
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Optimised synthesis and further structural diversity of ytterbium benzene-1,4-dicarboxylate MOFs†
Thomas W.
Chamberlain
ab,
Yasmine
c,
Claire T.
Coulthard
a,
Guy J.
Clarkson
 a,
Volkan
Degirmenci
b,
Yuni K.
Krisnandi
c and
Richard I.
Walton
*a
a,
Volkan
Degirmenci
b,
Yuni K.
Krisnandi
c and
Richard I.
Walton
*a
aDepartment of Chemistry, University of Warwick, Coventry, CV4 7AL, UK. E-mail: r.i.walton@warwick.ac.uk
bSchool of Engineering, University of Warwick, Coventry, CV4 7AL, UK
cDepartment of Chemistry, Universitas Indonesia, Depok 16424, Indonesia
First published on 14th September 2023
Abstract
The optimisation of the crystallisation of the hydrothermally-stable metal–organic framework Yb6-MOF (Yb6(BDC)7(OH)4(H2O)4) to provide a reproducible one-step synthesis is achieved by use of the sodium salt of benzene-1,4-dicarboxylate (Na2BDC) as ligand precursor and control of pH with aqueous NaOH at 190 °C over 3 days. Phase purity is confirmed using powder X-ray diffraction (PXRD) and thermogravimetric analysis (TGA). During exploration of synthesis conditions from the same set of chemical reagents, three further ytterbium benzene-1,4-dicarboxylates have been isolated and structurally characterised using single-crystal X-ray diffraction, with phase purity assessed by PXRD and TGA. UOW-3 (Yb2(H2O)6(BDC)3) crystallises by lowering pH, and has a relatively dense three-dimensionally connected structure with no Yb–O–Yb linkages but dimers of Yb bridged by BDC linkers lying in the ab plane with a pseudo, pillared-layered structure, where BDC connects along c. UOW-4 (Yb4(BDC)6(H2O)6) forms under the same chemical conditions but upon lowering the temperature to 100 °C, and this material again contains no Yb–O–Yb linkages, but chains of BDC-bridged Yb centres cross-linked to give a dense three-dimensional structure. Upon increasing pH of the synthesis mixture, the material UOW-5 forms, Yb5O(OH)8(BDC)2(HBDC), consisting of dense inorganic layers of ytterbium oxyhydroxide, cross linked by BDC and HBDC pillars. The formulation is supported by infrared spectroscopy, which provides evidence for the HBDC monoanion, and also the presence of a short O–O distance indicative of hydrogen bonding between a carboxylate OH and an oxide anion of the inorganic layer. UOW-3 and UOW-4 both convert to Yb6-MOF upon heating in water above their synthesis temperature, whereas UOW-5 is hydrothermally stable at 240 °C. The structures of the new materials are discussed in terms of ligand binding modes, and connectivity of metal centres, with comparison to other reported Yb-BDC phases in order to relate structural chemistry to their synthesis conditions and the hydrothermal stability of the materials.
Introduction
Metal–organic frameworks (MOFs) are a subclass of coordination polymers, now well known for their potential for extremely high porosity and large surface areas, compared to other porous materials such as zeolites.1 Their structures, consisting of metal ions or clusters connected infinitely in two- or three-dimensions by organic linkers, leads to ease of functionalisation and has led to a huge range of MOF materials with precisely tuned properties being described since their first reports almost 30 years ago in 1995.2 MOFs and other coordination polymers have numerous potential practical applications in fields such as molecular capture and separation that come from their porosity,3 and in composites for energy storage applications such as in batteries and supercapacitors.4 There are also considerable applications in heterogeneous catalysis where solid-state acidity and redox properties may be present, and these may be exploited even in the absence of porosity since the surface chemistry may provide unique properties.5Despite their tuneability and ease of functionality, the long-term stability of MOF materials, particularly towards water, has stifled their widespread adoption in fields such as catalysis.6,7 Relatively few MOF materials are currently suited for practical applications. Recently, several groups have attempted to identify the most suitable chemical compositions and structural features needed for water stability and for thermal stability.8–10 It can be concluded from these works that the MOF structures that offer the greatest stability towards temperature or solvent consist of high valent metal cations bonded by oxide or hydroxide in clusters or infinitely connected motifs, and bridged by ligands with a rigid aromatic core, often polycarboxylates or azolates. Previously, in our search for water-stable MOFs with acidic functionality, we demonstrated that the ytterbium benzene-1,4-dicarboxylate (BDC) Yb6(BDC)7(OH)4(H2O)4 (Yb6-MOF) offered the required hydrothermal stability (up to at least 200 °C) to allow for it to be an active heterogeneous catalyst in water, making use of both Lewis and Brønsted acidity.11 The use of Yb3+ as the metal centre stems from the use of ytterbium triflate as a well-known Lewis acid used for various organic transformations, and the use of YbCl3 in solution as a homogeneous Lewis acid catalyst.11 Yb6-MOF contains hexanuclear clusters containing three distinct eight-coordinate YbIII ions bridged by μ3-hydroxyl groups with four of the Yb centres coordinated by terminal water molecules, Fig. 1a. The hexameric clusters are held together by five distinct BDC linkers, two tridentate which bind through two μ2-η2:η1 modes each, Fig. 1b, and three bidentate which bind through two Z,Z-μ2-η1:η1 modes, Fig. 1c, to form a structure containing one dimensional channels of occluded water, Fig. 1d.
