DOI:
10.1039/D3CE00745F
(Paper)
CrystEngComm, 2023,
25, 5316-5323
Chalcogen bonding interactions in a series of aromatic selenocyanates†
Received
26th July 2023
, Accepted 30th August 2023
First published on 31st August 2023
Abstract
Selenium atoms in aromatic selenocyanates are characterized by the occurrence of two σ-holes, a stronger one in the prolongation of the NC–Se bond and a weaker one in the prolongation of the Ar–Se bond. The crystal structures of several bis(selenocyanato) derivatives, prepared by a method originally developed for ortho bis-substituted derivatives, illustrate very well this difference, with a short NC–Se⋯NC ChB interaction organizing the molecules into 1D motifs with reduction ratio (RR) values in the range of 0.87–0.91 (RR is defined as the interatomic distance over the van der Waals contact), complemented with Ar–Se⋯NC ChB interactions between antiparallel chains involving the second σ-hole, with larger RR values in the range of 0.92–0.94. Comparison with benzylic selenocyanates shows that this secondary lateral interaction involving the weaker σ-hole is notably enhanced in these novel aromatic compounds. The structure of co-crystals of the para derivative (1,4-diselenocyanatobenzene) with (E)-1,2-di(4-pyridyl)ethylene (bpen) is characterized by an extremely short NC–Se⋯Nbpen distance at 2.693(2) Å, i.e. a RR value of 0.78, complemented with interaction of the second σ-hole with the π system of the double bond of bpen.
Introduction
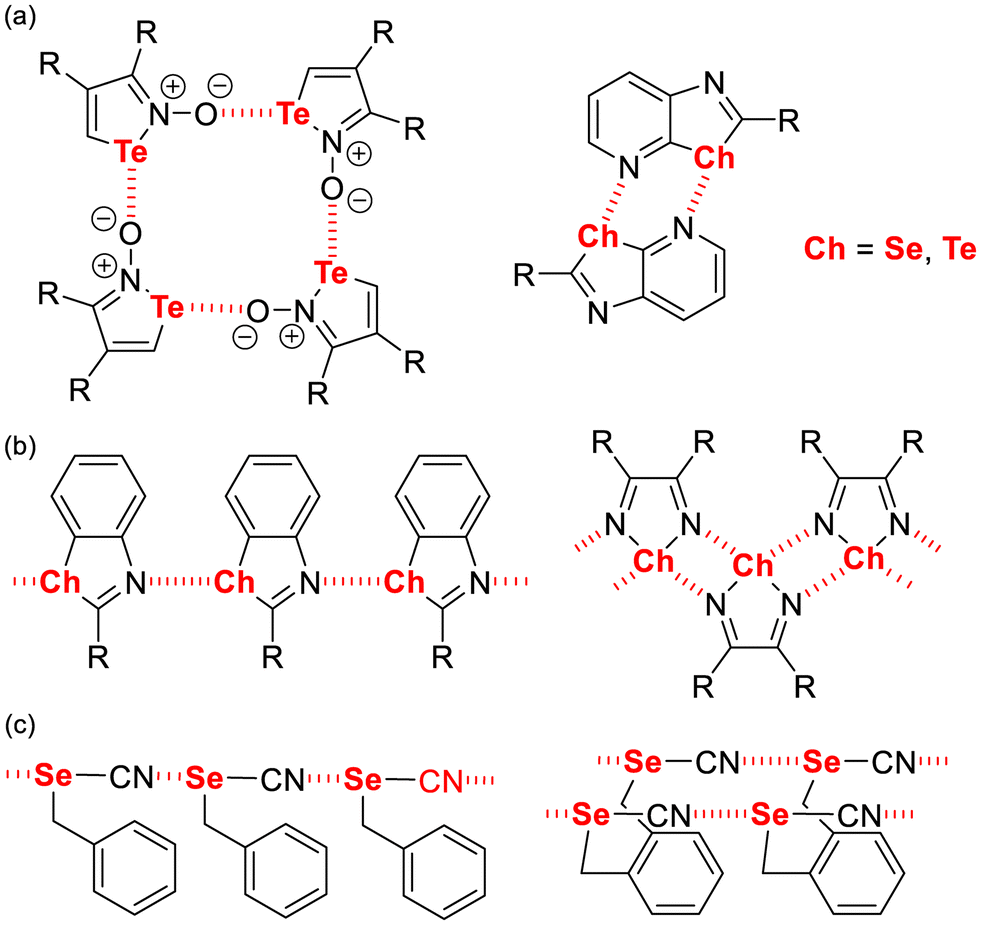
Following the intense efforts of the last thirty years conducted on the investigation of halogen bonding in crystal engineering strategies,1,2 this prototypical σ-hole interaction has by now been found in many other groups of the entire periodic table,3 and particularly in the closely related group 16 of the chalcogens.4 Chalcogen bonding has been accordingly defined as the interaction between a positively polarized chalcogen atom and a Lewis-base (LB)5 and has been the subject of several review papers in the recent years.6 Many reported systems are based on self-complementary molecules, i.e. molecules bearing simultaneously a chalcogen bond (ChB) donor and a ChB acceptor groups, as illustrated in Scheme 1.7–9
 |
| | Scheme 1 Examples of self-associated ChB donors into (a) cyclic motifs, (b and c) infinite 1D motifs. | |
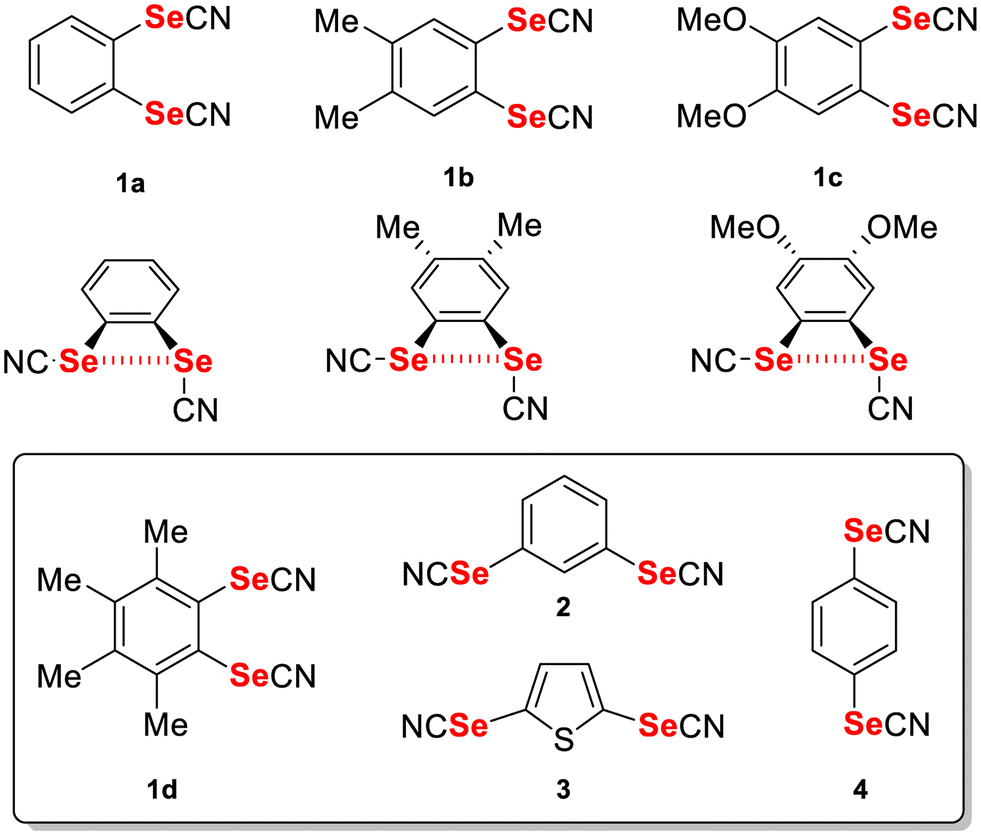
Among them, organic selenocyanates (Scheme 1c) have been shown to provide a recurrent one-dimensional infinite motif where the strongly activated σ-hole in the prolongation of the NC–Se bond interacts with the nitrogen lone pair of a neighbouring molecule.10 Extensive series of benzylic selenocyanates, with one or several SeCN groups, have been reported because of their ease of preparation from the corresponding benzyl halides.11 On the other hand, the number of aromatic selenocyanates is more limited, and restricted essentially to mono selenocyanate derivatives.12 We have recently developed a synthetic procedure toward aromatic bis(selenocyanate) derivatives13 and shown that the ortho-substituted bis(selenocyanato)benzene derivatives such as 1a–c (Scheme 2) crystallise with an intramolecular NC–Se⋯Se ChB. Co-crystallization with (E)-1,2-di(4-pyridyl)ethylene (bpen) led to the formation of rectangular motifs stabilized by NC–Se⋯Nbpen chalcogen bonding interactions, where the face-to-face association of bpen molecules favoured their photocyclization to cyclobutane adducts through single-crystal-to-single-crystal [2 + 2] photodimerization. In this paper, we want to extend further the crystal chemistry of such bis(selenocyanato)benzene derivatives to the more sterically constrained ortho derivative (1d) as well as to the meta (2, 3) and para (4) analogues. In all these situations where the intramolecular ChB is not possible anymore, the crystal structures reveal the effects of the presence of two sizeable σ-holes on each selenium atom, both able to participate in ChB interactions, as demonstrated below. Furthermore, cocrystals of both 2 and 4 with bpen were also obtained and analysed, showing particularly strong NC–Se⋯Nbpen chalcogen bonds.
 |
| | Scheme 2 Known ortho bis(selenocyanate) derivatives and the target compounds 1d, 2–4. | |
Results and discussion
Syntheses
The preparation of simple aromatic selenocyanates is usually based on the reaction of KSeCN with either diazonium salts14 or more electrophilic diaryliodonium salts,15 as reported for para-substituted derivative 4.12c,16 It has been recently reported that the ipso-functionalization of arylboronic acids affords arylselenocyanates in good yields, using either Se powder with TMS–CN,17 or SeO2 and malononitrile.18 We have recently adapted the last procedure, originally reported only for mono boronic acids,19 for the preparation of disubstituted ortho-bis(selenocyanato)benzene derivatives 1a–c by performing the selenocyanation reaction directly on boronic esters such as the ortho-bis(pinacol) boranes.13 As shown in Scheme 3, we have here extended this procedure with four other dibromoaromatic derivatives, which have been transformed through a Miyaura borylation reaction with bis(pinacolato)diborane to give the four corresponding bis(pinacol) boranes 5–8 in 76-87% yields, except with the sterically congested ortho-dibromotetramethylbenzene (7% yield). The following selenocyanation reactions of 5–8 with SeO2 and malononitrile successfully afforded 1d and 2–4 in 60–73% yields.
 |
| | Scheme 3 Synthetic path to 1d, 2–4. i: bis(pinacolato)diborane, KOAc, [(dppf)PdCl2]cat in DMF, 80 °C, 16 h; ii: bis(pinacolato)diborane, KOAc, [Pd(PPh3)4]cat in dioxane, 90 °C, 48 h; iii: SeO2, malononitrile in DMSO, 60 °C, 16 h. | |
Electrostatic surface potentials
The ChB donor ability of 1d, 2–4 has been first evaluated based on the calculated values of the extrema of the electrostatic potential (ESP) surfaces – the larger the positive ESP value, the more important the σ-hole at the chalcogen atom and therefore the ChB donor ability. As shown in Fig. 1 (see calculation details in ESI†), two σ-holes are systematically observed on the Se atoms, the strongest one in the prolongation of the NC–Se bond, the weaker one in the prolongation of the CAr–Se bond. Notable differences are observed between the four compounds. In the sterically constrained tetramethyl benzene derivative 1d, one SeCN moiety partly interacts with the neighbouring Se atom, leading to decreased VS,max values (VS,max is the most positive surface electrostatic potential). On the other hand, in both meta and para derivatives 2 and 4, geometry optimization afforded planar structures with the SeCN moieties lying within the aromatic plane. Under these circumstances, the strongest σ-hole on the Se atoms merges with the electron-depleted area of the neighbouring aromatic H atom, leading to an overall enhancement of the strongest VS,max values up to 41.5 and 38.2 kcal mol−1 in 2 and 4, respectively. The situation differs in the thiophene derivative 3 where the SeCN moieties are rotated out of the aromatic plane. In that respect, the VS,max values found in 3 are representative of a SeCN moiety with limited interaction with the aromatic system. A specific feature here is the proximity of the VS,max values of both σ-holes, at 35.8 kcal mol−1 along the NC–Se bond and 29.6 kcal mol−1 along the CAr–Se bond. Also, comparison with the mono-substituted phenyl selenocyanate shows that the disubstitution (as in 2 and 4) increases both σ-holes on the Se atoms.
 |
| | Fig. 1 Computed electrostatic potential on the 0.002 au isodensity surface of 1d, 2–4 and the model compound PhSeCN. Potential scale ranges from −37.7 kcal mol−1 (in red) to +43.9 kcal mol−1 (in blue). VS,max values for the two σ-holes (in blue) on the selenium atoms are calculated on the 0.001 au isodensity surface and are given in kcal mol−1. | |
A striking difference with the extensive series of benzylic selenocyanates reported to date is the strength of this second σ-hole, which differs from the strongest one by only 10 kcal mol−1, while a 20 kcal mol−1 difference was recurrently observed in the benzylic selenocyanates.10 The question then arises if the directing effect of the strongest σ-hole, clearly identified in the structures of benzylic selenocyanates to favour ⋯NC–(R)Se⋯NC–(R)Se⋯ chain-like motifs,10,11 will be maintained in these aromatic systems.
Crystal structures
Tetramethyl-1,2-bis(selenocyanato)benzene 1d crystallizes in the monoclinic system, space group P21/c, with two crystallographically independent molecules, both in general position. At variance with the other reported ortho-bis(selenocyanato)benzene derivatives 1a–c, the methyl substitution α to the SeCN moieties hinders the planarization of one of these groups and the formation of an intramolecular ChB illustrated in Scheme 2. Indeed, as shown in Fig. 2, the two SeCN groups of each molecule point now out of the benzene ring, with the Se atoms interacting with the electron-rich π system of the other independent molecule, forming dimeric entities with short NC–Se⋯CAr bonds. Structural characteristics of these ChB are collected in Table 1. Other ChB interactions can be identified between the dyads, involving the secondary σ-hole of the Se atoms, and the nitrogen atoms of neighbouring dyads. Surprisingly, these interactions involving the weaker σ-hole of the Se atoms are quite short with the Se2⋯N1 distance for example at 3.149(5) Å, i.e. a reduction ratio (RR) value of 0.91.
 |
| | Fig. 2 Details of the ChB interactions in 1d. ChBs in the prolongation of the NC–Se bonds are indicated as orange dotted lines, those in the prolongation of the CAr–Se bonds as blue dotted lines. | |
Table 1 Structural characteristics of ChBs in 1d. The RR values are calculated on the basis of Bondi's van der Waals radii, giving contact distances of Se⋯C 3.60 Å and Se⋯N 3.45 Å
| ChB |
ChB dist. (Å) |
RR |
ChB ang (°) |
|
i −x, 0.5 + y, 0.5 − z, ii 1 − x, −0.5 + y, 1.5 − z. |
| NC–Se1⋯C18 |
3.583(5) |
0.99 |
154.04(12) |
| NC–Se2⋯C15 |
3.516(6) |
0.98 |
148.40(12) |
| NC–Se2⋯C16 |
3.379(4) |
0.94 |
165.43(12) |
| NC–Se3⋯C5 |
3.630(4) |
1.00 |
168.57(12) |
| NC–Se4⋯C3 |
3.475(6) |
0.96 |
172.75(12) |
| NC–Se4⋯C4 |
3.391(4) |
0.94 |
157.03(12) |
| CAr–Se2⋯N1i |
3.149(5) |
0.91 |
172.71(12) |
| CAr–Se3⋯N4ii |
3.199(4) |
0.93 |
164.95(12) |
| CAr–Se4⋯N3ii |
3.510(7) |
1.02 |
140.89(11) |
The structures of compounds 2 and 3 are very closely related. They both crystallize in the orthorhombic system, space group Pnma, with the molecule containing a crystallographic mirror plane and the SeCN groups pointing out of the aromatic plane, with a torsion angle of 73° in 2 and 57° in 3. As shown in Fig. 3 for the compound 3 (cf. Fig. S1† for 2), the shortest ChB involves the strongest σ-hole in the prolongation of the NC–Se bond, leading to very short Se⋯N distances, 3.128(7) and 3.045(2) Å in 2 and 3, respectively, i.e. RR values of 0.91 and 0.88, and leading to the formation of chain motifs running along the a axis. Lateral CAr–Se⋯N ChB interactions between chains involve the second σ-hole and are only slightly longer (Table 2).
 |
| | Fig. 3 Details of the solid-state organization for 3. ChBs in the prolongation of the NC–Se bonds are indicated as orange dotted lines, those in the prolongation of the CAr–Se bond as blue dotted lines. | |
Table 2 Structural characteristics of ChB interactions in 2 and 3. The RR values are calculated on Se⋯N contact distances of 3.45 Å
| Compound |
ChB |
ChB dist. (Å) |
RR |
ChB ang (°) |
|
i 0.5 + x, y, 1.5 − z, ii 0.5 − x, 1 − y, −0.5 + z, iiix, 0.5 − y, 0.5 + z, iv −x, −0.5 + y, 0.5 − z. |
|
2
|
NC–Se1⋯N1i |
3.128(7) |
0.91 |
168.36(29) |
| CAr–Se1⋯N1ii |
3.223(7) |
0.93 |
173.37(25) |
|
3
|
NC–Se1⋯N1i |
3.045(2) |
0.88 |
175.52(6) |
| CAr–Se1⋯N1ii |
3.169(2) |
0.92 |
167.12(6) |
|
4
|
NC–Se1⋯N1iii |
3.060(2) |
0.87 |
176.09(7) |
| CAr–Se1⋯N1iv |
3.229(6) |
0.94 |
167.33(6) |
The structure of the para-substituted compound 4 has been already reported in the literature12c and has been redetermined here, improving significantly the precision on bond distances and angles (Fig. 4). It crystallizes in the monoclinic system, space group P21/c, with one independent molecule located on inversion centre. At variance with the structure optimization in gas phase (see above in Fig. 1), the SeCN moieties are not coplanar with the aromatic ring but make an angle of 43°. As shown in Fig. 4, the Se atom participates in two ChB interactions, the strongest one in the prolongation of the NC–Se bond and the weaker but sizeable one in the prolongation of the CAr–Se bond (cf.Table 2).
 |
| | Fig. 4 Details of the solid-state organization for 4. ChBs in the prolongation of the NC–Se bonds are indicated as orange dotted lines, those in the prolongation of the CAr–Se bond as blue dotted lines. | |
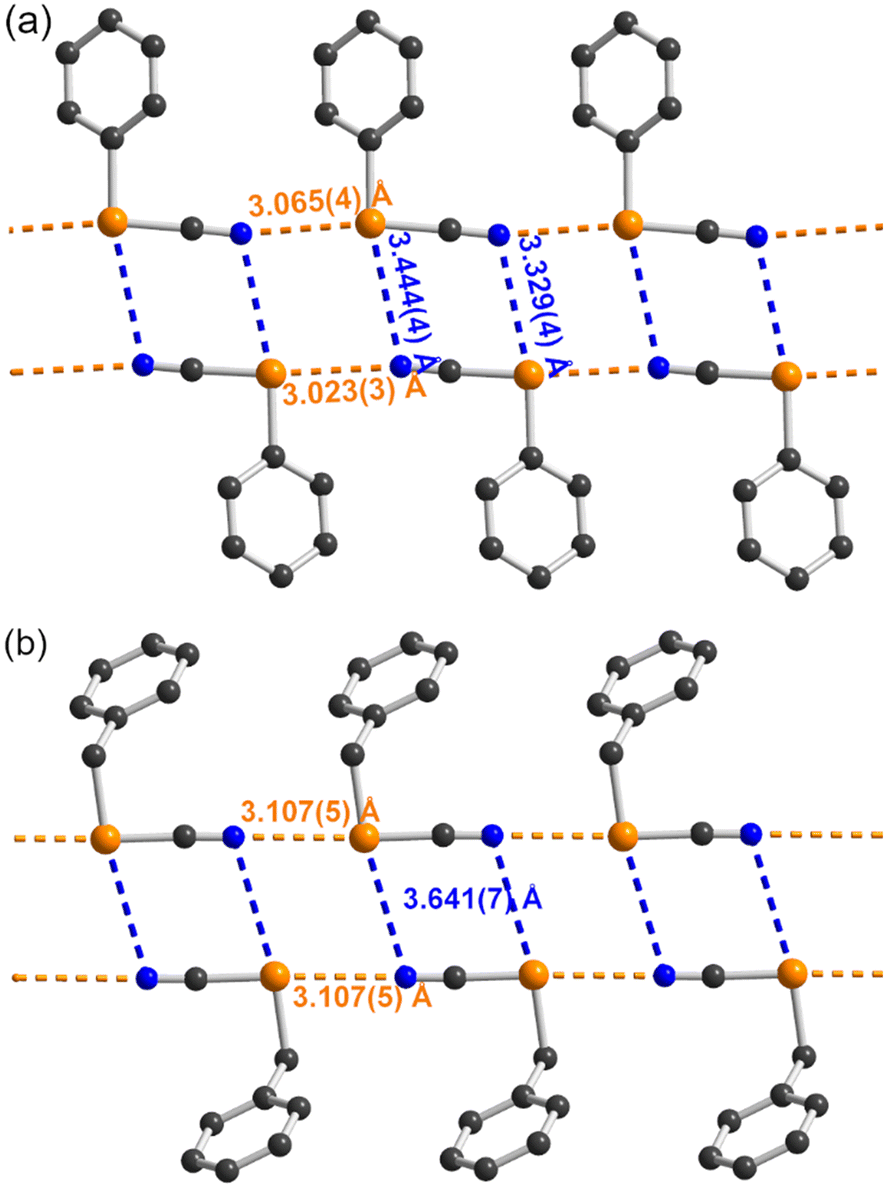
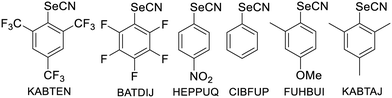
It is interesting at this stage to compare the systems described here with other aromatic selenocyanates as well as the extensive series of benzylic selenocyanates reported earlier. We have collected in Table 3 the structural characteristics of aromatic selenocyanates reported in the literature, excluding those exhibiting intramolecular ChB. In all derivatives as well as in 2–4, the strongest ChB involves the σ-hole in the prolongation of the NC–Se bond, while the secondary CAr–Se⋯N interaction along the CAr–Se bond is weaker (RR = 0.91–1.02) but most often below the van der Waals contact distance, particularly in 2–4 (RR = 0.92–0.93). These observations confirm the analysis of the ESP maps and the correlation between the values of the VS,max extrema and the strength of the ChB interactions associated with the Se⋯N distance. This point is further illustrated by the comparison with benzylic selenocyanates. They are characterized indeed by a comparable σ-hole in the prolongation of the NC–Se bond but a notably weaker σ-hole in the prolongation of the ArCH2–Se bond.10 As a consequence, this secondary ArCH2–Se⋯N interaction has been rarely mentioned, and when present, is observed at much longer distances, above the van der Waals contact distances (3.45 Å). This point is illustrated in Fig. 5 where the structures of the simplest phenyl selenocyanate and benzyl selenocyanate are compared.
Table 3 Details of the ChB interactions around selenium atom in structurally characterized aromatic selenocyanates (excluding those exhibiting intramolecular ChB)
|
|
| Compound |
Along NC–Se bond |
Along CAr–Se bond |
Ref. |
| Interaction |
RR |
Interaction |
RR |
|
iTwo crystallographically independent molecules in the unit cell. iiC6: centroid of the benzene ring. |
| KABTENi |
Se⋯N |
0.83 |
Se⋯N |
0.91 |
12d
|
| Se⋯N |
0.86 |
— |
— |
| BATDIJ |
Se⋯N |
0.86 |
Se⋯N |
1.00 |
12b
|
| HEPPUQ |
Se⋯O |
0.96 |
Se⋯N |
0.97 |
20
|
| CIBFUP |
Se⋯N |
0.89 |
Se⋯N |
0.96 |
12a
|
| FUHBUI |
Se⋯O |
0.94 |
Se⋯N |
1.00 |
21
|
| KABTAJ |
Se⋯(C6)ii |
0.95 |
Se⋯N |
1.02 |
12d
|
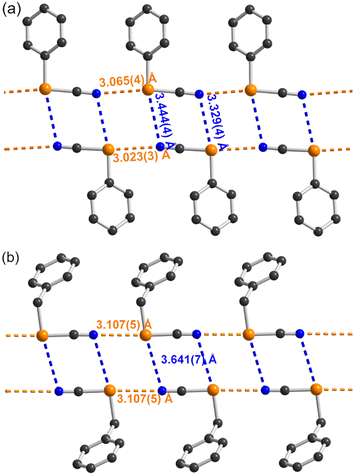 |
| | Fig. 5 Comparison of the ChB interactions between (a) the aromatic PhSeCN selenocyanate (CCDC: CIBFUP)12a and (b) the benzylic BzSeCN selenocyanate (CCDC: CIGGOO).11b ChBs in the prolongation of the NC–Se bonds are indicated as orange dotted lines, those in the prolongation of the CAr–Se bond as blue dotted lines. | |
Adducts with 1,2-bis(4-pyridyl)ethylene (bpen)
The bpen adducts with the ortho derivatives 1a–c have been reported to form tetramolecular associations into rectangular boxes with a short face-to-face interaction between bpen molecules, allowing for [2 + 2] cycloaddition under UV irradiation.13 We considered similar co-crystallization experiments of 1d, 2, 3 and 4 with bpen but could obtain co-crystals only with 2 and 4, as described below.
The meta derivative 2 co-crystallizes with bpen in the triclinic system, space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) , with one molecule 2 in general position, one bpen molecule disordered on two positions on an inversion centre and one bpen molecule, also located on an inversion centre, but with 0.5 occupancy (Fig. 6, see also Fig. S2† for disorder model). This corresponds to a (2)2(bpen_A)(bpen_B)0.5 complex formulation. Molecule bpen_B is not engaged in ChB interactions while molecule bpen_A interacts primarily with two molecules of 2 through two short NC–Se1⋯N3A [2.97(1) Å, RR = 0.86] and NC–Se1⋯N3B [3.03(1) Å, RR = 0.88] chalcogen bonds. The second σ-hole on Se1 as well as the two σ-holes on Se2 interact with nitrogen atoms of neighbouring molecules with slightly weaker interactions (Table 4). The overall structure does not lead to a face-to face organization of bpen molecules which could favour [2 + 2] cycloadditions.
, with one molecule 2 in general position, one bpen molecule disordered on two positions on an inversion centre and one bpen molecule, also located on an inversion centre, but with 0.5 occupancy (Fig. 6, see also Fig. S2† for disorder model). This corresponds to a (2)2(bpen_A)(bpen_B)0.5 complex formulation. Molecule bpen_B is not engaged in ChB interactions while molecule bpen_A interacts primarily with two molecules of 2 through two short NC–Se1⋯N3A [2.97(1) Å, RR = 0.86] and NC–Se1⋯N3B [3.03(1) Å, RR = 0.88] chalcogen bonds. The second σ-hole on Se1 as well as the two σ-holes on Se2 interact with nitrogen atoms of neighbouring molecules with slightly weaker interactions (Table 4). The overall structure does not lead to a face-to face organization of bpen molecules which could favour [2 + 2] cycloadditions.
 |
| | Fig. 6 Details of the ChB interaction in (2)2(bpen_A)(bpen_B)0.5. Molecule bpen_B is not shown. Molecule bpen_A is disordered on inversion centre and only one of the two components is shown (black C atoms with N3A, grey C atoms with N3B). ChB interactions in the prolongation of the (NC)–Se bonds are indicated as orange dotted lines, those in the prolongation of the CAr–Se bond as blue dotted lines. | |
Table 4 Structural characteristics of ChB interactions in (2)2(bpen_A)(bpen_B)0.5. The RR values are calculated on Se⋯N contact distances of 3.45 Å
| ChB |
ChB dist. (Å) |
RR |
ChB ang (°) |
|
i −1 + x, −1 + y, z. ii 1 − x, −y, −z. iiix, −1 + y, z. iv 2 − x, −y, −z. |
| NC–Se1⋯N3Ai |
2.968(13) |
0.86 |
165.9(2) |
| NC–Se1⋯N3Bi |
3.030(13) |
0.88 |
169.2(2) |
| CAr–Se1⋯N2ii |
3.937(12) |
1.14 |
162.13(8) |
| NC–Se2⋯N3Aiii |
3.435(18) |
0.99 |
168.0(2) |
| NC–Se2⋯N3Biii |
3.443(17) |
1.00 |
166.1(2) |
| CAr–Se2⋯N1iv |
3.185(11) |
0.92 |
168.16(8) |
The adduct of the para derivative 4 with bpen crystallizes in the monoclinic system, space group P21/c, with both 4 and bpen on inversion centre, corresponding to a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 stoichiometry (Fig. 7). As in the structure of compound 4 alone (see above), the SeCN moieties are not coplanar with the aromatic ring but make an angle of 47°. A chain-like motif is formed through the interaction of the stronger σ-hole of the Se atom with the pyridinic nitrogen atom, at an extremely short distance of 2.693(2) Å, i.e. a RR value of 0.78. The second σ-hole of the Se atom is engaged in a weaker but directional interaction with the π cloud of the central double bond of a neighbouring bpen molecule, with the shortest Se⋯Cπ distance at 3.531(7) Å, i.e. a RR value of 0.98, with a CAr–Se1⋯Cπ angle at 165.49(6)°. Here again, the solid-state organization does not favour [2 + 2] cycloaddition.
:1 stoichiometry (Fig. 7). As in the structure of compound 4 alone (see above), the SeCN moieties are not coplanar with the aromatic ring but make an angle of 47°. A chain-like motif is formed through the interaction of the stronger σ-hole of the Se atom with the pyridinic nitrogen atom, at an extremely short distance of 2.693(2) Å, i.e. a RR value of 0.78. The second σ-hole of the Se atom is engaged in a weaker but directional interaction with the π cloud of the central double bond of a neighbouring bpen molecule, with the shortest Se⋯Cπ distance at 3.531(7) Å, i.e. a RR value of 0.98, with a CAr–Se1⋯Cπ angle at 165.49(6)°. Here again, the solid-state organization does not favour [2 + 2] cycloaddition.
 |
| | Fig. 7 Details of the chain-like motif in the (4)(bpen) co-crystal. | |
Conclusions
We have demonstrated here that the synthetic access to bis(selenocyanato)benzene derivatives, reported earlier for ortho-substituted compounds, could be successfully extended to meta (2) and para (4) derivatives as well as the α,α′-disubstituted thiophene analogue 3. Electrostatic potential (ESP) calculations show that selenium atoms in such aromatic selenocyanates are characterized by the occurrence of two σ-holes, a stronger one in the prolongation of the NC–Se bond and a weaker one in the prolongation of the Ar–Se bond. Their crystal structures illustrate very well this difference, with a short NC–Se⋯NC ChB interaction organizing the molecules into 1D motifs (RR values in the range of 0.87–0.91), complemented with Ar–Se⋯NC ChB interactions between antiparallel chains involving the second σ-hole, with larger RR values in the range of 0.92–0.94. On the other hand, in the sterically constrained tetramethyl-1,2-bis(selenocyanato)benzene ortho-derivative 1d, an intramolecular ChB interaction is not possible (at variance the other ortho-bis-selenocyanates reported earlier) and both strong σ-holes in the prolongation of the two NC–Se bonds point toward the electron-rich π-cloud of a neighbouring molecule acting here as ChB acceptor. Comparison with benzylic selenocyanates shows that the secondary lateral interaction involving the weaker σ-hole is notably enhanced in the aromatic derivatives. Co-crystals of the para derivative 4 with bpen is characterized by an extremely short NC–Se⋯Nbpen distance at 2.693(2) Å, i.e. a RR value of 0.78, complemented with interaction of the second σ-hole with the π system of the double bond of bpen acting here also as ChB acceptor. Further developments will involve the elaboration of 1,3,5-trisubstituted derivatives and their co-crystals, with neutral as well as anionic (halides) Lewis bases acceptors, expanding this even broader series of ChB systems based on such selenocyanates derivatives.
Experimental section
General considerations
Oxygen- and moisture-sensitive experiments were carried out under a dry oxygen-free argon atmosphere using standard Schlenk techniques. Solvents were dried by standard methods. The NMR spectra were recorded on Bruker spectrometers (300 MHz) referenced to residual solvent signals as internal standards. Elemental analyses were performed at Centre Regional de Mesures Physiques de l'Ouest (CRMPO) with Elementar Vario/Perkin Elmer 2400 series. (E)-1,2-di(4-pyridyl)ethylene (bpen) was obtained from Aldrich and used as received.
General procedure for the Miyaura borylation reactions (derivatives 5, 6, 8)
To bis(pinacolato)diborane (635 mg, 2.5 mmol), anhydrous potassium acetate (589 mg, 6 mmol) and a dibromo derivative (1 mmol), 3.5 ml of anhydrous DMF was added and the mixture was degassed with argon. [1,1′-Bis(diphenylphosphino)ferrocene]palladium(II) dichloride (73 mg, 0.1 mmol) was added and the mixture was stirred in argon atmosphere at 80 °C overnight. After cooling to room temperature, the mixture was poured to a separatory funnel, diluted with water and extracted with AcOEt (50 ml). The aqueous layer was discarded and the organic layer was washed with water (twice) and brine and then dried over MgSO4. Volatiles were removed on a rotovap and the crude product was purified by flash chromatography (petroleum ether:AcOEt = 9:1) to give pure diboronic acid pinacol diesters 5, 6 and 8.
Compound 5.
Yield: 7%. White solid. 1H NMR (300 MHz, CDCl3): δ 2.39 (s, 6H), 2.20 (s, 6H), 1.40 (s, 24H). 11B NMR (96 MHz, CDCl3): δ 31.51.
Compound 6.
Yield: 87%. White solid. 1H NMR (300 MHz, CDCl3): δ 8.31 (td, J = 1.4, 0.7 Hz, 1H), 7.93 (dd, J = 7.4, 1.4 Hz, 2H), 7.40 (td, J = 7.4, 0.8 Hz, 1H), 1.37 (s, 24H). 11B NMR (96 MHz, CDCl3): δ 31.08. 13C NMR (75 MHz, CDCl3): δ 141.24, 137.63, 127.03, 83.72, 24.87.
Compound 8.
Yield: 76%. White solid. 1H NMR (300 MHz, CDCl3): δ 7.83 (s, 4H), 1.37 (s, 24H). 11B NMR (96 MHz, CDCl3): δ 32.09. 13C NMR (75 MHz, CDCl3): δ 133.88, 83.82, 24.87.
Compound 7.
To bis(pinacolato)diborane (635 mg, 2.5 mmol), anhydrous potassium acetate (589 mg, 6 mmol) and 2,5-dibromothiophene (242 mg, 1 mmol) 3.5 ml of anhydrous dioxane was added and the mixture was degassed with argon. Tetrakis(triphenylphosphine)palladium(0) (58 mg, 0.05 mmol) was added and the mixture was stirred in argon atmosphere at 90 °C for 48 h. After cooling to room temperature, the mixture was diluted with DCM and filtered through celite. Solvents were removed on a rotovap and the crude product was purified by flash chromatography (petroleum ether:DCM = 1:1) to give pure 7. Yield: 76%. White solid. 1H NMR (300 MHz, CDCl3): δ 7.69 (s, 2H), 1.36 (s, 24H). 11B NMR (96 MHz, CDCl3): δ 28.96. 13C NMR (75 MHz, CDCl3): δ 137.64, 84.10, 24.75.
General procedure for the synthesis of the diselenocyanatoarenes18
To SeO2 (666 mg, 6 mmol) and malononitrile (132 mg, 2 mmol) 2 ml of anhydrous DMSO was added in argon atmosphere and the mixture was stirred for 30 min at room temperature. To the resulting orange solution diboronic acid pinacol diester 5–8 (1 mmol) was added and the mixture was stirred at 60 °C overnight. After cooling to room temperature water (10 ml) was added and the mixture was extracted with DCM (3 × 20 ml). The organic layers were combined, dried with MgSO4 and volatiles were removed on a rotovap. The crude product was purified by column chromatography (petroleum ether:DCM = 1:1 or petroleum ether:AcOEt = 9:1) to give pure diselenocyanatoarenes 1d, 2–4.
Compound 1d.
Yield: 60%. Pale yellow needles, mp 150–151 °C. 1H NMR (300 MHz, DMSO-d6): δ 2.66 (s, 6H), 2.31 (s, 6H). 13C NMR (75 MHz, DMSO-d6): δ 139.91, 139.88, 131.78, 105.54, 24.13, 18.34. 77Se NMR (76 MHz, DMSO-d6): δ 335.86. Elem. Anal. Calcd. for C12H12N2Se2: C, 42.12; H, 3.54; N, 8.19. Found: C, 41.84; H, 3.37; N, 8.09. Crystals for X-ray diffraction were obtained as pale yellow needles after dissolution in small amount of hot AcOEt and slow cooling to RT and layering with petroleum ether.
Compound 2.
Yield: 73%. Light yellow solid, mp 89–90 °C. 1H NMR (300 MHz, DMSO-d6): δ 8.06 (t, J = 1.7 Hz, 1H), 7.80 (dd, J = 7.9, 1.8 Hz, 2H), 7.52 (dd, J = 8.3, 7.5 Hz, 1H). 13C NMR (75 MHz, DMSO-d6): δ 137.35, 134.23, 131.96, 126.44, 105.55. 77Se NMR (76 MHz, DMSO-d6): δ 341.99. Elem. Anal. Calcd. for C8H4N2Se2: C, 33.59; H, 1.41; N, 9.79. Found: C, 33.34; H, 1.57; N, 9.16. Crystals for X-ray diffraction were obtained as yellow rods from CHCl3 solution by concentration.
Compound 3.
Yield: 60%. Yellow solid, mp 148–153 °C. 1H NMR (300 MHz, DMSO-d6): δ 7.49 (s, 4H). 13C NMR (75 MHz, DMSO-d6): δ 138.61, 124.49, 105.94. 77Se NMR (76 MHz, DMSO-d6): δ 273.47. Elem. Anal. Calcd. for C6H2N2SSe2: C, 24.67; H, 0.69; N, 9.59; S, 10.98. Found: C, 25.05; H, 0.85; N, 9.04; S, 11.15. Crystals for X-ray diffraction were obtained as yellow rods after dissolution in small amount of hot AcOEt and slow cooling to +5 °C.
Compound 4.
Yield: 73%. Pale yellow solid, mp 156–158 °C (reported 156°).12c1H NMR (300 MHz, DMSO-d6): δ 7.77 (s, 4H). 13C NMR (75 MHz, DMSO-d6): δ 134.84, 126.23, 105.49. 77Se NMR (76 MHz, DMSO-d6): δ 334.46. Elem. Anal. Calcd. for C8H4N2Se2: C, 33.59; H, 1.41; N, 9.79. Found: C, 34.01; H, 1.51; N, 9.55. Crystals for X-ray diffraction were obtained as yellow rods after dissolution in small amount of hot AcOEt and slow cooling to RT and layering with petroleum ether.
Co-crystallization experiments with bpen
With 2: in a test tube, 14 mg (0.05 mmol) of 2 and 9 mg (0.05 mmol) of bpen were dissolved in 0.5 ml of CHCl3. On top of the solution, pentane was added slowly to prevent mixing of the solvents and the test tube was sealed. After three days yellow crystals appeared (mp 81–82 °C). Elemental analysis was hampered by the systematic presence of starting materials co-precipitating with the product.
With 4: in a test tube, 14 mg (0.05 mmol) of 4 and 9 mg (0.05 mmol) of bpen were dissolved in 1 ml of acetonitrile and left for slow evaporation of the solvent. After three days light brown crystals appeared (mp 188–190 °C). Elem. Anal. Calcd. for (C8H4N2Se2)·(C12H10N2): C, 51.30; H, 3.01; N, 11.96 found: C, 51.36; H, 2.81; N, 11.86.
Crystallography.
Data collections at RT were performed on an APEXII Bruker-AXS diffractometer equipped with a CCD camera and data collections at 150 K on a D8 VENTURE Bruker AXS diffractometer. Structures were solved by direct methods using the SIR97 program,22 and then refined with full-matrix least-square methods based on F2 (SHELXL-2014/7)23 with the aid of the WINGX program.24 All non-hydrogen atoms were refined with anisotropic atomic displacement parameters. H atoms were finally included in their calculated positions. Details about data collection and solution refinement are given in Table S1.† The crystallographic data for all compounds were deposited in the Cambridge Crystallographic Data Centre (CCDC) with the CCDC no. 2280515–2280520.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
We thank ANR (Paris) for financial support under grant ANR-17-CE07-0025-02. This work has been also supported by the Integrated Development Program of the Gdańsk University of Technology program No. POWR.03.05.00-00.Z044/17 funded by the European Social Fund under the Knowledge Education Development Operational Program. Computations were carried out using the computers of Centre of Informatics Tricity Academic Supercomputer & Network.
Notes and references
- P. Metrangolo and G. Resnati, Chem. – Eur. J., 2001, 7, 2511–2519 CrossRef CAS PubMed.
- P. Metrangolo, H. Neukirch, T. Pilati and G. Resnati, Acc. Chem. Res., 2005, 38, 386–395 CrossRef CAS PubMed.
- G. Cavallo, P. Metrangolo, T. Pilati, G. Resnati and G. Terraneo, Cryst. Growth Des., 2014, 14, 2697–2702 CrossRef CAS.
-
(a) D. B. Werz, R. Gleiter and F. Rominger, J. Am. Chem. Soc., 2002, 124, 10638–10639 CrossRef CAS PubMed;
(b) J. S. Murray, P. Lane, T. Clark and P. Politzer, J. Mol. Model., 2007, 13, 1033–1038 CrossRef CAS PubMed.
- E. Arunan, G. R. Desiraju, R. A. Klein, J. Sadlej, S. Scheiner, I. Alkorta, D. C. Clary, R. H. Crabtree, J. J. Dannenberg, P. Hobza, H. G. Kjaergaard, A. C. Legon, B. Mennucci and D. J. Nesbitt, Pure Appl. Chem., 2011, 83, 1637–1641 CrossRef CAS.
