
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis and characterisation of new coordination polymers by combining 2-pyridyl oximes or alcohols with functionalised terephthalic acid analogues†
Foteini
Dimakopoulou
a,
Constantinos G.
Efthymiou
a,
Andreas
Kourtellaris
b,
Ciaran
O'Malley‡
a,
Lamis
Alaa Eldin Refat
 a,
Anastasios
Tasiopoulos
b,
Patrick
McArdle
a and
Constantina
Papatriantafyllopoulou
*a
a,
Anastasios
Tasiopoulos
b,
Patrick
McArdle
a and
Constantina
Papatriantafyllopoulou
*a
aSchool of Biological and Chemical Sciences, College of Science and Engineering, University of Galway, H91 TK33 Galway, Ireland. E-mail: constantina.papatriantafyllopo@universityofgalway.ie; Tel: +353 91 493462
bDepartment of Chemistry, University of Cyprus, 1678 Nicosia, Cyprus
First published on 4th October 2023
Abstract
Isolation of mixed-ligand compounds has attracted considerable research interest as the synergy between ligands results in species with interesting technological, environmental and biomedical applications. In this study, initial employment of 2-pyridyl oximes or 2-pyridyl alcohols in combination with 2-hydroxyterephthalic acid (H3MHBDC) and 2,5-dihydroxyterephthalic acid (H4DHBDC) has yielded a new family of 1D coordination polymers and a metal cluster, namely [M(H2pyaox)2(H2DHBDC)]n (M = Zn, 1; Mn, 2), [Co3(mpko)4(Hmpko)2](H2DHBDC)2·2DMF (3·2DMF), [M(Hhmp)2(H2DHBDC)]n·DMF (M = Ni, 4·DMF; Zn, 5·DMF), [Ni(H2pyaox)2(HMHBDC)]n·2DMF (6·2DMF), and [Zn(Hmpko)2(HMHBDC)]n·DMF (7·DMF), where H2pyaox = pyridine-2-amidoxime and Hmpko = 2-methyl pyridyl ketoxime. 1–7 are the first compounds bearing a 2-pyridyl oxime or 2-pyridinemethanol (Hhmp) with H3MHBDC or H4DHBDC in their neutral or anionic form. Notably, 4 exhibits a high capacity for Co2+ uptake (900 mg Co2+ g−14), which can be attributed to the presence of free coordination sites in the carboxylate linker.
Introduction
The synthesis and characterisation of novel coordination polymers (CPs) remain highly relevant due to their wide range of technological, biomedical, industrial, and environmental applications.1–6 CPs consist of mononuclear or low nuclearity inorganic units that are linked by organic ligands through coordination bonds, forming networks with well-defined crystalline structures. From a topological point of view, CPs can be classified as chain (1D) and framework (2D/3D) structures, while the structures of 1D chains can be described as linear, zigzag, ribbon, ladder, or helical.1–3,7,8 It is worth mentioning that as the dimensionality of CPs increases, induced porosity can be combined with other physical properties (e.g. magnetism, photoluminescence, etc.), leading to the development of hybrid multifunctional materials. Furthermore, a synergistic effect between two different properties can be observed, which often enhances the performance of such materials. For instance, CPs that combine porosity with magnetism and/or photoluminescence can be used in theranostic applications for the diagnosis and treatment of diseases, as MRI contrast agents and magnetism-based sensors, and in the medical device industry,9–13 while in the case of photoluminescence, porosity can affect the emission properties of a photoluminescent material, allowing for tailored emission wavelengths, intensities, and lifetimes.14–16A class of 2D/3D CPs that has attracted immense research interest in the last two decades is metal–organic frameworks (MOFs), which are crystalline porous materials built from inorganic secondary building units (SBUs) that are connected through polytopic organic linkers.5,17–19 MOFs have expanded the range of applications of CPs to include gas storage, gas separation, imaging, energy storage and conversion, optics, heterogeneous catalysis, and other fields.3–5,20,21 The discovery of MOFs in 1995 was a significant milestone in the field of CPs and materials chemistry; however, the use of 1D chains in applications remains of special interest because they can offer advantages compared to their framework analogues. This is merely due to their simple topology8 and repeating unit diversity, while their relatively high solubility facilitates their processability in industrial applications and manufacturing.
The configuration and properties of 1D chains are strongly influenced by the nature of the metal ions and the organic linkers present in their structure. For example, 1D CPs of paramagnetic metal ions can exhibit single chain magnetism (SCM) behavior, i.e. they can display slow relaxation of magnetization due to strong intrachain exchange interactions between high spin structural building units along the chain.22–25 SCM is an excellent candidate for applications in high-density information storage, molecular spintronics, quantum computation, etc.26,27 In addition, 1D chains can form multidimensional supramolecular networks through non-covalent (interchain) interactions, including hydrogen bonding and π–π stacking; this yields flexible porous materials that, in combination with their high surface area, are suitable for use in catalysis and sensing applications, as molecular sieves and separation membranes, drug carriers, etc.1–3,8
The versatility of structures and applications of 1D CPs, as well as the tunability of their properties, necessitates the development of efficient synthetic approaches that will provide an element of control, leading to compounds with tailored structures, porosity, and functionality. To this end, numerous synthetic approaches towards these species have been developed, including one-pot processes, two-pot processes, etc.;28 in all cases, the identity of the isolated products is affected by the choice of metals and ligands, as well as reaction conditions such as the metal–ligand ratio, counterions, solvent, pH environment, and temperature. A recently developed approach involves the incorporation of two organic ligands, resulting in mixed-ligand or multi-variant species.29–32 The latter had a profound impact on the field of CPs as these materials display properties that may not arise from the linear combination of their discrete components, but rather result from the synergy between different ligands.33 Furthermore, the inclusion of diverse ligands in the structure creates additional opportunities for functionalization and customization of its properties.
In the context of the mixed-ligand approach, our group has been investigating the use of 2-pyridyl oximes or alcohols in combination with polycarboxylates (e.g. benzene-1,4-dicarboxylic acid, benzene-1,3,5-tricarboxylic acid, benzene-1,2,4,5-tetracarboxylic acid, etc.) to isolate mixed-ligand CPs and MOFs. These ligands can bridge a large number of metal ions, promoting the presence of ferromagnetic interactions; as a result, multifunctional species that combine porosity with interesting magnetic properties can be isolated. Preliminary results indicate that new CPs or MOFs can be synthesized using ligands such as pyridine-2-amidoxime, 2-methyl pyridyl ketoxime, 2-pyridinemethanol, and pyridine-2,6-dimethanol.34–37 Some of the isolated MOFs have novel framework topology and demonstrate good performance in metal ion encapsulation.34
With the above in mind, we wished to investigate further this approach for the synthesis of mixed-ligand CPs and MOFs, and in particular we were prompted to employ organic linkers with additional functional groups beyond carboxylates. To this end, we introduced 2-hydroxyterephthalic acid (H3MHBDC) and 2,5-dihydroxyterephthalic acid (H4DHBDC, Scheme 1) to the reaction system aiming at investigating the effect of the OH groups on the structures and physical properties of the isolated compounds. Both linkers have been used in the past for the synthesis of CPs and MOFs;38–42 however, their combination with oximic or alkoxy-based ligands has not yet been explored. Herein, we report the synthesis, structural characterisation, and physical properties of seven new compounds, including a metal cluster and six CPs, which represent the first examples that combine H3MHBDC or H4DHBDC with 2-pyridyl oximes or Hhmp.