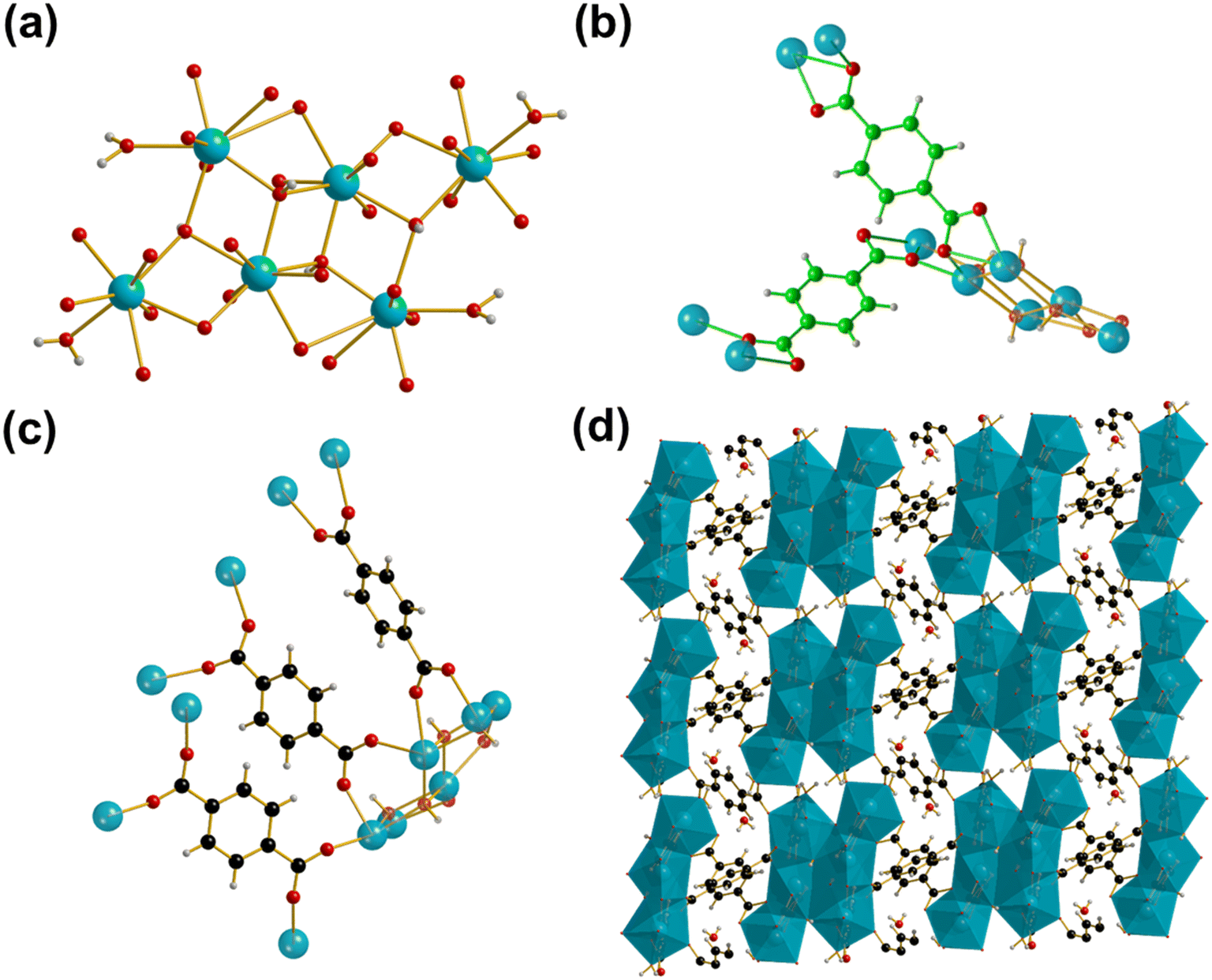 | ||
| Fig. 1 The structure of Yb6-MOF with ytterbium ions shown in blue, carbon in black and green, oxygen in red and hydrogen in white. (a): A single hexanuclear cluster Yb6(OH)4(H2O)4 with the μ3-OH and terminally bound water molecules visible. Note that the metal-linker bonds are depicted as dangling oxygens for clarity. Ytterbium is shown in grey, oxygen in red and hydrogen in white. (b): A hexameric cluster of Yb6-MOF with the two distinct tridentate BDC linkers which bind through two μ2-η2:η1 modes each shown in green. (c): A hexameric cluster of Yb6-MOF with the three distinct bidentate BDC linkers which bind through two Z,Z-μ2-η1:η1 modes each shown in black. (d): The overall structure of Yb6-MOF viewed along the 100 direction with the hexameric clusters shown as blue polyhedra along with the BDC linkers. The narrow channels containing occluded water molecules are also visible. | ||
Yb6-MOF was first reported by Weng et al. where the synthesis of three isostructural materials containing Yb, Y and Er, and the photoluminescence properties of a heterometallic Yb/Er form was described.12 The Yb analogue was later investigated by our group as a bifunctional dual acid (Lewis and Brønsted) catalyst for the conversion of glucose into the platform chemical 5-hydroxymethylfurfural (HMF), with moderate conversion of glucose but excellent selectivity towards HMF.11 Yb6-MOF was noted for its extremely high stability, both thermally (ca. 500 °C) and with no loss in crystallinity observed after heating hydrothermally in water at 240 °C for multiple days. The tuneability of Yb6-MOF, whereby it can be synthesised with a variety of lanthanide ions, combined with its unusually high stability, then led to its use as a thermometer.13 Here, the Y analogue was doped with a precise ratio of europium and terbium ions to afford a photoluminescent material whose optical response varied with temperature. To-date, (Y0.89Tb0.10Eu0.01)6-MOF has the largest temperature detection range (up to at least 573 K) of any MOF thermometer reported.
The original synthesis of Yb6-MOF and other rare earth analogues described by Weng et al. required pre-hydrolysis of the precursor lanthanide salt (either nitrate or chloride) for an unspecified time followed by precise adjustment of the pH using an unspecified base, which made the synthesis difficult to reproduce.12 We previously devised a reliable synthetic route for the material, but this required a two-step solvothermal synthesis via the precursor material Yb2(BDC)3(DMF)2(H2O)2 which itself was made in a mixed DMF/water solution and needed a subsequent hydrothermal treatment.14 Later, we reported the synthesis of (Y0.89Tb0.10Eu0.01)6-MOF, requiring hydrothermal heating of the lanthanide salts with the disodium salt of the organic linker at a precise pH.13 Hydrothermally washing the mixture at 190 °C for multiple days resulted in crystallisation of the (Y0.89Tb0.10Eu0.01)6-MOF phase as large micron-sized crystals free of any impurities.
A large number of coordination polymers and MOFs have been reported by combining lanthanides with the BDC ligand and Kitamura et al. have recently surveyed the reported materials and used this to identify unexplored regions of chemical reaction space to investigate the discovery of novel frameworks.15 In their extensive literature review, they listed 10 distinct Yb-BDC materials (including Yb6-MOF), some of which are polymorphs of each other, and some of which are also found for other lanthanide cations. Furthermore, for the other smaller lanthanoid cations (Ho–Lu, and Y) some unique phases are reported, which might also be anticipated for ytterbium. The structural diversity arising from a single cation and ligand combination is noteworthy and draws attention to the difficulty in predicting the synthesis of new MOF materials from a given combination of ligand and metal precursor. We have therefore explored the synthesis further, initially with the aim of developing a one-step route to Yb6-MOF, in view of its interesting applications. Herein, we describe a novel synthetic route to Yb6-MOF using only water as the solvent which drastically reduces the synthesis time of the MOF. In developing this, three other phases were also discovered, with single crystals being produced which allowed for their structures to be determined. Conclusions are drawn between the precise synthesis conditions of each MOF and their resulting structures which may help to direct the future discovery of ytterbium and other lanthanide-based carboxylate coordination polymers and MOFs.
