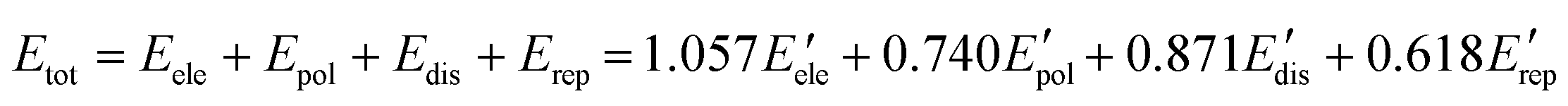DOI:
10.1039/D3CE00608E
(Paper)
CrystEngComm, 2023,
25, 4798-4811
Virtual assessment achieved two binary cocrystals based on a liquid and a solid pyridine derivative with modulated thermal stabilities†
Received
16th June 2023
, Accepted 21st July 2023
First published on 4th August 2023
Abstract
The rational design of cocrystals triggered by the control of recurrent H-bonded patterns referred to as supramolecular synthons enabled the correlation between their structure and properties, which has been a topic of interest owing to the possibility to modulate them depending on the selected components. Accordingly, melting point has been one of the most studied properties, providing materials with enhanced thermal stability for specific applications. Within this frame, in this work we have selected a liquid and a solid pyridine derivative (dPy), namely 4-acetylpyridine (4-Acpy) and 2-hydroxypyridine (2-OHpy) to combine with carboxylic acids to obtain a pair of cocrystals. An initial virtual screening of some carboxylic acids based on the positive and negative critical points of the molecular electrostatic potential (MEP) surfaces was performed to evaluate the feasibility of cocrystal formation. This enabled us to select 1,3-benzodioxole-5-carboxylic acid (piperonylic acid, HPip) to combine with 4-Acpy and α-acetamidocinnamic acid (HACA) with 2-OHpy. Then, we have obtained the corresponding cocrystal experimentally by means of liquid-assisted grinding (LAG), and their crystal structures were elucidated, revealing the formation of (HPip)(4-Acpy) (1) and (HACA)(Pdon) (2) (Pdon = 2-pyridone), observing the tautomerization of 2-OHpy to Pdon. Both cocrystals were characterized by analytical and spectroscopic techniques. In addition, a Cambridge Structural Database (CSD) survey of 4-Acpy and Pdon in cocrystal systems was performed and the observed preferences regarding their preferable synthons and dimensionalities were shown. Finally, their melting points have been determined, and the resulting values have been correlated with the crystal packing of the compounds, supported by Hirshfeld surface analysis and energy frameworks.
Introduction
Over the past decades, the study of organic multicomponent crystalline materials has attracted the attention of researchers owing to their promising applications in the fields of medicine,1,2 agriculture,3,4 and explosives,5,6 among others.7,8 In particular, the design of cocrystals has been a hot topic due to their ability to improve the physicochemical properties of pharmaceuticals without altering the pharmacological properties of the selected active pharmaceutical ingredients (APIs)9,10 as well as their opportunities to overcome patent protection of existing APIs.11 Interestingly, recent studies used this approach to stabilize liquid molecules in the solid state,12,13 providing a better handling of pesticides14 or medicines.15 We emphasize herein that these materials should also be regarded as cocrystals despite one of their former components being a liquid at room temperature, which has been subjected to debate during the past two decades.16 Therefore, researchers have focused their efforts on the control of intermolecular interactions towards the rational design of these materials. Accordingly, the emergence of the concept of supramolecular synthon17 not only led the way to the preparation of cocrystals based on predictable H-bonded patterns (i.e. acid⋯pyridine,18 acid⋯amide,19 and alcohol⋯pyridine,20 among others21) but also permitted the understanding of their structure–property relationships.22,23
Within this frame, melting point has been an extensively studied physical property of solids.24–26 Its modulation has enabled the enhancement of the thermal stability and processability of the determined molecules through the formation of cocrystals.27–29 Indeed, our group has previously studied the formation of cocrystals based on the acid⋯amide30 and acid⋯pyridine heterosynthons31 and the correlation of the crystal packing with their melting point values.31 Therefore, aiming to extend our knowledge on the structure–property relationship of cocrystals focusing on the melting point, we have selected a liquid and a solid pyridine derivative (dPy), namely 4-acetylpyridine (4-Acpy) and 2-hydroxypyridine (2-OHpy), respectively. It should be noted that 2-OHpy exists in equilibrium with its keto form (2-pyridone, Pdon), which in the solid state is preferentially shifted to Pdon.32 Then, we evaluated the most promising carboxylic acids over a group of available components in our laboratory as well as the feasibility of cocrystal formation of the chosen combinations using a virtual assessment based on the maximum and minimum critical points from their corresponding MEP surfaces. This enabled us to select the combinations of 4-Acpy with 1,3-benzodioxole-5-carboxylic acid (piperonylic acid, HPip), and 2-hydroxypyridine (2-OHpy) with α-acetamidocinnamic acid (HACA), which led us to successfully obtain a pair of cocrystals by means of liquid-assisted grinding (LAG) (Scheme 1). The elucidation of their crystal structures showing the formulas (HPip)(4-Acpy) (1) and (HACA)(Pdon) (2) enabled an exhaustive study of their intermolecular interactions and their resulting crystal packings supported by Hirshfeld surface analysis and energy frameworks. Moreover, a Cambridge Structural Database (CSD)33 survey of the selected dPy in cocrystal systems was performed and their preferable synthons and dimensionalities have been compiled. Finally, the structural features of both cocrystals have been correlated with their melting point values.
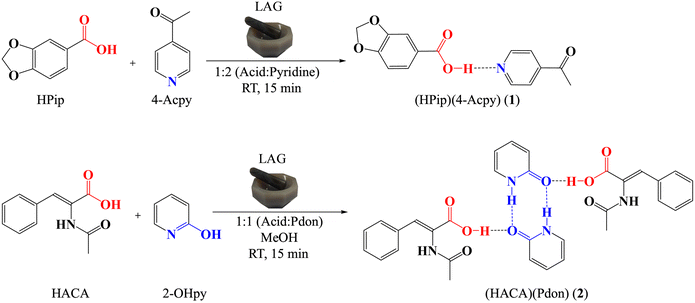 |
| | Scheme 1 Outline of the preparation of cocrystals 1 and 2. | |
Experimental section
Materials and general methods
1,3-Benzodioxole-5-carboxylic acid (piperonylic acid, HPip), α-acetamidocinnamic acid (HACA), 4-acetylpyridine (4-Acpy), 2-hydroxypyridine (2-OHpy), and methanol (MeOH) as solvent were purchased from Sigma-Aldrich. Deuterated methanol (CD3OD) was used for the NMR experiments and was purchased from Eurisotop. All of them were used without further purification. All the reactions and manipulations were carried out in air at room temperature (RT). Powder X-ray diffraction (PXRD) patterns were measured with a Panalytical X'Pert PRO MPD apparatus using monochromatic CuKα radiation with λ = 1.5406 Å. All of them were recorded from 2θ = 5° to 30° with a step scan of 0.01671°. Melting point (m.p.) was measured on a Stuart melting point apparatus SMP30 using a 2.0 °C min−1 step rate from RT to 200 °C. Elemental analyses (EAs) were carried out on a Thermo Scientific Flash 2000 CHNS analyzer. FTIR-ATR spectra were recorded on a Perkin Elmer spectrometer equipped with an attenuated total reflectance (ATR) accessory model MKII Golden Gate with a diamond window in the range 4000–500 cm−1. 1H, 13C{1H} and DEPT-135 NMR spectra were recorded on a Bruker Ascend 300 MHz spectrometer in CD3OD solution at RT. All the chemical shifts (δ) are given in ppm relative to tetramethylsilane (Me4Si) as internal standard. Simultaneous thermogravimetric/differential thermal analysis (TG/DTA) determinations were carried out using 69.30 mg (1) and 52.30 mg (2), in a Netzsch STA 409 instrument with an aluminium oxide powder (Al2O3) crucible and heating at 5 °C min−1 from 25 to 350 °C, under a nitrogen atmosphere with a flow rate of 80 mL min−1. Al2O3 (Perkin-Elmer 0419-0197) was used as standard.
Synthesis of cocrystal 1
Cocrystal 1 was prepared by placing in an agate mortar 200 mg (1.20 mmol) of HPip and 268 μL (2.42 mmol) of 4-Acpy and then grinding for about 15 minutes until a homogeneous powder was obtained. Then, the phase purity of the sample was verified by PXRD (ESI,† Fig. S1). Single crystals suitable for X-ray diffraction were obtained by recrystallization of the powder in MeOH and allowing the resulting solution to evaporate at RT for seven days.
1
.
Isolated yield: 293 mg (84.6%). M.p. 110–111 °C. Elemental analysis calc (%) for C15H13NO5 (287.27): C 62.71; H 4.56; N 4.88; found: C 62.48; H 4.39; N 4.62. FTIR-ATR (wavenumber, cm−1): 3078–3040(w) [ν(C–H)ar+alk], 2993–2799(w) [ν(C–H)al], 2691–2156(br) [ν(O–H)]HPip, 2135–1757(w) [ν(O–H⋯N)], 1693(br) [ν(C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O)]4-Acpy + [ν(COOH)]HPip, 1605(w) [ν(CC/CN)], 1560(w), 1502(w), 1493(w), 1439(m), 1412(m) [δ(CC/CN)], 1364(w), 1286(m), 1257(s), 1232(s), 1213(m), 1165(m) [ν(C–O–C)], 1119(m), 1086(m), 1063(m), 1032(s), 1013(s), 959(w), 930(m), 916(m) [δip(C–H)], 883(m), 845(w), 822(s), 768(s) [δoop(C–H)], 748(w), 719(w), 667(w), 658(w), 588(s), 532(m). 1H NMR (300 MHz, CD3OD; Me4Si; 298 K): δ = 9.99 [2H, d, 3J = 5.9 Hz, o-H4-Acpy], 9.08 [2H, dd, 3J = 4.5 Hz, 4J = 1.7 Hz, m-H4-Acpy], 8.86 [1H, dd, 3J = 8.2 Hz, 4J = 1.7 Hz, HOOC–C–CH–CHHPip], 8.63 [1H, d, 4J = 1.7 Hz, HOOC–C–CH–COHPip], 8.12 [1H, d, 3J = 8.2 Hz, HOOC–C–CH–CHHPip], 5.71 [2H, s, O–CH2–OHPip], 3.86 [3H, s, CH3,4-Acpy]. 13C{1H} NMR (75 MHz, CD3OD; Me4Si; 298 K): δ = 198.8 [CO4-Acpy], 168.4 [COOHHPip], 152.9 [HOOC–C–CH–CH–CHPip], 151.5 [o-C4-Acpy], 149.0 [HOOC–C–CH–CHPip], 144.6 [H3C–CO–C4-Acpy], 128.8 [HOOC–CHPip], 126.4 [HOOC–C–CH–CHHPip], 122.8 [m-C4-Acpy], 110.2 [HOOC–C–CH–COHPip], 108.9 [HOOC–C–CH–CHHPip], 103.2 [O–CH2–OHPip], 26.9 [CH3,4-Acpy]. DEPT-135 NMR (75 MHz, CD3OD; Me4Si; 298 K): δ = 151.5 [o-C4-Acpy], 126.4 [HOOC–C–CH–CHHPip], 122.9 [m-C4-Acpy], 110.2 [HOOC–C–CH–COHPip], 108.9 [HOOC–C–CH–CHHPip], 103.2 [O–CH2–OHPip], 26.9 [CH3,4-Acpy].