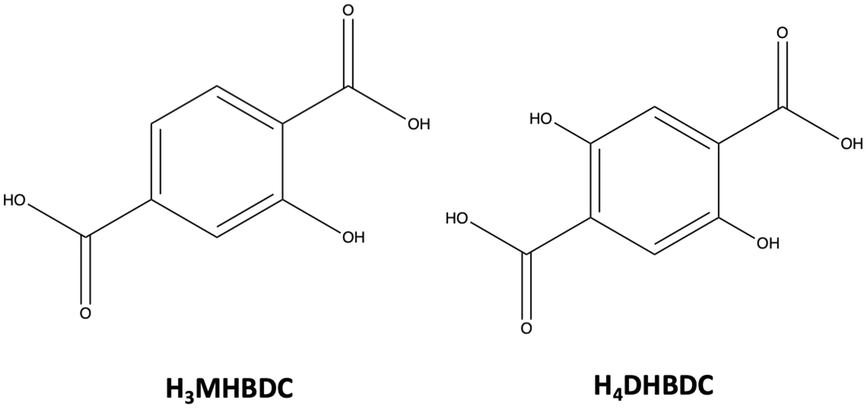 | ||
| Scheme 1 Schematic representation of 2-hydroxyterephthalic acid (H3MHBDC, left) and 2,5-dihydroxyterephthalic acid (H4DHBDC). | ||
Experimental
All manipulations were performed under aerobic conditions using materials (reagent grade) and solvents as received. WARNING: perchlorate and nitrate salts are potentially explosive; such compounds should be used in small quantities and treated with the utmost care at all times.Elemental analysis (C, H, N) was performed at the in-house facilities of the University of Galway, School of Biological and Chemical Sciences. IR spectra (4000–400 cm−1) were recorded on a Perkin-Elmer Spectrum 400 FT-IR spectrometer. TGA experiments were performed on an STA625 thermal analyzer from Rheometric Scientific (Piscataway, New Jersey). The heating rate was kept constant at 10 °C min−1, and all runs were carried out between 20 and 600 °C. The measurements were performed in open aluminum crucibles, nitrogen was purged in ambient mode, and calibration was performed using an indium standard.
Synthesis of [Zn(H2pyaox)2(H2DHBDC)]n·3DMF (1·3DMF)
Zn(NO3)2·6H2O (0.057 g, 0.30 mmol) and H2pyaox (0.042 g, 0.30 mmol) were dissolved in DMF (5 ml). The resultant solution was placed in an oven and heated at 110 °C for 1 h. Then, H4DHBDC (0.013 g, 0.06 mmol) was added and the vial was placed in the oven at 80 °C. After 5 days, yellow block crystals were formed. The crystals were collected with filtration, washed with cold MeCN (2 ml) and Et2O (2 × 5 ml), and dried in air. Yield 50%. Anal. calcd (found) for 1·3DMF: C, 46.13 (46.51%); H, 5.21 (5.39%); N, 16.70 (17.06%). IR data: ν (cm−1) = 3424 w, 3327 w, 3277 w, 3195 w, 3058 w, 2928 w, 2853 w, 2566 w, 1656 s, 1627 w, 1597 m, 1577 m, 1488 m, 1436 s, 1409 s, 1387 m, 1355 w, 1331 s, 1301 w, 1236 s, 1168 w, 1091 s, 1060 w, 1019 s, 1000 m, 973 w, 910 w, 858 w, 840 w, 812 w, 780 s, 749 w, 689 m, 659 m.Synthesis of [Mn(H2pyaox)2(H2DHBDC)]n·2DMF (2·2DMF)
Synthesis of [Co3(mpko)4(Hmpko)2](H2DHBDC)2·2DMF (3·2DMF)
Co(CH3CO2)2·4H2O (0.049 g, 0.20 mmol) and Hmpko (0.073 g, 0.53 mmol) were dissolved in a mixture of H2O/DMF (5/5 ml) in a scintillation vial. The resultant solution was placed in an oven at 130 °C for 1 h. H4DHBDC (0.020 g, 0.10 mmol) was added and the vial was returned to the oven. After three days, X-ray quality dark orange needles started to form. The crystals were collected by filtration, washed with MeCN (2 ml) and Et2O (2 × 5 ml) and dried in air. Yield: 65%. Anal. calcd for 3·2DMF: C, 26.24 (25.99%); H, 2.20 (2.44%); N, 5.40 (5.21%). IR data: 3376 m, 3230 w, 3063 m, 1777 w, 1668 w, 1600 s, 1577 w, 1527 m, 1475 s, 1431 s, 1377 w, 1361 s, 1325 w, 1299 w, 1246 m, 1177 s, 1112 s, 1084 m, 1053 w, 1023 w, 998 w, 894 w, 854 w, 816 w, 780 s, 750 w, 691 s, 655 m.Synthesis of [Ni(Hhmp)2(H2DHBDC)]n·DMF (4·DMF)
Synthesis of [Zn(Hhmp)2(H2DHBDC)]n·DMF (5·DMF)
Synthesis of [Ni(H2pyaox)2(HMHBDC)]n·2DMF (6·2DMF)
H3MHBDC (0.018 g, 0.10 mmol) was dissolved in a mixture of H2O/DMF (15/15 ml) in a scintillation vial. The resulting solution was sonicated and then placed in an oven at 80 °C. H2pyaox (0.028 g, 0.20 mmol) and CH3ONa (0.011 g, 0.20 mmol) were added to the solution which was then stirred for 30 min. Ni(ClO4)2·6H2O (0.073 g, 0.20 mmol) was added to the solution and the vial was placed in the oven at 100 °C for two days, after which time, the formation of X-ray quality, purple, rhombic crystals was observed. The crystals were collected by filtration, washed with MeCN (2 ml) and Et2O (2 × 5 ml) and dried in air. Yield: 60%. Anal. calcd (found) for 6·3DMF: C, 47.56 (47.81%); H, 5.37 (5.08%); N, 17.21 (16.91%). IR data: 3444 w, 3380 w, 3320 w, 3277 w, 3211 w, 3071 w, 2928 w, 2888 w, 2741 w, 2579 w, 1655 s, 1600 m, 1571 w, 1544 m, 1489 m, 1431 m, 1382 s, 1397 s, 1355 s, 1334 s, 1262 w, 1288 w, 1242 m, 1222 w, 1179 w, 1162 w, 1144 w, 1091 m, 1027 s, 953 w, 931 w, 905 w, 847 m, 824 m, 788 s, 774 s, 751 m, 691 m, 659 m.Synthesis of [Zn(Hmpko)2(HMHBDC)]n·DMF (7·DMF)
Hmpko (0.027 g, 0.20 mmol) and H3MHBDC (0.018 g, 0.10 mmol) were dissolved in DMF (5 ml) in a scintillation vial. Zn(CH3CO2)2·2H2O (0.022 g, 0.10 mmol) was added to the solution and the vial was placed in an oven at 110 °C for one day. The resultant solution turned cloudy, filtered and placed back in the oven at 110 °C. After 5 days, X-ray quality yellow block crystals were formed. Yield: 55%. Anal. calcd (found): C, 50.82 (51.02%); H, 4.61 (4.17%); N, 11.85 (12.03%). IR data: 3075 w, 2930 w, 2522 w, 1846 w, 1663 s, 1597 m, 1551 s, 1493 w, 1478 w, 1433 w, 1402 m, 1373 w, 1359 w, 1295 m, 1242 s, 1222 w, 1140 m, 1096 m, 1063 m, 1047 m, 1014 w, 972 w, 954 w, 899 w, 862 w, 826 m, 786 s, 778 s, 749 w, 701 w, 686 m, 658 m.X-ray crystallography
Single crystal diffraction data for 1–3 and 5 were collected on an Oxford-Diffraction SuperNova A diffractometer using Cu Kα radiation (λ = 1.54184 Å). Crystallographic data for 4, 6, and 7 were collected on an Oxford Diffraction Xcalibur CCD diffractometer using Mo Kα radiation (λ = 0.71073 Å). Empirical absorption corrections were applied using the CrysAlis RED software, based on multi-scan symmetry-related measurements. The structures were solved using SHELXT,43 embedded in the OSCAIL software.44 Non-H atoms were treated anisotropically, whereas hydrogen atoms of aromatic rings were placed in calculated ideal positions and refined as riding on their respective carbon atoms. Molecular graphics were produced using DIAMOND.45 The program SQUEEZE,46 a part of the PLATON package crystallographic software, was used to remove the contribution of the highly disordered DMF molecules in 2, 3, and 5.Table S1† lists the unit cell data and structure refinement details for 1–7.
