DOI:
10.1039/D3CE00526G
(Paper)
CrystEngComm, 2023,
25, 3904-3915
Triple-armed aliphatic tricarboxylic acids as sources of ligands for uranyl ion: influence of bridgehead functionalization†
Received
23rd May 2023
, Accepted 12th June 2023
First published on 13th June 2023
Abstract
The two triple-armed species tris(2-carboxyethyl)nitromethane (H3tcenm) and tris(2-carboxyethyl)phosphine (H3tcep) have been used to synthesize seven uranyl ion complexes under (solvo)-hydrothermal conditions and in the presence of various structure-directing cations. The three carboxylate groups chelate three different cations and the nitro group is uncoordinated in [H2NMe2][UO2(tcenm)]·3H2O (1), [C(NH2)3][UO2(tcenm)]·0.5H2O (2) and [PPh3Me][UO2(tcenm)] (3), which crystallize as diperiodic coordination polymers with the hcb topology and minor variations in shape depending on the counterion. The two isomorphous complexes [UO2(tcenm)M(bipy)2][UO2(tcenm)]·3H2O, with M = Ni (4) or Cu (5) and bipy = 2,2′-bipyridine, display the same arrangement, with a M(bipy)22+ group bridging two adjoining carboxylate donors in one uranyl equatorial plane. [(UO2)2(tcenm)2Cu(R,S-Me6cyclam)]·2H2O (6), where R,S-Me6cyclam = 7(R),14(S)-5,5,7,12,12,14-hexamethyl-1,4,8,11-tetraazacyclotetradecane, is the only triperiodic framework in the series, with the tcs topology resulting from CuII pillaring diperiodic, uranyl-based sql networks. H3tcep is oxidized in situ to give the phosphine oxide H3tcepo, which is partially deprotonated in [UO2(Htcepo)] (7); the phosphine oxide and the two carboxylate groups are coordinated, and the diperiodic, three-dimensional network formed has the point symbol {66} and the vertex symbol 62·62·63·66·64·64, with the rings involved in Hopf links formation. Only complexes 3 and 7 are significantly emissive in the solid state, with photoluminescence quantum yields of 9%, and the emission maxima positions are in agreement with the number of uranyl equatorial donors.
Introduction
Although tripodal ligands possessing a hydrogen bond donor bridgehead able to interact with one uranyl oxo group were proposed long ago for selective uranyl recognition through so-called stereognostic coordination,1 and other tripodal ligands have been investigated for use in uranyl solvent extraction,2 they are not the most obvious choice for the synthesis of uranyl-based coordination polymers of frameworks.3 These known species are ligands with long and often rather rigid arms intended to be convergent about a single uranyl ion and thus to give mononuclear, discrete complexes; as such, they provide little prospect for use in uranyl-based coordination polymer synthesis. Of other ligands which can be viewed as tripodal due to their capacity to adopt a conformation displaying three convergent functional groups, most are of small size, such as the conformationally restricted tricarboxylate derived from Kemp's triacid (cis,cis-1,3,5-trimethylcyclohexane-1,3,5-tricarboxylic acid), and this is a ligand known to give a wide range of discrete polynuclear or polymeric species.4 A more commonly encountered, flexible tripodal ligand, with cation encapsulation ability and a potentially coordinating bridgehead, is nitrilotriacetate,5 for which, besides cases in which the central nitrogen atom is protonated5a,b or coordinated to an additional nickel(II) cation,5cONO-chelation of uranyl is known,5d with however the third carboxylate arm being diverted away so as to give a coordination polymer, the size of the tripodal cavity being definitely too small for uranyl ion encapsulation.1a A semi-rigid, tripodal tricarboxylate ligand based on the triazine platform has also been used to synthesize various cage-like or polymeric, diperiodic complexes,6 and a very recent publication has shown that a large coordination cage can be obtained with a calix[3]arene-carboxylate tripod rigidified by uranyl ion coordination as a bridgehead.7 Recently, several larger tripodal, zwitterionic tricarboxylates have been investigated,8 and 1,1′,1′′-[(2,4,6-trimethylbenzene-1,3,5-triyl)tris(methylene)]tris(pyridin-1-ium-4-carboxylate) in particular was shown to retain its convergent shape when encapsulating a bromide ion, while a more irregular conformation yielded a triperiodic uranyl-based framework.8d To further explore these diverse indications of competition between encapsulation and polymer formation, we have now synthesized a series of seven complexes from the triple-armed species tris(2-carboxyethyl)nitromethane (H3tcenm; 4-(2′-hydroxycarbonylethyl)-4-nitro-heptane-1,7-dioic acid; Scheme 1) and tris(2-carboxyethyl)phosphine (H3tcep; 4-(2′-hydroxycarbonylethyl)-4-phospha-heptane-1,7-dioic acid), which have been characterized by their crystal structure and emission spectrum in the solid state. Under the solvo-hydrothermal conditions used for the syntheses, the phosphine group in H3tcep is oxidized to give H3tcepo (Scheme 1), and the nitromethane and phosphine oxide bridgeheads appear to have very different effects on the charge of the bound ligand and the structures of the complexes formed, another instance of structural control through internal functionality of the bridging ligands.9
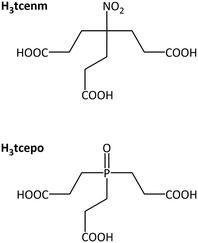 |
| | Scheme 1 The ligands H3tcenm and H3tcepo. | |
Experimental
Synthesis
Caution! Uranium is a radioactive and chemically toxic element, and uranium-containing samples must be handled with suitable care and protection. Small quantities of reagents and solvents were employed to minimize any potential hazards arising both from the presence of uranium and the use of pressurized vessels for the syntheses.
[UO2(NO3)2(H2O)2]·4H2O (RP Normapur, 99%) and Ni(NO3)2·6H2O were purchased from Prolabo. Tris(2-carboxyethyl)nitromethane (H3tcenm), tris(2-carboxyethyl)phosphine hydrochloride (H3tcep·HCl), and Cu(NO3)2·2.5H2O were from Sigma-Aldrich, 2,2′-bipyridine (bipy) was from Fluka, and guanidinium nitrate was from Alfa-Aesar. R,S-Me6cyclam (meso isomer of 7(R),14(S)-5,5,7,12,12,14-hexamethyl-1,4,8,11-tetraazacyclotetradecane) was prepared as described in the literature,10 and (7(R),14(S)-5,5,7,12,12,14-hexamethyl-1,4,8,11-tetraazacyclotetradecane)copper(II) dinitrate, [Cu(R,S-Me6cyclam)](NO3)2, was synthesized as previously reported.11 Elemental analyses were performed by MEDAC Ltd. For all syntheses, the solutions were placed in 10 mL tightly closed glass vessels (Pyrex culture tubes with SVL15 stoppers and Teflon-coated seals, provided by VWR) and heated at 140 °C in a sand bath (Harry Gestigkeit ST72). The crystals were grown in the hot, pressurized solutions and not as a result of a final return to ambient conditions.
[H2NMe2][UO2(tcenm)]·3H2O (1).
H3tcenm (28 mg, 0.10 mmol) and [UO2(NO3)2(H2O)2]·4H2O (50 mg, 0.10 mmol) were dissolved in a mixture of water (0.5 mL) and N,N-dimethylformamide (0.2 mL). Yellow crystals of complex 1 were obtained within three days (15 mg, 23%). Anal. Calcd for C12H26N2O13U: C, 22.37; H, 4.07; N, 4.35. Found: C, 22.65; H, 3.75; N, 4.20%.
[C(NH2)3][UO2(tcenm)]·0.5H2O (2).
H3tcenm (28 mg, 0.10 mmol), [UO2(NO3)2(H2O)2]·4H2O (35 mg, 0.07 mmol), and guanidinium nitrate (24 mg, 0.20 mmol) were dissolved in a mixture of water (0.6 mL) and acetonitrile (0.2 mL). Yellow crystals of complex 2 were obtained within four days (13 mg, 30%). Anal. Calcd for C22H38N8O21U2: C, 21.54; H, 3.12; N, 9.13. Found: C, 21.43; H, 3.26; N, 9.16%.
[PPh3Me][UO2(tcenm)] (3).
H3tcenm (28 mg, 0.10 mmol), [UO2(NO3)2(H2O)2]·4H2O (35 mg, 0.07 mmol), and PPh3MeBr (36 mg, 0.10 mmol) were dissolved in a mixture of water (0.6 mL) and acetonitrile (0.2 mL). Yellow crystals of complex 3 were obtained within two weeks (45 mg, 78%). Anal. Calcd for C29H30NO10PU: C, 42.40; H, 3.68; N, 1.70. Found: C, 42.23; H, 3.66; N, 1.74%.
[UO2(tcenm)Ni(bipy)2][UO2(tcenm)]·3H2O (4).
H3tcenm (28 mg, 0.10 mmol), [UO2(NO3)2(H2O)2]·4H2O (35 mg, 0.07 mmol), Ni(NO3)2·6H2O (15 mg, 0.05 mmol), and 2,2′-bipyridine (24 mg, 0.15 mmol) were dissolved in a mixture of water (0.5 mL) and acetonitrile (0.2 mL). Pale pink crystals of complex 4 were obtained within three days (31 mg, 59%). Anal. Calcd for C40H46N6NiO23U2: C, 31.74; H, 3.06; N, 5.55. Found: C, 31.92; H, 2.93; N, 5.75%.
[UO2(tcenm)Cu(bipy)2][UO2(tcenm)]·3H2O (5).
H3tcenm (28 mg, 0.10 mmol), [UO2(NO3)2(H2O)2]·4H2O (35 mg, 0.07 mmol), Cu(NO3)2·2.5H2O (12 mg, 0.05 mmol), and 2,2′-bipyridine (24 mg, 0.15 mmol) were dissolved in a mixture of water (0.5 mL) and acetonitrile (0.2 mL). Blue-purple crystals of complex 5 were obtained within three days (21 mg, 40%). Anal. Calcd for C40H46CuN6O23U2: C, 31.64; H, 3.05; N, 5.53. Found: C, 31.69; H, 2.88; N, 5.61%.
[(UO2)2(tcenm)2Cu(R,S-Me6cyclam)]·2H2O (6).
H3tcenm (28 mg, 0.10 mmol), [UO2(NO3)2(H2O)2]·4H2O (35 mg, 0.07 mmol), and [Cu(R,S-Me6cyclam)(NO3)2] (24 mg, 0.05 mmol) were dissolved in a mixture of water (0.6 mL) and acetonitrile (0.2 mL). Purple crystals of complex 6 were obtained within three days (16 mg, 31%). The elemental analysis results indicate the presence of approximately one additional acetonitrile molecule, in keeping with the presence of voids containing disordered solvent molecules in the structure (see below), although the number of acetonitrile molecules determined from elemental analysis is smaller than that estimated from the structure determination, probably due to the loss of part of the solvent upon drying. Anal. Calcd for C36H64CuN6O22U2 + CH3CN: C, 30.15; H, 4.46; N, 6.48. Found: C, 30.52; H, 4.18; N, 6.78%.
[UO2(Htcepo)] (7).
H3tcep·HCl (15 mg, 0.05 mmol) and [UO2(NO3)2(H2O)2]·4H2O (25 mg, 0.05 mmol) were dissolved in water (0.5 mL). Yellow crystals of complex 7 were obtained within three days (17 mg, 64%). Anal. Calcd for C9H13O9PU: C, 20.24; H, 2.45. Found: C, 19.80; H, 2.42%.
Crystallography
Data collections were performed at 100(2) K on a Bruker D8 Quest diffractometer using an Incoatec Microfocus Source (IμS 3.0 Mo) and a PHOTON III area detector, and operated with APEX3.12 The data were processed with SAINT,13 and empirical absorption corrections were made with SADABS.14 The structures were solved by intrinsic phasing with SHELXT,15 and refined by full-matrix least-squares on F2 with SHELXL,16 using the ShelXle interface.17 When possible, hydrogen atoms bound to oxygen and nitrogen atoms were retrieved from residual electron density maps and they were refined either freely or with geometric restraints. The hydrogen atoms of the guanidinium cations in 2 and all carbon-bound hydrogen atoms in all compounds were introduced at calculated positions and treated as riding atoms with an isotropic displacement parameter equal to 1.2 times that of the parent atom (1.5 for CH3). For compound 2, the SQUEEZE18 software was used to subtract the contribution of other, disordered solvent molecules to the structure factors; about 28 electrons were found in the unit cell, which could correspond to about 1.5 additional water molecules per formula unit. Use of SQUEEZE for compound 6 gave 40 electrons, corresponding to about 2 acetonitrile molecules per formula unit. For compound 2, the ADDSYM software19 indicates the possible space group P21/c with 86% fit, but no satisfying refinement could be made in this space group. Crystal data and structure refinement parameters are given in Table 1. Drawings were made with ORTEP-3 (ref. 20) and VESTA,21 and topological analyses were performed with ToposPro.22
Table 1 Crystal data and structure refinement details
|
|
1
|
2
|
3
|
4
|
5
|
6
|
7
|
| Chemical formula |
C12H26N2O13U |
C22H38N8O21U2 |
C29H30NO10PU |
C40H46N6NiO23U2 |
C40H46CuN6O23U2 |
C36H64CuN6O22U2 |
C9H13O9PU |
|
M/g mol−1 |
644.38 |
1226.66 |
821.54 |
1513.60 |
1518.43 |
1472.53 |
534.19 |
| Crystal system |
Monoclinic |
Monoclinic |
Monoclinic |
Monoclinic |
Monoclinic |
Triclinic |
Monoclinic |
| Space group |
P21/n |
P21 |
P21/c |
Cc
|
Cc
|
P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) |
P21/c |
|
a/Å |
11.6052(7) |
11.6822(6) |
11.4827(4) |
12.4106(7) |
12.4847(7) |
11.3872(4) |
7.3820(5) |
|
b/Å |
15.1797(10) |
15.3912(8) |
14.2168(5) |
17.2410(9) |
17.0888(11) |
11.5224(4) |
23.3063(18) |
|
c/Å |
12.7681(13) |
11.7959(5) |
17.7990(6) |
23.5189(14) |
23.5455(16) |
12.7894(5) |
8.5496(7) |
|
α/° |
90 |
90 |
90 |
90 |
90 |
114.7971(13) |
90 |
|
β/° |
116.773(3) |
115.5602(16) |
92.2934(15) |
102.782(2) |
103.131(2) |
98.3735(14) |
113.584(2) |
|
γ/° |
90 |
90 |
90 |
90 |
90 |
110.7704(13) |
90 |
|
V/Å3 |
2008.1(3) |
1913.37(16) |
2903.31(17) |
4907.7(5) |
4892.1(5) |
1335.95(9) |
1348.07(18) |
|
Z
|
4 |
2 |
4 |
4 |
4 |
1 |
4 |
| Reflections collected |
71![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 359 359 |
46015 |
89833 |
60642 |
39817 |
53010 |
44893 |
| Independent reflections |
3814 |
7198 |
7497 |
9277 |
8866 |
6907 |
3476 |
| Observed reflections [I > 2σ(I)] |
3589 |
7097 |
6879 |
9090 |
8727 |
6523 |
3338 |
|
R
int
|
0.052 |
0.056 |
0.060 |
0.068 |
0.058 |
0.052 |
0.060 |
| Parameters refined |
269 |
485 |
380 |
650 |
650 |
313 |
184 |
|
R
1
|
0.019 |
0.029 |
0.021 |
0.036 |
0.029 |
0.019 |
0.022 |
| wR2 |
0.046 |
0.075 |
0.055 |
0.093 |
0.071 |
0.042 |
0.052 |
|
S
|
1.049 |
1.040 |
1.054 |
1.069 |
1.044 |
1.074 |
1.096 |
| Δρmin/e Å−3 |
−0.65 |
−0.72 |
−1.58 |
−1.15 |
−0.50 |
−0.89 |
−1.56 |
| Δρmax/e Å−3 |
1.18 |
1.59 |
1.43 |
1.19 |
0.74 |
0.83 |
1.37 |
| Flack parameter |
|
0.416(11) |
|
−0.001(10) |
−0.003(7) |
|
|
Luminescence measurements
Emission spectra were recorded on solid samples using an Edinburgh Instruments FS5 spectrofluorimeter equipped with a 150 W CW ozone-free xenon arc lamp, dual-grating excitation and emission monochromators (2.1 nm mm−1 dispersion; 1200 grooves per mm) and an R928P photomultiplier detector. The powdered compounds were pressed to the wall of a quartz tube, and the measurements were performed using the right-angle mode in the SC-05 cassette. An excitation wavelength of 420 nm was used in all cases and the emission was monitored between 450 and 600 nm. The quantum yield measurements were performed by using a Hamamatsu Quantaurus C11347 absolute photoluminescence quantum yield spectrometer and exciting the samples between 300 and 400 nm.
Results and discussion
Synthesis
It is quite usual to find that in solvothermal syntheses of uranyl ion complexes of carboxylates there is no need to add a base to deprotonate the parent carboxylic acids, so that the trianionic form of the ligand found to be present in complexes 1–6 is no surprise. Incomplete deprotonation, however, is not unknown and given that this is seen in the presently unique example of the dicarboxylato species seen in complex 7, it cannot be attributed great significance, except in that this is the only complex of the present series obtained from pure water alone. The organic solvents, N,N-dimethylformamide and acetonitrile, used as co-solvents in the preparation of 1–6 both undergo hydrolysis under solvothermal conditions, a reaction which must lead to buffering of the medium at near-neutral pH, so that this is a factor which should be recognized as yet another influence on the composition of the crystalline deposits. A further influence of solubility is possibly reflected in the identical composition (and structure) of complexes 4 and 5, where differences were expected due to the unlike preferences of the two metal ions NiII and CuII for tris- and bis-(2,2′-bipyridine) binding and where sufficient 2,2′-bipyridine was added to form [Ni(bipy)3]2+,23 but only the Ni(bipy)22+ unit was present in the deposited crystals. Given the sensitivity of alkylphosphines in general to oxidation and the common observations of oxalate formation by oxidation of ligands during solvothermal syntheses of uranyl ion complexes, the presence of the phosphine oxide in complex 7 was perhaps to be expected, though it is of interest as a possible example of the simplest step in uranyl ion catalyzed oxidation of an organic substrate. This complex was repeatedly obtained from several different attempts at synthesis involving different additional reagents, [Cu(R,S-Me6cyclam)](NO3)2 or the couples NiII/bipy and ZnII/phen, all with acetonitrile as cosolvent. While the same attempts were made with both H3tcenm and H3tcep, only complex 7 resulted in the latter case, most syntheses giving amorphous precipitates or no solid whatever, which points to the particular stability and/or insolubility of this complex.
Crystal structures
The complex [H2NMe2][UO2(tcenm)]·3H2O (1) includes dimethylammonium counterions formed from hydrolysis of the organic cosolvent N,N-dimethylformamide, as very commonly observed,24 and the 1:1 metal/ligand stoichiometry used during the synthesis is retained in the solid state. The uranyl ion is tris-κ2O,O′-chelated by three carboxylate groups from three tcenm3− ligands [U–O(oxo), 1.763(3) and 1.769(3) Å; U–O(carboxylate), 2.440(2)–2.496(2) Å] (Fig. 1). The ligand tcenm3− has a conformation completely lacking in symmetry and is therefore a source of chirality but both enantiomers are present since the complex crystallizes in a centrosymmetric space group. Both metal and ligand are 3-coordinated (3-c) nodes in the diperiodic network formed, which is parallel to (001) and has the {63} point symbol and the hcb topological type, as commonly found in tris-chelated uranyl carboxylate complexes. When viewed down [100], the homochiral layers have a zigzag profile and the nitro groups are located near the peaks and troughs and thus alternate in their orientation relative to the mean plane. The nitro C–N bonds lie at an angle near 60° to the mean plane and the nitrogen atom lies at 7.127(3)–7.532(3) Å of the three UVI centres bound to the same ligand, although the shortest N⋯U separations of 4.509(3) and 5.813(3) Å involve two centres in one adjacent sheet. The H2NMe2+ cation forms bifurcated hydrogen bonds with two carboxylate, one nitro and one water oxygen atoms [N⋯O distances, 2.779(6)–2.911(5) Å; N–H⋯O angles, 113(4)–154(5)°], thus linking two sheets, while the water molecule for which the hydrogen atoms could be located also links two sheets through hydrogen bonding to one oxo and one carboxylate oxygen donors. Due to the inability to locate the hydrogen atoms of the other water molecules, the hydrogen bonding network cannot be defined in complete detail but the identifiable bonds unite the sheets into a triperiodic assembly. The packing is quite compact, as indicated by the Kitaigorodsky packing index (KPI, evaluated with PLATON19) of 0.70.
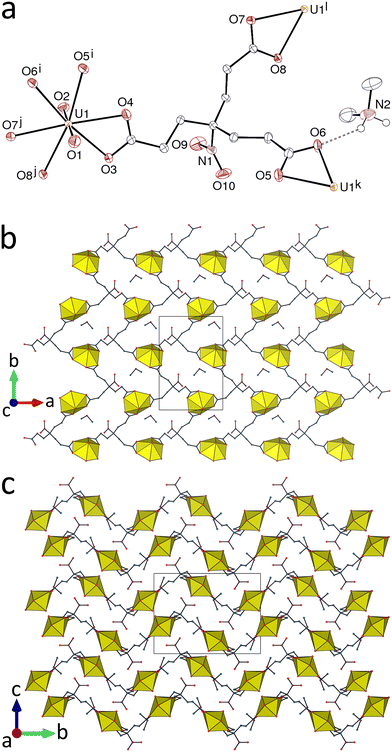 |
| | Fig. 1 (a) View of complex 1 with displacement ellipsoids shown at the 50% probability level. Solvent molecules and carbon-bound hydrogen atoms are omitted, and the hydrogen bond is shown as a dashed line. Symmetry codes: i = 3/2 − x, y + 1/2, 3/2 − z; j = x − 1, y, z; k = 3/2 − x, y − 1/2, 3/2 − z; l = x + 1, y, z. (b) View of the diperiodic assembly showing uranium coordination polyhedra. (c) Packing with layers viewed edge-on. | |
The complex involving the guanidinium counterion, [C(NH2)3][UO2(tcenm)]·0.5H2O (2), crystallizes in the Sohncke group P21 with two independent uranyl cations in the asymmetric unit, both in the same environment as in complex 1 [U–O(oxo), 1.763(6)–1.793(7) Å; U–O(carboxylate), 2.416(12)–2.501(6) Å] (Fig. 2). The anionic sheets formed, parallel to (10) and here also with the hcb topology, have more a square-wave profile than those in 1 when viewed down [101], with the uranyl equatorial planes roughly parallel to the sheet mean plane instead of being tilted as in 1. The ligand nitro groups again project in alternation to each side of the sheets, with the C–N bonds at an angle of close to 75° to the mean plane. The sheets are tightly packed so as to form channels parallel to [001], with a section of ∼5 Å × ∼10 Å, occupied by the guanidinium counterions and water molecules. There are two slightly different minimum U⋯NO2 separations of 4.812(8) and 4.720(8) Å, both slightly longer than the shorter one in 1 but again between and not within sheets. In both sheets, the ligand has a pseudo-mirror plane containing one of the carboxylate arms and the nitro group, and it is thus achiral (Cs symmetry). The chirality of the crystal appears to result from the cation–water associates within the channels of the structure. The guanidinium cations form either eight or six simple or bifurcated hydrogen bonds involving carboxylate, nitro or water acceptors [N⋯O distances, 2.910(14)–3.171(15) Å; N–H⋯O angles, 125–159°]. Pairs of guanidinium cations bridged by hydrogen bonding to a water molecule form units which, considered alone, have C2 symmetry and are therefore chiral, so that the chirality of the crystal can be considered a result of the fact that within a given crystal all these units have the same configuration. Further hydrogen bonding interactions of the water molecule and both cations, mostly involving carboxylate acceptors, reduce the actual symmetry to C1 but only one cation of a given unit interacts with a nitro group, producing the inequivalence of adjacent sheets. The KPI amounts to only 0.66, some voids being occupied by unresolved solvent molecules (see Experimental).
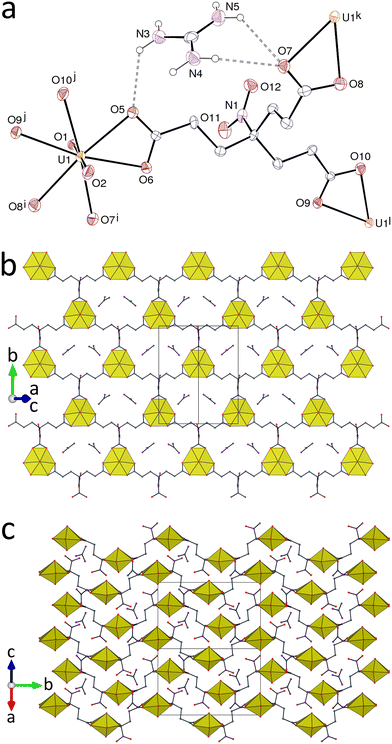 |
| | Fig. 2 (a) View of one of the two independent complex units in compound 2 with displacement ellipsoids shown at the 30% probability level. Solvent molecules and carbon-bound hydrogen atoms are omitted, and hydrogen bonds are shown as dashed lines. Symmetry codes: i = 1 − x, y + 1/2, 2 − z; j = x + 1, y, z + 1; k = 1 − x, y − 1/2, 2 − z; l = x − 1, y, z − 1. (b) View of the diperiodic assembly showing uranium coordination polyhedra. (c) Packing with layers viewed edge-on. | |
Use of the bulky methyltriphenylphosphonium cation yields the complex [PPh3Me][UO2(tcenm)] (3), in which the uranium atom environment is the same as in 1 and 2 [U–O(oxo), 1.7721(17) and 1.7773(18) Å; U–O(carboxylate), 2.4345(18)–2.4930(17) Å] (Fig. 3). Here also, the network formed, parallel to (001), has the hcb topology, and its shape is close to that found in 1. The packing of sheets is however different, as indicated by the shortest intersheet U⋯N contact at 5.823(3) Å, much larger than in 1 as a result of both the larger spacing of the sheets and a lateral displacement resulting in the uranium atoms being arranged in layers parallel to (010). The PPh3Me+ counterions occupy the interlayer spaces, with one aromatic ring crossing a hexagonal cell, and they are too far away from one another for phenyl-embrace or π-stacking interactions, all centroid⋯centroid distances being larger than 5 Å. The KPI of 0.70 indicates that no significant void is present.
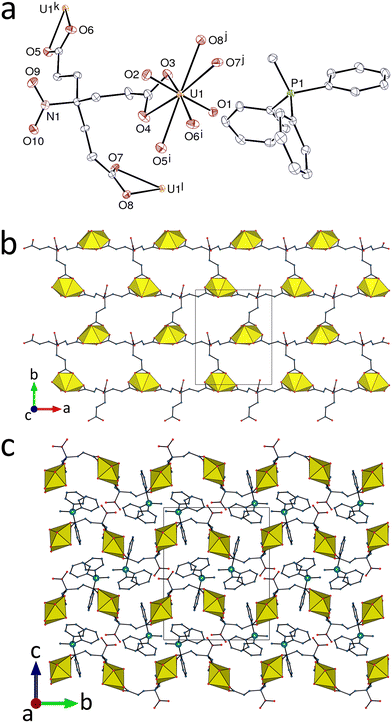 |
| | Fig. 3 (a) View of compound 3 with displacement ellipsoids shown at the 50% probability level and hydrogen atoms omitted. Symmetry codes: i = x + 1, y, z; j = 1 − x, y + 1/2, 3/2 − z; k = x − 1, y, z; l = 1 − x, y − 1/2, 3/2 − z. (b) The diperiodic assembly with uranium coordination polyhedra yellow. (c) Packing with layers viewed edge-on. | |
The two complexes [UO2(tcenm)M(bipy)2][UO2(tcenm)]·3H2O with M = Ni (4) or Cu (5) are isomorphous, despite anticipation that the different stereochemical preferences of NiII and CuII might have been of influence, and they crystallize in the non-centrosymmetric space group Cc. As shown in Fig. 4 in the case of complex 4, the crystal contains two independent, cationic and anionic units which are hetero- and homometallic, respectively. In both units, the uranium environment is that found in the previous complexes [U–O(oxo), 1.762(10)–1.787(10) Å; U–O(carboxylate), 2.424(7)–2.515(10) Å, including both compounds], and, as in 2, the tcenm3− ligand has either pseudo-mirror symmetry in the cation, or is not far from it in the anion (with the nitro group slightly tilted). The transition metal cation is bound to two carboxylate oxygen atoms, thus forming a four-membered UMO2 ring, and to two chelating bipy ligands, its environment being approximately octahedral, with slightly more distorsion with Cu than with Ni [Ni–N, 2.030(10)–2.061(10) Å; Ni–O, 2.130(8) and 2.156(9) Å; Cu–N, 1.953(7)–2.076(8) Å; Cu–O, 2.362(7) and 2.450(6) Å]. The transition metal achieves 6-coordination by binding to UO2(carboxylate)3− units in a manner similar to that long known in the case of Na[UO2(CH3CO2)3],25 where the uranate anion behaves as a metalloligand via chelation of Na+ by O–U–O units derived from two adjacent acetates. Both cation and anion form uranyl-based hcb networks parallel to (001), with the M(bipy)22+ groups forming an additional link between two tcenm3− nodes in the cation (Fig. 4d). Viewed down [001], the triuranacyclic units have a near-rectangular form very similar to that in complex 2, but when viewed down [100] both sheets have a flattened sawtooth profile in which all nitro groups lie to one side of their sheet. The nitro groups of the cationic sheet confront those of the anionic, so that an alternative view of the structure is as of one where neutral double sheets stack down [001]. U⋯N(nitro) separations are considerably longer than in complexes 1 and 2, with the shortest being U2⋯N1 of 6.037(13) Å in 4 and 6.104(9) Å in 5, again an intersheet contact, all the others being larger than 6.7 Å. This may in part be a consequence of the fact that while the nitro groups retain their capacity to become involved in hydrogen bonding with water molecules, there is also an insertion of one nitro group of the anionic sheet into the space between bipy ligands in an adjacent cationic sheet producing O19⋯π interactions [O⋯centroid distances, 3.062(19) and 3.353(19) Å in 4, 2.904(12) Å in 5; N–O⋯centroid angles, 157 and 126° in 4, 155° in 5]; these interactions appear as exceeding dispersion on the Hirshfeld surface (HS) calculated with CrystalExplorer.26 The 6-coordinate MII unit is chiral and each cationic sheet is enantiomerically pure but the chirality alternates from one sheet to the next. The M(bipy)22+ substituents largely occupy the space defined by the triuranacycles, so that although the projection of sheets onto one another is very similar to that in complex 2, no channels are apparent because they are blocked by the M(bipy)22+ attachments. This does mean, however, that, because the anionic sheets have cyclic units occupied only by presumably replaceable water molecules, the complete structure can be regarded as containing cavities with caps defined by enantiomeric M(bipy)2(O)2 entities at a M⋯M separation of 11.8301(8) and 11.8006(8) Å in 4 and 5, respectively. With KPIs of 0.68 and 0.69, the packings in 4 and 5 do not contain significant free spaces.
 |
| | Fig. 4 (a) View of compound 4 with displacement ellipsoids shown at the 50% probability level. Solvent molecules and hydrogen atoms are omitted. Symmetry codes: i = x + 1/2, y + 1/2, z; j = x + 1, y, z; k = x − 1/2, y − 1/2, z; l = x − 1, y, z; m = x − 1/2, y + 1/2, z; n = x + 1/2, y − 1/2, z. (b) The heterometallic diperiodic assembly with uranium coordination polyhedra yellow and those of nickel green. (c) Packing with layers viewed edge-on. (d) Nodal representation of the heterometallic network (yellow, uranium nodes; blue, tcenm3− nodes; green, nickel edges; same orientation as in b). | |
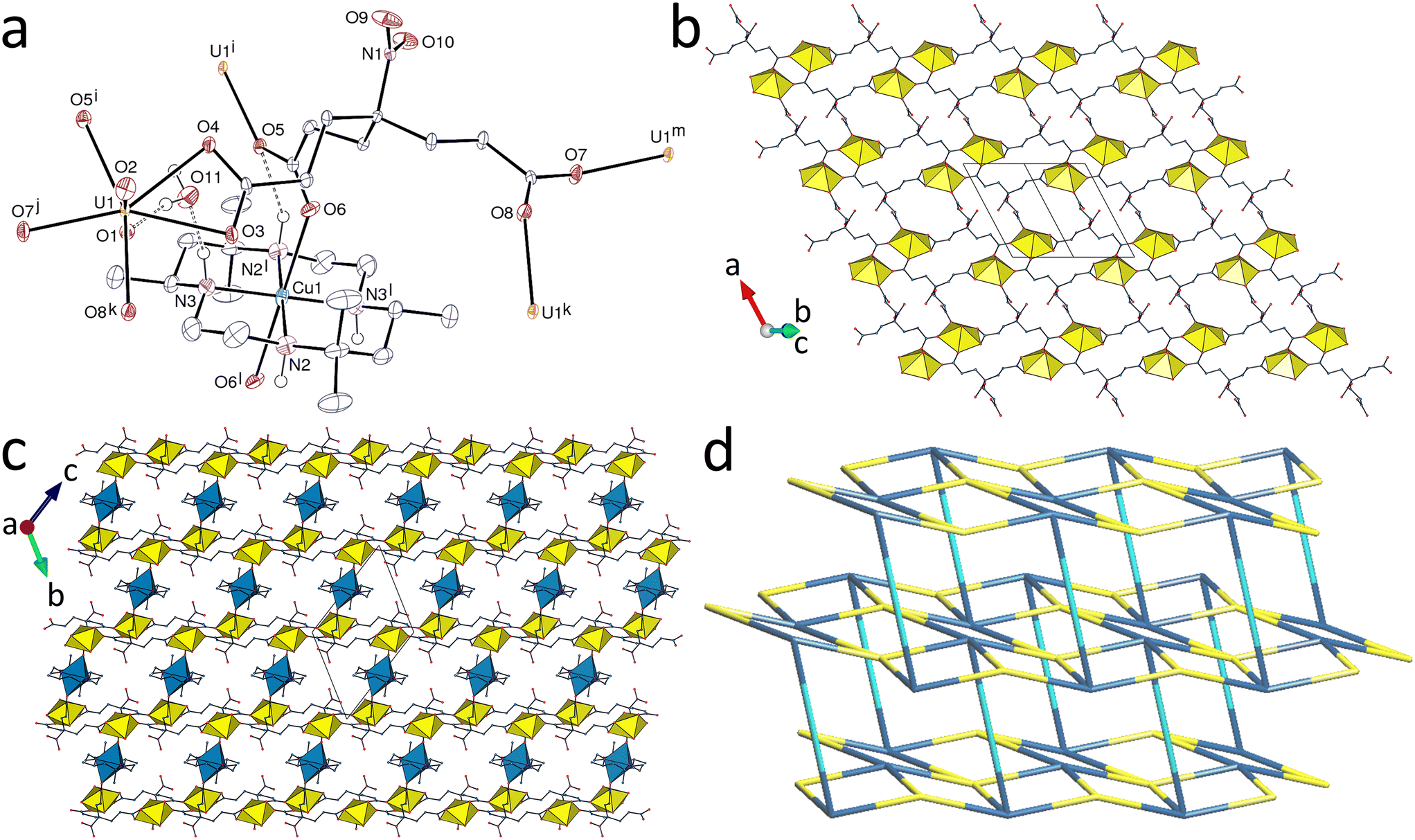
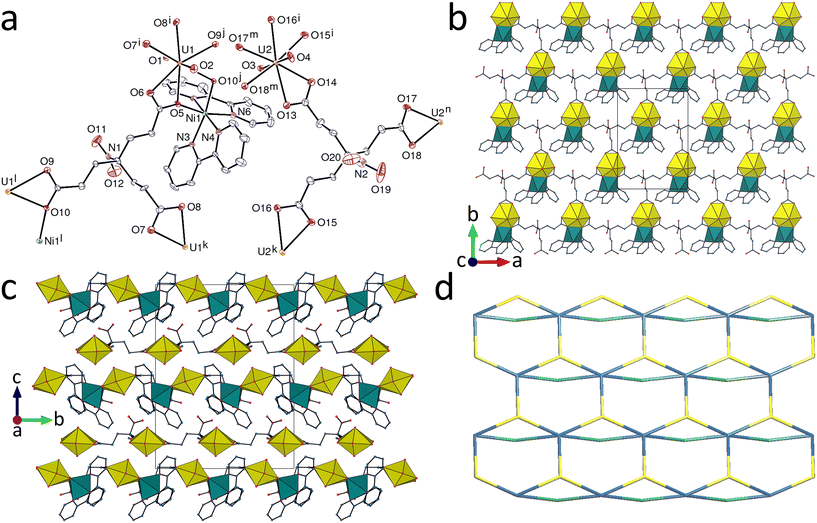
While complexes 1–5 have structures based on a common hcb network topology, introduction of the Cu(R,S-Me6cyclam)2+ species results in formation of a heterometallic complex with a different connectivity, [(UO2)2(tcenm)2Cu(R,S-Me6cyclam)]·2H2O (6). The unique uranium atom is here in a pentagonal-bipyramidal environment, being chelated by one carboxylate group and bound to three more oxygen donors from three different ligands [U–O(oxo), 1.7803(18) and 1.7899(17) Å; U–O(carboxylate), 2.4560(17) and 2.4850(16) Å for the chelating group and 2.3149(17)–2.3269(17) Å for the others] (Fig. 5). The tcenm3− ligand has thus one κ2O,O′-chelating, one μ2-κ1O:κ1O′-bridging and one monodentate carboxylate groups in its coordination to uranyl. If only uranium and tcenm3− are considered, both are 4-c nodes and the coordination polymer formed is diperiodic and parallel to (01), having the {44·62} point symbol and the common sql topological type. With respect to the ideal square lattice, two kinds of cells, either 8- or 20-membered, are however present here. These subunits are further linked through CuII bridges, the transition metal cation being bound to the four nitrogen atoms of the centrosymmetric azamacrocycle and to two axial carboxylate oxygen atoms to give an axially elongated octahedral environment [Cu–N, 2.020(2) and 2.061(2) Å; Cu–O, 2.5581(18) Å]. The tcenm3− ligand thus becomes a 5-c node, with one chelating and two μ2-κ1O:κ1O′-bridging carboxylate groups. The resulting assembly is a 4,5-c binodal triperiodic framework with the {44·62}{44·66} point symbol (first symbol for U and second for tcenm3−) and the tcs (ThCr2Si2) topological type, and it is to the best of our knowledge the first occurrence of this somewhat uncommon topology27 in uranyl chemistry. As usually found when transition metal azamacrocyclic complexes are used as structure-directing species in uranyl cation coordination polymers, one ammonium group is hydrogen bonded to the carboxylate group bound to the transition metal cation, while the other is linked to the water molecule, itself hydrogen bonded to oxo and carboxylato groups [N⋯O distances, 2.979(3)–3.373(3) Å; N–H⋯O angles, 115–161°; O⋯O distances, 2.914(3) and 2.919(3) Å; O–H⋯O angles, 161(3) and 165(3)°]. The shortest U⋯N(nitro) contact is here between atoms pertaining to the same diperiodic subunit, at 5.987(2) Å, due to the large intersheet separation associated with the CuII links. The KPI is 0.63 only, some voids being occupied by disordered acetonitrile molecules (see Experimental).
 |
| | Fig. 5 (a) View of compound 6 with displacement ellipsoids shown at the 50% probability level. Carbon-bound hydrogen atoms are omitted and hydrogen bonds are shown as dashed lines. Symmetry codes: i = 1 − x, 1 − y, 1 − z; j = x, y + 1, z + 1; k = 2 − x, 1 − y, 1 − z; l = 1 − x, −y, 1 − z; m = x, y − 1, z − 1. (b) The uranyl-based diperiodic subunit. (c) View of the triperiodic framework with uranium coordination polyhedra yellow and those of copper blue. (d) Nodal representation of the framework (yellow, uranium; dark blue, tcenm3− nodes; light blue, copper edges). | |
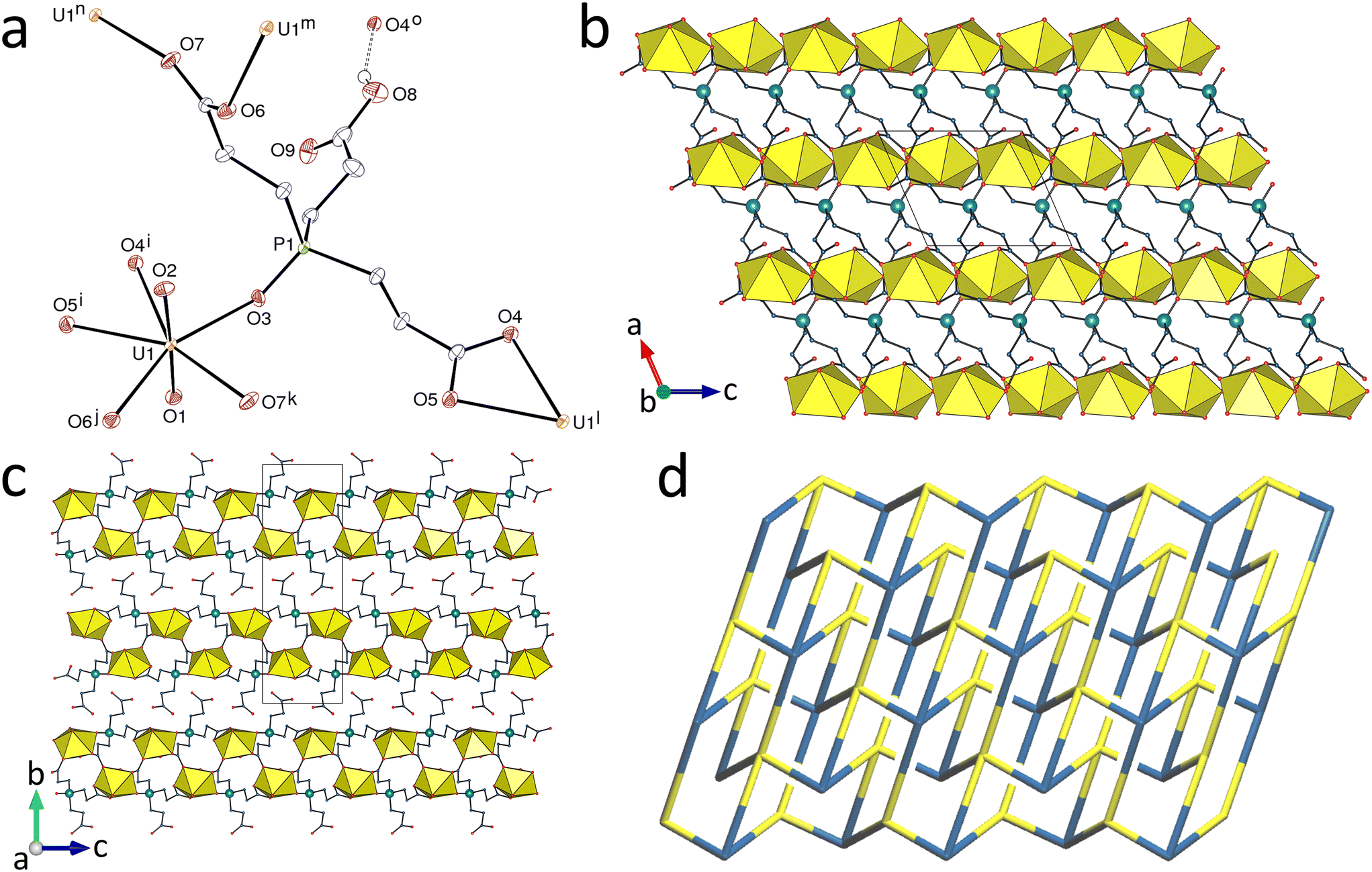
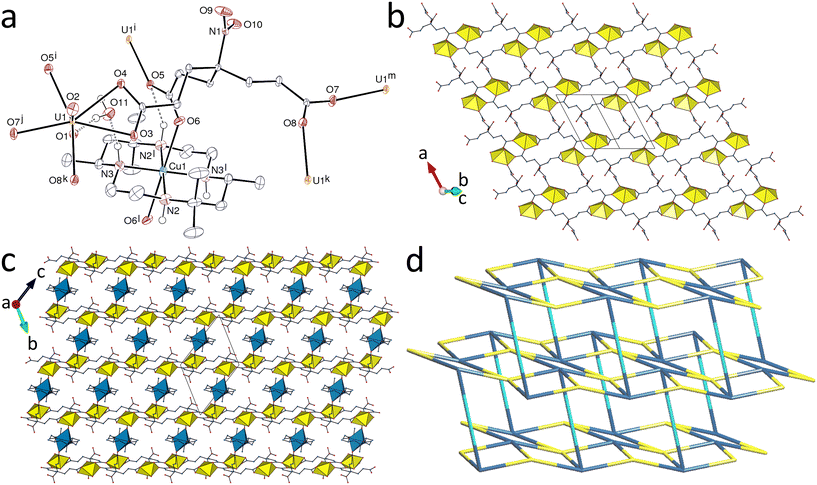
Replacing H3tcenm by H3tcep has remarkable effects on the complexes produced. H3tcep is oxydized in situ to give the corresponding phosphine oxide, which retains one of its carboxylic protons in the complex [UO2(Htcepo)] (7). The asymmetric unit contains a single uranium atom in a pentagonal-bipyramidal environment resulting from chelation by one carboxylate group and bonding to two more carboxylate donors and one oxide group from three different ligands [U–O(oxo), 1.774(3) and 1.778(3) Å; U–O(carboxylate), 2.439(3) and 2.544(3) Å for the chelating group, 2.337(3) and 2.349(3) Å for the others; U–O(phosphine oxide), 2.302(3) Å] (Fig. 6). 98 crystal structures of complexes with uranyl cations bound to phosphine oxides are reported in the Cambridge Structural Database (CSD, Version 5.43),28 with U–O(P) bond lengths in the range of 2.257–2.462 Å [mean value, 2.36(4) Å]. The uranium pentagonal-bipyramidal coordination found here contrasts with that in the complexes of tcenm3−, where hexagonal-bipyramidal coordination predominates, though with one example of pentagonal-bipyramidal coordination in 6, so no real conclusion can be drawn from what is seen in 7, even if it is tempting to believe that the strong donor capacity of PO may be a factor favouring a lower coordination number. Although H3tcep and its anions have previously been used as ligands, with 14 crystal structures of complexes reported in the CSD, H3tcepo or its deprotonated forms are unknown as ligands, and only the crystal structure of the uncomplexed molecule has been described.29 The Htcepo2− ligand has one κ2O,O′-chelating and one μ2-κ1O:κ1O′-bridging carboxylate groups, and one uncoordinated carboxylic group, and it is thus a 4-c node, as is also the uranium atom. The uninodal diperiodic network formed, parallel to (010), has the point symbol {66}. This point symbol is the same as that of the triperiodic dia topology, but the vertex symbols30 are different, 62·62·63·66·64·64 here and 62·62·62·62·62·62 for the dia net (and also the less common lon net). Instead of containing adamantane-like units as found in the dia network, there are here crossings giving Hopf links with a multiplicity of 2 (Fig. 6d). The sheets formed display two layers of uranyl ions and the carboxylic groups are directed outward on each side, giving a width of ∼14 Å between the planes defined by the outermost oxygen atoms, the sheets being slightly interdigitated. The carboxylic group makes an intersheet hydrogen bond with the uranium-bound carboxylate oxygen atom O4 [O⋯O distance, 2.783(4) Å; O–H⋯O angle, 150(6)°], which induces a further level of organization leading to formation of an intricate and compact (KPI, 0.72) 4,5-c triperiodic network with the point symbol {66}{69·8} (with hydrogen bonds considered as edges between Htcepo2− nodes) and the presence of eight crossings (Hopf links).
 |
| | Fig. 6 (a) View of compound 7 with displacement ellipsoids shown at the 50% probability level. Carbon-bound hydrogen atoms are omitted and the hydrogen bond is shown as a dashed line. Symmetry codes: i = x, y, z − 1; j = x − 1, 3/2 − y, z − 1/2; k = x − 1, y, z; l = x, y, z + 1; m = x + 1, 3/2 − y, z + 1/2; n = x + 1, y, z; o = 2 − x, 1 − y, 2 − z. (b) The diperiodic assembly with uranium coordination polyhedra yellow. (c) Packing with layers viewed edge-on. (d) Nodal representation of the diperiodic network (yellow, uranium; blue, Htcepo2− nodes). | |
Discussion of the crystal structures
While the role of metal ions in coordination polymers and frameworks can be purely structural, particular properties of the metal ions such as magnetism, redox activity, electronic absorption, luminescence, stereochemical preferences, and catalytic activity can also be critical in regard to any applications, as is evident in numerous recent research articles and reviews.31 A further tool of structural control can arise from internal functionality of the bridging ligands,9 simple examples being where an additional donor atom provides a site for chelation32 or, more commonly, where stacking arrays33 are formed by small aromatic units,34 although once again such functionality in more sophisticated forms,35,36 has been exploited to engender special properties in the structures formed. In the particular case of tripodal ligands, the nature of the bridgehead is of prime importance,37 as shown in the present case. The ligand Htcepo2− produced by oxidation of the reactant H3tcep under the reaction conditions and found in the neutral crystalline complex 7 provides a significant contrast with the similar tripod ligand tcenm3− in its mode of coordination. While valence bond representations of both NO2 and PO units mean that they can both be considered as “vicinal zwitterions”,38 and thus as sources of negatively charged oxygen donors, the known coordination chemistry of phosphine oxides is vastly more extensive39 than that of nitroalkanes, which are commonly considered as rather poor ligands,40 although nitro coordination and chelation are well-known for functionalized nitroarenes.41 The present work confirms an apparently major difference in donor capacity of the two units in that only in 7 is coordination other than of carboxylate donors observed. More significantly, the κ1O U–OP bond length of 2.302(3) Å is shorter than either of those of the κ1O U–O(carboxylate) bonds of 2.337(3) and 2.349(3) Å. This may explain why one carboxylic acid group of H3tcepo retains its proton and does not bind to UVI, though such a conclusion must be qualified by recognizing that what is observed in the solid state is determined to some extent by solubility and does not necessarily represent the dominant species in solution. Coordination of the bridgehead unit of Htcepo2− does not produce a ligand conformation which is radically different from that adopted by tcenm3− and both can be considered as flattened, extended tripods where each of the donor units binds to a separate UVI centre. It has been estimated that a minimum of 6-atom spacing (in addition to the bonding groups) is required for coordination to one uranyl centre of the three carboxylate groups of a tripodal ligand with a tertiary ammonium bridgehead hydrogen bonded to one uranyl oxo group.1a As with nitrilotriacetate,5 the two present ligands fall short of this limit and it is thus unsurprising that the coordinating carboxylate groups in both tcenm3− and Htcepo2− are divergently oriented and bound to different cations. The non-coordinating nature of the nitro group in the first case leads in most cases (complexes 1–5) to threefold uranyl κ2O,O′-chelation (with additional bridging to transition metal cations in 4 and 5) and formation of hcb networks. Only in complex 6 is a different connectivity of tcenm3− observed, with carboxylate axial bonding to copper(II) disrupting the previous pattern and making the ligand a κ2O,O′;bis(μ2-κ1O:κ1O′)-bonded node; a diperiodic uranyl coordination polymer, here of sql topology, is however formed here also, with triperiodic extension ensured by the CuII pillars. Bridgehead coordination with the Htcepo2− ligand, although it does not increase much the overall number of bonded cations due to the carboxylic group being uncoordinated, yields nevertheless a diperiodic network which is not quasi-planar or able to be embedded into a plane, but displays crossings of rings and can thus be considered as three-dimensional diperiodic.42
Luminescence properties
Emission spectra of complexes 1–7 have been measured in the solid state under excitation at 420 nm. Complexes 2 and 4–6 are non-emissive, unsurprisingly for the 3d metal cation-containing complexes 4–6, for which this is usual and probably results from either preferential absorption by the transition metal cation or energy transfer and non-radiative relaxation,43 but less so for 2, although weak emission in guanidinium-containing species has previously been observed.44 Although emission from 1 was detectable with maxima very close to those of complex 3, it was particularly weak and showed an anomalous sharp peak at 514 nm possibly due to an impurity. The spectra of complexes 3 and 7, which both have a photoluminescence quantum yield (PLQY) of 9%, indicating no special efficacity of either the nitro or PO substituent as an antenna for UVI excitation, are shown in Fig. 7 (U⋯N(nitro) separations in the tcenm3− complexes were estimated (see above) to see if they might correlate with PLQY values and indicate a significant role as an antenna for the nitro group but this was clearly not the case). These spectra are well-resolved and show the typical vibronic progression due to the S11 → S00 and S10 → S0ν (ν = 0–4) transitions of the uranyl ion.45 The main maxima positions for 3 (482, 502, 523 and 547 nm) and 7 (492, 514, 537 and 562 nm) are in agreement with the red-shift usually associated with the passage from six- to five-coordinated equatorial environment in carboxylate uranyl ion complexes.46
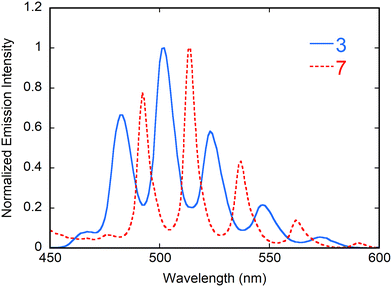 |
| | Fig. 7 Emission spectra of complexes 3 and 7 in the crystalline state upon excitation at 420 nm. | |
Conclusions
Two triple-armed ligands, H3tcenm and H3tcep, the latter oxidized in situ into the corresponding phosphine oxide H3tcepo, have been used to synthesize seven uranyl ion complexes under (solvo)-hydrothermal conditions and in the presence of various structure-directing cations. The tricarboxylate tcenm3− appears to be much more sensitive to the presence of these additional species than the dicarboxylate Htcepo2− since six of the complexes involve the former, while only one complex could be obtained, under different conditions, with the latter. Five of the six tcenm3− complexes share however the common feature of threefold κ2O,O′-chelation of three uranyl ions to give hcb networks, some geometric variety being introduced by the different counterions or additional coordinated transition metal cations, with the nitro substituent being involved in various hydrogen bonding interactions (O⋯HN and O⋯HC) presumably of some influence. Pillaring of sql networks by Cu(R,S-Me6cyclam)2+ provides the only triperiodic framework in the series, which has the tcs topology. In no case does the nitro group participate in complexation, in contrast to the phosphine oxide group which is bound to uranyl in the Htcepo2− complex, yielding a diperiodic, three-dimensional network with rings forming Hopf links. Broad considerations of the donor capacity of phosphine-oxide and carboxylate towards UVI indicate that the former is the more effective,47 explaining the unsymmetrical bonding and incomplete ionization of Htcepo, while the poor coordinating ability of an aliphatic nitro group explains the action of tcenm3− as a tricarboxylate donor. Both these triple-armed ligands are too small to encompass a single uranyl cation bound to all three carboxylate groups, and indeed adopt conformations describable as rather flattened tripods, so that they act instead as divergent ligands with either an uncoordinated or a coordinated bridgehead. The conformation restrictions due to the sophisticated design of triple-armed ligands forming mononuclear uranyl ion complexes2 therefore appear to have been critical in enforcing a convergent, tripodal form.
Conflicts of interest
There are no conflicts of interest to declare.
References
-
(a) T. S. Franczyk, K. R. Czerwinski and K. N. Raymond, J. Am. Chem. Soc., 1992, 114, 8138 CrossRef CAS;
(b) P. H. Walton and K. N. Raymond, Inorg. Chim. Acta, 1995, 240, 593 CrossRef CAS.
- M. Sawicki, J. M. Siaugue, C. Jacopin, C. Moulin, T. Bailly, R. Burgada, S. Meunier, P. Baret, J. L. Pierre and F. Taran, Chem. – Eur. J., 2005, 11, 3689 CrossRef CAS PubMed.
-
(a) K. X. Wang and J. S. Chen, Acc. Chem. Res., 2011, 44, 531 CrossRef CAS PubMed;
(b) M. B. Andrews and C. L. Cahill, Chem. Rev., 2013, 113, 1121 CrossRef CAS PubMed;
(c) T. Loiseau, I. Mihalcea, N. Henry and C. Volkringer, Coord. Chem. Rev., 2014, 266–267, 69 CrossRef CAS;
(d) J. Su and J. S. Chen, Struct. Bonding, 2015, 163, 265 CrossRef CAS;
(e) P. Thuéry and J. Harrowfield, Dalton Trans., 2017, 46, 13660 RSC;
(f) K. Lv, S. Fichter, M. Gu, J. März and M. Schmidt, Coord. Chem. Rev., 2021, 446, 214011 CrossRef CAS.
-
(a) P. Thuéry, Cryst. Growth Des., 2014, 14, 901 CrossRef;
(b) P. Thuéry, Cryst. Growth Des., 2014, 14, 2665 CrossRef;
(c) J. Harrowfield and P. Thuéry, Eur. J. Inorg. Chem., 2020, 749 CrossRef CAS;
(d) P. Thuéry and J. Harrowfield, Inorg. Chem., 2021, 60, 1683 CrossRef PubMed;
(e) P. Thuéry and J. Harrowfield, Dalton Trans., 2021, 50, 11021 RSC.
-
(a) M. S. Grigoriev, C. Den Auwer, D. Meyer and P. Moisy, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 2006, 62, m163 CrossRef PubMed;
(b) P. Thuéry, Inorg. Chem. Commun., 2007, 10, 423 CrossRef;
(c) P. Thuéry and J. Harrowfield, Cryst. Growth Des., 2014, 14, 1314 CrossRef;
(d) Y. Atoini, J. Harrowfield, Y. Kim and P. Thuéry, J. Inclusion Phenom. Macrocyclic Chem., 2021, 100, 89 CrossRef CAS.
-
(a) X. L. Zhang, K. Q. Hu, L. Mei, Y. B. Zhao, Y. T. Wang, Z. F. Chai and W. Q. Shi, Inorg. Chem., 2018, 57, 4492 CrossRef CAS PubMed;
(b) L. W. Zeng, K. Q. Hu, Z. W. Huang, L. Mei, X. H. Kong, K. Liu, X. L. Zhang, Z. H. Zhang, Z. F. Chai and W. Q. Shi, Dalton Trans., 2021, 50, 4499 RSC;
(c) X. H. Kong, K. Q. Hu, L. Mei, A. Li, K. Liu, L. W. Zeng, Q. Y. Wu, Z. F. Chai, C. M. Nie and W. Q. Shi, Inorg. Chem., 2021, 60, 11485 CrossRef CAS PubMed.
- J. C. Wu, E. C. Escudero-Adán, M. Martínez-Belmonte and J. de Mendoza, Front. Chem., 2023, 11, 1163178 CrossRef CAS PubMed.
-
(a) L. Liang, R. Zhang, N. S. Weng, J. Zhao and C. Liu, Inorg. Chem. Commun., 2016, 64, 56 CrossRef CAS;
(b) Z. Bai, Y. Wang, Y. Li, W. Liu, L. Chen, D. Sheng, J. Diwu, Z. Chai, T. E. Albrecht-Schmitt and S. Wang, Inorg. Chem., 2016, 55, 6358 CrossRef CAS PubMed;
(c) Y. Meng, F. Niu, X. Zhang, D. Liu, Q. Lan and Y. Yang, Indian J. Chem., Sect. A: Inorg., Bio-inorg., Phys., Theor. Anal. Chem., 2021, 60, 1409 Search PubMed;
(d) S. Kusumoto, Y. Atoini, S. Masuda, J. Y. Kim, S. Hayami, Y. Kim, J. Harrowfield and P. Thuéry, Inorg. Chem., 2022, 61, 15182 CrossRef CAS PubMed.
- W. Lu, Z. Wei, Z. Y. Gu, T. F. Liu, J. Park, J. Park, J. Tian, M. Zhang, Q. Zhang, T. Gentle III, M. Bosch and H. C. Zhou, Chem. Soc. Rev., 2014, 43, 5561 RSC.
-
A. M. Tait and D. H. Busch, Inorganic Syntheses, ed. B. E. Douglas, John Wiley & Sons, New York, 1978, ch. 1.2, vol. 18, p. 10 Search PubMed.
- P. Thuéry and J. Harrowfield, Cryst. Growth Des., 2018, 18, 5512 CrossRef.
-
APEX3, ver. 2019.1–0, Bruker AXS, Madison, WI, 2019.
-
SAINT, ver. 8.40A, Bruker Nano, Madison, WI, 2019.
-
(a)
SADABS, ver. 2016/2, Bruker AXS, Madison, WI, 2016;
(b) L. Krause, R. Herbst-Irmer, G. M. Sheldrick and D. Stalke, J. Appl. Crystallogr., 2015, 48, 3 CrossRef CAS PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Adv., 2015, 71, 3 CrossRef PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3 Search PubMed.
- C. B. Hübschle, G. M. Sheldrick and B. Dittrich, J. Appl. Crystallogr., 2011, 44, 1281 CrossRef.
- A. L. Spek, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 9 CrossRef CAS PubMed.
- A. L. Spek, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2009, 65, 148 CrossRef CAS.
-
(a)
M. N. Burnett and C. K. Johnson, ORTEPIII, Report ORNL-6895, Oak Ridge National Laboratory, TN, 1996 Search PubMed;
(b) L. J. Farrugia, J. Appl. Crystallogr., 2012, 45, 849 CrossRef CAS.
- K. Momma and F. Izumi, J. Appl. Crystallogr., 2011, 44, 1272 CrossRef CAS.
- V. A. Blatov, A. P. Shevchenko and D. M. Proserpio, Cryst. Growth Des., 2014, 14, 3576 CrossRef CAS.
- C. Ruiz-Pérez, P. A. Lorenzo Luis, F. Lloret and M. Julve, Inorg. Chim. Acta, 2002, 336, 131 CrossRef.
- J. Harrowfield, Y. Atoini and P. Thuéry, CrystEngComm, 2022, 24, 1475 RSC.
- W. H. Zachariasen and H. A. Plettinger, Acta Crystallogr., 1959, 12, 526 CrossRef CAS.
- P. R. Spackman, M. J. Turner, J. J. McKinnon, S. K. Wolff, D. J. Grimwood, D. Jayatilaka and M. A. Spackman, J. Appl. Crystallogr., 2021, 54, 1006 CrossRef CAS PubMed.
-
(a) S. V. Rosokha, J. Lu, T. Y. Rosokha and J. K. Kochi, Chem. Commun., 2007, 3383 RSC;
(b) C. Borel, M. Ghazzali, V. Langer and L. Öhrström, Inorg. Chem. Commun., 2009, 12, 105 CrossRef CAS;
(c) X. C. