
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Supporting coordination through hydrogen bonding in lanthanide complexes of 7-azaindole-N-oxide†
Oskar G.
Wood
 a,
Leanne
Jones
b and
Chris S.
Hawes
*a
a,
Leanne
Jones
b and
Chris S.
Hawes
*a
aSchool of Chemical and Physical Sciences, Keele University, Keele ST55BG, UK. E-mail: c.s.hawes@keele.ac.uk
bFaculty of Natural Sciences, Keele University, Keele ST55BG, UK
First published on 1st November 2023
Abstract
Ligands capable of simultaneous metal coordination and hydrogen bond donation provide useful structural features to enhance cooperativity and favour specific geometries within the coordination sphere. Here we present the first structurally characterised examples of coordination compounds containing protonated 7-azaindole-N-oxide HL, bearing a neutral oxo donor capable of terminal or bridging coordination modes adjacent to a convergent hydrogen bond donor. The ligand itself shows a strong tendency for dimeric assembly in the solid state, but is easily deprotonated to give the chelate complex [CuL2] 1. In the presence of lanthanide ions, however, four new complexes [Eu(NO3)3(HL)3] 2, [Gd(NO3)3(HL)3] 3, [Eu2(μ2-HL)2(HL)4Cl6] 4 and [YbCl3(HL)3][YbCl(HL)5OH2]2Cl 5 were prepared and crystallographically characterised. All four species show strong tendencies for hydrogen bonding from the ligand to impact their overall structures, including a C3 propellor-like macrocyclic motif in 2 and 3 and a combination of intramolecular N–H⋯Cl contacts, and intermolecular tridentate anion binding in 5. Solution studies including HRMS and phosphorescence emission spectroscopy reveal persistence of the europium complex 2 in solution, despite the multiple possible binding modes of this ligand, hinting at a degree of cooperativity in these systems. These results show the utility of hydrogen bonding within the coordination sphere for influencing structural outcomes, relevant to the construction of stable higher-order crystalline assemblies.
Introduction
In the design of supramolecular and metallo-supramolecular architectures, reversible and directional interactions such as hydrogen bonding provide a convenient intermediate level of structural influence, weaker than covalent and coordination bonds but more directional than other intermolecular forces.1 A wide variety of extended networks have been prepared using hydrogen bonding as the main supramolecular driving force,2 and intramolecular hydrogen bonds can also be used effectively within coordination assemblies to influence geometry, rigidity, and to help protect against ligand displacement.3 This approach can be particularly effective in tuning the behaviour of metal binding pockets, for example in enhancing selectivity of metal extractions.4 While amides and ureas are popular choices of hydrogen bond donors for supramolecular assemblies,5 heteroaromatic N–H functionalities provide a particularly rigid and directional hydrogen bond donor when incorporated into molecular assemblies.6Where hydrogen bonding is desired alongside coordination bonding, pyrazole is a useful building block which combines a coordinating imine-like nitrogen atom directly attached to a pyrrole-like hydrogen bond donor.7 N–H pyrazoles and indazoles have been widely used in engineering coordination polymers and MOFs supported by intra- or intermolecular hydrogen bonding,8 with N–H⋯O contacts in particular playing a key role in mixed-ligand networks.9 Beyond 1,2-diazoles however, endocyclic N–H donor functionality adjacent to a heterocyclic coordination site and with a sufficiently high pKa to remain protonated under typical reaction and crystallisation conditions is less common. 7-Azaindole is one such example, containing a pyrrolic N–H donor fused to a pyridine ring as shown in Fig. 1. While well known in photophysical studies for its excited-state proton transfer behaviour and as a DNA base analogue,10 the coordination chemistry of 7-azaindole and its derivatives has been somewhat less explored. Nonetheless, a small number of structurally characterised examples have shown the potential for this skeleton to form coordination compounds and multinuclear assemblies supported by coordination bonding, hydrogen bonding or both.11
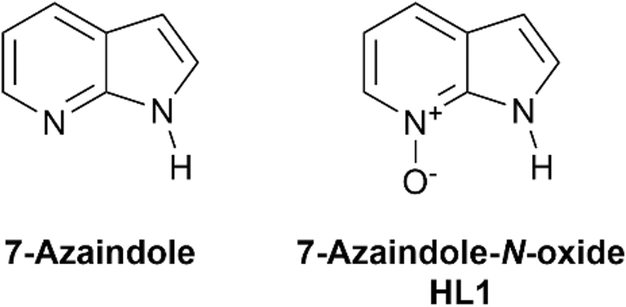 | ||
| Fig. 1 The structures of 7-azaindole and 7-azaindole-N-oxide (HL1). | ||
Given the tendencies for heterocyclic N-oxides to form intricate high-connectivity coordination assemblies,12 replacing the pyridine donor with a pyridine N-oxide would be anticipated to significantly diversify the possible coordination modes of 7-azaindole. If so, structure direction from ancillary hydrogen bonding would be an important asset in providing some level of structural control. Surprisingly, however, to date the only report examining the structural chemistry of 7-azaindole-N-oxide, either as an organic building block itself or in any coordination compounds, has been a contemporary report by Das and co-workers examining its role in copper-catalysed N-arylation reactions (vide infra).13 Here, we explore 7-azaindole-N-oxide HL as a ligand capable of simultaneously coordinating to oxophilic lanthanide ions while facilitating hydrogen bonding to support the coordination sphere and influence the local and extended structure.