Metal uptake kinetic and thermodynamic studies
The metal adsorption capacity of compound 4 was investigated as follows: Co(NO3)2·6H2O was added to a glass vial containing 10 mL of EtOH and stirred until all solids were dissolved. Then, 0.200 g of solid 4 was added, and the mixture was left stirring at room temperature. For the kinetic study, small volumes of aliquots were taken at designated time intervals and centrifuged, and the metal content in the supernatant solution was determined by spectroscopic (UV-vis) techniques. For the thermodynamic study, the same procedure was repeated with varying 4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :metal ratios. The mixture was stirred for 40 hours and filtered, and the filtrate was analyzed for its metal content. To further confirm the metal uptake capacity of 4, EDX studies were conducted on the crystalline material of 4 before and after the metal uptake.
:metal ratios. The mixture was stirred for 40 hours and filtered, and the filtrate was analyzed for its metal content. To further confirm the metal uptake capacity of 4, EDX studies were conducted on the crystalline material of 4 before and after the metal uptake.
Results and discussion
Synthesis
Several experiments were performed to thoroughly study the effect of synthetic parameters such as the presence/absence or type of base, metal ratio of reactants, metal sources, etc. To ensure coordination of both the oximic or alkoxy ligand and the dicarboxylic acid, the solution containing the metal and the oxime or Hhmp was left to stir or placed in an oven for at least 1 hour before adding the carboxylic acid. It is worth noting that the experimental ratio that produced most of the mixed-ligand species was either 2:2:1 or 1:2:1 (metal source/oxime or Hhmp/carboxylic acid).
The reaction between a metal salt (Zn(NO3)2·6H2O, 1; Mn(CH3CO2)2·4H2O or Mn(NO3)2·4H2O, 2), H2pyaox and H4DHBCD in DMF at high temperature provided access to crystals of [M(H2pyaox)2(H2DHBDC)]n·DMF (M = ZnII, 1·DMF; MnII, 2·DMF) in good yield. Using a similar synthetic approach to the one that yielded compounds 1·DMF and 2·DMF, but by employing Hmpko instead of H2pyaox, the trinuclear cluster [CoIII2CoII(mpko)4(Hmpko)2](H2DHBDC)2·2DMF (3·2DMF) was isolated. The stoichiometric equations of the reactions that led to the formation of 1–3 are shown in eqn (1) and (2):
| M(NO3)2·xH2O + 2H2pyaox + H4DHBDC → [M(H2pyaox)2(H2DHBDC)]n + 2HNO3 + xH2O(M = ZnII, 1; MnII, 2) | (1) |
| 3Co(CH3CO2)2·4H2O + 6Hmpko + 2H4DHBDC + 1/2O2 → [CoIII2CoII(mpko)4(Hmpko)2](H2DHBDC)2 + 6CH3CO2H + 13H2O (3) | (2) |
Subsequently, the impact of the functional group of the chelate ligand on the structural features of the isolated products was investigated. To achieve this goal, oximic ligands were replaced with Hhmp in the reaction mixture. In particular, The reaction of a metal salt (Ni(CH3CO2)2·4H2O, 4; Zn(ClO4)2·6H2O or Zn(CH3CO2)2·6H2O or Zn(NO3)2·6H2O, 5), Hhmp and H4DHBCD in DMF at high temperature yielded crystals of [M(Hhmp)2(H2DHBDC)]n·DMF (M = NiII, 4·DMF; ZnII, 5·DMF) as shown in eqn (3):
| M(CH3CO2)2·xH2O + 2Hhmp + H4DHBDC → [M(Hhmp)2(H2DHBDC)]n + 2CH3CO2H + xH2O(M = NiII, 4; ZnII, 5) | (3) |
This reaction system favours the formation of 1D chains, which is also the case when using H3MHBDC instead of H4DHBDC. Hence, the reaction mixture of Ni(ClO4)2·6H2O, H2pyaox and H3MHBDC in DMF yielded the 1D chain [Ni(H2pyaox)2(HMHBDC)]n·DMF (6·DMF), while the use of Hmpko provided access to [Zn(Hmpko)2(HMHBDC)]n·DMF (7·DMF) (eqn (4) and (5)).
| Ni(ClO4)2·6H2O + 2H2pyaox + H3MHBDC + DMF → [Ni(H2pyaox)2(HMHBDC)]n·DMF + 2HClO4 + 6H2O (6·DMF) | (4) |
| Zn(CH3CO2)2·2H2O + 2Hmpko + H3MHBDC + DMF → [Zn(Hmpko)2(HMHBDC)]n·DMF + 2CH3COOH + 2H2O (7·DMF) | (5) |
It is worth mentioning that the metal source used does not affect the identity of the isolated products. Acetate salts of metals speed up the synthesis process, likely by acting as weak bases. When the same experiments were conducted using metal nitrates, the same product was isolated, but the crystallisation process lasted longer. The addition of a strong base in the reaction mixture did not affect the identity of the product or resulted in amorphous precipitates that could not be further characterized.
The same synthesis method was followed for the aforementioned CPs, which is in situ with the experimental ratio of 1:1 between the metal and chelate linker, respectively. The dicarboxylate analogue was added in an amount equal to or less than half of the other two reactants. When using extreme reagent ratios that include an excess of oxime/Hhmp or dicarboxylate linker, compounds were formed with only one linker present, as indicated by IR analysis (Fig. S20†).
Description of structures
Representations of the molecular structures of 1–7 are shown in Fig. 1–7. The oxidation state of the metal ions and the protonation level of the ligands were confirmed by considering charge balance and bond-valence sum (BVS) calculations. | ||
| Fig. 1 Representation of the repeating unit (top) and a part of the 1D zigzag chain of 1·3DMF (bottom). Color code: ZnII, yellow; O, red; N, blue; C, grey. The hydrogen atoms and lattice DMF molecules are omitted for clarity. | ||
Compounds 1·3DMF and 2·2DMF have similar structures with their main difference being the type of the metal ion (1, ZnII; 2, MnII) present in the structure. Therefore, only the structure of 1·3DMF will be discussed in detail. 1·3DMF crystallizes in the triclinic space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) and its structure (Fig. 1) is based on the repeating unit [Zn(H2pyaox)2(H2DHBDC)] and lattice DMF molecules. The neighbouring units are linked through H2DHBDC2− ligands, resulting in the formation of 1D zigzag chains. The coordination sphere of ZnII is completed by two terminal, chelate H2pyaox ligands. Each metal ion is six coordinated with slightly distorted octahedral geometry due to the relatively small bite angle of the H2pyaox ligand (N1–Zn1–N2 = 74.3(2)°).
and its structure (Fig. 1) is based on the repeating unit [Zn(H2pyaox)2(H2DHBDC)] and lattice DMF molecules. The neighbouring units are linked through H2DHBDC2− ligands, resulting in the formation of 1D zigzag chains. The coordination sphere of ZnII is completed by two terminal, chelate H2pyaox ligands. Each metal ion is six coordinated with slightly distorted octahedral geometry due to the relatively small bite angle of the H2pyaox ligand (N1–Zn1–N2 = 74.3(2)°).