O)]4-Acpy + [ν(COOH)]HPip, 1605(w) [ν(CC/CN)], 1560(w), 1502(w), 1493(w), 1439(m), 1412(m) [δ(CC/CN)], 1364(w), 1286(m), 1257(s), 1232(s), 1213(m), 1165(m) [ν(C–O–C)], 1119(m), 1086(m), 1063(m), 1032(s), 1013(s), 959(w), 930(m), 916(m) [δip(C–H)], 883(m), 845(w), 822(s), 768(s) [δoop(C–H)], 748(w), 719(w), 667(w), 658(w), 588(s), 532(m). 1H NMR (300 MHz, CD3OD; Me4Si; 298 K): δ = 9.99 [2H, d, 3J = 5.9 Hz, o-H4-Acpy], 9.08 [2H, dd, 3J = 4.5 Hz, 4J = 1.7 Hz, m-H4-Acpy], 8.86 [1H, dd, 3J = 8.2 Hz, 4J = 1.7 Hz, HOOC–C–CH–CHHPip], 8.63 [1H, d, 4J = 1.7 Hz, HOOC–C–CH–COHPip], 8.12 [1H, d, 3J = 8.2 Hz, HOOC–C–CH–CHHPip], 5.71 [2H, s, O–CH2–OHPip], 3.86 [3H, s, CH3,4-Acpy]. 13C{1H} NMR (75 MHz, CD3OD; Me4Si; 298 K): δ = 198.8 [CO4-Acpy], 168.4 [COOHHPip], 152.9 [HOOC–C–CH–CH–CHPip], 151.5 [o-C4-Acpy], 149.0 [HOOC–C–CH–CHPip], 144.6 [H3C–CO–C4-Acpy], 128.8 [HOOC–CHPip], 126.4 [HOOC–C–CH–CHHPip], 122.8 [m-C4-Acpy], 110.2 [HOOC–C–CH–COHPip], 108.9 [HOOC–C–CH–CHHPip], 103.2 [O–CH2–OHPip], 26.9 [CH3,4-Acpy]. DEPT-135 NMR (75 MHz, CD3OD; Me4Si; 298 K): δ = 151.5 [o-C4-Acpy], 126.4 [HOOC–C–CH–CHHPip], 122.9 [m-C4-Acpy], 110.2 [HOOC–C–CH–COHPip], 108.9 [HOOC–C–CH–CHHPip], 103.2 [O–CH2–OHPip], 26.9 [CH3,4-Acpy].
Synthesis of cocrystal 2
Cocrystal 2 was prepared using the same methodology as for the synthesis of 1 with 200 mg (0.975 mmol) of HACA, 92.7 mg (0.975 mmol) of 2-OHpy, and 100 μL of MeOH. The phase purity of the sample was verified by PXRD (ESI† Fig. S2). Single crystals suitable for X-ray diffraction were obtained by recrystallization of the powder in MeOH and allowing the resulting solution to evaporate at RT for five days.
2
.
Isolated yield: 278 mg (95.1%). M.p. 156–157 °C. Elemental analysis calc (%) for C16H16N2O4 (300.31): C 63.99; H 5.37; N 9.33; found: C 63.74; H 5.18; N 9.12. FTIR-ATR (wavenumber, cm−1): 3263(m) [ν(N–H)]HACA, 3167–2604(br) [ν(N–H)]Pdon + [ν(C–H)ar+alk+al], 2556–2075(br) [ν(O–H)]HACA, 2029–1746(br) [ν(O–H⋯O)], 1703(w) [ν(CO)]Pdon, 1636(br) [ν(COOH)]HACA + [ν(CO)]HACA, 1609(s) [ν(CC/CN)], 1518(m), 1492(w), 1475(w), 1424(m) [δ(CC/CN)], 1368(m), 1333(w), 1319(w), 1309(w), 1286(w), 1265(m), 1237(s), 1215(s), 1156(m), 1138(m), 1072(w), 1038(w), 1021(w), 991(m), 985(m), 917(m) [δip(C–H)], 889(w), 863(w), 851(w), 791(s) [δoop(C–H)], 761(m), 735(w), 712(w), 683(s), 619(w), 606(m), 577(s), 550(w), 521(s), 511(s). 1H NMR (300 MHz, CD3OD; Me4Si; 298 K): δ = 7.58 [3H, m, OC–CH–CHPdon + o-HHACA], 7.48 [1H, s, HOOC–C–CHHACA], 7.39 [4H, m, HN–CHPdon + m-HHACA + p-HHACA], 6.54 [1H, m, HN–CH–CHPdon], 6.41 [1H, td, 3J = 6.6 Hz, 4J = 1.1 Hz, OC–CHPdon], 2.10 [3H, s, CH3,HACA]. 13C{1H} NMR (75 MHz, CD3OD; Me4Si; 298 K): δ = 173.2 [HN–COHACA], 168.2 [COOHHACA], 165.8 [HN–COPdon], 143.8 [OC–CH–CHPdon], 136.0 [HN–CHPdon], 135.5 [HOOC–C–CHHACA], 135.0 [HOOC–C–CH–CHACA], 130.9 [o-CHACA], 130.6 [p-CHACA], 129.7 [m-CHACA], 127.0 [HOOC–CHACA], 120.8 [OC–CHPdon], 108.5 [HN–CH–CHPdon], 22.5 [CH3,HACA]. DEPT-135 NMR (75 MHz, CD3OD; Me4Si; 298 K): δ = 143.7 [OC–CH–CHPdon], 136.0 [HN–CHPdon], 135.5 [HOOC–C–CHHACA], 130.8 [o-CHACA], 130.6 [p-CHACA], 129.6 [m-CHACA], 120.8 [OC–CHPdon], 108.5 [HN–CH–CHPdon], 22.5 [CH3,HACA].
X-ray crystallographic data
Yellowish prism-like (1) and colorless needle-like (2) specimens were used for the X-ray crystallographic analysis. The X-ray intensity data were measured on a D8 Venture system equipped with a multilayer monochromator (λ = 0.71073 Å). For both compounds, the frames were integrated using the Bruker SAINT software package (version-2018/3). The integration of the data with 0.70 Å (1) and 0.73 Å (2) resolution, of which 3980 (1) and 3952 (2) reflections were independent, gave an average redundancy of 8.677 (1) and 9.327 (2), completeness of 99.8% (1) and 99.7% (2), Rsig of 1.85% (1) and 3.35% (2), presenting 3380 (84.92%) (1) and 3010 (76.16%) (2) reflections greater than 2σ(|F|2).