Results and discussion
Synthesis and structural characterisation
Based on the expected tendency for the ligand HL to coordinate a neutral N-oxide donor to one or more metal ions through the oxo group, we set out to build a picture of how coordination sphere hydrogen bonding impacts the local and extended structures of the resulting complexes. Firstly, single crystals of the free ligand were grown by slow evaporation from acetonitrile, to gauge the geometry and hydrogen bonding tendency in the absence of metal ions, and to compare with the ubiquitous 7-azaindole parent heterocycle.The diffraction data were solved and refined in the monoclinic space group P21/c, revealing four crystallographically independent HL residues alongside three water molecules. Most likely, these water molecules arise from exposure to ambient air during evaporation. The N–O distances of all four residues are equivalent (1.3342(15)–1.3364(15) Å), comparable to that observed in typical pyridine N-oxides14 and suggesting a similar donor strength. As shown in Fig. 2, while the hydrogen bonding interactions between molecules of HL take the form of dimers supported by R22(10) rings, similar to the R22(8) dimerization mode of substituted 7-azaindoles,15 the lone pair geometry of the oxygen acceptors forces a cleft shape to the two molecules of each dimer. The mean interplanar angle of the two HL molecules involved in each dimer falls in the range 111–116°, while the N⋯O distances in the range 2.7018(16)–2.7714(16) imply relatively strong neutral hydrogen bonding interactions in these species. The lattice water molecules also engage in hydrogen bonding interactions. As well as H2O⋯H2O interactions, all four unique N-oxide oxygen atoms accept a second hydrogen bond from the lattice water molecules, although at a wider range of O⋯O distances (2.6845(15)–2.8146(15) Å). This tendency for N-oxide oxygen atoms to act as polytopic electron donors, capable of accepting up to three hydrogen bonds (or bridging up to three metal ions) is a key element of their importance as supramolecular tectons which differentiates and diversifies them from their parent heterocycles.16 Recently, Das and co-workers reported the room-temperature structure of a triclinic hydrate of HL crystallised from dichloromethane/hexane, which exhibited similar angled hydrogen-bonded dimers between HL units.13 Despite the less hydrophilic crystallisation solvent, association of water was still observed in that case, giving a hemihydrate HL·0.5H2O.
 | ||
| Fig. 2 Structure of a hydrogen-bonded dimer in the structure of HL, with heteroatom labelling scheme and hydrogen atoms shown in pink. Lattice solvent molecules and selected hydrogen atoms are omitted for clarity and ADPs are rendered at the 50% probability level. | ||
The ligand HL was then combined with copper(II) acetate, initially with the aim of generating coordination compounds with a strongly hydrogen bond accepting anion. The only isolable phases from this reaction contained the deprotonated L− anion coordinating as a chelating ligand, indicating sufficient acidity from the N–H group to undergo deprotonation under these conditions. A recent study by Das and co-workers also observed direct deprotonation of HL on reaction of copper(II) fluoride in methanol in the absence of base.13 The predominant phase [CuL2] 1 is crystallised as green plates from methanol after combining with methanolic copper acetate at room temperature. In some batches, a minute quantity of a second blue phase was obtained, with the formula [Cu2(OMe)2L2] (ESI†), but this phase was difficult to reproduce; both PXRD and elemental analysis confirmed that 1 is formed essentially exclusively under these conditions.
The diffraction data for 1 were solved and refined in the monoclinic space group P21/n with the asymmetric unit containing the complex in its entirety, as shown in Fig. 3. The copper ion adopts a square planar, trans configuration with two L anions chelating. The Cu–N distances are shorter than the Cu–O distances (1.918(2) and 1.933(2) Å versus 1.997(2) and 2.011(2) Å, respectively), and the ligand enforces an average bite angle of 84.7°. On one face, a long axial Cu⋯O contact is evident at a distance of 2.5703(19) Å; adjacent complexes dimerise through these contacts which are supported by offset face-to-face π⋯π interactions. The shortest interatomic distance in these interactions is 3.075(3) Å for N4⋯N1, though the two rings are bent outwards at an interplanar angle of 14.67(3)°. A second π⋯π contact is observed between parallel rings at a mean interplanar distance of 3.39 Å on the outer faces of the dimers, by which the dimers form a close-packed layer in the ab plane. Das and co-workers found that these dimeric species tend to be solvent dependent, and tended to observe a discrete CuL2 complex crystallised from methanol (using copper(II) fluoride) and selective crystallisation of the axially-coordinated dimer 1 only when recrystallised from dichloromethane/hexane.13 In our hands, using copper acetate as the copper source we observed only the axially bridged dimer and minute quantities of the methoxide bridged species.