Results
Synthesis of Yb6-MOF
In the new synthetic route to Yb6-MOF, an aqueous solution of ytterbium chloride was prepared and to this the disodium salt of BDC, Na2BDC, was added. 440 mg of 2 M NaOH solution was then added to adjust the concentration of solution to 0.08 M, see Experimental section. Precise determination of the pH was not possible due to gradual dissolution of the organic linker which reduced the pH over time. The amount of NaOH solution added to the reaction mixture was found to be vital as either an increase or decrease in the pH was found to produce side products or different products entirely. The precursors were heated hydrothermally to produce a white crystalline powder and Pawley fitting against the PXRD data using the reported single-crystal lattice parameters of Yb6-MOF showed good agreement and confirmed the successful synthesis of the material (Fig. 2a and Table S1, ESI†). Thermogravimetric analysis (TGA) confirmed the purity of the material (Fig. 2b). The observed mass loss at ∼400 °C, which is attributed to the loss of the terminally bound water molecules, of 5% is comparable to the theoretical loss of 4.6%. The combustion of the organic linker at 500–700 °C resulted in a total mass loss of 51%, which is in good agreement with the theoretical loss of 50%.11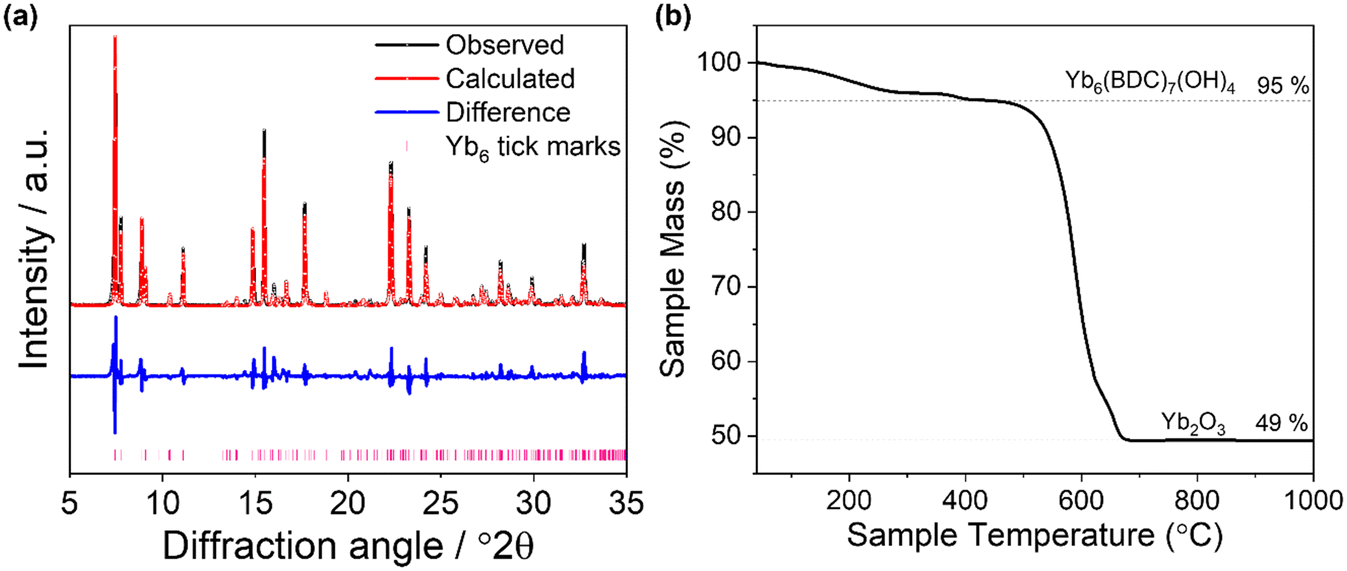 | ||
| Fig. 2 (a) Pawley fit of powder XRD pattern of as made sample of Yb6-MOF. The data points are shown in black, the fitted pattern in red, the difference curve in blue and the allowed tick positions in pink. The refined lattice parameters are shown in Table S1.† (b) Thermogravimetric analysis of Yb6-MOF. | ||
In optimising the synthesis conditions of Yb6-MOF to allow for the formation of a single phase, several other materials were discovered. The synthesis of three of these new materials was then optimised to produce single crystals of sufficient size to allow for the determination of their structures by single crystal diffraction. The optimised synthesis for each of the materials including Yb6-MOF is given in the Experimental section, and their structures described in the following sections.
Synthesis and structure of UOW-3, Yb2(BDC)3(H2O)6
The material Yb2(BDC)3(H2O)6 which we denote as UOW-3 (University of Warwick 3, following from UOW-1 and UOW-2 that are yttrium carboxylates reported elsewhere16), was synthesised by a hydrothermal method, using a reduced basicity of 0.045 M NaOH compared to 0.08 M in the synthesis of Yb6-MOF but the same molar ratios of Yb and linker precursors and the same temperature of 190 °C. Single crystal X-ray structural analysis revealed the crystals to be a Yb-BDC coordination polymer, the structure of which has been reported independently by two previous groups.17,18 Feng used a complex reagent mixture of AgNO3, 2-pyrazinecarboxylic acid, with Yb2O3 as Yb source and benzene-1,4-dicarboxylic acid as linker precursor with pH adjusted to 2 by addition of HClO4, which was heated in water at 170 °C for 6 days, but only the crystal structure was reported and phase purity was not assesed.17 Zehnder et al. used the same metal and ligand precursors as Feng, but in an aqueous solution of glutaric acid and sodium chloride heated at 170 °C for 4 weeks, and again phase purity was not assessed.18 It should also be noted that Er (ref. 19) and Lu (ref. 20) analogues have also been reported.UOW-3 crystallises in the triclinic space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) with chemical formula Yb2(BDC)3(H2O)6. It contains one eight coordinate YbIII centre, two distinct benzene-1,4-dicarboxylate linkers and three terminal water molecules per YbIII centre. Within the structure, each YbIII centre is directly bound to one benzene-1,4-dicarboxylate linker which connects to one additional YbIII through two η2 binding modes. The water molecules are all bound directly to Yb. The closest Yb centres are not linked directly by oxygen, but by Yb–O–C–O–Yb bridges form dimers with the closest Yb–Yb distance of 4.905 Å. A second type of benzene-1,4-dicarboxylate is connected unsymmetrically through an η1 binding mode at one end and a Z,Z-μ2-η1:η1 binding mode at the other (Fig. 3a). The uncoordinated oxygen of the η1 binding mode is too distant from any Yb centres (>4 Å) to be considered as a taking part in a bonding interaction. There is evidence for hydrogen bonding between the Yb-bound water molecules and oxygens of carboxylates (O16–O11 = 2.701 Å, O17–O9 = 2.712 Å, O18–O2 = 2.714 Å), and also between the water molecules (O18–O17 = 2.752 Å). The dimers of Yb lie in the ab plane (Fig. 3b) and the BDC linkers cross connect these to give a pseudo, pillared-layered structure (Fig. 3c).
with chemical formula Yb2(BDC)3(H2O)6. It contains one eight coordinate YbIII centre, two distinct benzene-1,4-dicarboxylate linkers and three terminal water molecules per YbIII centre. Within the structure, each YbIII centre is directly bound to one benzene-1,4-dicarboxylate linker which connects to one additional YbIII through two η2 binding modes. The water molecules are all bound directly to Yb. The closest Yb centres are not linked directly by oxygen, but by Yb–O–C–O–Yb bridges form dimers with the closest Yb–Yb distance of 4.905 Å. A second type of benzene-1,4-dicarboxylate is connected unsymmetrically through an η1 binding mode at one end and a Z,Z-μ2-η1:η1 binding mode at the other (Fig. 3a). The uncoordinated oxygen of the η1 binding mode is too distant from any Yb centres (>4 Å) to be considered as a taking part in a bonding interaction. There is evidence for hydrogen bonding between the Yb-bound water molecules and oxygens of carboxylates (O16–O11 = 2.701 Å, O17–O9 = 2.712 Å, O18–O2 = 2.714 Å), and also between the water molecules (O18–O17 = 2.752 Å). The dimers of Yb lie in the ab plane (Fig. 3b) and the BDC linkers cross connect these to give a pseudo, pillared-layered structure (Fig. 3c).
 | ||
| Fig. 3 The structure of UOW-3. (a): Coordination geometry around the blue YbIII centre depicting the three partially uncoordinated ‘type 2’ BDC linkers in black, the fully coordinated η2 symmetrical BDC linker in green and each of the directly connected YbIII centres shown with their complete coordination spheres. (b): The overall structure of UOW-3 viewed along the [001] direction, with isolated YbIII ions depicted as blue polyhedra, and narrow channels with carboxylates depicted protruding into the pores. (c): The structure of UOW-3 viewed along the [100] direction showing the pseudo, pillared layered structure. | ||
Powder X-ray diffraction of the bulk material shows agreement with the simulated pattern of UOW-3 although with clear preferred orientation effects resulting from the highly crystalline material, Fig. 4a. The powder pattern also indicated the presence of at least one obvious impurity, with notable peaks at 8.97 and 16.51° 2θ, and the low angle diffraction peaks of this secondary phase suggest the presence of another Yb-BDC MOF although did not match any of the Yb-BDC-based materials reported to date. TGA shows a mass loss of 8% to 300 °C, attributed to the loss of the six bound water molecules in Yb2(BDC)3(H2O)6 compared to a theoretical value of 11%, Fig. 4b. The thermal combustion of the linker at ∼500–600 °C resulted in a total mass loss of 55% compared to a theoretical value of 60% to yield Yb2O3. The deviations from the theoretical mass losses are attributed to the presence of Yb-BDC based impurities within the as-made sample.
 | ||
| Fig. 4 (a): Powder X-ray diffraction pattern of the bulk sample from which single crystals of UOW-3 were obtained along with the simulated diffraction pattern of UOW-3. (b): TGA trace of as made UOW-3 with the observed mass losses on the thermal decomposition of Yb2(BDC)3(H2O)6 shown. | ||
Synthesis and structure of UOW-4, Yb4(BDC)6(H2O)6
On lowering the synthesis temperature from 190 °C which gave UOW-3, to 100 °C while keeping all other synthesis conditions the same, a further highly crystalline phase was produced, and single crystals were isolated. Single crystal X-ray analysis revealed this to be a three-dimensional structure consisting of YbIII ions connected by benzene-1,4-dicarboxylate linkers to give the formula Yb4(BDC)6(H2O)6. This material has been named UOW-4 and crystallises in the orthorhombic Pbca space group and adopts a three-dimensionally connected framework structure containing channels into which ytterbium-bound water molecules protrude, Fig. 5a. The structure contains four distinct YbIII centres that are not directly connected by oxygens, but bridged by Yb–O–C–O–Yb connections to give four distinct chains running along the material in the a direction. There are two chains of alternating 7-coordinate Yb1 and Yb2, one consisting of solely 8-coordinate Yb3, and the other 7-coordinate Yb4 ions, Fig. 5b. The material also contains six distinct terminally bound water molecules, one coordinated to each of Yb1 and Yb2 which project into the channels within the material, and two coordinated to each of Yb3 and Yb4, Fig. 5c. There are five distinct benzene-1,4-dicarboxylate linkers which coordinate to the four YbIII ions each through two Z,Z-μ2-η1:η1 binding modes, and one which binds through one carboxylate in the same Z,Z-μ2-η1:η1 binding mode and through the other carboxylate to a single Yb ion in a η1 fashion leaving an uncoordinated carboxylate, Fig. 5d. This uncoordinated carboxylate group is distant from any Yb centres (>3.7 Å), but is involved with two hydrogen bonding interactions. It acts as a proton acceptor to two hydrogen atoms of the two water molecules terminally bound to Yb4. | ||
| Fig. 5 The structure of Yb4(BDC)6(H2O)6, UOW-4. The YbIII ions are shown in blue, oxygen in red, hydrogen in white and carbon atoms in black and various colours in (d). (a): The overall structure of the UOW-4, with the four chains of YbIII ions visible and the geometry of the benzene-1,4-dicarboxylate linkers to give a porous material with channels containing terminally bound water molecules. (b): The locations of the four distinct YbIII centres within UOW-4, and their relation to each other. On the left: two chains of alternating Yb1 and Yb2 ions and, on the right: two chains consisting of solely Yb3 (bottom) and Yb4 ions (top). (c): View of the four distinct YbIII centres with the six terminally bound water molecules clearly visible. Note that the external benzene-1,4-dicarboxylate linkers have been omitted for clarity. (d): Binding geometries of each of the six distinct linker molecules. Each linker has been coloured differently and their protons removed for clarity. The hydrogen bonding between the carboxylate of the blue linker molecule and the two terminally bound waters of Yb4 is indicated. | ||
The structure of UOW-4 has been previously reported for an erbium analogue, which was synthesised solvothermally in a water/ethanol system.21 The purity of the bulk sample from which crystals of UOW-4 were obtained was investigated by a combination of powder XRD, and TGA, Fig. 6. A Rietveld refinement of the crystal structure against the powder diffraction pattern (with atom positions fixed at values from single crystal diffraction) showed good agreement with the expected lattice parameters of UOW-4 (see ESI†), with only small impurity peaks present, Fig. 6a. The impurities remain unidentified but are likely be other Yb-benzene-1,4-dicarboxylate materials, since their Bragg diffraction peaks are seen at low angles. The TGA of the bulk sample showed an initial mass loss below 200 °C, likely due to loss of water, followed by a large mass loss due to combustion of the organic linker observed at 400–600 °C, Fig. 6b. The observed percentage mass losses were found to be in good agreement with the expected values of 6% for the loss of bound water and 59% on linker combustion based on the thermal decomposition of Yb4(BDC)6(H2O)6. The first observed mass loss, which is attributed to the thermal removal of the water from the MOF, can be seen to occur in two distinct steps, which is consistent with the two water environments within the material, four terminally bound water molecules, and water that is hydrogen bonded to the carboxylate of the BDC linker and would be expected to be released at a higher temperature.
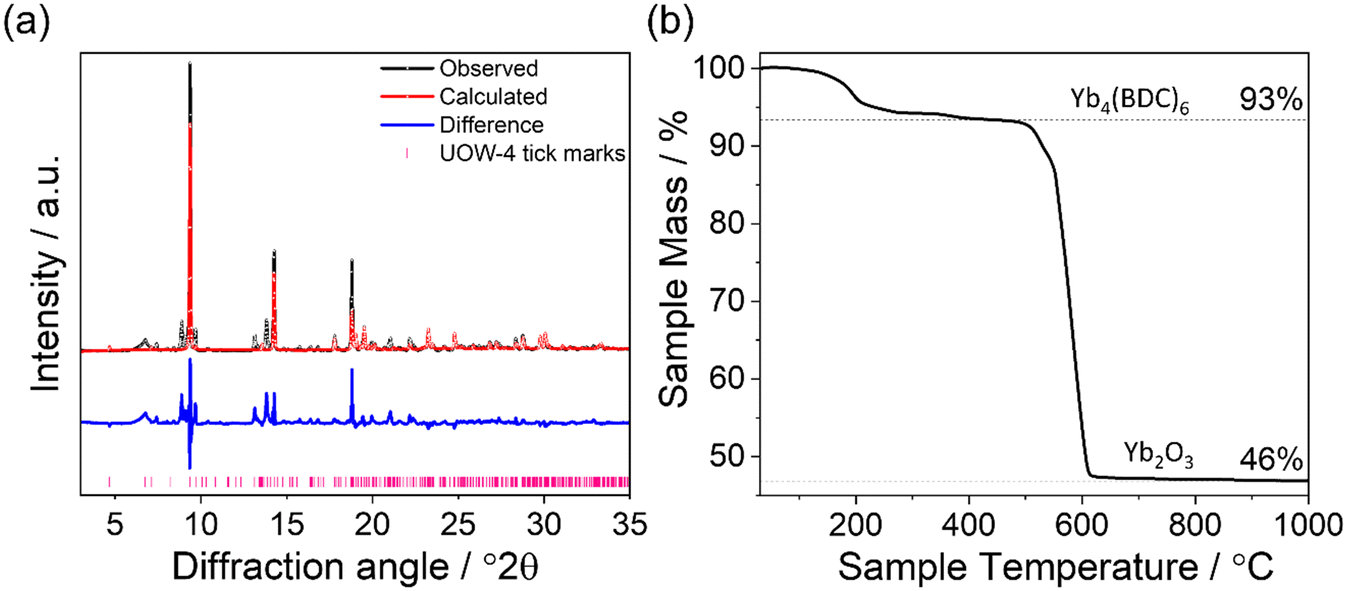 | ||
| Fig. 6 (a): Rietveld refinement of the structure of a sample of UOW-4 from which single crystals were obtained compared to the simulated pattern of UOW-4. The data points are shown in black, the fitted pattern in red, the difference curve in blue and the allowed tick positions in pink. Fitted lattice parameters are shown in Table S2.† (b): TGA trace of as made UOW-4 with expected mass losses based on the decomposition of the desolvated material Yb4(BDC)6(H2O)6 compared to that observed. Deviations from the expected mass losses are attributed to the impurities within the bulk sample. | ||
Synthesis and structure of UOW-5, Yb5O(OH)8(BDC)2(HBDC)
On increasing the NaOH concentration in the synthesis to 0.18 M compared to 0.08 M used for Yb6-MOF and whilst keeping the other conditions the same, another highly crystalline material was produced, which we have named UOW-5. Single crystal X-ray analysis revealed that UOW-5 crystallises in the monoclinic P21/c space group and has the chemical formula Yb5O(OH)8(BDC)2(HBDC). It adopts a pillared-layered-type structure with two-dimensional layers of Yb5O(OH)8 connected by three distinct benzene-1,4-dicarboxylate linkers. Two linkers are completely coordinated BDC which adopt Z,Z-μ2-η1:η1 binding modes through both carboxylates, and one is a monoprotonated HBDC which binds through the carboxylate in a Z,Z-μ2-η1:η1 mode and through the acid in a η1 mode (Fig. 7a). The layers of Yb5O(OH)8 contain three distinct eight coordinate and two distinct seven coordinate YbIII centres bridged by μ3 OH groups and μ2 oxides (Fig. 7b). This is evidenced by the longer Yb–μ3-OH bond lengths of (average) = 2.30 Å compared to μ2-O = 2.23 Å and with one of the BDC ligand remaining protonated, this gives an overall charged-balanced structure. Although the HBDC proton cannot be located by X-ray diffraction, its presence is deduced since there is evidence for a hydrogen bond between the carboxylic acid group and a bridging μ2-oxide (Fig. 7c) with the distance O(209)–O3 of 2.775 Å. Furthermore, the FTIR spectrum show the presence of a carboxylic O–H vibration, but with a significant shift in the stretching frequency compared to H2BDC (1610 vs. 1670 cm−1) with the weaker O–H vibrational frequency indicating a longer O–H bond (Fig. 7d). This feature is missing in the IR spectrum of UOW-4 (Fig. S1, ESI†), as expected for a material in which the BDC ligand is fully deprotonated, which further confirms the assignment of the band. The O–H region of the IR spectrum also provides evidence for O–H stretching band of carboxylic group, distinct from those of the hydroxides (Fig. S2, ESI†).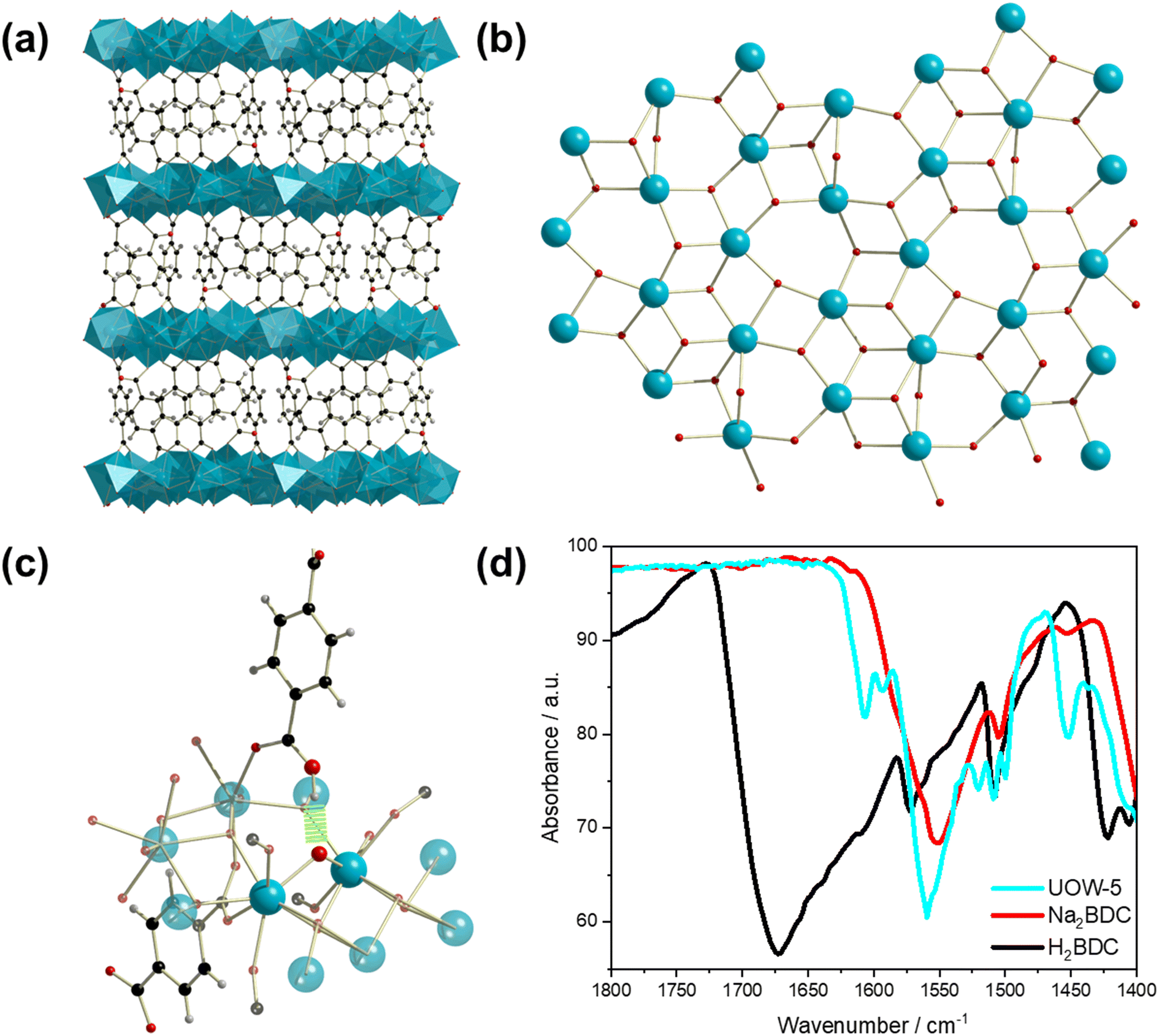 | ||
| Fig. 7 The structure of UOW-5. (a): The overall pillared-layered type structure of UOW-5, with layers of YbIII ions depicted as blue polyhedra along with the BDC linker pillars. (b): The two-dimensional structure of Yb5O(OH)8 layers in UOW-5 with five distinct YbIII centres, μ3 OH ligands, and the μ2 oxides are clearly visible. (c): Hydrogen bonding between the protonated carboxylic acid of the HBDC linker and the μ2 oxide of the Yb5O(OH)8 layers highlighted in green. The surrounding atoms have been shown but faded for clarity. (d): FTIR spectra of H2BDC with carbonyl stretching frequency of 1673 cm−1, Na2BDC with carbonyl stretching frequency of 1551 cm−1, and UOW-5 with two carbonyl stretching frequencies of 1607 and 1560 cm−1. The band at 1560 cm−1 is attributed to the carbonyl stretches of the BDC linkers in UOW-5, whilst the stretch at 1607 cm−1, which is attributed to the protonated HBDC carbonyl stretch, suggests hydrogen bonding between the carboxylic acid of HBDC and the μ2 oxide of the Yb5O(OH) layers. | ||
Powder XRD of the bulk sample of UOW-5 showed that the material was pure, Fig. 8a. A Rietveld refinement against the powder pattern was performed using the crystal structure obtained by single crystal diffraction, and this showed good agreement with the bulk sample, with preferred orientation resulting from the highly crystalline nature of the sample. A small impurity phase is evidenced by the weak additional peaks at low angles, and this is likely to be another Yb-BDC material that is formed along with UOW-5. TGA of the material showed two distinct mass losses (Fig. 8b). The initial mass loss at approximately 100–200 °C is attributed to the loss of water from the surface and pores of the material. The subsequent mass loss at between 400 and 600 °C is attributed to the combustion of the linker from UOW-5 to leave Yb2O3 as the final product at 1000 °C. The observed final mass of Yb2O3 of 61% was in good agreement with the theoretical value of 63% based on the complete combustion of Yb5O(OH)8(BDC)2(HBDC). The small deviation from the theoretical value is attributed to the presence of minor impurity phases.