Strong inter- and intrachain interactions stabilise the crystal structure of 1·3DMF. In particular, intrachain hydrogen bonding interactions are formed between the OH groups of the H2DHBDC2− ligand (O8; donor) and the carboxylate group of the same ligand (O5; acceptor), as well as between the oximic group (O1; donor) and the carboxylate group of the H2DHBDC2− ion (O5; acceptor). Furthermore, the –NH2 group (N6, donor) of the oximic ligand forms strong interchain hydrogen bonds with the oximic groups (O1, O7; acceptors) of neighboring chains. Hence, O1 acts as both a donor and acceptor in intra- and interchain hydrogen bonding interactions. Information regarding distances and angles of hydrogen bonding interactions in 1·3DMF can be found in Table S2.†
Compound 3 crystallises in the triclinic space group P. Its crystal structure consists of [CoIII2CoII(mpko)4(Hmpko)2]4+ cations, H2DHBDC2− counterions, and DMF solvate molecules (Fig. 2). The cationic complex is composed of two Co(III) ions and one Co(II) ion that are held together through four anionic η1:η1:η1:μ mpko− and two neutral η1:η1:η1:μ Hmpko ligands. The structural core of the complex consists of a linear [Co3(NO)6]8+ unit, where the six diatomic NO bridges originate from six different oximate groups. The CoIII ions are external, while the CoII is internal. The CoIII ions have distorted octahedral coordination spheres, completed by six N donor atoms, three pyridyl, and three oximic groups in a facial (fac-) arrangement.
 | ||
| Fig. 2 Representation of the molecular structure (top) and the metal core of compound 3·2DMF (bottom). Color code: CoII, magenta pink; CoIII, dark red; O, red; N, blue; C, grey. The hydrogen atoms, lattice DMF molecules and DHBDC4− ions are omitted for clarity. | ||
The coordination geometry around CoII is octahedral, with the two Co–O bonds corresponding to the neutral Hmpko (O1, O6) being 2.13 Å and the remaining, belonging to the anionic mpko-ligands, being below 2.09 Å. The intracationic intermetallic distances are Co1⋯Co2 = 3.450(1) Å and Co1⋯Co3 = 6.880(1) Å. Upon close inspection, there are no significant inter- or intracationic hydrogen bonding interactions, and the cations are well-isolated, with the metal⋯metal separation between the neighboring Co3 units being 8.168(2) Å (Co3⋯Co3).
Compounds 4·DMF and 5·DMF are isostructural differing merely on the type of the metal ion (4, NiII; 5, ZnII) present in the structure; hence, only the structure of 4·DMF will be discussed in detail. 4·DMF crystallizes in the triclinic space group P and its structure is based on the repeating unit [Ni(Hhmp)2(H2DHBDC)] and lattice DMF molecules (Fig. 3). Hhmp is neutral and adopts a terminal chelating coordination mode. The neighbouring units are held together through H2DHBDC2− ligands, forming 1D zigzag chains. The NiII ion is six coordinated with slightly distorted octahedral geometry due to the relatively small bite angle of the Hhmp ligand (N2–Ni1–O7 = 79.5(2)°).
 | ||
| Fig. 3 Representation of the repeating unit (top) and a part of the 1D zigzag chain of 4·DMF(bottom). Color code: NiII, green; O, red; N, blue; C, grey. The hydrogen atoms and lattice DMF molecules are omitted for clarity. | ||
The crystal structure of compound 4·DMF is stabilized by strong interactions, which result in the formation of a 3D-supramolecular framework (Fig. 4). In particular, intrachain hydrogen bonding interactions occur between the OH groups of the H2DHBDC2− ligand (O3, O6; donors) and the carboxylate groups of the same ligand (O2, O5; acceptors), as well as between the OH group of Hhmp (O7, O8; donors) and the carboxylate group of the H2DHBDC2− ion (O1, O5; acceptors). Information about the distances and angles in these interactions can be found in Table S3.† The aromatic rings of the Hhmp ligands of neighbouring chains in 4·DMF interact further through strong π–π stacking interactions; the distance between the centroids is 3.6 Å.
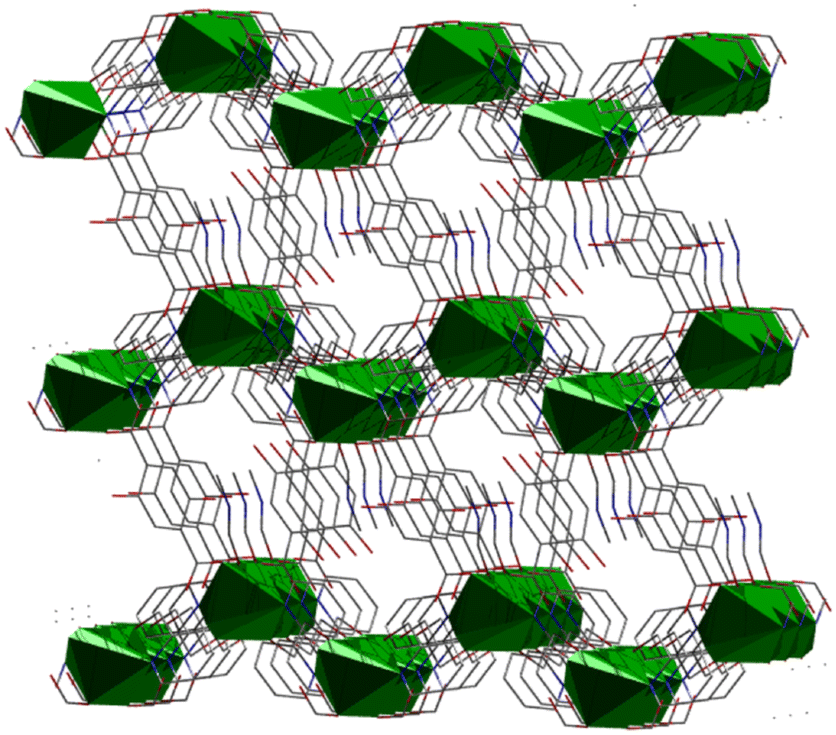 | ||
| Fig. 4 Representation of the 3D network of 4·DMF; green polyhedra: coordination spheres of NiII. | ||
The supramolecular network of compound 4·DMF is composed of parallel zigzag two-dimensional layers, which are separated by a maximum distance of 10.640 Å. The arrangement of these layers creates a system of interconnected channels, each of which accommodates/encapsulates one DMF molecule (Fig. 5, top). Upon the removal of DMF, the channel network is capable of enclosing small host molecules such as metal salts, solvent molecules, etc. The topological arrangement of 4 can be described using two distinct interconnected nodes, which can form 1D zigzag chains. One node corresponds to the inorganic SBU {Ni(Hhmp)2}2+, and the other node corresponds to the organic linker H2DHBDC. The connectivity is (2,2), with each node connected to two neighboring nodes, forming a 2-c net of 2C1 topology (Fig. 5, bottom).47,484·DMF undergoes decomposition in multiple steps (Fig. S1†), as expected from the presence of different ligands in its structure. A mass loss of ∼9% occurs below 150 °C, which is attributed to lattice DMF. A plateau is observed from 300–350 °C, which is then followed by a step at 375 °C corresponding to the breakdown of the polymer.