 | ||
| Fig. 3 (a) The structure of [Cu(L)2] 1 with heteroatom labelling scheme. Hydrogen atoms are omitted for clarity and ADPs are rendered at the 50% probability level. (b) Contacts between adjacent complexes in the structure of 1, where the oxo ligands weakly bridge via the axial coordination site of the adjacent copper ion. | ||
While crystallisations were attempted with less basic anions of copper(II) in an attempt to preserve the acidic N–H functionality within the coordination sphere, we were unable to reproducibly isolate any other structurally-identifiable copper(II) complexes by this approach, in either methanol or acetonitrile as an aprotic alternative. Instead, we elected to focus our efforts on lanthanide ions, using chloride and nitrate anions and favouring acetonitrile except where this was impractical on solubility grounds for some lanthanide halides. Reaction of HL with europium(III) nitrate hexahydrate in acetonitrile at room temperature gave colourless crystals of [Eu(NO3)3(HL)3] 2 after diffusion of diethyl ether vapour. The diffraction data, solved and refined in the triclinic space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) , revealed a 9-coordinate tricapped trigonal prismatic geometry for the europium ion with three monodentate HL species occupying one face and the remaining sites occupied by three chelating nitrato ligands, shown in Fig. 4. The HL oxygen atoms form shorter bonds to the europium ion (2.3295(11)–2.3566(13) Å) than the nitrato oxygen atoms (2.4522(13)–2.5321(13) Å), and all three show N–O–Eu angles in the range 132.61(8)–142.50(11)°.
, revealed a 9-coordinate tricapped trigonal prismatic geometry for the europium ion with three monodentate HL species occupying one face and the remaining sites occupied by three chelating nitrato ligands, shown in Fig. 4. The HL oxygen atoms form shorter bonds to the europium ion (2.3295(11)–2.3566(13) Å) than the nitrato oxygen atoms (2.4522(13)–2.5321(13) Å), and all three show N–O–Eu angles in the range 132.61(8)–142.50(11)°.
 | ||
| Fig. 4 The structure of [Eu(NO3)3(HL)3] 2 with labelling scheme for selected heteroatoms and hydrogen atoms shown in pink. Selected hydrogen atoms are omitted for clarity, and ADPs are rendered at the 50% probability level. | ||
Interestingly, the assembly of three HL species on the same face of the europium ion leads to a unique inner-sphere hydrogen bonding motif. As shown in Fig. 5, all three HL ligands accept and donate hydrogen bonds to adjacent ligands, forming a 15-membered R33(15) crown with a threefold propellor-like shape around the europium core. The N⋯O distances, in the range 2.7696(15)–2.822(2) Å are only marginally longer than those observed in the structure of the free ligand, indicating the acceptor capability of the N-oxide group is not significantly hindered by coordination. A similar hydrogen-bonded threefold propellor motif is observed in the Gd3+ complex of a 2-(N-amido)pyrimidine N-oxide ligand, although in that case the steric bulk of a tetrathiafulvalene substituent forces much longer N⋯O distances (>2.98 Å).17
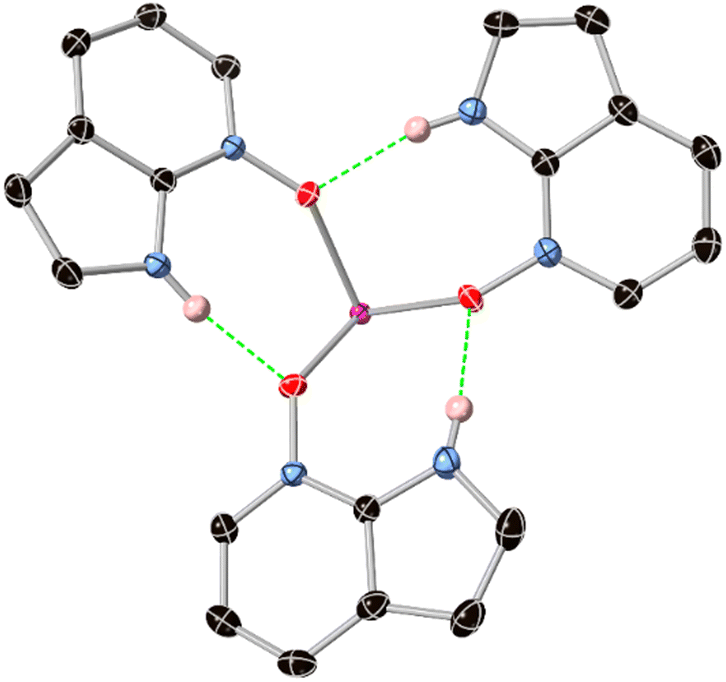 | ||
| Fig. 5 Enlarged view of the M(HL)3 assembly (M = Eu3+, Gd3+) in complexes 2 and 3 showing the hydrogen bonding fragment of the 15-membered ring surrounding one face of the metal ion. Selected hydrogen atoms are omitted for clarity; the ADPs are generated from the complex 2 model. | ||
With all classical hydrogen bond donors occupied within the macrocyclic assembly, adjacent complexes of 2 associate through partially overlapped π⋯π interactions (although these are restricted somewhat by the propellor shape of the aromatic face of the complex) and more diffuse C–H⋯O contacts. Reaction of HL with gadolinium(III) nitrate in place of europium gave an isostructural complex [Gd(NO3)3(HL)3] 3, whose structural metrics were essentially equivalent. We were unable to generate crystalline material for structural analysis under the same conditions with either ytterbium or lanthanum nitrates, suggesting a cation size influence which may particularly favour intermediate lanthanide(III) ions for this assembly. If this apparent selectivity is maintained in the solution phase, such selectivity may contribute to a use for these complexes in lanthanide extraction and separation applications.