For 1 and 2, the final cell constants and volumes are based upon refinement of the XYZ-centroids of reflections above 20σ(I). Data were corrected for absorption effects using the Multi-Scan method (SADABS). Crystal data and additional details of structure refinement for 1 and 2 are reported in Table 1. Complete information about the crystal structure and molecular geometry is available in CIF format via CCDC 2267801 (1), and 2267802 (2). Molecular graphics were generated using Mercury 4.3.1 software34 with the POV-Ray image package.35 The color codes for all the molecular graphics are as follows: red (O), light blue (N), gray (C), and white (H). The topological analysis was done using the ToposPro 5.3.3.4 program.36
Table 1 Crystal data and structure refinement for 1 and 2
|
|
1
|
2
|
| Empirical formula |
C15H13NO5 |
C16H16N2O4 |
| Formula weight |
287.26 |
300.31 |
|
T (K) |
100(2) |
100(2) |
| Wavelength (Å) |
0.71073 |
0.71073 |
| System, space group |
Triclinic, P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) |
Monoclinic, P21/c |
| Unit cell dimensions |
|
|
|
a (Å) |
7.4176(5) |
17.3245(7) |
|
b (Å) |
7.5344(5) |
4.8311(2) |
|
c (Å) |
13.4049(9) |
18.3867(8) |
|
α (°) |
94.585(2) |
90 |
|
β (°) |
101.837(2) |
107.778(2) |
|
γ (°) |
115.880(2) |
90 |
|
V (Å3) |
647.36(8) |
1465.41(11) |
|
Z
|
2 |
4 |
|
D
calc (g cm−3) |
1.474 |
1.361 |
|
μ (mm−1) |
0.112 |
0.099 |
|
F (000) |
300 |
632 |
| Crystal size (mm) |
0.362 × 0.158 × 0.078 |
0.180 × 0.060 × 0.040 |
|
hkl ranges |
−10 ≤ h ≤ 10, |
−23 ≤ h ≤ 23, |
| −10 ≤ k ≤ 10, |
−6 ≤ k ≤ 6, |
| −19 ≤ l ≤ 19 |
−25 ≤ l ≤ 25 |
|
θ range (°) |
3.062 to 30.571 |
2.277 to 29.154 |
| Reflections collected/unique/[Rint] |
34![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 534/3980/0.0319 534/3980/0.0319 |
36859/3952/0.0664 |
| Completeness to θ (%) |
99.9 |
99.7 |
| Absorption correction |
Semi-empirical from equivalents |
Semi-empirical from equivalents |
| Max. and min. transmission |
0.7461 and 0.7246 |
0.7458 and 0.6929 |
| Refinement method |
Full-matrix least-squares on |F|2 |
Full-matrix least-squares on |F|2 |
| Data/restrains/parameters |
3980/0/191 |
3952/0/202 |
| Goodness of fit on F2 |
1.045 |
1.058 |
| Final R indices [I > 2σ(I)] |
R
1 = 0.0392 |
R
1 = 0.0412 |
| wR2 = 0.1092 |
wR2 = 0.0896 |
|
R indices (all data) |
R
1 = 0.0484 |
R
1 = 0.0646 |
| wR2 = 0.1154 |
wR2 = 0.1035 |
| Extinction coefficient |
n/a |
n/a |
| Largest diff. peak and hole (e Å−3) |
0.492 and −0.278 |
0.295 and −0.247 |
Computational details
The virtual screening method proposed by Hunter et al.37,38 was used to evaluate if the combinations of the available carboxylic acids and the selected dPy are likely to overcome the energy of the packings of the single components. The carboxylic acids used for the virtual screening were 1,3-benzodioxole-5-carboxylic acid (piperonylic acid, HPip), α-acetamidocinnamic acid (HACA), trans-cinnamic acid (HCinn), and 2-furoic acid (2-FA), which were available in our laboratory. This methodology utilized the local maxima and minima of the molecular electrostatic potential (MEP) surfaces of the initial components of the cocrystals to identify surface site interaction points (SSIPs), which are used to extract the corresponding hydrogen bond donor and acceptor interaction site parameter (α and β) values.37 These parameters were used to calculate the interaction site pairing energies (E) from eqn (1), which were used to obtain ΔE values from eqn (2),| |  | (1) |
where E1, E2, and ECC are the interaction site pairing energies of the initial components and the cocrystal, and n and m stand for the proportion of initial components, which has been considered as 1:1 for all the combinations owing to the use of monopyridines and monocarboxylic acids which only allow the formation of a single main synthon. ΔE < 0 indicates the feasibility of cocrystal formation, while ΔE > 0 suggests that the formation of the cocrystal is unlikely to occur. Noteworthily, Pdon has been considered as the initial component for the formation of cocrystal 2 owing to its strong preference over 2-OHpy in the solid state as stated in the Introduction section.32 All the structures were energy-minimized using the COMPASS II force field in Materials Studio.39,40 Then, geometry optimizations were done using density functional theory (DFT) with the B3LYP/6-31G(+) theory level with Gaussian09 software version D.01.41 Since it has been shown that different conformations of the same components do not change significantly the α and β values unless intramolecular interactions are possible to occur,42 only the most stable conformation in the gas phase has been considered for each component. The local minima and maxima from the MEP surfaces were extracted using Multiwfn software,43 while their visualization and rendering were done in the VMD program.44
Hirshfeld surface analysis and energy frameworks of 1 and 2 have been performed with CrystalExplorer 17.5.45 The Hirshfeld surfaces of each component of the cocrystals have been calculated independently using an isovalue of 0.5 e au−3. Moreover, both cocrystals have been analyzed with energy frameworks with TONTO,46 using a scale factor of 150, and the CE-B3LYP/6-31G(d,p) energy model, starting from the .cif files obtained from the single-crystal X-ray diffraction data. Each of the molecules of the cocrystals has been confined in a cluster of 20 Å in the unit cell, including those that are crystallographically independent. The contribution of all the molecular pairs around the selected cluster has been considered following a previously reported methodology.44 In addition, the calculation of the total energy for each interaction has been done using eqn (3),47,48 while the obtention of the lattice energy (Elatt) has been done with eqn (4).49
| | 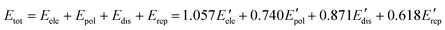 | (3) |
| | 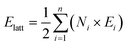 | (4) |
Results and discussion
Synthesis of cocrystals 1 and 2
Before starting the experimental trials with the selected dPy, we performed a virtual screening considering the carboxylic acids available in our laboratory to determine the probabilities of success. From the ΔE extracted using the potential combinations, it is shown that the formation of cocrystals with 4-Acpy is generally more favorable than the formation of cocrystals with Pdon, which should be attributed to the stronger crystal packing of Pdon with respect to 4-Acpy (Table 2). In addition, combinations with similar ΔE values to those proposed by us have been successfully found experimentally.38,50 The screening using 4-Acpy showed the HPip:4-Acpy combination as the more favorable, and thus it was selected for the experimental trials obtaining cocrystal 1. Otherwise, all the combinations using Pdon displayed similar ΔE values, and therefore we envisage the potential feasibility to successfully achieve all of them. However, we have focused the study on the combination of HACA:Pdon, leading to cocrystal 2.
Table 2 Calculated energies and energetic differences in interaction site pairing energies (kJ mol−1) of the screened components and their potential cocrystal outcome considering a 1:1 molar ratio
| Component combinations |
E (acid) |
E (dPy) |
E (cocrystal) |
ΔE |
| HPip + 4-Acpy |
−24.5 |
−12.7 |
−40.4 |
−3.2 |
| 2-FA + 4-Acpy |
−23.2 |
−37.8 |
−1.9 |
| HCinn + 4-Acpy |
−21.4 |
−35.0 |
−0.9 |
| HACA + 4-Acpy |
−39.0 |
−52.0 |
−0.3 |
| HPip + Pdon |
−24.5 |
−24.6 |
−50.1 |
−1.0 |
| 2-FA + Pdon |
−23.1 |
−48.7 |
−1.0 |
| HACA + Pdon |
−39.0 |
−64.1 |
−0.5 |
| HCinn + Pdon |
−21.4 |
−46.5 |
−0.5 |
According to Etter's rule,51,52 it is expected that for the HPip:4-Acpy combination, the acid⋯pyridine heterosynthon stands out as its main synthon (Fig. 1a). In addition, for HACA:Pdon, the carboxylic and amide moieties presented similar α values (Fig. 1b), which should compete or cooperate towards H-bond formation with the carbonyl moiety from Pdon, which presents the highest β value.
 |
| | Fig. 1 MEP representations for the components of cocrystals (a) 1 and (b) 2 with their corresponding α (red) and β (blue) values. | |
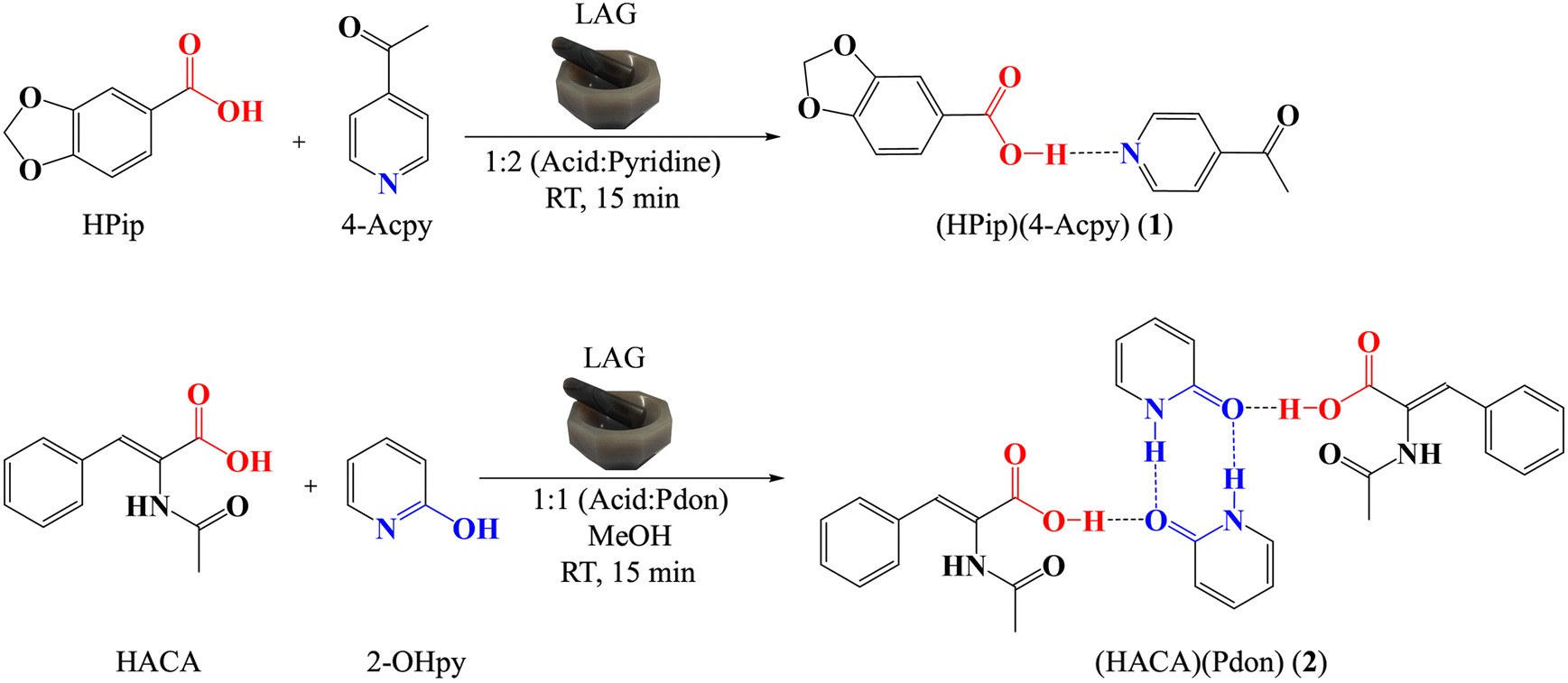
Neat grinding has been established as an efficient and greener approach to obtain cocrystals compared with conventional solution methods. However, it has been demonstrated that the addition of small quantities of solvent during the grinding process, referred to as liquid-assisted grinding (LAG), improves significantly the kinetics of the reactions, allowing a better molecular diffusion, while keeping the high yields commonly achieved through mechanochemical methods.53 Hence, we selected this methodology for the preparation of both cocrystals. Cocrystal 1 was afforded using a 1:2 (HPip:4-Acpy) molar ratio, where the excess of 4-Acpy served as the medium for the LAG synthesis. Instead, for cocrystal 2 a small amount of MeOH promoting a more displaced shifting towards Pdon formation was employed (Scheme 2),54 using a 1:1 (HACA:2-OHpy) molar ratio.
