- J. P. Patterson, E. G. Kelley, R. P. Murphy, A. O. Moughton, M. P. Robin, A. Lu, O. Colombani, C. Chassenieux, D. Cheung, M. O. Sullivan, T. H. Epps and R. K. O’Reilly, Macromolecules, 2013, 46, 6319–6325 CrossRef CAS PubMed.
- H. Fukuda, A. Goto and T. Imae, Langmuir, 2002, 18, 7107–7114 CrossRef CAS.
- C. Malardier-Jugroot, T. G. M. van de Ven, T. Cosgrove, R. M. Richardson and M. A. Whitehead, Langmuir, 2005, 21, 10179–10187 CrossRef CAS PubMed.
- H. Kumari, S. R. Kline, D. A. Fowler, A. V. Mossine, C. A. Deakyne and J. L. Atwood, Chem. Commun., 2014, 50, 109–111 RSC.
- H. Kumari, S. R. Kline, N. J. Schuster, C. L. Barnes and J. L. Atwood, J. Am. Chem. Soc., 2011, 133, 18102–18105 CrossRef CAS PubMed.
- J. van Esch, R. M. Kellogg and B. L. Feringa, Tetrahedron Lett., 1997, 38, 281–284 CrossRef CAS.
- J. van Esch, S. De Feyter, R. M. Kellogg, F. De Schryver and B. L. Feringa, Chem. – Eur. J., 1997, 3, 1238–1243 CrossRef CAS.
- M. Yamanaka, J. Inclusion Phenom. Macrocyclic Chem., 2013, 77, 33–48 CrossRef CAS.
- J. W. Steed, Chem. Soc. Rev., 2010, 39, 3686–3699 RSC.
- E. Yashima, N. Ousaka, D. Taura, K. Shimomura, T. Ikai and K. Maeda, Chem. Rev., 2016, 116, 13752–13990 CrossRef CAS PubMed.
- P. Terech, D. Pasquier, V. Bordas and C. Rossat, Langmuir, 2000, 16, 4485–4494 CrossRef CAS.
- P. Terech, A. Coutin and A. M. Giroud-Godquin, J. Phys. Chem. B, 1997, 101, 6810–6818 CrossRef CAS.
- M. George, G. P. Funkhouser, P. Terech and R. G. Weiss, Langmuir, 2006, 22, 7885–7893 CrossRef CAS PubMed.
- P. Terech, E. Ostuni and R. G. Weiss, J. Phys. Chem., 1996, 100, 3759–3766 CrossRef CAS.
- R. A. Hule, R. P. Nagarkar, A. Altunbas, H. R. Ramay, M. C. Branco, J. P. Schneider and D. J. Pochan, Faraday Discuss., 2008, 139, 251–264 RSC.
- A. R. Hirst, J. F. Miravet, B. Escuder, L. Noirez, V. Castelletto, I. W. Hamley and D. K. Smith, Chem. – Eur. J., 2009, 15, 372–379 CrossRef CAS PubMed.
- A. R. Hirst, I. A. Coates, T. R. Boucheteau, J. F. Miravet, B. Escuder, V. Castelletto, I. W. Hamley and D. K. Smith, J. Am. Chem. Soc., 2008, 130, 9113–9121 CrossRef CAS PubMed.
- I. W. Hamley, S. Burholt, J. Hutchinson, V. Castelletto, E. Rodrigo da Silva, W. Alves, P. Gutfreund, L. Porcar, R. Dattani, D. Hermida-Merino, G. Newby, M. Reza, J. Ruokolainen and J. Stasiak, Biomacromolecules, 2017, 18, 141–149 CrossRef CAS PubMed.
- P. Terech, I. Furman and R. G. Weiss, J. Phys. Chem., 1995, 99, 9558–9566 CrossRef CAS.
- A. Dawn, M. Mirzamani, C. D. Jones, D. S. Yufit, S. Qian, J. W. Steed and H. Kumari, Soft Matter, 2018, 14, 9489–9497 RSC.
- H. Kumari, S. E. Armitage, S. R. Kline, K. K. Damodaran, S. R. Kennedy, J. L. Atwood and J. W. Steed, Soft Matter, 2015, 11, 8471–8478 RSC.
- H. Kumari, S. R. Kline, S. R. Kennedy, C. Garvey, C. L. Raston, J. L. Atwood and J. W. Steed, Chem. Commun., 2016, 52, 4513–4516 RSC.
- W. T. Heller, M. J. Cuneo, L. M. Debeer-Schmitt, C. Do, L. He, L. Heroux, K. C. Littrell, S. V. Pingali, S. Qian, C. B. Stanley, V. S. Urban, B. Wu and W. Bras, J. Appl. Crystallogr., 2018, 51, 1–7 CrossRef.
- O. Arnold, J. C. Bilheux, J. M. Borreguero, A. Buts, S. I. Campbell, L. Chapon, M. Doucet, N. Draper, R. Ferraz Leal, M. A. Gigg, V. E. Lynch, A. Markvardsen, D. J. Mikkelson, R. L. Mikkelson, R. Miller, K. Palmen, P. Parker, G. Passos, T. G. Perring, P. F. Peterson, S. Ren, M. A. Reuter, A. T. Savici, J. W. Taylor, R. J. Taylor, R. Tolchenov, W. Zhou and J. Zikovsky, Nucl. Instrum. Methods Phys. Res., Sect. A, 2014, 764, 156–166 CrossRef CAS.
- S. R. Kline, J. Appl. Crystallogr., 2006, 39, 895–900 CrossRef CAS.
- M. Doucet, J. H. Cho, G. Alina, Z. Attala, J. Bakker, W. Bouwman, P. Butler, K. Campbell, T. Cooper-Benun, C. Durniak, L. Forster, M. Gonzales, R. Heenan, A. Jackson, S. King, P. Kienzle, J. Krzywon, T. Nielsen, L. O'Driscoll, W. Potrzebowski, S. Prescott, R. Ferraz Leal, P. Rozycko, T. Snow and A. Washington, Zenodo, 2020 DOI:10.5281/zenodo.3930098.
- X. Yu, L. Chen, M. Zhang and T. Yi, Chem. Soc. Rev., 2014, 43, 5346–5371 RSC.
- E. Fukushima and S. B. Roeder, Experimental Pulse NMR: A Nuts and Bolts Approach, Avalon Publishing, 1993 Search PubMed.
- A. C. Dona, G. Pages, R. G. Gilbert, M. Gaborieau and P. W. Kuchel, Biomacromolecules, 2009, 10, 638–644 CrossRef CAS PubMed.
- A. C. Dona, G. Pages, R. G. Gilbert and P. W. Kuchel, Carbohydr. Polym., 2011, 83, 1775–1786 CrossRef CAS.
- F. S. Schoonbeek, J. H. v Esch, R. Hulst, R. M. Kellogg and B. Feringa, Chem. – Eur. J., 2000, 6, 2633–2643 CrossRef CAS.
- J. Teixeira, J. Appl. Crystallogr., 1998, 21, 781–785 CrossRef.
- I. Livsey, J. Chem. Soc., Faraday Trans. 2, 1987, 83, 1445–1452 RSC.
- A. E. Whitten, S. Cai and J. Trewhella, J. Appl. Crystallogr., 2008, 41, 222–226 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: 1H-NMR of 1 in DMSO-d6, the ESI mass spectra of 1, molecular length calculation of hypothetically extended 1, and details of the SANS fitting results and scattering length density calculations. See DOI: https://doi.org/10.1039/d2cp01826h |
| ‡ These authors contributed equally. |
| This journal is © the Owner Societies 2023 |