Carrying out a similar reaction between HL and europium(III) chloride in methanol also yielded colourless crystals following diffusion of diethyl ether vapour. The diffraction data, solved and refined in the triclinic space group P, revealed a centrosymmetric dimer of the form [Eu2(μ2-HL)2(HL)4Cl6], as shown in Fig. 6. The two equivalent europium ions adopt 7-coordinate pentagonal bipyramidal coordination geometries with chlorido ligands occupying the two axial positions and one equatorial site. The remaining four coordination sites are occupied by oxygen atoms from HL, two in terminal positions and two bridging between the two europium ions. The Eu–Eu distance of 4.2727(5) Å is consistent with typical doubly N-oxido bridged Eu3+ species,18 and unsurprisingly the bridging ligands show longer Eu–O distances than those in the terminal positions (2.430(2) and 2.509(2) Å, versus 2.268(2) and 2.327(2) Å). The latter are shorter than those observed in complex 2, due to the lower coordination number in 4. The two bridging HL units exhibit disorder across two positions related by a 180° rotation about the vector bisecting the bridging Eu–O–Eu angle (ESI,† Fig. S8); similar disorder modes are regularly observed for ligands with mixed 6/5-membered rings and fused-ring systems.19
 | ||
| Fig. 6 Structure of complex [Eu2(μ2-HL)2(HL)4Cl6] 4 with labelling scheme for unique heteroatoms and hydrogen atoms shown in pink. Selected hydrogen atoms are omitted for clarity and ADPs are rendered at the 50% probability level. Disorder on the O3 fragments is omitted for clarity (ESI,† Fig. S8). | ||
As all six HL units remain protonated, each acts as a hydrogen bond donor within the two coordination spheres. Unlike complexes 2 and 3, however, only the chlorido ligands act as hydrogen bond acceptors. This can be rationalised by the bridging coordination mode of the central ligands occupying two of the three possible acceptor positions of these oxygen atoms, and the nearly opposite orientations of the two terminal ligands (with O1–Eu1–O2 angle of 168.1(3)°) restricting the possibility for inner-sphere N–H⋯O contacts. For the terminal sites, the N⋯Cl distances of 3.363(3) and 3.541(3) Å are typical for N–H⋯Cl contacts, though the smaller than ideal N–H⋯Cl angles of 167(4) and 137(3)°, respectively, suggests a mismatch in the bond lengths and angles within the rest of the R11(7) rings for ideal hydrogen bonding. The N–H groups of the two bridging HL species bisect two equivalent axial chlorido acceptors, with the second disordered orientation facing the equivalent situation on the opposite face of the molecule. Presumably, the presence of four chemically equivalent acceptors, and the lack of any significant directional interaction beyond these, relates to the tendency for disorder at this site. Adjacent complexes of 4 interact through various face-to-face π⋯π interactions in the ac plane, with face-to-face and edge-to-face interactions involving all six of the HL species from each complex. While X-ray powder diffraction shows the formation of a pure phase, the crystals are hygroscopic and showed slight association of atmospheric water on standing in air, exhibiting a tendency to clump together after standing in air, and displaying elemental analysis results more consistent with a partial hydrate.
Although a crystalline Yb(NO3)3 complex of HL was not forthcoming in our hands, reaction of HL with a sonicated dispersion of ytterbium chloride hexahydrate in acetonitrile followed by diethyl ether vapour diffusion gave colourless crystals of complex 5. The diffraction data were solved and refined in the triclinic space group P, and revealed a salt of the formula [YbCl3(HL)3][YbCl(HL)5OH2]2Cl, shown in Fig. 7. The asymmetric unit contains two unique ytterbium sites. Yb1 is a seven-coordinate pentagonal bipyramidal species with five HL ligands occupying the equatorial positions, and axial positions filled by one chlorido and one aqua ligand. The HL ligands adopt a fivefold paddlewheel-like geometry around the central core, with three of the N–H groups on the same face as the aqua ligand and the remaining two facing the chlorido ligand. Yb2 adopts a fac-octahedral geometry defined by three chlorido ligands and three HL ligands. Two further non-coordinating chloride anions are present within the asymmetric unit, and although (as with the EuCl3 complex 4) the crystals appeared hygroscopic visually and by elemental analysis, no additional ordered lattice solvent was detected crystallographically, although the material gradually lost crystallinity after removal from the mother liquor.
 | ||
| Fig. 7 The two Yb3+ environments in the structure of 5, showing (a) the pentagonal bipyramidal coordination of Yb1 and (b) the octahedral environment of Yb2 and binding of a lattice chloride ion by tridentate hydrogen bonding from HL. Heteroatom labels are shown and hydrogen atoms are shown in pink. Selected hydrogen atoms and lattice anions are omitted for clarity. ADPs are rendered at the 50% probability level. | ||
Interestingly, and in contrast to complexes 2–4, inner-sphere interactions are not the dominant mode of hydrogen bonding for the HL species in 5. Only the two HL residues on Yb1 oriented towards the chlorido ligand donate hydrogen bonds within the coordination sphere, with N⋯Cl distances 3.305(7) and 3.908(8) Å similar to those in 4. The other three HL species attached to Yb1, alongside the aqua ligand, donate hydrogen bonds to two symmetry equivalent sites of lattice chloride ion Cl6. The N⋯Cl distances in these interactions (3.195(9)–3.322(7) Å) are significantly shorter than the inner-sphere contacts, indicating a beneficial lack of ring strain from the unbound chloride ion. The facially coordinated HL ligands attached to Yb2 each donate hydrogen bonds in a notable tris-chelate fashion to the other lattice chloride ion Cl5. The N⋯Cl distances in these interactions are shorter still, in the range 3.106(8)–3.149(6) Å, somewhat short for (sp2) R2N–H⋯Cl interactions with neutral donors and more comparable with charge-assisted contacts.20 This suggests a potential for facially-coordinated HL species to act as anionophores, where similar chelate-type interactions have been put to good use in the outer coordination sphere for capturing anions.21 Beyond classical hydrogen bonding, adjacent complexes interact through similar modes of π⋯π interaction as those observed in the other lanthanide complexes 2–4.