 |
| | Scheme 2 Tautomeric equilibrium between 2-OHpy and Pdon in the presence of MeOH. | |
Characterization of cocrystals 1 and 2
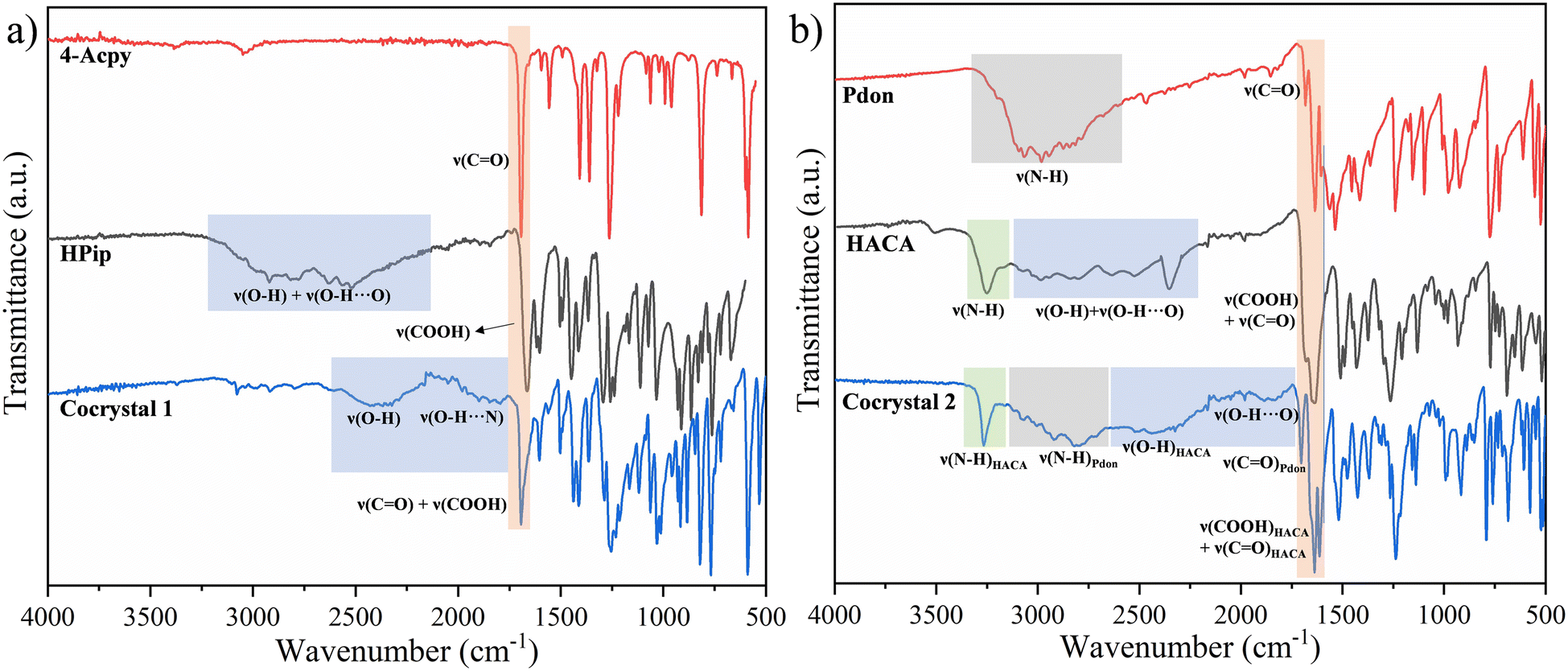
Cocrystals 1 and 2 were characterized by powder X-ray diffraction (PXRD), elemental analysis (EA), FTIR-ATR, 1H, 13C{1H}, and DEPT-135 NMR spectroscopies, and single crystal X-ray diffraction. The phase purity of the ground samples was verified by PXRD (ESI† Fig. S1 and S2). The EA agrees with the proposed formulas. The FTIR-ATR spectra of both cocrystals displayed broad bands attributable to ν(O–H) of the carboxylic acids in the 2691–2156 cm−1 (1) and 2556–2075 cm−1 (2) regions. These bands are shifted to lower wavenumbers compared with the free carboxylic acids due to the formation of the acid⋯pyridine heterosynthons (Fig. 2). Additional broad bands at 2135–1757 cm−1 (1) and 2029–1746 cm−1 (2) have also been assigned to the ν(O–H⋯N/O) vibrations of the acid⋯pyridine (1) and acid⋯amide (2) heterosynthons, while the ν(CO) (1693 cm−1 (1); 1703 cm−1 (2)), and ν(COOH) (1693 cm−1 (1); 1636 cm−1 (2)) vibrations have also been identified. In addition, the spectrum of cocrystal 2 also presents two additional bands in the upper region, displaying a sharp signal at 3263 cm−1 and another broad band between 3167 and 2604 cm−1, which are assigned to ν(N–H) from HACA and Pdon, respectively. Accordingly, these bands suggest that for cocrystal 2 various synthons are responsible for the assembly of the cocrystal, showing the strongest strength for the interactions involving the H donors in the order NHHACA > NHPdon > COOHHACA, respectively (Fig. 2).19,55 The complete assignation is provided in the Experimental section and the ESI† (Fig. S3 and S4).
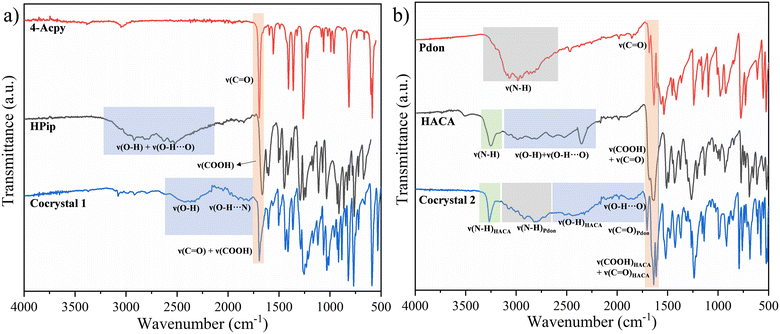 |
| | Fig. 2 Comparative FTIR-ATR spectra of the former components and the resulting cocrystal in (a) 1 and (b) 2. | |
The 1H, 13C{1H} and DEPT-135 NMR spectra of cocrystals 1 and 2 have been recorded in CD3OD solution. The spectra show the signals attributable to the corresponding ligands with an acid:dPy ratio of 1:1 for both compounds (ESI† Fig. S5 and S6). Furthermore, the 13C{1H} NMR spectra present all the bands of the corresponding ligands (ESI† Fig. S7 and S8). Both 1H and 13C{1H} spectra of cocrystal 2 do not show signals corresponding to 2-OHPy, which combined with the presence of three signals in the carbonyl region of its 13C{1H} NMR spectrum (173.2–165.8 ppm) suggest the full conversion of 2-OHpy to Pdon. Further details about the proton and carbon assignations are provided in the Experimental section and the ESI† (Fig. S5 and S8).
Crystal and extended structure of cocrystal 1
Cocrystal 1 belongs to the triclinic P space group. It consists of a binary cocrystal formed by one HPip and one 4-Acpy molecules held together by an acid⋯pyridine heterosynthon (O(1)–H(1O)⋯N(1)) (Fig. 3a), in line with the MEP surface prediction. This synthon represents 3.9% of the HPip and 6.5% of the 4-Acpy area in their 2D fingerprint plots with distances notably shorter compared with the rest of the interactions (Table 3), standing out as the main interaction of the basic structural motif (BSM) (ESI† Fig. S9 and S10),56 which is also shown in its Hirshfeld surfaces as well as its associated interaction energy (Table 3). In addition, the presence of the C(4)–H(4)⋯O(2) association completes the construction of a supramolecular ring with R44(20) as the graph-set descriptor (Table 3 and Fig. 3a). Then, the C(11)–H(11)⋯O(4), C(12)–H(12)⋯O(5) and C(6)–H(6A)⋯O(5) interactions lead to the formation of a R23(8) ring, while the latter interaction is also involved in the formation of a R22(7) ring conjointly with the C(16)–H(16A)⋯O(3) association (Fig. 3b). The 2D expansion along the (4![[4 with combining macron]](https://www.rsc.org/images/entities/char_0034_0304.gif) 0) plane is completed by a R33(16) motif formed by the C(11)–H(11)⋯O(4), C(16)–H(16A)⋯O(3) and C(4)–H(4)⋯O(2) interactions (Fig. 3b). These associations define two orthogonal layers connected by C–H⋯π interactions between a methyl H atom from 4-Acpy and a HPip ring (C(16)–H(16C)⋯Cg(1)), assembling a 2D bilayer with 56.22° of slippage. Furthermore, reciprocal C–H⋯O associations between a dioxole H atom and the carbonyl oxygen atoms from HPip (C(6)–H(6B)⋯O(2)) supported the bilayer formation, shaping an R22(17) ring (Fig. 3b). Finally, the bilayers interact between them by weak π⋯π forces between 4-Acpy ligands, forming a 3D network presenting a {39·422·55} point symbol corresponding to a nci underlying topology (Fig. 3c). Detailed information on the intermolecular interactions and their associated interaction energies is provided in Table 3. The complete topological analysis is supplied in the ESI† (Fig. S13).
0) plane is completed by a R33(16) motif formed by the C(11)–H(11)⋯O(4), C(16)–H(16A)⋯O(3) and C(4)–H(4)⋯O(2) interactions (Fig. 3b). These associations define two orthogonal layers connected by C–H⋯π interactions between a methyl H atom from 4-Acpy and a HPip ring (C(16)–H(16C)⋯Cg(1)), assembling a 2D bilayer with 56.22° of slippage. Furthermore, reciprocal C–H⋯O associations between a dioxole H atom and the carbonyl oxygen atoms from HPip (C(6)–H(6B)⋯O(2)) supported the bilayer formation, shaping an R22(17) ring (Fig. 3b). Finally, the bilayers interact between them by weak π⋯π forces between 4-Acpy ligands, forming a 3D network presenting a {39·422·55} point symbol corresponding to a nci underlying topology (Fig. 3c). Detailed information on the intermolecular interactions and their associated interaction energies is provided in Table 3. The complete topological analysis is supplied in the ESI† (Fig. S13).