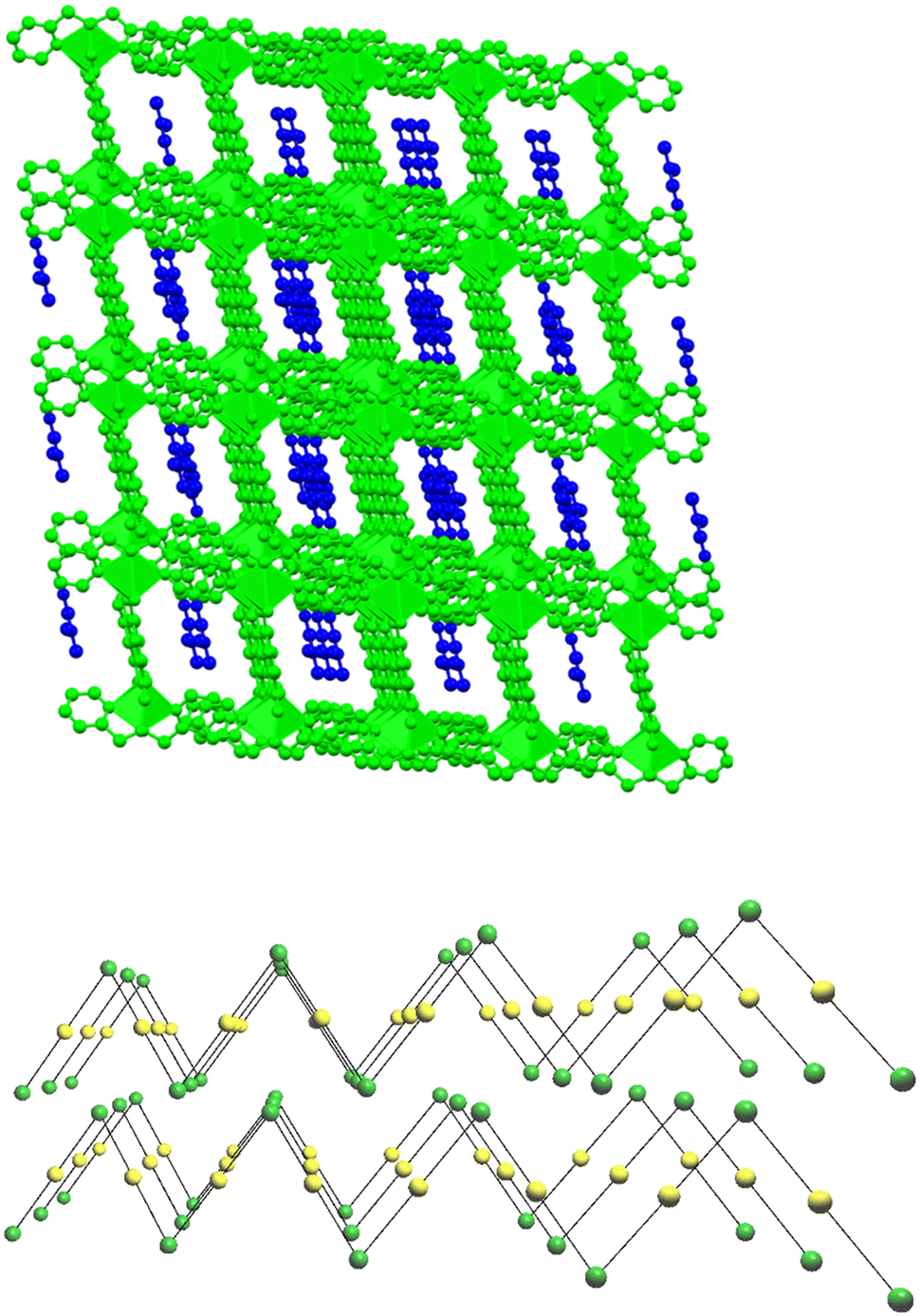 | ||
| Fig. 5 Top: The supramolecular network of 4 (green), along with the system of channels where DMF guest molecules (blue) are enclosed. The Ni metal centers of the polymers are represented as polyhedra. Bottom: The 2-c network of 4 along the [100] plane ({Ni(Hhmp)2}2+: green and H2DHBDC: yellow nodes). | ||
Compound 6·2DMF crystallises in the monoclinic space group P21/c. Its structure consists of 1D zigzag chains based on the repeating unit [Ni(H2pyaox)2(HMHBDC)] and DMF lattice molecules (Fig. 6). The repeating units are linked through HMHBDC2− ligands to form a CP. Each NiII ion is six-coordinated, and its coordination sphere is completed by two terminal, chelate H2pyaox ligands. The structure of 6·2DMF is stabilised by strong intra- and interchain hydrogen bonding interactions, resulting in the formation of a 3D network (Fig. 7). Intermolecular interactions are formed between: a) the hydroxyl group of the HMHBDC2− ligand (O3, donor) and the carboxylate group of the same ligand (O3, acceptor), b) the oximic group (O6, donor) and the carboxylate group (O5, acceptor), and c) the NH2 group of H2pyaox as the donor (N2, N6) and the solvate DMF molecules as acceptors (O8, O9). The chains are held together by hydrogen bonds between the amino oximic group (N6, donor) and the hydroxyl group (O3) of HMHBDC2− of a neighbouring chain. A list of the distances and angles of hydrogen bonding interactions in 6·2DMF is provided in Table S4.†6·2DMF decomposes via multiple steps (Fig. S2†). There is a mass loss of ∼20% at 150 °C, which is followed by a plateau with the step at 350 °C corresponding to the chain breakdown.
 | ||
| Fig. 6 Representation of the repeating unit (top) and a part of the 1D zigzag chain of 6·2DMF (bottom). Colour code: NiII, green; O, red; N, blue; C, grey. The hydrogen atoms and lattice DMF molecules are omitted for clarity. | ||
 | ||
| Fig. 7 Representation of the 3D network of compound 6·2DMF. Colour code: green polyhedra: coordination spheres of NiII. | ||
Compound 7·DMF consists of a [Zn(Hmpko)2(HMHBDC)] chain and DMF solvate molecules; hence, it has a similar formula to 6·2DMF, with two main differences being the types of metals (Ni, 6; Zn; 7) and oximic ligands present in the structures (H2pyaox, 6; Hmpko, 7). The coordination modes of the ligands are the same in both compounds (Fig. 8). Notably, the structures of 6·2DMF and 7·DMF crystallize in different space groups (P21/c, 6; Ia, 7), which results in a closer packing of the 1D chains in 7. This also affects the value of the closest metal⋯metal interchain distance, which is decreased from 10.063 Å in 6 to 9.678 Å in 7. The crystal structure of 7·DMF is stabilized by strong hydrogen bonding interactions, similar to 6·2DMF (Table S5†). In addition, π–π interactions occur between the aromatic rings of the oximic ligands of neighboring chains, providing further stability; the distance between the centroids of the aromatic rings is 4.2 Å.
 | ||
| Fig. 8 Representation of the repeating unit (top) and a part of the 1D zigzag chain of 7·DMF (bottom). Color code: ZnII, yellow; O, red; N, blue; C, grey. The hydrogen atoms and the lattice DMF molecules are omitted for clarity. | ||
1–7 are the first reported examples of compounds bearing 2-pyridyl oxime or Hhmp and H3MHBDC or H4DHBDC. Compound 3 is a linear trinuclear CoIICoIII2 cluster with a common topology, which has been reported in the past in homo- and heterometallic compounds with 2-pyridyl oximes.49–51 Compounds 1, 2 and 4–7 are 1D zigzag chains, forming supramolecular structures through intermolecular interactions. All CPs in this work form a 2-c net of 2C1 topology (Fig. S3 and S4†). The purity of the reported CPs has been further confirmed by PXRD studies (Fig. S5–S10 in the ESI†).