Solution studies
The prevalence of inner-sphere hydrogen bonding in the complexes 2–4 prompted an investigation into the persistence of these species in solution, to establish whether these interactions furthered their stability beyond that expected of simple monodentate complexes. Electrospray ionisation mass spectrometry revealed that the [M(NO3)3(HL)3] motif (M = Eu, Gd) remains detectable in acetonitrile solution at low concentrations as the sodiated cations, and we could not identify any intermediate [M(NO3)3(HL)2] species. Similarly compound 1 was also readily detected as the protonated cation. We were unable to convincingly detect complex 4 or any likely fragments, while only the neutral octahedral species [YbCl3(HL)3] from complex 5 was detected as the monosodiated cation; we were unable to detect the pentagonal bipyramidal dication or any likely fragments.As the mass spectrometry results hinted at persistence of complexes 1–3 in solution, we employed photophysical measurements to probe the solution stability and likely speciation distribution of the complexes. We focused on the europium complexes 2 and 4, based on the useful photophysical handle provided by the europium(III) phosphorescence emission which is readily sensitised by absorption from coordinated aromatic ligands.22 The free ligand exhibits several absorbances below 350 nm in both acetonitrile and methanol, corresponding to various n → π* and/or π → π* transitions. Unlike the parent 7-azaindole,23 only a single weak fluorescence band was observed with no significant solvent dependence, implying that the ESIPT processes involved in that system likely do not complicate interpretation of the spectra of the N-oxide complexes. The addition of aliquots of methanolic europium(III) chloride hexahydrate to a solution of HL in methanol failed to effect significant changes in the absorption spectrum, and no emergence of the characteristic Eu3+ phosphorescence bands was observed, suggesting minimal formation of complex 4 at photophysically-relevant concentrations in a competitive solvent and consistent with the HRMS results. However, titration of europium nitrate solution caused immediately visible changes in the absorbance profile of HL in acetonitrile, as shown in Fig. 8. The low energy ligand bands at 326 and 317 nm diminished with a stationary point slightly above 300 nm indicating growth of a new band in this region. The broad bands at 288 and 256 nm and the strong band at 227 nm also diminished with concurrent growth of a shoulder around 240 nm, and monotonic growth of a higher energy band at 203 nm corresponding to the free metal salt in solution.
 | ||
| Fig. 8 Absorbance (top) and phosphorescence emission (bottom, λex = 304 nm) spectra following sequential additions of Eu(NO3)3(H2O)6 aliquots (0 → 0.7 eq.) to HL (35 μM) in acetonitrile at room temperature. | ||
In the phosphorescence emission spectra (Fig. 8), growth of the 5D0 → 7FJ (J = 0–4) bands were observed with additions of Eu3+, which gradually slowed after the 1M![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3L ratio was reached. The typically weak 5D0 → 7F0 peak is clearly observed at 580 nm at 28% the intensity of the next 5D0 → 7F1 band, fully consistent with the expected C3 symmetry.24 Global fitting of both the absorbance and phosphorescence spectra to a 1:3 binding model using Reactlab Equilibrium25 gave an overall stability constant logβ3 of 13.7(2), which gave good fits to both sets of data without the need to model any other absorptive or emissive species in solution (ESI,† Fig. S11 and S12†). This value for the overall stability constant suggests only slightly lower stepwise stability constants for the HL1-Eu3+ system than for some Eu3+ complexes with neutral bidentate donors such as the well-known pyridyl-benzimidazole systems, at least in their mononuclear forms.26 The phosphorescence decay profile was readily fitted to a monoexponential function with observed lifetime τobs = 0.24 ms (ESI,† Fig. S9). These data further support the existence of only a single sensitized Eu complex predominating in solution. The phosphorescence is relatively short-lived, consistent with the presence of multiple N–H oscillators within the inner coordination sphere,27 particularly as these are engaged in hydrogen bonding with coordinated oxygen atoms. Based on the combined structural, HRMS and photophysical data, we suggest the pseudo-macrocyclic motif observed crystallographically is also predominant in solution for complex 2, which seems to exhibit a cooperative binding process supported by hydrogen bonding to favour the 1M:3L complex.