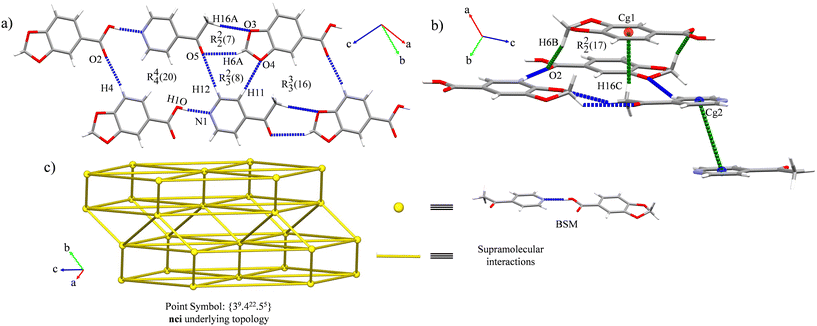 |
| | Fig. 3 (a) General view of the 2D layers along the (40) plane in 1. (b) View of the 3D expansion of cocrystal 1 by weak interactions highlighted in green. (c) Schematic representation of the topology of cocrystal 1. | |
Table 3 Selected intermolecular interactions for cocrystal 1
| D–H⋯A (Å) |
D–H (Å) |
H⋯A (Å) |
D⋯A (Å) |
>D–H⋯A (°) |
Associated energy (kJ mol−1) |
Number of interactionsa |
| O(1)–H(1O) ⋯N(1) |
0.84 |
1.78 |
2.622(1) |
174 |
−46.2 |
1 |
| C(4)–H(4)⋯O(2) |
0.95 |
2.38 |
3.324(1) |
173 |
−9.1 |
1 |
| C(11)–H(11)⋯O(4) |
0.95 |
2.48 |
3.393(1) |
162 |
−11.7 |
1 |
| C(12)–H(12)⋯O(5) |
0.95 |
2.58 |
3.521(1) |
171 |
−7.8 |
1 |
| C(16)–H(16A)⋯O(3) |
0.98 |
2.51 |
3.449(1) |
160 |
−13.8 |
1 |
| C(6)–H(6A)⋯O(5) |
0.99 |
2.61 |
3.409(1) |
138 |
| C(6)–H(6B)⋯O(2) |
0.99 |
2.54 |
3.380(1) |
143 |
−28.6 |
2 |
| X–H⋯Cg(J) |
H⋯Cg(J) (Å) |
H-Perpb (Å) |
γ
(°) |
X⋯Cg(J) (Å) |
X–H, Pid (°) |
Associated energy (kJ mol−1) |
Number of interactionsa |
| C(16)–H(16C)⋯Cg(1) |
2.77 |
2.76 |
4.80 |
3.5418(13) |
50 |
−29.4 |
1 |
| Cg(I)⋯Cg(J) |
d
Cg⋯Cg
(Å) |
α
(°) |
β, γg (°) |
d
plane⋯plane
(Å) |
d
offset
(Å) |
Associated energy (kJ mol−1) |
Number of interactionsa |
|
Number of interactions encompassed in each associated total energy.
Perpendicular distance of H to ring plane J.
Angle between Cg(J)–H vector and ring J normal.
Angle of the X–H bond with the Pi plane (Pi stands for plane of the aromatic ring; perpendicular = 90°, parallel = 0°).
Cg⋯Cg = distance between ring centroids.
α = dihedral angle between planes I and J.
Offset angles: β = angle Cg(I)–Cg(J) and normal to plane I and γ = angle Cg(I)–Cg(J) and normal to plane J (β = γ, when α = 0°).
Perpendicular distance of Cg(I) on plane J and perpendicular distance of Cg(J) on plane I (equal when α = 0°).
Slippage = horizontal displacement between Cg(I) and Cg(J) (equal for both centroids when α = 0°). Cg(1) = C(2) C(3) C(4) C(5) C(7) C(8); Cg(2) = N(1) C(10) C(11) C(12) C(14) C(15).
|
| Cg(2)⋯Cg(2) |
3.9732(7) |
0.00(5) |
30.0 |
3.4423(5) |
1.984 |
−11.1 |
1 |
A search in the CSD33 of multicomponent solids containing 4-Acpy revealed a total of 5 salts57–61 and 10 cocrystals.62–69 Details about the components forming the cocrystals as well as their dimensionalities are provided in the ESI† (Table S1). Within the 10 cocrystal structures, their BSMs were held together by alcohol⋯pyridine (2 hits),62,63 halogen⋯pyridine (2 hits),64 and acid⋯pyridine (6 hits) heterosynthons.65–69 Focusing on the latter type of cocrystals, they contained monoacids (4 hits),65–67 diacids (1 hit),68 and triacids (1 hit),69 all of them presenting similar bond lengths and angles of their H-bonds compared with cocrystal 1. Analysis of the dimensionality of these structures showed 3 hits with layered arrangements connected by weak interactions leading to 3D nets,67–69 while the remaining 3 hits consisted of different types of 3D networks,70,71 probably attributed to the presence of bidirectional H-donor groups (e.g. amine groups) able to tilt neighboring molecules towards the formation of recurrent synthons such as the amine⋯acid or the amine⋯amine. Thomas et al. found that in the 4-Acpy:3-fluorobenzoic acid system, the ability to form layered arrays was ascribed to the orientation of the acetyl moiety, showing that small changes completely altered their resulting crystal packings.67 Thus, it is important to highlight the need to incorporate an additional H-acceptor group in the acid⋯4-Acpy-based cocrystals that orients the molecules in a planar fashion by a recurrent synthon or by a medium/weak interaction unable to tilt the molecules out of the plane.
In cocrystal 1, the presence of the dioxole group of HPip provides a donor (H6A) and an acceptor (O3) atom that interact with the nearby 4-Acpy, ordering the molecules in a proper HPip⋯4-Acpy⋯HPip pattern towards the layered arrangement (Fig. 3a). These interactions leave only one H atom from the dioxole unit (H6B) to connect two adjacent layers that are oppositely aligned in a way that both dispose their free H atom towards the formation of a slipped bilayer through the formation of an R22(17) ring (Fig. 3b), bringing this combination of acid⋯pyridine synthons and weak interactions of 1 as an adequate example for the formation of layered arrangements.
Crystal and extended structure of cocrystal 2
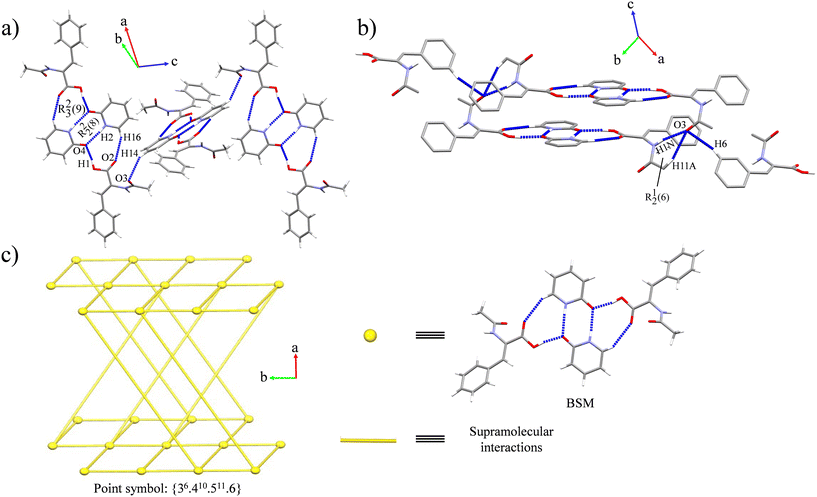
Cocrystal 2 belongs to the monoclinic P21/c space group. It consists of a binary cocrystal formed by two HACA and two Pdon molecules. The BSM of the cocrystal is held together by an R22(8) ring between Pdon molecules forming a reciprocal amide⋯amide homosynthon (N(2)–H(2)⋯O(4)), being the strongest contribution, as the Hirshfeld surface of Pdon and its associated interaction energy indicate (Table 4 and Fig. S11 and S12, ESI†). Moreover, this motif is supported by two H-bonds between two carboxylic acid moieties with two carbonyl groups from Pdon (O(1)–H(1)⋯O(4)), forming two acid⋯amide heterosynthons, which combined with two C–H⋯O interactions between the carboxylate oxygen atoms and a H atom from Pdon (C(16)–H(16)⋯O(2)) lead to a robustly connected tetrameric array (Fig. 4a). This arrangement is in line with the observed α and β values of the MEP predictions, which also follows Etter's rule.51,52 The tetramers are ordered along the [001] direction in a crossed fashion with a torsion angle of 84.60° between BSMs owing to the C(14)–H(14)⋯O(3) interaction (Fig. 4a). Finally, the amide⋯amide homosynthons between HACA molecules (N(1)–H(1N)⋯O(3)), supported by the C(11)–H(11A)⋯O(3) interactions propagate the structure along the [010] direction, while the C(6)–H(6)⋯O(3) association extends the array along the [100] direction, forming a 3D net with {36·410·511·6} as point symbol (Fig. 4b and c).72 Remarkably, the associated interaction energies of the three recurrent synthons driving the packing of 2 (Pdon⋯Pdon, acid⋯amide and amide⋯amide) are in line with the order of strengths suggested by its FTIR-ATR spectrum. Detailed information on the intermolecular interactions and their associated interaction energies is provided in Table 4. The complete topological analysis is supplied in the ESI† (Fig. S14).
Table 4 Selected intermolecular interactions for cocrystal 2a
| D–H⋯A (Å) |
D–H (Å) |
H⋯A (Å) |
D⋯A (Å) |
>D–H⋯A (°) |
Associated energy (kJ mol−1) |
Number of interactionsa |
|
Number of interactions encompassed in each associated total energy.