 | ||
| Fig. 8 (a): Rietveld refinement of the bulk sample of as made UOW-5 compared to the simulated pattern of UOW-5 obtained by single crystal diffraction. Atom positions and occupancies were not fitted. The data points are shown in black, the fitted pattern in red, the difference curve in blue and the allowed tick positions in pink. The refined lattice parameters are given in Table S3.† (b): TGA trace of as made UOW-5 showing three distinct mass losses, with the second being attributed to linker decomposition in a small impurity phase to leave Yb5O(OH)8(BDC)2(HBDC) and Yb2O3. The final mass loss is attributed to linker combustion in UOW-5 to leave Yb2O3 as the final product. | ||
Hydrothermal stability of Yb-BDC materials
The hydrothermal stability of the materials was tested, as this relates directly to their potential utility in a range of applications such as catalysis where stability in water is particularly desirable. Yb6-MOF has previously been shown to be extremely water stable, even hydrothermally, where it can be heated to 240 °C for multiple days with no loss in crystallinity.13 Each of the three Yb-BDC frameworks reported here was heated in water at various temperatures for 24 hours after which powder XRD was used to determine whether decomposition of the MOFs had occurred, Fig. 9. UOW-4, which was synthesised at the lowest temperature, was found to be the least stable of the three materials. It completely decomposed after 24 hours at 120 °C to yield Yb6-MOF as the sole crystalline product. UOW-3, which was synthesised at 190 °C was found to be stable up to around 150 °C in pure water, but at 190 °C, complete decomposition of UOW-3 was observed, and Yb6-MOF had formed with a considerable preferred orientation effects evident by the relative peak intensities of the PXRD pattern. UOW-5, which was synthesised at 190 °C and high pH was found to be the most hydrothermally stable of the three materials, with the sample showing no change after heating for 24 hours at 240 °C.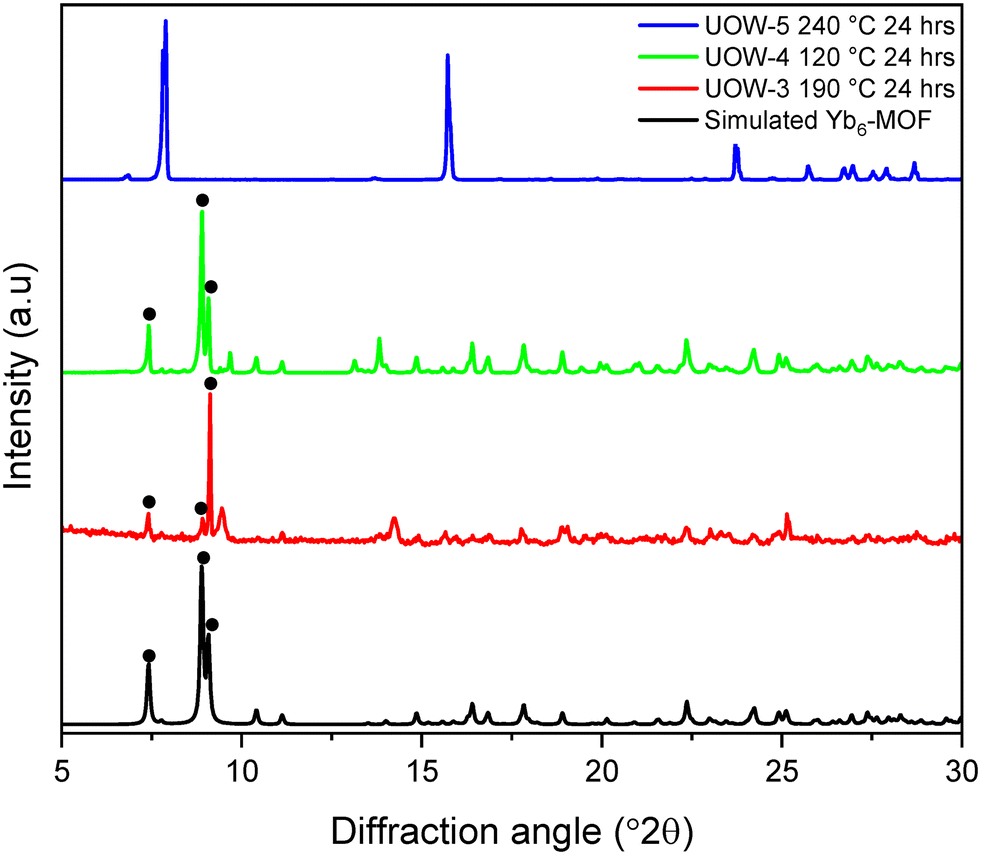 | ||
| Fig. 9 Powder XRD measured after heating UOW-3, UOW-4 and UOW-5 in water, compared to the simulated pattern of Yb6-MOF. Characteristic Bragg peaks of Yb6-MOF are indicated by •. | ||
Discussion
The materials we have described demonstrate the diversity in benzene-1,4-dicarboxylates of ytterbium and are formed simply by altering the pH employed in their syntheses and by lowering the temperature in the case of UOW-4. As noted above, the literature already contains at least 10 reported Yb-BDC materials, as well as other structures for other small lanthanoid cations.15 In an attempt to correlate the structures of the materials to the conditions employed in their syntheses, we categorise our materials in terms of their YbIII coordination numbers, Yb-BDC binding modes, oxygen connectivity, and overall structural connectivity (using the ImOn notation of Cheetham et al.22), Table 1.| Material | NaOH Conc./M (temperature/°C) | ImOn | Yb CN | BDC binding modes | Number of uncoordinated BDC oxygens per total Yb | n(Yb–O–Yb) per YbOx (n/x) |
|---|---|---|---|---|---|---|
| UOW-3 | 0.045 (190) | I0O3 | 8 | Types 1 + 2 | 1/1 | 0/8 |
| UOW-4 | 0.045 (100) | I0O3 | 7 + 8 | Types 2 + 3 | 1/4 | 0/7 (Yb1) |
| 0/7 (Yb2) | ||||||
| 0/8 (Yb3) | ||||||
| 0/7 (Yb4) | ||||||
| Yb6-MOF | 0.08 (190) | I0O3 | 8 | Types 3 + 4 | 0/3 | 2/8 (Yb1) |
| 3/8 (Yb2) | ||||||
| 4/8 (Yb3) | ||||||
| UOW-5 | 0.18 (190) | I2O1 | 7 + 8 | Types 2 + 3 | 1/5 | 6/8 (Yb1) |
| 5/7 (Yb2) | ||||||
| 5/8 (Yb3) | ||||||
| 5/7 (Yb4) | ||||||
| 5/7 (Yb5) |
The materials all contain Yb in coordination number 7 or 8 and there is no evident trend in the distribution of these coordination number types with the either the overall structural features of the material or with the synthesis conditions. Within the set of these four materials, there are four distinct Yb-BDC binding modes describing the YbIII to BDC binding environments and these are denoted Types 1–4 and depicted in Fig. 10. Comparing these binding modes to the NaOH concentration used in their synthesis, at the lowest NaOH concentration of 0.045 M BDC Types 1, 2 and 3 are observed. Increasing the NaOH concentration to 0.08 M led to the formation of Yb6-MOF which contains BDC Types 3 and 4, but at the highest concentration of 0.18 M, BDC Types 2 and 3 were again observed. Interestingly, the BDC binding modes Types 2 and 3 were observed in both UOW-4 and UOW-5 which were synthesised at the two extreme NaOH concentrations. This suggests that the pH employed in the synthesis of the ytterbium-terephthalates does not direct the BDC binding modes.