:3L ratio was reached. The typically weak 5D0 → 7F0 peak is clearly observed at 580 nm at 28% the intensity of the next 5D0 → 7F1 band, fully consistent with the expected C3 symmetry.24 Global fitting of both the absorbance and phosphorescence spectra to a 1:3 binding model using Reactlab Equilibrium25 gave an overall stability constant logβ3 of 13.7(2), which gave good fits to both sets of data without the need to model any other absorptive or emissive species in solution (ESI,† Fig. S11 and S12†). This value for the overall stability constant suggests only slightly lower stepwise stability constants for the HL1-Eu3+ system than for some Eu3+ complexes with neutral bidentate donors such as the well-known pyridyl-benzimidazole systems, at least in their mononuclear forms.26 The phosphorescence decay profile was readily fitted to a monoexponential function with observed lifetime τobs = 0.24 ms (ESI,† Fig. S9). These data further support the existence of only a single sensitized Eu complex predominating in solution. The phosphorescence is relatively short-lived, consistent with the presence of multiple N–H oscillators within the inner coordination sphere,27 particularly as these are engaged in hydrogen bonding with coordinated oxygen atoms. Based on the combined structural, HRMS and photophysical data, we suggest the pseudo-macrocyclic motif observed crystallographically is also predominant in solution for complex 2, which seems to exhibit a cooperative binding process supported by hydrogen bonding to favour the 1M:3L complex.
Experimental
Synthesis of [CuL2] 1
To a solution of HL (10 mg, 75 μmol) in methanol (5 mL) was added a solution of copper(II) acetate hydrate (15 mg, 75 μmol) in methanol (5 mL). The resulting green solution was sealed and allowed to stand undisturbed for 48 hours, depositing dark green crystals of the product which were isolated by filtration and washed with cold methanol. A trace quantity of the blue phase [Cu2(OMe)2L2] (ESI†) is occasionally observed in this preparation but bulk characterisation shows this phase is not present in significant quantities. Yield 7 mg (21 μmol, 57%). MP 263–265 °C (decomp.); found C, 50.60; H, 3.07; N, 16.76%; calculated for C14H10N4O2Cu C, 50.99; H, 3.06, N, 16.99%; m/z 330.0168 ([M + H]+, calculated for C14H11CuN4O2 330.0178); νmax (ATR, cm−1) 3078m sh, 1596m, 1489s, 1469m, 1445m, 1384m, 1343s, 1312m, 1251m, 1222s, 1176m, 1152s, 1040m, 947m, 860m, 794m, 781s, 720s, 689s.Synthesis of [Eu(NO3)3(HL)3] 2
To a solution of HL (20 mg, 150 μmol) in acetonitrile (5 mL) was added a solution of europium(III) nitrate hexahydrate (24 mg, 54 μmol) in acetonitrile (5 mL). The resulting solution was briefly sonicated to ensure full dissolution, and then subjected to diffusion of diethyl ether vapour. After 1 week, colourless single crystals of the product were recovered by filtration. Yield 17.9 mg (24 μmol, 48%). MP 214–217 °C; found C, 33.96; H, 2.43; N, 17.06%; calculated for C21H18N9O12Eu C, 34.07; H, 2.45; N, 17.03%; m/z 764.0213 ([M + Na]+, calculated for C21H18EuN9O12Na 764.0185); νmax (ATR, cm−1) 3149w, 3098m sh, 2927w sh, 1626w, 1499s, 1461s, 1424m, 1349m, 1331m, 1283s, 1246s, 1227s, 1171m, 1109w, 1046m, 1025m, 958w, 897w, 860m, 789s, 698s sh.Synthesis of [Gd(NO3)3(HL)3] 3
The preparation of the title compound was equivalent to compound 2, using gadolinium(III) nitrate hexahydrate (24 mg, 53 μmol) in place of europium. Yield 26.8 mg (36 μmol, 72%). MP 217–219 °C; found C, 33.92; H, 2.42; N, 16.62%; calculated for C21H18N9O12Gd C, 33.83; H, 2.43; N, 16.91%; m/z 769.0227 ([M + Na]+, calculated for C21H18GdN9O12Na 769.0214); νmax (ATR, cm−1) 3139w, 3098m sh, 2925w sh, 1622w, 1461s sh, 1349s, 1283s, 1246s, 1228s, 1173m, 1108m, 1062w, 1044m, 1024m, 961w, 859m, 788s, 741m, 700s sh, 614m, 597m.Synthesis of [Eu2(μ2-HL)2(HL)4Cl6] 4
To a solution of HL (20 mg, 150 μmol) in methanol (3 mL) was added a solution of europium(III) chloride hexahydrate (22 mg, 60 μmol) in methanol (3 mL). The solution was briefly sonicated and subjected to diffusion of diethyl ether vapour, yielding colourless crystals of the title compound within one week which were isolated by filtration. The crystals readily associated with atmospheric water on standing in air. Yield 10.7 mg (8.1 μmol, 32%). MP 266–271 °C (decomp.); found C, 36.85; H, 2.68; N, 11.91%; calculated for C42H36N12O6Cl6Eu2 + 3H2O C, 36.67; H, 3.08; N, 12.22%; νmax (ATR, cm−1) 3193m br, 3093m, 1622m br, 1497s, 1457s, 1423m, 1350s, 1332s, 1288m, 1243s, 1225m, 1169m, 1099w, 1037s, 964w, 858m, 788s, 712s.Synthesis of [YbCl3(HL)3][YbCl(HL)5OH2]2Cl 5