|
| N(2)–H(2)⋯O(4) |
0.88 |
1.88 |
2.754(1) |
175 |
−91.6 |
2 |
| O(1)–H(1)⋯O(4) |
0.920(19) |
1.657(19) |
2.561(1) |
166.7(13) |
−44.7 |
1 |
| C(16)–H(16)⋯O(2) |
0.95 |
2.49 |
3.344(2) |
150 |
−12.1 |
1 |
| C(14)–H(14)⋯O(3) |
0.95 |
2.60 |
3.445(1) |
148 |
−22.6 |
1 |
| N(1)–H(1 N)⋯O(3) |
0.88 |
2.00 |
2.866(1) |
169 |
−53.6 |
1 |
| C(11)–H(11A)⋯O(3) |
0.98 |
2.55 |
3.412(2) |
147 |
| C(6)–H(6)⋯O(3) |
0.95 |
2.82 |
3.592(2) |
139 |
−30.5 |
2 |
 |
| | Fig. 4 Propagation of the BSMs of cocrystal 2 along the (a) [001] direction and (b) (110) plane. (c) Schematic representation of the topology of cocrystal 2. | |
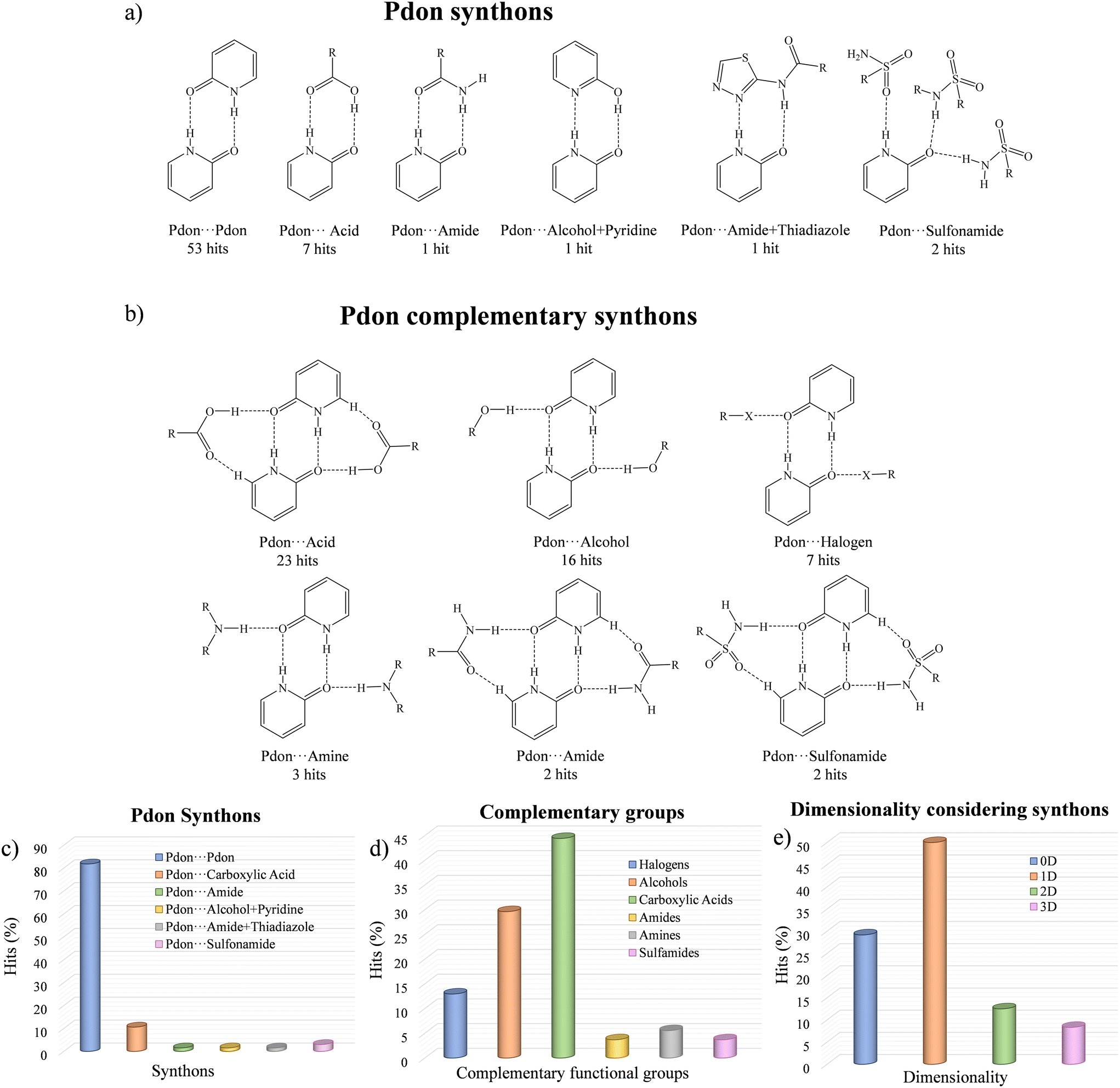
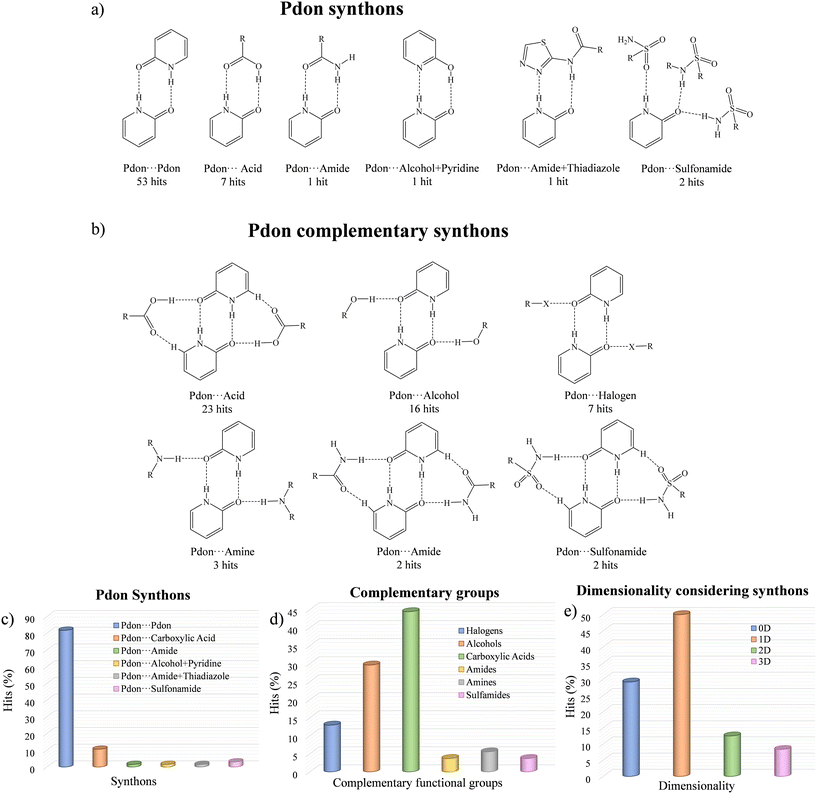
A search in the CSD33 of cocrystals containing Pdon/2-OHpy molecules showed 73 hits, whose main information is summarized in Fig. 5 and detailed in the ESI† (Table S2). It was observed that the Pdon form promotes the formation of cocrystals (65 hits), while the 2-OHpy form resulted in molecular salts (8 hits). Within the cocrystal arrangements, the Pdon⋯Pdon homosynthon stands out as the most abundant primary synthon, appearing in 81.54% of the hits, while in the rest of the dispositions it is disrupted by heterosynthons involving carboxylic acids (10.77%), or even combinations of various sulfonamides (3.08%), amides (1.54%), alcohols and pyridines (1.54%), or amides and thiadiazoles (1.54%) (Fig. 5a and c). In addition, the disposition of the carbonyl group in the Pdon⋯Pdon motif allows the presence of a secondary synthon able to stabilize a tetrameric array through carboxylic acids (43.40%), alcohols (30.19%), halogens (13.21%), amines (5.66%), amides (3.77%), or sulfonamides (3.77%) (Fig. 5b and d). This supramolecular motif brings the potential possibility to form ternary cocrystals where different components could be located forming asymmetric secondary synthons with Pdon. However, to the best of our knowledge they have not been found so far, and thus, only the 1:1 proportion is expected in this type of cocrystal arrays. The overall packing considering the synthons resulted in 0D tetramers (7 hits), 1D chains (12 hits), 2D layers (2 hits) and 3D nets (2 hits), whose scaffolds are extended by weak interactions (i.e. C–H⋯O, C–H⋯π, and π⋯π interactions) leading to 1D chains (5 hits), 2D layers (8 hits), and 3D nets (10 hits) (Fig. 5e and Table S2, ESI†). Among them, the family of aliphatic diacids has been extensively studied showing 13 of the hits, leading to tetrameric arrays forming 1D chains assisted by primary Pdon⋯Pdon and secondary Pdon⋯acid synthons in most of the examples,73–78 while in a few cases there is a competition between the Pdon⋯Pdon and the acid⋯acid interactions as primary synthons.78 Remarkably, the obtention of 2D/3D arrangements using recurrent synthons has been achieved only with sulfonamides,79,80 alcohols,74 or cocrystallized water molecules.73 Nevertheless, these examples evinced the low reliability of these groups, which presented different behaviors within the same crystal structure. However, in cocrystal 2 the different orientations of the acid and amide from HACA moieties combined with the synthon reliability between the amide moieties of Pdon lead to the formation of strongly arranged 2D layers, being the first example of this kind of disposition through consistent supramolecular synthons, which defines this combination of carboxylic acid and amide moieties with this specific disposition as a promising system to be further applied to the design of cocrystals of higher complexity.
 |
| | Fig. 5 (a) Types of primary supramolecular synthons involving Pdon. (b) Types of complementary groups involved in the secondary Pdon synthons. Outline of the results of the CSD study regarding (c) types of primary Pdon synthons, (d) complementary groups involved in the secondary Pdon synthons, and (e) dimensionality considering recurrent synthons. | |
Thermal properties of cocrystals 1 and 2
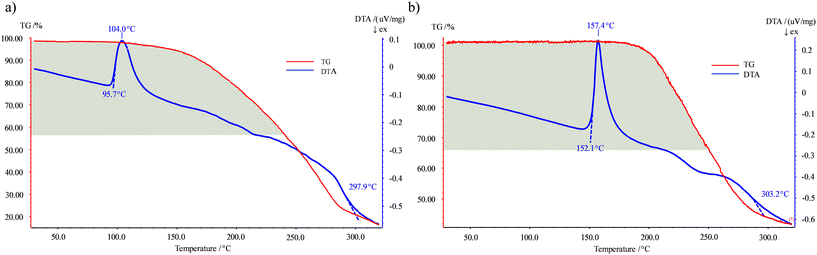
The thermal behavior of cocrystals 1 and 2 was studied by TG-DTA. For both cocrystals a single endothermic event is observed involving melt degradation of the products. The melting point peak of cocrystal 1 presents a Tonset = 95.7 °C and a Tpeak = 104.0 °C followed by the continuous loss of mass of all its components and its further decomposition at 297.9 °C (Fig. 6a). However, the DTA curve showed a change in the thermal events around 215.0 °C, which approximately fits with the mass of 4-Acpy (exp. 43.0%; calc. 42.2%) and could indicate that the loss of mass in cocrystal 1 started with the pyridine component followed by carboxylic acid. Otherwise, for cocrystal 2 the melting point appeared at a higher temperature, displaying a Tonset = 152.1 °C and a Tpeak = 157.4 °C, followed again by the continuous loss of mass of all its components and its further decomposition at 303.2 °C (Fig. 6b). Likewise, the DTA curve revealed a change around 210.0 °C, which could be associated with the mass of Pdon (exp. 33.8%; calc. 31.7%) and could indicate that the loss of mass followed the same trend as in 1. In addition, the values obtained using the melting point apparatus (110–111 °C, 1; 156–157 °C, 2) were in line with those observed in the thermograms and lie in between the melting point values of their former molecules, as obtained in most of the reported examples.81 However, the formation of cocrystal 1 represents an interesting example of how a liquid (4-Acpy) can be stabilized in the solid state using the cocrystal approach, which is pursued in terms of stability and processability.12,15
 |
| | Fig. 6 TG/DTA of cocrystal (a) 1 and (b) 2 between 25 °C and 350 °C. The regions marked in green are suggested as the areas where the first change after the melting point event is observed in the DTA curves, which potentially fit with the loss of mass of the corresponding dPy of cocrystals 1 and 2. | |
Structure–property relationship in cocrystals 1 and 2
The relationship between the crystal packing of cocrystals 1 and 2 and their melting points was investigated using energy framework calculations.82 To this aim, the different contribution energies for both cocrystals were analyzed, observing a higher influence of the electrostatic factors rather than the dispersion effects (ESI† Fig. S15). Detailed values of the contribution of each energetic type are provided in the ESI† (Table S3).