 | ||
| Fig. 10 Overview of the ytterbium-terephthalate binding modes within the materials UOW-3, −4 and −5 and Yb6-MOF (Types 1–4). Type 1 consists of two η2 modes, Type 2 one Z,Z-μ2-η1:η1 and one η1 mode, Type 3 has two Z,Z-μ2-η1:η1 modes and Type 4 two μ2-η2:η1 binding modes. Note that in Type 2, the uncoordinated oxygen may be protonated as in UOW-5. Types 5 and 6 are seen in other Yb-BDC materials reported in the literature (see text). | ||
The overall structural connectivity is based on solely organic linkages for UOW-3, UOW4 and Yb6-MOF (I0O3) but for UOW-5 there is inorganic connectivity, here extending in two-dimensions (I2O1), and this material notably was prepared at the highest pH. The lack of oxygen connectivity between Yb centres in UOW-3 and UOW-4 reflects the fact that they contain Yb centres not directly linked by oxygens, and for these materials the synthesis pH and temperature is lowest, and on subsequent hydrothermal treatment they collapse into the material Yb6. For Yb6-MOF there are direct Yb–O–Yb connections, via hydroxide ions, reflecting the clusters present in the material, while for UOW-5 the Yb–O–Yb connectivity is greatest with bridging oxide and hydroxide, leading to infinite inorganic sheets; these two materials are hydrothermally stable at 240 °C. It is noteworthy that Yb6 is the only material here in which the BDC linkers are completely coordinated to Yb centres, i.e. it contains no Type 2 BDC (or Types 5 and 6), and this conceivably also contributes to its hydrothermal stability. UOW-5 also has a high proportion of fully coordinated carboxylate oxygens, and the Type 2 ligand is in fact protonated HBDC.
While the synthesis conditions that we have used have been varied systematically around a consistent set of chemical reagents, it is difficult to make comparisons with the materials already reported in the literature. This is because in some cases, as noted above for UOW-3, the previously reported analogues were prepared using a complex chemical mixture, including salts that provide no components for the resulting structure, and gave samples whose bulk phase purity was not tested. In other cases, Yb2O3 was used as a precursor instead of a more soluble salt such as the nitrate or chloride. When a nonaqueous solvent, such as N,N-dimethylformamide is used, the solvent itself is found as part of the structure formed, or may decompose on extended reaction to yield formate which is then included in the structure.14 Two other Yb-BDC structures reported by Zehnder et al. are worth commenting upon, even though both were prepared in the presence of salts or other additives in the hydrothermal synthesis.18 The material Yb2(BDC)3(H2O)2 was isolated at shorter reactions times (5 days) from the same chemical composition of reagents and temperature used to prepare their analogue of UOW-3 (4 weeks). Yb2(BDC)3(H2O)2 contains Type 2 and Type 3 BDC, so is another example of a material that contains uncoordinated BDC oxygens, and is clearly metastable with respect to another structure. On the other hand, the material Yb2(BDC)3(H2O)8·(H2O)2 was prepared at neutral pH (only NaCl in solution) and this has a structure in which all BDCs contain an uncoordinated oxygen (Type 5 and Type 6 in Fig. 10). We suggest that this material is likely to be hydrothermally unstable on further heating.
Conclusions
We have demonstrated the complexity in the synthesis of coordination polymers, where from a combination of one metal cation and one organic linker numerous open framework materials may crystallise depending on small variations in reaction conditions, including temperature and pH. The structural variety seen for hybrid structures based on Yb3+ (and other small lanthanoids) may be in part driven by the asymmetric coordination environment of the cation, typically 7 coordinate, but sometimes 8, which will inevitably lead to various possible modes of connection of carboxylate ligands. The various binding modes of one the simplest ligands used to construct coordination polymers is also noteworthy. In terms of hydrothermal stability of the materials, a combination of directly connected Yb centres (Yb–O–Yb linkages with oxide or hydroxide) and fully coordinated BDC ligands emerge as the key structural features for providing the most robust materials. Given the interest in such materials in applications such as heterogeneous catalysis, these considerations provide some design principles for prediction of novel structures. More broadly, most practical applications of any material require stability towards ambient conditions and the presence of humidity, and the effect of water on structural integrity is a key consideration. The connection between structural features needed for stability and synthesis conditions of a particular phase is important to map out, as it will provide valuable information for computational design of new stable materials. This could be used as input for machine learning, which is currently being explored for prediction of synthesis conditions of new structures with desired properties with on reported experimental reaction conditions as input data.Experimental section
Synthesis of Yb6(BDC)7(OH)4(H2O)4 (Yb6-MOF)
Ytterbium chloride hexahydrate (490 mg, 1.26 mmol, Merck), was added to a 23 ml PTFE autoclave liner. To this, disodium terephthalate (315 mg, 1.5 mmol, Alfa Aesar) was added followed by 10 ml deionised water and finally 440 mg of 2 M aqueous sodium hydroxide solution. The synthesis mixture was stirred for 5 min and then sealed and heated to 190 °C for 72 hours. After cooling, the powder was collected by vacuum filtration and washed twice with water and twice with methanol before being dried in air at 70 °C (Table 2 shows the reaction conditions used, along with those for the other materials reported herein).| YbCl3·6H2O | Na2BDC | Water | 2 M NaOH | Temp | Time | ||||
|---|---|---|---|---|---|---|---|---|---|
| Material | Mass/mg | Mols/mmol | Mass/mg | Mols/mmol | Mass/mg | Mass/mg | Concentration/mmol | °C | Hours |
| Yb6-MOF | 490 | 1.26 | 315 | 1.5 | 10 | 440 | 0.08 | 190 | 72 |
| UOW-3 | 490 | 1.26 | 315 | 1.5 | 10 | 250 | 0.045 | 190 | 72 |
| UOW-4 | 490 | 1.26 | 315 | 1.5 | 10 | 250 | 0.045 | 100 | 24 |
| UOW-5 | 490 | 1.26 | 315 | 1.5 | 10 | 1000 | 0.18 | 190 | 72 |
Synthesis of Yb2(H2O)6(BDC)3 (UOW-3)
Ytterbium chloride hexahydrate (490 mg, 1.26 mmol, Merck), was added to a 23 ml PTFE autoclave liner. To this, disodium terephthalate (315 mg, 1.5 mmol, Alfa Aesar) was added followed by 10 ml deionised water and finally 250 mg of 2 M aqueous sodium hydroxide solution. The synthesis mixture was stirred for 5 min and then sealed and heated to 190 °C for 72 hours. After cooling, the crystalline powder was collected by vacuum filtration and washed twice with water and twice with methanol before being dried in air at 70 °C resulting in white crystals of around 100–500 μm in size.Synthesis of Yb4(BDC)6(H2O)6 (UOW-4)
Ytterbium chloride hexahydrate (490 mg, 1.26 mmol, Merck), was added to a 23 ml PTFE autoclave liner. To this, disodium terephthalate (315 mg, 1.5 mmol, Alfa Aesar) was added followed by 10 ml deionised water and finally 250 mg of 2 M aqueous sodium hydroxide solution. The synthesis mixture was stirred for 5 min and then sealed and heated to 100 °C for 24 hours. After cooling, the powder was collected by vacuum filtration and washed twice with water and twice with methanol before being dried in air at 70 °C.Synthesis of Yb5O(OH)8(BDC)2(HBDC) (UOW-5)
Ytterbium chloride hexahydrate (490 mg, 1.26 mmol, Merck), was added to a 23 ml PTFE autoclave liner. To this, disodium terephthalate (315 mg, 1.5 mmol, Alfa Aesar) was added followed by 10 ml deionised water and finally 1000 mg of 2 M aqueous sodium hydroxide solution. The synthesis mixture was stirred for 5 min and then sealed and heated to 190 °C for 72 hours. After cooling, the powder was collected by vacuum filtration and washed twice with water and twice with methanol before being dried in air at 70 °C.Characterisation
Single crystal diffraction data were recorded from suitable crystals and mounted on a glass fibre with Fomblin oil and placed on a Rigaku Oxford Diffraction Synergy-S diffractometer with a dual source equipped with a Hybrid pixel array detector. The crystal was kept at 100(2) K during data collection. Using Olex2,23 the structure was solved with the SHELXT24 structure solution program using Intrinsic Phasing and refined with the SHELXL25 refinement package using least squares minimisation. Powder XRD patterns were measured at room temperature using a Panalytical Empyrean diffractometer operating with Cu Kα1/2 radiation. The diffraction profile was fitted using the GSAS suite of software26 to obtain lattice parameters via the Pawley method or the Rietveld method. Thermogravimetric analysis was performed using a Mettler-Toledo TGA/DSC1 under an air atmosphere with a heating rate of 5 °C. FT-IR spectra were recorded using a Bruker ALPHA FTIR ATR spectrometer.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are grateful to the British Council for award of a Newton Fund Institutional Links project (527290660), and the BOPTN Scheme 2020–2021 (NKB-294/UKN.RST/HKP.05.00/2020 and NKB-250/UKN.RST/HKP.05.00/2021) for matched funding. Yasmine thanks the Royal Society of Chemistry for the award of an International Student Research Bursary. Some of the equipment used in this work was provided by the University of Warwick's Research Technology Platforms. For the purpose of open access, the author has applied a Creative Commons Attribution (CC-BY) licence to any Author Accepted Manuscript version arising from this submission. CCDC 2256799, 2280621 and 2280622 contain the supplementary crystallographic data for this paper.References
- A. Kirchon, L. Feng, H. F. Drake, E. A. Joseph and H. C. Zhou, Chem. Soc. Rev., 2018, 47, 8611–8638 RSC.
- O. M. Yaghi and H. L. Li, J. Am. Chem. Soc., 1995, 117, 10401–10402 CrossRef CAS.
- C. Jiang, X. Wang, Y. Ouyang, K. Lu, W. Jiang, H. Xu, X. Wei, Z. Wang, F. Dai and D. Sun, Nanoscale Adv., 2022, 4, 2077–2089 RSC.
- Y. Peng, J. Xu, J. Xu, J. Ma, Y. Bai, S. Cao, S. Zhang and H. Pang, Adv. Colloid Interface Sci., 2022, 307, 102732 CrossRef CAS PubMed.
- A. Bavykina, N. Kolobov, I. S. Khan, J. A. Bau, A. Ramirez and J. Gascon, Chem. Rev., 2020, 120, 8468–8535 CrossRef CAS PubMed.
- K. K. Gangu and S. B. Jonnalagadda, Front. Chem., 2021, 9, 747615 CrossRef CAS PubMed.
- F. Gao, R. Yan, Y. Shu, Q. Cao and L. Zhang, RSC Adv., 2022, 12, 10114–10125 RSC.
- S. Yuan, L. Feng, K. Wang, J. Pang, M. Bosch, C. Lollar, Y. Sun, J. Qin, X. Yang, P. Zhang, Q. Wang, L. Zou, Y. Zhang, L. Zhang, Y. Fang, J. Li and H.-C. Zhou, Adv. Mater., 2018, 30, 1704303 CrossRef PubMed.
- A. Howarth, Y. Liu, P. Li, Z. Li, T. C. Wang, J. T. Hupp and O. K. Farha, Nat. Rev. Mater., 2016, 1, 15018 CrossRef CAS.
- M. Ding, X. Cai and H. Jiang, Chem. Sci., 2019, 10, 10209 RSC.
- D. L. Burnett, R. Oozeerally, R. Pertiwi, T. W. Chamberlain, N. Cherkasov, G. J. Clarkson, Y. K. Krisnandi, V. Degirmenci and R. I. Walton, Chem. Commun., 2019, 55, 11446–11449 RSC.
- D. F. Weng, X. J. Zheng and L. P. Jin, Eur. J. Inorg. Chem., 2006, 4184–4190 CrossRef CAS.
- T. W. Chamberlain, R. V. Perrella, T. M. Oliveira, P. C. de Sousa Filho and R. I. Walton, Chem. – Eur. J., 2022, 28, e202200410 CrossRef CAS PubMed.
- M. I. Breeze, T. W. Chamberlain, G. J. Clarkson, R. P. de Camargo, Y. Wu, J. F. de Lima, F. Millange, O. A. Serra, D. O'Hare and R. I. Walton, CrystEngComm, 2017, 19, 2424–2433 RSC.
- Y. Kitamura, Y. Nakamura, K. Sugimoto, H. Yoshikawa and D. Tanaka, Chem. Commun., 2022, 58, 11426–11429 RSC.
- S. Das, J. Zhang, T. W. Chamberlain, G. J. Clarkson and R. I. Walton, Inorg. Chem., 2022, 61, 18536–18544 CrossRef CAS PubMed.
- S. Feng, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2010, 66, m33 CrossRef CAS PubMed.
- R. A. Zehnder, R. A. Renn, E. Pippin, M. Zeller, K. A. Wheeler, J. A. Carr, N. Fontaine and N. C. McMullen, J. Mol. Struct., 2011, 985, 109–119 CrossRef CAS.
- C. Daiguebonne, N. Kerbellec, K. Bernot, Y. Gerault, A. Deluzet and O. Guillou, Inorg. Chem., 2006, 45, 5399–5406 CrossRef CAS PubMed.
- S. Xie, B. Xie, X. Tang, N. Wang and S. Yue, Z. Anorg. Allg. Chem., 2008, 634, 842–844 CrossRef CAS.
- L. Pan, N. Zheng, Y. Wu, S. Han, R. Yang, X. Huang and J. Li, Inorg. Chem., 2001, 40, 828–830 CrossRef CAS.
- A. K. Cheetham, C. N. R. Rao and R. K. Feller, Chem. Commun., 2006, 4780–4795 RSC.
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, J. Appl. Crystallogr., 2009, 42, 339–341 CrossRef CAS.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Adv., 2015, 71, 3–8 CrossRef PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3–8 Search PubMed.
- B. H. Toby and R. B. Von Dreele, J. Appl. Crystallogr., 2013, 46, 544–549 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2256799, 2280621 and 2280622. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3ce00865g |
| This journal is © The Royal Society of Chemistry 2023 |