A solution of HL (20 mg, 150 μmol) in acetonitrile (4 mL) was added to a suspension of ytterbium(III) chloride hexahydrate (12 mg, 31 μmol) dispersed in acetonitrile (6 mL). The resulting suspension was sonicated for 5 minutes and filtered, and the filtrate was subjected to diffusion of diethyl ether vapour. After one week, colourless crystals were isolated by filtration. Similarly to complex 4, this solid also readily adsorbs atmospheric water, and loses crystallinity on extended air exposure. Yield 7.6 mg (4.6 μmol, 30%). MP 186–190 °C (decomp.); found C, 37.06; H, 3.09; N, 11.99%; calculated for C56H50N16O9Cl6Yb2 + 8H2O C, 37.49; H, 3.71; N, 12.49%; m/z 703.9787 (octahedral species [M + Na]+, calculated for C21H18Cl3N6O3YbNa 703.9792); νmax (ATR, cm−1) 3190s br, 3082m, 1621m br, 1499s, 1456s, 1423m, 1349s, 1330s, 1285w, 1244s sh, 1222m, 1169m, 1105w, 1035m, 891w, 858m, 790s, 712s.Conclusions
The 7-azaindole-N-oxide ligand HL provides a convenient and pre-organized source of hydrogen bonding to structurally influence the inner coordination sphere in lanthanide complexes. While this group is readily deprotonated in the presence of copper(II) salts, in lanthanide complexes these hydrogen bonds appear to buttress the coordination environment. In complexes 2 and 3, a 15-membered C3-symmetric cyclic hydrogen bonding motif encircles the metal ion, and spectroscopic evidence suggests that this species forms cooperatively and persists in solution. Complexes 4 and 5 show diverse coordination and hydrogen bonding modes of the HL ligand. Both bridging and terminal coordination behaviour is seen in complex 4, while the mixed coordination numbers of the two Yb3+ fragments in complex 5 also exhibit both inter- and intra-molecular hydrogen bonding in the crystalline phase. These results indicate that combined coordination and hydrogen bonding assemblies of 7-azaindole-N-oxide and its analogues may be powerful structural motifs for designing higher order crystalline assemblies, using hydrogen bonding as a handle for reinforcing the otherwise labile coordination behaviour of N-oxide ligands.Author contributions
OGW: conceptualization, investigation, writing. LJ: investigation, formal analysis, writing. CSH: conceptualization, investigation, supervision, writing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors gratefully acknowledge the School of Chemical and Physical Sciences, Keele University, for a summer research studentship for OGW and instrument funding support, and CPS-WRITE for support with manuscript preparation.References
- L. J. Karas, C.-H. Wu, R. Das and J. I.-C. Wu, WIREs Comput. Mol. Sci., 2020, 10, e1477 CrossRef CAS PubMed.
- R.-B. Lin and B. Chen, Chem, 2022, 8, 2114–2135 Search PubMed; D. A. Cullen, M. G. Gardiner and N. G. White, Chem. Commun., 2019, 55, 12020–12023 RSC; Y.-L. Li, E. V. Alexandrov, Q. Yin, L. Li, Z.-B. Fang, W. Yuan, D. Proserpio and T.-F. Liu, J. Am. Chem. Soc., 2020, 142, 7218–7224 CrossRef CAS PubMed; P. Li, M. R. Ryder and J. F. Stoddart, Acc. Mater. Res., 2020, 1, 77–87 CrossRef.
- C. S. Hawes, Dalton Trans., 2021, 50, 6034–6049 RSC.
- R. S. Forgan, B. D. Roach, P. A. Wood, F. J. White, J. Campbell, D. K. Henderson, E. Kamenetzky, F. E. McAllister, S. Parsons, E. Pidcock, P. Richardson, R. M. Swart and P. A. Tasker, Inorg. Chem., 2011, 50, 4515–4522 CrossRef CAS PubMed; J. R. Turkington, P. J. Bailey, J. B. Love, M. Wilson and P. Tasker, Chem. Commun., 2013, 49, 1891–1899 RSC.
- C. Kulkarni, E. W. Meijer and A. R. A. Palmans, Acc. Chem. Res., 2017, 50, 1928–1936 CrossRef CAS PubMed; A. E. Hooper, S. R. Kennedy, C. D. Jones and J. W. Steed, Chem. Commun., 2016, 52, 198–201 RSC; O. Kotova, R. Daly, C. M. G. dos Santos, M. Boese, P. E. Kruger, J. J. Boland and T. Gunnlaugsson, Angew. Chem., Int. Ed., 2012, 51, 7208–7212 CrossRef PubMed; R. Wechsel, M. Žabka, J. W. Ward and J. Clayden, J. Am. Chem. Soc., 2018, 140, 3528–3531 CrossRef PubMed.
- D. A. Leigh, C. C. Robertson, A. M. Z. Slawin and P. I. T. Thomson, J. Am. Chem. Soc., 2013, 135, 9939–9943 CrossRef CAS PubMed; G. I. Vargas-Zúñiga and J. L. Sessler, Coord. Chem. Rev., 2017, 345, 281–296 CrossRef PubMed.
- M. A. Halcrow, Dalton Trans., 2009, 2059–2073 RSC.
- S. Milan, G. Makhloufi and C. Janiak, Z. Anorg. Allg. Chem., 2019, 645, 893–899 CrossRef CAS; S. Sengupta, S. Ganguly, A. Goswami, S. Bala, S. Bhattacharya and R. Mondal, CrystEngComm, 2012, 14, 7428–7437 RSC; C. S. Hawes and P. E. Kruger, Dalton Trans., 2014, 43, 16450–16458 RSC.