To select the more representative interactions between each molecular pair, the associated total energy of the interactions between each cocrystal molecule have been plotted with respect to the radius around a central cocrystal molecule, showing that the main interactions that held together the crystal packing of 1 and 2 present associated interaction energies below −20 kJ mol−1 (Fig. 7a and b). Thus, a cut-off energy of −20 kJ mol−1 was used for the energy framework representations of Fig. 7c–h. For cocrystal 1, the acid⋯pyridine heterosynthon was highlighted as the unique electrostatic component above the cut-off energy, forming discrete BSM entities (Fig. 7c). Remarkably, the dispersion energy represented in Fig. 7d stands out as the main contributor for the connection of the BSMs mainly through the interactions responsible for the bilayer formation (C(6)–H(6B)⋯O(2)) and (C(16)–H(16C)⋯Cg(1)), leading to 2D layers (Fig. 7e). In addition, for cocrystal 2 a strong binding of its BSM through the Pdon⋯Pdon and Pdon⋯acid synthons and the interactions responsible for the expansion along the [010] direction (N(1)–H(1N)⋯O(3) + C(11)–H(11A)⋯O(3)) are the more representative electrostatic contributions (Fig. 7f), while the dispersion forces do not play a key role such as in 1 (Fig. 7g), leading to 3D nets (Fig. 7h).
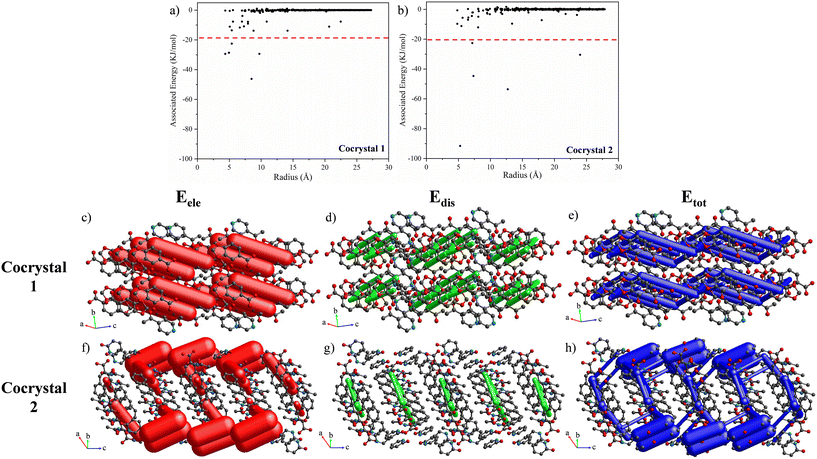 |
| | Fig. 7 Relationship between the molecular pair radius and the associated interaction energies in (a) cocrystal 1 and (b) cocrystal 2. Energy frameworks (Eele, Edis, Etot) for cocrystals (c–e) 1 and (f–h) 2. All the diagrams use the same energy cylinder scale factor of 150 and an energy cut-off of −20 kJ mol−1 within a 2 × 2 × 2 (1) and a 1 × 2 × 2 (2) unit cell. | |
From these data, it is inferred that the 2D layers assembled by weakly dispersive forces of 1 resulted in a lower melting point than the 3D nets connected by strong electrostatic interactions of 2, showing a difference of 53.4 °C. This is also supported by their corresponding lattice energies, with the packing of 2 being significantly more stable than that of 1, which also correlates with their crystal densities (Table 5).83,84 Therefore, the extrapolation of these combinations of interactions to other systems bearing relevant molecules could be a useful way to modulate the melting points of either liquid or solid components.
Table 5 Structural parameters and melting point values of cocrystals 1 and 2
| Cocrystal |
Melting point (°C) |
Crystal density (g cm−3) |
E
latt (kJ mol−1) |
|
1
|
104.0 |
1.474 |
−211.0 |
|
2
|
157.4 |
1.361 |
−274.2 |
Conclusions
Two binary cocrystals have been successfully predicted using the critical points from the MEP surfaces of the initial components and then prepared by LAG and fully characterized. Their compositions include a liquid (4-Acpy) and a solid (Pdon) dPy combined with two different carboxylic acids (HPip and HACA, respectively). The elucidation of their crystal structures revealed that their BSMs are constructed from the acid⋯pyridine heterosynthon (1) and the Pdon⋯Pdon homosynthon supported by secondary Pdon⋯acid synthons (2), leading to a dimeric (1) and a tetrameric (2) BSMs. The study of their crystal packings showed the formation of bilayers connected via weak π⋯π interactions (1) and a 3D network where the BSM is arranged by recurrent amide⋯amide interactions between HACA ligands supported by other weak C–H⋯O associations (2). Furthermore, a CSD study of the cocrystals containing 4-Acpy evinced the challenge that supposes the formation of cocrystals using liquid components, with 1 as one of the few examples containing 4-Acpy. Otherwise, the CSD search of cocrystals bearing Pdon showed a great versatility of this system, displaying a high affinity for the Pdon⋯Pdon homosynthon in the presence of different competitor groups, and tunable lateral positions where mostly carboxylic acids, alcohols and halogens were attached. However, to the best of our knowledge, 2 is the first reported cocrystal forming 2D layers by recurrent supramolecular synthons involving Pdon. Finally, the melting points of 1 and 2 have been determined and the resulting values have been correlated with their structures using energy frameworks, observing 2D layers based on dispersive interactions where the BSMs are connected by the acid⋯pyridine heterosynthon for 1, and a 3D net connected by stronger electrostatic interactions for 2, which results in a remarkably higher melting point of 2 (157.4 °C) compared with 1 (104.0 °C). This work provides new examples of binary cocrystals and correlates the structural features with their thermal stability as well as collects some tendencies regarding the preferred behavior of 4-Acpy and Pdon in the formation of cocrystals, which can be helpful for further research in the design of new cocrystals.
Author contributions
Conceptualization, J. P. Methodology, D. E. Software, D. E. Validation, J. P. and T. C. Formal analysis, D. E. and M. F.-B. Investigation, D. E. Resources, J. P. and T. C. Data curation, D. E. and M. F.-B. Writing—original draft preparation, D. E. Writing—review and editing, J. P. Visualization, D. E. Supervision, J. P. Project administration, J. P. Funding acquisition, J. P. All authors have read and agreed to the published version of the manuscript.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
J. P. acknowledges financial support from the CB615921 project, the CB616406 project from “Fundació La Caixa”, and the 2021SGR00262 project from the Generalitat de Catalunya. D. E. acknowledges the PIF pre-doctoral fellowship from the Universitat Autònoma de Barcelona.
References
- N. K. Duggirala, M. L. Perry, Ö. Almarsson and M. J. Zaworotko, Chem. Commun., 2016, 52, 640–655 RSC.
- M. A. E. Yousef and V. R. Vangala, Cryst. Growth Des., 2019, 19, 7420–7438 CrossRef CAS.
- Y. Xiao, C. Wu, L. Zhou, Q. Yin and J. Yang, Green Chem., 2022, 24, 8088–8099 RSC.
- F. Montisci, P. P. Mazzeo, C. Carraro, M. Prencipe, P. Pelagatti, F. Fornari, F. Bianchi, M. Careri and A. Bacchi, ACS Sustainable Chem. Eng., 2022, 10, 8388–8399 CrossRef CAS.
- C. Zhang, F. Jiao and H. Li, Cryst. Growth Des., 2018, 18, 5713–5726 CrossRef CAS.
- J. C. Bennion and A. J. Matzger, Acc. Chem. Res., 2021, 54, 1699–1710 CrossRef CAS PubMed.
- P. R. Laity, G. M. Day and W. Jones, Adv. Mater., 2009, 21, 3905–3909 CrossRef.
- J. Christopherson, F. Topic, C. J. Barrett and T. Friščic, Cryst. Growth Des., 2018, 18, 1245–1259 CrossRef CAS.
- O. N. Kavanagh, D. M. Croker, G. M. Walker and M. J. Zaworotko, Drug Discovery Today, 2019, 24, 796–804 CrossRef CAS PubMed.
- G. Bolla, B. Sarma and A. K. Nangia, Chem. Rev., 2022, 122, 11514–11603 CrossRef CAS PubMed.
- Ö. Almarsson, M. L. Peterson and M. Zaworotko, Pharm. Pat. Anal., 2012, 1, 313–327 CrossRef PubMed.
- C. B. Aakeröy, T. K. Wijethunga, J. Benton and J. Desper, Chem. Commun., 2015, 51, 2425–2428 RSC.
- D. Capucci, D. Balestri, P. P. Mazzeo, P. Pelagatti, K. Rubini and A. Bacchi, Cryst. Growth Des., 2017, 17, 4958–4964 CrossRef CAS.
- P. P. Mazzeo, C. Carraro, A. Monica, D. Capucci, P. Pelagatti, F. Bianchi, S. Agazzi, M. Careri, A. Raio, M. Carta, F. Menicucci, M. Belli, M. Michelozzi and A. Bacchi, ACS Sustainable Chem. Eng., 2019, 7, 17929–17940 CrossRef CAS.
- A. Bacchi, D. Capucci, M. Giannetto, M. Mattarozzi, P. Pelagatti, N. Rodriguez-Hornedo, K. Rubini and A. Sala, Cryst. Growth Des., 2016, 16, 6547–6555 CrossRef CAS.
- A. D. Bond, CrystEngComm, 2007, 9, 833–834 RSC.
- G. R. Desiraju, Angew. Chem., Int. Ed. Engl., 1995, 34, 2311–2327 CrossRef CAS.
- T. R. Shattock, K. K. Arora, P. Vishweshwar and M. J. Zaworotko, Cryst. Growth Des., 2008, 8, 4533–4545 CrossRef CAS.
- S. Saha and G. R. Desiraju, J. Am. Chem. Soc., 2018, 140, 6361–6373 CrossRef CAS PubMed.
- L. R. MacGillivray, G. S. Papaefstathiou, T. Friščić, T. D. Hamilton, D.-K. Bučar, Q. Chu, D. B. Varshney and I. G. Georgiev, Acc. Chem. Res., 2008, 41, 280–291 CrossRef CAS PubMed.
- M. K. Corpinot and D.-K. Bučar, Cryst. Growth Des., 2019, 19, 1426–1453 CrossRef CAS.
- R. Bhowal, S. Biswas, A. Thumbarathil, A. L. Koner and D. Chopra, J. Phys. Chem. C, 2019, 123, 9311–9322 CrossRef CAS.
- A. M. Abeysekera, B. B. Averkiev, A. S. Sinha and C. B. Aakeröy, Chem. Commun., 2022, 58, 9480–9483 RSC.
- M. K. Stanton and A. Bak, Cryst. Growth Des., 2008, 8, 3856–3862 CrossRef CAS.
- J. B. Nanubolu and K. Ravikumar, CrystEngComm, 2016, 18, 1024–1038 RSC.
- S. Tothadi and A. Phadkule, CrystEngComm, 2019, 21, 2481–2484 RSC.
- O. Bolton and A. J. Matzger, Angew. Chem., 2011, 123, 9122–9125 CrossRef.
- H. Qu, S. Jin, J. Gong, S. Du, L. Jia and S. Wu, Cryst. Growth Des., 2020, 20, 7356–7367 CrossRef CAS.
- S. Majodina, L. Ndima, O. O. Abosede, E. C. Hosten, C. M. A. Lorentino, H. F. Frota, L. S. Sangenito, M. H. Branquinha, A. L. S. Santos and A. S. Ogunlaja, CrystEngComm, 2021, 23, 335–352 RSC.
- F. Sánchez-Férez, D. Ejarque, T. Calvet, M. Font-Bardia and J. Pons, Molecules, 2019, 24, 4169 CrossRef PubMed.
- D. Ejarque, T. Calvet, M. Font-Bardia and J. Pons, Crystals, 2021, 11, 191 CrossRef CAS.
- N. Bedeković, L. Fotović, V. Stilinović and D. Cinčić, Cryst. Growth Des., 2022, 22, 987–992 CrossRef PubMed.
- C. R. Groom, I. J. Bruno, M. P. Lightfoot and S. C. Ward, Acta Crystallogr., Sect. B: Struct. Sci., Cryst. Eng. Mater., 2016, 72, 171–179 CrossRef CAS PubMed.
- C. F. MacRae, I. Sovago, S. J. Cottrell, P. T. A. Galek, P. McCabe, E. Pidcock, M. Platings, G. P. Shields, J. S. Stevens, M. Towler and P. A. Wood, J. Appl. Crystallogr., 2020, 53, 226–235 CrossRef CAS PubMed.
- Persistence of Vision Pty. Ltd. Persistence of Vision (TM) Raytracer, Williamstown, Australia, 2004, Available online: https://www.povray.org/, (Accessed 17 July 2023).
- V. A. Blatov, A. P. Shevchenko and D. M. Proserpio, Cryst. Growth Des., 2014, 14, 3576–3586 CrossRef CAS.
- D. Musumeci, C. A. Hunter, R. Prohens, S. Scuderi and J. F. McCabe, Chem. Sci., 2011, 2, 883–890 RSC.
- T. Grecu, C. A. Hunter, E. J. Gardiner and J. F. McCabe, Cryst. Growth Des., 2014, 14, 165–171 CrossRef CAS.
- H. Sun, Z. Jin, C. Yang, R. L. C. Akkermans, S. H. Robertson, N. A. Spenley, S. Miller and S. M. Todd, J. Mol. Model., 2016, 22, 47 CrossRef PubMed.
-
Dassault Systemes, BIOVIA Materials Studio, version 8.0.0.843, Dassault Systemes, San Diego, 2014 Search PubMed.
-
M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery, Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, T. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 09, Revision D.01, Gaussian, Inc., Wallingford CT, 2016 Search PubMed.
- M. Khalaji, M. J. Potrzebowski and M. K. Dudek, Cryst. Growth Des., 2021, 21, 2301–2314 CrossRef CAS.
- T. Lu and F. Chen, J. Comput. Chem., 2012, 33, 580–592 CrossRef CAS PubMed.
- W. Humphrey, A. Dalke and K. Schulten, J. Mol. Graphics, 1996, 14, 33–38 CrossRef CAS PubMed.
-
M. J. Turner, J. J. McKinnon, S. K. Wolff, D. J. Grimwood, P. R. Spackman, D. Jayatilaka and M. A. Spackman. CrystalExplorer17, University of Western Australia, 2017 Search PubMed.
-
D. Jayatilaka and D. Grimwood, in Computational Science – ICCS 2003, ed. P. A. Sloot, D. Abramson, A. Bogdanov, Y. Gorbachev, J. Dongarra and A. Zomaya, Springer Berlin Heidelberg, 2003, vol. 2660, pp. 142–151 Search PubMed.
- C. F. Mackenzie, P. R. Spackman, D. Jayatilaka and M. A. Spackman, IUCrJ, 2017, 4, 575–587 CrossRef CAS PubMed.
- S. P. Thomas, P. R. Spackman, D. Jayatilaka and M. A. Spackman, J. Chem. Theory Comput., 2018, 14, 1614–1623 CrossRef CAS PubMed.
- M. A. Spackman, CrystEngComm, 2018, 20, 5340–5347 RSC.
- A. O. Surov, A. P. Voronin, N. A. Vasilev, A. B. Ilyukhin and G. L. Perlovich, New J. Chem., 2021, 45, 3034–3047 RSC.
- M. C. Etter, Acc. Chem. Res., 1990, 23, 120–126 CrossRef CAS.
- M. C. Etter, J. Phys. Chem., 1991, 95, 4601–4610 CrossRef CAS.
- D. Braga, L. Maini and F. Grepioni, Chem. Soc. Rev., 2013, 42, 7638–7648 RSC.
- N. Tsuchida and S. Yamabe, J. Phys. Chem. A, 2005, 109, 1974–1980 CrossRef CAS PubMed.
- A. Mukherjee, S. Tothadi, S. Chakraborty, S. Ganguly and G. R. Desiraju, CrystEngComm, 2013, 15, 4640–4654 RSC.
- O. V. Shishkin, R. I. Zubatyuk, S. V. Shishkina, V. V. Dyakonenko and V. V. Medviediev, Phys. Chem. Chem. Phys., 2014, 16, 6773–6786 RSC.
- A. Kochel, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2005, 61, o926–o927 CrossRef CAS.
- X. Fu, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2009, 65, o1804 CrossRef CAS PubMed.
- X. Fu, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2009, 65, o2385 CrossRef CAS PubMed.
- J. Xu and X. Fu, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2010, 66, o1628 CrossRef CAS PubMed.
- A. F. Crespi, P. N. Zomero, V. M. Sánchez, A. L. Pérez, C. D. Brondino, D. Vega, E. Rodríguez-Castellón and J. M. Lázaro-Martínez, ChemPlusChem, 2022, 87, e202200169 CrossRef CAS PubMed.
- I. Majerz, Z. Malarski and W. Sawka-Dobrowolska, J. Mol. Struct., 1991, 249, 109–116 CrossRef CAS.
- A. Bacchi, M. Carcelli, T. Chiodo, G. Cantoni, C. De Filippo and S. Pipolo, CrystEngComm, 2009, 11, 1433–1441 RSC.
- N. Bedeković, T. Piteša, M. Eraković, V. Stilinović and D. Cinčić, Cryst. Growth Des., 2022, 22, 2644–2653 CrossRef PubMed.
- M. Singh, S. Anthal, P. J. Srijana, B. Narayana, B. K. Sarojini, U. Likhitha, Kamal and R. Kant, J. Mol. Struct., 2022, 1262, 133061 CrossRef CAS.
- T. Daisy Rani, M. Rajkumar and A. Chandramohan, Mater. Lett., 2018, 222, 118–121 CrossRef CAS.
- L. H. Thomas, G. A. Craig, M. J. Gutmann, A. Parkin, K. Shankland and C. C. Wilson, CrystEngComm, 2011, 13, 3349–3354 RSC.
- K. Xu, B.-Y. Zhang, J.-J. Nie and D.-J. Xu, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2009, 65, o1467–o1467 CrossRef CAS PubMed.
- S. Muniappan, S. Lipstman and I. Goldberg, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 2007, 63, o395–o399 CrossRef CAS PubMed.
- A. G. P. Maloney, P. A. Wood and S. Parsons, CrystEngComm, 2014, 16, 3867–3882 RSC.
- D. Li, M. Kong, J. Li, Z. Deng and H. Zhang, CrystEngComm, 2018, 20, 5112–5118 RSC.
- V. Tudor, G. Marin, V. Kravtsov, Y. A. Simonov, M. Julve, F. Lloret and M. Andruh, Rev. Roum. Chim., 2006, 51, 367–371 CAS.
- C. Aakeröy, A. Beatty and M. Zou, Cryst. Eng., 1998, 1, 225–241 CrossRef.
- T. Fukunaga, S. Kashino and H. Ishida, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 2004, 60, o718–o722 CrossRef PubMed.
- S. Kashino, T. Fukunaga, H. Izutsu and S. Miyashita, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 2001, 57, 627–631 CrossRef CAS PubMed.
- S. Mittapalli, M. K. C. Mannava, R. Sahoo and A. Nangia, Cryst. Growth Des., 2019, 19, 219–230 CrossRef CAS.
- C. B. Aakeröy, A. M. Beatty, M. Nieuwenhuyzen and M. Zou, Tetrahedron, 2000, 56, 6693–6699 CrossRef.
- M. R. Edwards, W. Jones and W. D. S. Motherwell, Cryst. Eng., 2002, 5, 25–36 CrossRef CAS.
- G. Bolla and A. Nangia, Chem. Commun., 2015, 51, 15578–15581 RSC.
- S. Allu, G. Bolla, S. Tothadi and A. Nangia, Cryst. Growth Des., 2017, 17, 4225–4236 CrossRef CAS.
- G. L. Perlovich, CrystEngComm, 2015, 17, 7019–7028 RSC.
- M. J. Turner, S. P. Thomas, M. W. Shi, D. Jayatilaka and M. A. Spackman, Chem. Commun., 2015, 51, 3735–3738 RSC.
- P. Vishweshwar, A. Nangia and V. M. Lynch, Cryst. Growth Des., 2003, 3, 783–790 CrossRef CAS.
- A. Nayak and V. R. Pedireddi, Cryst. Growth Des., 2016, 16, 5966–5975 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2023 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  a,
Teresa
Calvet
b,
Mercè
Font-Bardia
c and
Josefina
Pons
a,
Teresa
Calvet
b,
Mercè
Font-Bardia
c and
Josefina
Pons