It is worth mentioning that the isolation of a higher dimensionality CP or MOF was not achieved, possibly due to steric or electronic hindrance from the presence of additional functional groups in the organic linker. However, the presence of functional groups within the structures serves a crucial purpose as they enable the framework to interact effectively with guest species, particularly metal ions, through covalent or electrostatic interactions.
Metal encapsulation studies
The presence of free coordination sites in 1–6 prompted us to investigate their ability to adsorb or remove metal ions from solutions. To this end, 4 was chosen as a representative example because of its pseudo-porous structure formed through strong intermolecular interactions, as well as its low cost and high yield of production. Metal encapsulation studies for 4 were conducted by immersing the polymer crystals in EtOH solutions of Co(NO3)2·6H2O. To investigate the metal encapsulation, the solutions were stirred and aliquots were measured using UV-vis spectroscopy at designated times. It is worth mentioning that the crystals of 4 changed colour upon Co2+ encapsulation, turning from blue to yellow.The adsorption of Co2+ by 4 exhibits slow kinetics. As shown in Fig. 8, the adsorption capacity increases smoothly over time and reaches a plateau after 40 hours. No metal adsorption is observed after this time. To better understand the adsorption mechanism, experimental kinetic data were fitted to a theoretical model. To this end, pseudo-first order and pseudo-second order kinetic models were used, according to eqn (6) and (7):52
| ln(qe − qt) = lnqe − k1t | (6) |
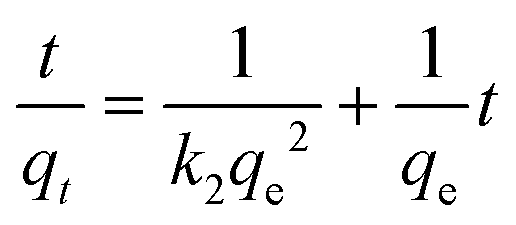 | (7) |
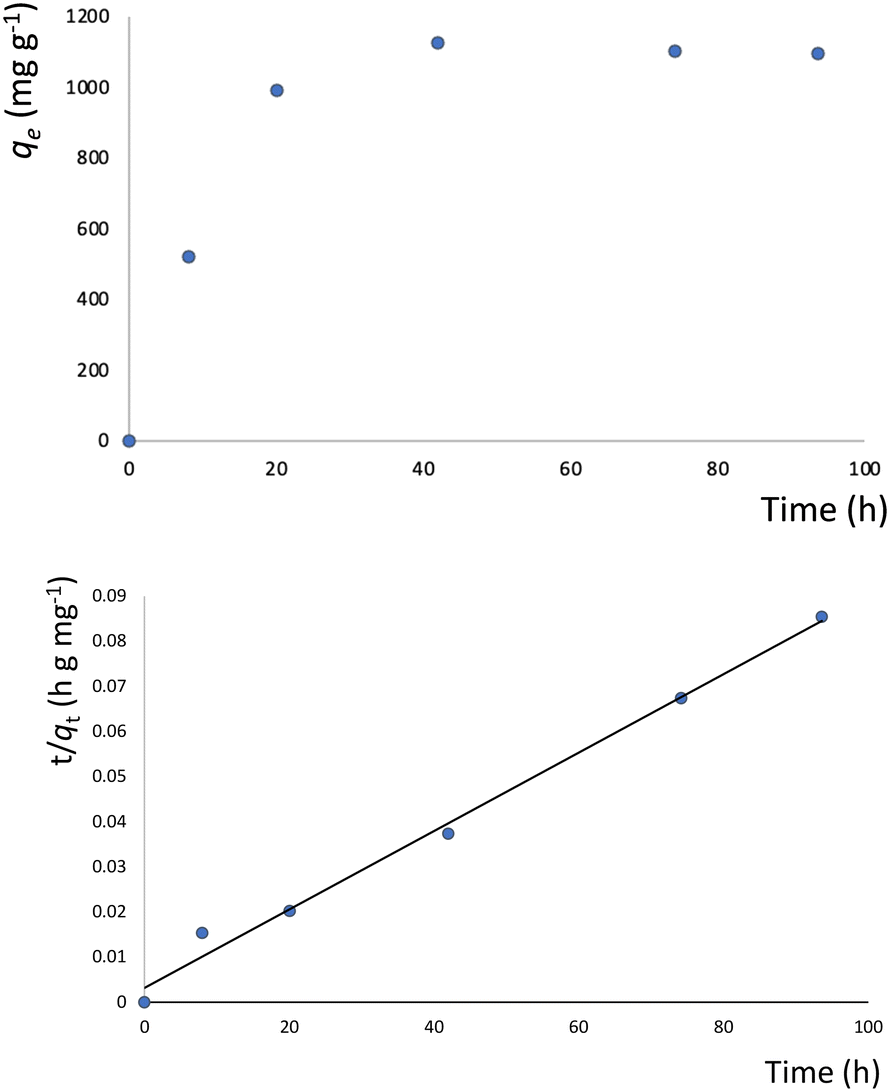 | ||
| Fig. 9 Top: Metal adsorption capacity (mg g−1) versus time (h) plot; bottom: simulation of the experimental data to the pseudo-second order kinetic model. The solid line represents the fitting of the data. | ||
Fig. 10 shows the metal adsorption equilibrium data. The Langmuir model53 provides the best fit for the data by considering a monolayer adsorption with a finite number of homogeneous and equivalent active sites, as given by eqn (8):
 | (8) |
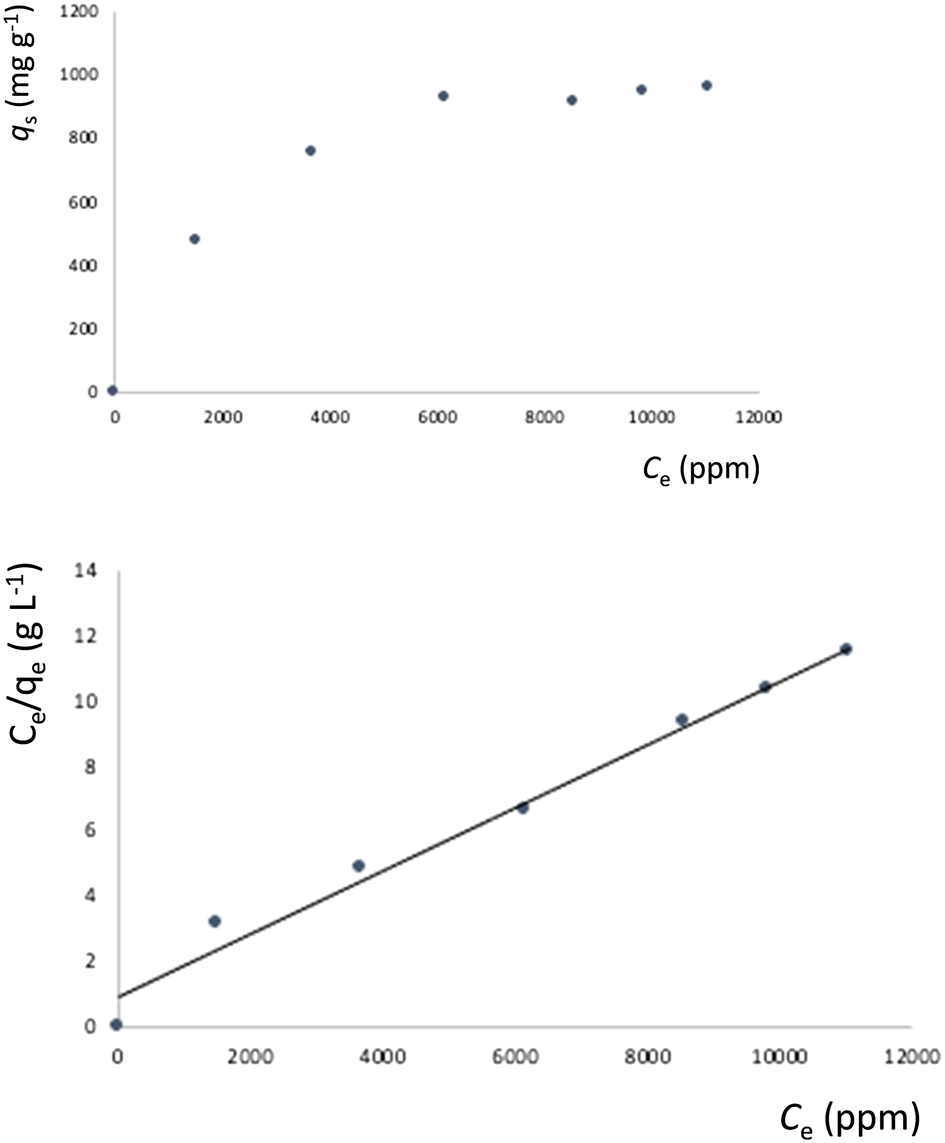 | ||
| Fig. 10 Top: Equilibrium data for the metal adsorption by 4 (contact time: 40 h); bottom: fitting of the metal adsorption data to the Langmuir model. | ||
The capacity of 4 to uptake Co2+ was also confirmed through EDX studies. Fig. 11 displays the EDX spectra of 4 and Co@4. Both samples were found to contain Ni, C, N, and O. The second sample also displayed an additional peak of Co2+ that had been adsorbed, hence confirming the metal uptake. It is noteworthy that the possibility of regenerating and reusing 4 was thoroughly investigated using a variety of methods, including treatment with a 0.2 M EDTA solution. However, it was found that regeneration of the initial compound is not feasible, which supports further the chemisorption mechanism of the interaction between 4 and the guest metal ion.
 | ||
| Fig. 11 EDX spectra of 4 (left) and Co@4. | ||
Conclusions