- T. Basu, H. A. Sparkes, M. K. Bhunia and R. Mondal, Cryst. Growth Des., 2009, 9, 3488–3496 CrossRef CAS.
- R. S. Moog and M. Maroncelli, J. Phys. Chem., 1991, 95, 10359–10369 CrossRef CAS; A. V. Smirnov, D. S. English, R. L. Rich, J. Lane, L. Teyton, A. W. Schwabacher, S. Luo, R. W. Thornburg and J. W. Petrich, J. Phys. Chem. B, 1997, 101, 2758–2769 CrossRef.
- A. Domínguez-Martín, M. D. P. Brandi-Blanco, A. Matilla-Hernández, H. El Bakkali, V. M. Nurchi, J. M. González-Pérez, A. Castiñeiras and J. Niclós-Gutiérrez, Coord. Chem. Rev., 2013, 257, 2814–2838 CrossRef; A. B. Coles, O. G. Wood and C. S. Hawes, J. Serb. Chem. Soc., 2023 DOI:10.2298/JSC230623061C , In Press.
- Y. Xiong, Y.-Z. Fan, R. Yang, S. Chen, M. Pan, J.-J. Jiang and C.-Y. Su, Chem. Commun., 2014, 50, 14631–14634 RSC; S. J. Bullock, L. P. Harding, M. P. Moore, A. Mills, S. A. F. Piela, C. R. Rice, L. Towns-Andrews and M. Whitehead, Dalton Trans., 2013, 42, 5805–5811 RSC.
- K. Mondal, N. Mukhopadhyay, S. Patra, T. Roy and P. Das, ACS Catal., 2023, 13, 11977–11995 CrossRef CAS.
- D. Ülkü, B. P. Huddle and J. C. Morrow, Acta Crystallogr., Sect. B: Struct. Sci., Cryst. Eng. Mater., 1971, 27, 432–436 CrossRef.
- B. Marzyk-Ociepa, K. Dysz, I. Turowska-Tyrk and D. Michalska, J. Mol. Struct., 2015, 1101, 91–100 CrossRef.
- R. Puttreddy and P. J. Steel, CrystEngComm, 2014, 16, 556–560 RSC.
- F. Pointillart, Y. Le Gal, S. Golhen, O. Cador and L. Ouahab, Inorg. Chem., 2009, 48, 4631–4633 CrossRef CAS PubMed.
- Z. Wang, N. Liu, H. Li, P. Chen and P. Yan, Eur. J. Inorg. Chem., 2017, 2211–2219 CrossRef CAS; L. Armelao, D. B. Dell'Amico, L. Bellucci, G. Bottaro, S. Ciattini, L. Labella, G. Manfroni, F. Marchetti, C. A. Mattei and S. Samaritani, Eur. J. Inorg. Chem., 2018, 4421–4428 CrossRef; F. Pointillart, B. Le Guennic, O. Maury, S. Golhen, O. Cador and L. Ouahab, Inorg. Chem., 2013, 52, 1398–1408 CrossRef PubMed.
- A. T. Baker, N. J. Ferguson, H. A. Goodwin and A. D. Rae, Aust. J. Chem., 1989, 42, 623–638 CrossRef CAS; J. W. Goodwin, P. E. Kruger and C. S. Hawes, J. Coord. Chem., 2021, 74, 341–360 CrossRef.
- T. Steiner, Acta Crystallogr., Sect. B: Struct. Sci., Cryst. Eng. Mater., 1998, 54, 456–463 CrossRef.
- S. H. Hewitt, G. Macey, R. Mailhot, M. R. J. Elsegood, F. Duarte, A. M. Kenwright and S. J. Butler, Chem. Sci., 2020, 11, 3619–3628 RSC.
- B. Alpha, R. Ballardini, V. Balzani, J.-M. Lehn, S. Perathoner and N. Sabbatini, Photochem. Photobiol., 1990, 52, 299–306 CrossRef CAS.
- C. A. Taylor, M. A. El-Bayoumi and M. Kasha, Proc. Natl. Acad. Sci. U. S. A., 1969, 63, 253–260 CrossRef CAS PubMed; J. Konijnenberg, A. H. Huizer and C. A. G. O. Varma, J. Chem. Soc., Faraday Trans. 2, 1988, 84, 1163–1175 RSC.
- K. Binnemans, Coord. Chem. Rev., 2015, 295, 1–45 CrossRef CAS.
- ReactLab Equilibria, Jplus Consulting Ltd, Fremantle, Australia, 2018 Search PubMed.
- J. Hamacek, S. Blanc, M. Elhabiri, E. Leize, A. Van Dorsselaer, C. Piguet and A.-M. Albrecht-Gary, J. Am. Chem. Soc., 2003, 125, 1541–1550 CrossRef CAS PubMed.
- R. M. Supkowski and W. D. Horrocks Jr., Inorg. Chim. Acta, 2002, 340, 44–48 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Materials and methods section, X-ray powder diffraction figures, spectroscopic data, mass spectrometry figures, crystallographic data tables, and structure description and figures of [Cu2(OMe)2L2]. CCDC 2288060–2288066. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3ce00985h |
| This journal is © The Royal Society of Chemistry 2023 |