-
(a) L. Vogel, P. Wonner and S. M. Huber, Angew. Chem., Int. Ed., 2019, 58, 1880–1891 CrossRef CAS PubMed;
(b) K. T. Mahmudov, M. N. Kopylovich, M. F. C. Guedes da Silva and A. J. L. Pombeiro, Dalton Trans., 2017, 46, 10121–10138 RSC;
(c) M. Fourmigué and A. Dhaka, Coord. Chem. Rev., 2020, 403, 213084 CrossRef;
(d) N. Biot and D. Bonifaci, Coord. Chem. Rev., 2020, 413, 213243 CrossRef CAS;
(e) L. Brammer, A. Peuronen and T. M. Rosevaere, Acta Crystallogr., Sect. C: Struct. Chem., 2023, 79, 204–216 CrossRef CAS PubMed.
-
(a) P. C. Ho, P. Szydlowski, J. Sinclair, P. J. W. Elder, J. Kubel, C. Gendy, L. M. Lee, H. Jenkins, J. F. Britten, D. R. Morim and I. Vargas-Baca, Nat. Commun., 2016, 7, 11299 CrossRef CAS PubMed;
(b) J. Kübel, P. J. W. Elder, H. A. Jenkins and I. Vargas-Baca, Dalton Trans., 2010, 39, 11126–11128 RSC.
-
(a) A. F. Cozzolino, Q. Yang and I. Vargas-Baca, Cryst. Growth Des., 2010, 10, 4959–4964 CrossRef CAS;
(b) A. F. Cozzolino and I. Vargas-Baca, J. Organomet. Chem., 2007, 692, 2654–2657 CrossRef CAS.
- A. Kremer, A. Fermi, N. Biot, J. Wouters and D. Bonifazi, Chem. – Eur. J., 2016, 22, 5665–5675 CrossRef CAS PubMed.
- O. Jeannin, H.-T. Huynh, A. M. S. Riel and M. Fourmigué, New J. Chem., 2018, 42, 10502–10509 RSC.
-
(a) H.-T. Huynh, O. Jeannin and M. Fourmigué, Chem. Commun., 2017, 53, 8467–8469 RSC;
(b) K. Maartmann-Moe, K. A. Sanderud and J. Songstad, Acta Chem. Scand., Ser. A, 1984, 38, 187–200 CrossRef;
(c) S. L. W. McWhinnie, A. B. Brooks and I. Abrahams, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 1998, 54, 126–128 CrossRef;
(d) A. Lari, R. Gleiter and F. Rominger, Eur. J. Org. Chem., 2009, 2267–2274 CrossRef CAS;
(e) A. M. S. Riel, O. Jeannin, O. B. Berryman and M. Fourmigué, Acta Crystallogr., Sect. B: Struct. Sci., Cryst. Eng. Mater., 2019, 75, 34–38 CrossRef CAS PubMed;
(f) H.-T. Huynh, O. Jeannin and M. Fourmigué, New J. Chem., 2021, 45, 76–84 RSC;
(g) A. M. S. Riel, H.-T. Huynh, O. Jeannin, O. Berryman and M. Fourmigué, Cryst. Growth Des., 2019, 19, 1418–1425 CrossRef CAS.
-
(a) N. A. Barnes, S. M. Godfrey, R. T. A. Halton, I. Mushtaq, S. Parsons, R. G. Pritchard and M. Sadler, Polyhedron, 2007, 26, 1053–1060 CrossRef CAS;
(b) T. M. Klapötke, B. Krumm and K. Polborn, Eur. J. Inorg. Chem., 1999, 1359–1366 CrossRef;
(c) W. S. McDonald and L. D. Pettit, J. Chem. Soc. A, 1970, 2044–2046 RSC;
(d) T. M. Klapötke, B. Krumm, P. Mayer, H. Piotrowski and M. Vogt, Z. Anorg. Allg. Chem., 2003, 629, 1117–1123 CrossRef.
- J. Alfuth, O. Jeannin and M. Fourmigué, Angew. Chem., Int. Ed., 2022, 61, e202206249 CrossRef CAS PubMed.
- P. Nikolaienko and M. Rueping, Chem. – Eur. J., 2016, 22, 2620–2623 CrossRef CAS PubMed.
- Y. Guan and S. D. Townsend, Org. Lett., 2017, 19, 5252–5255 CrossRef CAS PubMed.
- W. H. H. Guenther and M. N. Salzman, Ann. N. Y. Acad. Sci., 1972, 192, 25–43 CrossRef CAS PubMed.
- X. Zhang, X.-B. Huang, Y.-B. Zhou, M.-C. Liu and H.-Y. Wu, Chem. – Eur. J., 2021, 27, 944–948 CrossRef CAS PubMed.
- S. Redon, A. R. O. Kosso, J. Broggi and P. Vanelle, Synthesis, 2019, 51, 3758–3765 CrossRef CAS.
-
(a) K. Durka, K. N. Jarzembska, R. Kaminski, S. Lulinski, J. Serwatowski and K. Wozniak, Cryst. Growth Des., 2013, 13, 4181–4185 CrossRef CAS;
(b) O. Seven, M. Bolte, H.-W. Lerner and M. Wagner, Organometallics, 2014, 33, 1291–1299 CrossRef CAS.
- H. Wang, J. Liu and W. Wang, Phys. Chem. Chem. Phys., 2018, 20, 5227–5234 RSC.
- J. Cui, M. Wei, L. Pang, J. Xiao, C. Gan, J. Guo, C. Xie, Q. Zhu and Y. Huang, Tetrahedron, 2020, 76, 130978 CrossRef CAS.
- A. Altomare, M. C. Burla, M. Camalli, G. Cascarano, C. Giacovazzo, A. Guagliardi, A. G. G. Moliterni, G. Polidori and R. Spagna, J. Appl. Crystallogr., 1999, 32, 115–119 CrossRef CAS.
- G. M. Sheldrick, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3–8 Search PubMed.
- L. G. Farrugia, J. Appl. Crystallogr., 2012, 45, 849–854 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Fig. S1 and S2, details on theoretical calculations and crystallographic data. CCDC 2280515–2280520. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3ce00745f |
|
| This journal is © The Royal Society of Chemistry 2023 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence ab,
Olivier
Jeannin
ab,
Olivier
Jeannin