The combination of 2-pyridyl oximes or 2-pyridinemethanol and a hydroxo-functionalized dicarboxylic acid (H3MHBDC or H4DHBDC) has provided access to seven new compounds, including CPs and a metal cluster. These compounds were isolated via a one-pot reaction between a metal source and the ligand blend at high temperature and are the first examples of compounds bearing this ligand combination. The CPs are zigzag chains based on mononuclear repeating units, while the metal cluster is a new addition in the family of linear trinuclear metal compounds containing a 2-pyridyl oxime and represents the first such example bearing H2DHBDC2− ions.It is worth mentioning that the additional hydroxo groups in the carboxylate ligand, in combination with the presence of bulky aromatic rings from the alkoxy or pyridyl ligands, have prevented the isolation of higher dimensionality materials; yet, strong intermolecular interactions between adjacent chains form a 3D supramolecular network. The latter, along with the presence of free coordination sites (OH groups from the carboxylate ligands), makes these materials suitable candidates for the removal of metal ions from the environment. To this end, compound 4 was studied in detail and found to uptake 900.0 mg Co2+ per g of 4.
CPs based on 2-pyridyl oximes or alcohols in combination with polycarboxylic acids have been shown to exhibit metal ion uptake ability.34,35 This work further establishes their capability and reveals that the presence of additional functional groups in the carboxylate ions substantially improves their metal ion uptake capability. This is further enhanced by the lower dimensionality of these species, which makes the free coordination sites more accessible to metal ions.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was funded by the University of Galway with a Hardiman Scholarship to FD.References
- M. J. Zaworotko, J. Am. Chem. Soc., 2010, 132, 7821 CrossRef CAS.
- G. H. Morritt, H. Michaels and M. Freitag, Chem. Phys. Rev., 2022, 3, 011306 CrossRef CAS.
- M. Tran, K. Kline, Y. Qin, Y. Shen, M. D. Green and S. Tongay, Appl. Phys. Rev., 2019, 6, 041311(1) Search PubMed.
- Z. Hu, B. J. Deibert and J. Li, Chem. Soc. Rev., 2014, 43, 5815 RSC.
- A. Ahmed, D. McHugh and C. Papatriantafyllopoulou, Molecules, 2022, 27, 6585 CrossRef CAS PubMed.
- L. Ma, J. M. Falkowski, C. Abney and W. Lin, Nat. Chem., 2010, 2, 838 CAS.
- E. Loukopoulos and G. E. Kostakis, J. Coord. Chem., 2017, 71, 371 CrossRef.
- J. Zhao, J. Yuan, Z. Fang, S. Huang, Z. Chen, F. Qiu, C. Lu, J. Zhu and X. Zhuang, Coord. Chem. Rev., 2022, 471, 214735 CrossRef CAS.
- X. Chen, F. Wo, Y. Jin, J. Tan, Y. Lai and J. Wu, ACS Nano, 2017, 11, 7938 CAS.
- T. Tieu, M. Alba, R. Elnathan, A. Cifuentes-Rius and N. H. Voelcker, Adv. Ther., 2019, 2(1), 1800095 CrossRef.
- W. Hatakeyama, T. J. Sanchez, M. D. Rowe, N. J. Serkova, M. W. Liberatore and S. G. Boyes, ACS Appl. Mater. Interfaces, 2011, 3, 1502 CrossRef CAS PubMed.
- F. D. Duman and R. S. Forgan, J. Mater. Chem. B, 2021, 9, 3423 RSC.
- M. Winterlich, C. G. Efthymiou, W. Papawassiliou, J. P. Carvalho, A. J. Pell, J. Mayans, A. Escuer, M. P. Carty, P. McArdle, E. Tylianakis, L. Morrison, G. Froudakis and C. Papatriantafyllopoulou, Mater. Adv., 2020, 1, 2248 RSC.
- A. S. Lenshin, Y. A. Peshkov, O. V. Chernousova, S. V. Kannykin and D. A. Minakov, Eur. Phys. J.: Appl. Phys., 2023, 98, 36 CrossRef CAS.
- M. Rudenko, N. Gaponenko, V. Litvivov, A. Ermachikhin, E. Chubenko, V. Borisenko, N. Mukhin, Y. Radyush, A. Tumarkin and A. Gagarin, Materials, 2020, 13, 5767 CrossRef CAS PubMed.
- S. N. Aisyiyah Jenie, S. Pace, B. Sciacca, R. D. Brooks, S. E. Plush and N. H. Voelcker, ACS Appl. Mater. Interfaces, 2014, 6, 12012 CrossRef PubMed.
- M. Safaei, M. M. Foroughi, N. Ebrahimpoor, S. Jahani, A. Omidi and M. Khatami, TrAC, Trends Anal. Chem., 2019, 118, 401 CrossRef CAS.
- H. Li, M. Eddaoudi, M. O'Keeffe and O. M. Yaghi, Nature, 1999, 402, 276 CrossRef CAS.
- O. M. Yaghi, M. J. Kalmutzki and C. S. Diercks, Introduction to Reticular Chemistry, Wiley, 2019 Search PubMed.
- C. Wang, D. Liu and W. Lin, J. Am. Chem. Soc., 2013, 135, 13222 CrossRef CAS PubMed.
- A. Ahmed, C. G. Efthymiou, R. Sanii, E. Patyk-Kazmierczak, Amir M. Alsharabasy, M. Winterlich, N. Kumar, D. Sensharma, W. Tong, S. Guerin, P. Farras, S. Hudson, D. Thompson, M. J. Zaworotko, A. J. Tasiopoulos and C. Papatriantafyllopoulou, J. Mater. Chem. B, 2022, 10, 1378 RSC.
- C. Coulon, H. Miyasaka and R. Clérac, Struct. Bonding, 2006, 122, 163 CrossRef CAS.
- T.-T. Wang, M. Ren, S.-S. Bao, B. Liu, L. Pi, Z.-S. Cai, Z.-H. Zheng, Z.-L. Xu and L.-M. Zheng, Inorg. Chem., 2014, 53, 3117 CrossRef CAS PubMed.
- C. Papatriantafyllopoulou, S. Zartilas, M. J. Manos, C. Pichon, R. Clérac and A. J. Tasiopoulos, Chem. Commun., 2014, 50, 14873 RSC.
- R. A. Allao Cassaro, S. G. Reis, T. S. Araujo, P. M. Lahti and M. A. Novak, Inorg. Chem., 2015, 54(19), 9381 CrossRef PubMed.
- L. Bogani and W. Wernsdorfer, Nat. Mater., 2008, 7, 179 CrossRef CAS PubMed.
- M. N. Leuenberger and D. Loss, Nature, 2001, 410, 789 CrossRef CAS PubMed.
- M.-L. Tong and X.-M. Chen, Modern Inorganic Synthetic Chemistry, ed. R. Xu and Y. Xu, Elsevier, 2nd edn, 2017, ch. 8, p. 189 Search PubMed.
- A. Schneemann, Y. Jing, J. D. Evans, T. Toyao, Y. Hijikata, Y. Kamiya, K. Shimizu, N. C. Burtch and S. Noro, Dalton Trans., 2021, 50, 10423 RSC.
- S. Noro, J. Mizutani, Y. Hijikata, R. Matsuda, H. Sato, S. Kitagawa, K. Sugimoto, Y. Inubushi, K. Kubo and T. Nakamura, Nat. Commun., 2015, 6, 5851 CrossRef CAS PubMed.
- S. Pullen and G. H. Clever, Acc. Chem. Res., 2018, 51, 3052 CrossRef CAS PubMed.
- Z. Yin, Y.-L. Zhou, M.-H. Zeng and M. Kurmoo, Dalton Trans., 2015, 44, 5258 RSC.
- A. Helal, Z. H. Yamani, K. E. Cordova and O. M. Yaghi, Natl. Sci. Rev., 2017, 4, 296 CrossRef CAS.
- I. Mylonas-Margaritis, A. Gerard, K. Skordi, J. Mayans, A. Tasiopoulos, P. McArdle and C. Papatriantafyllopoulou, Materials, 2020, 4084 CrossRef CAS PubMed.
- I. Mylonas-Margaritis, J. Mayans, P. McArdle and C. Papatriantafyllopoulou, Molecules, 2021, 26, 491 CrossRef CAS PubMed.
- I. Mylonas-Margaritis, J. Mayans, W. Tong, P. Farras, A. Escuer, P. McArdle and C. Papatriantafyllopoulou, CrystEngComm, 2021, 23, 5489 RSC.
- I. Mylonas-Margaritis, J. Mayans, C. G. Efthymiou, P. McArdle and C. Papatriantafyllopoulou, Eur. J. Inorg. Chem., 2022, e202200140 CrossRef CAS.
- J. F. Kurisingal, Y. Rachuri, Y. Gu, Y. Choe and D.-W. Park, Chem. Eng. J., 2020, 386, 121700 CrossRef CAS.
- M. Kubo, H. Hagi, A. Shimojima and T. Okubo, Chem. – Asian J., 2013, 8, 2801 CrossRef CAS PubMed.
- Y. Fu, Y. Yao, A. C. Forse, J. Li, K. Mochizuki, J. R. Long, J. A. Reimer, G. d. Paepe and X. Kong, Nat. Commun., 2023, 14, 2386 CrossRef CAS PubMed.
- H. Chun and D. Moon, Cryst. Growth Des., 2017, 17, 2140 CrossRef CAS.
- S. Cadot, L. Veyre, D. Luneau, D. Farrusseng and E. A. Quadrelli, J. Mater. Chem. A, 2014, 2, 17757 RSC.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Adv., 2015, 71, 3 CrossRef PubMed.
- P. McArdle, K. Gilligan, D. Cunningham, R. Dark and M. Mahon, CrystEngComm, 2004, 6, 303 RSC.
- K. Brandenburg, DIAMOND, Version 2003.2001d, Crystal Impact GbR, Bonn, Germany, 2006 Search PubMed.
- P. Van der Sluis and A. L. Spek, Acta Crystallogr., Sect. A: Found. Crystallogr., 1990, 46, 194 CrossRef.
- G. Mahmoudi, H. Chowdhuri, S. E. Lofland, B. Kumar Ghosh and A. M. Kirillov, J. Coord. Chem., 2017, 70, 1973 CrossRef CAS.
- A. Sadhukhan, P. Brandao, S. Saha, D. Mal and N. Sepay, J. Mol. Struct., 2023, 1272, 134204 CrossRef CAS.
- T. C. Stamatatos, A. Bell, P. Cooper, A. Terzis, C. P. Raptopoulou, S. L. Heath, R. E. P. Winpenny and S. P. Perlepes, Inorg. Chem. Commun., 2005, 8, 533 CrossRef CAS.
- X. M. Qiu, S. N. Wang, J. Lu, L.-Q. Kong, D.-C. Li and J. M. Dou, J. Coord. Chem., 2012, 65, 3308 CrossRef CAS.
- C. G. Efthymiou, I. Mylonas-Margaritis, S. Das Gupta, A. Tasiopoulos, V. Nastopoulos, G. Christou, S. P. Perlepes and C. Papatriantafyllopoulou, Polyhedron, 2019, 171, 330 CrossRef CAS.
- K.-D. Zhang, F.-C. Tsai, N. Ma, Y. Xia, H.-L. Liu, X.-Q. Zhan, X.-Y. Yu, X.-Z. Zeng, T. Jiang, D. Shi and C.-J. Chang, Materials, 2017, 10, 205 CrossRef PubMed.
- I. Langmuir, Part I. Solids, J. Am. Chem. Soc., 1915, 38, 102 Search PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Tables with X-ray data and hydrogen bonding interactions; thermal analysis graphs. CCDC 2282116–2282122. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3ce00727h |
| ‡ Current address: Department of Physics, Bernal Institute, University of Limerick, Limerick, Republic of Ireland. |
| This journal is © The Royal Society of Chemistry 2023 |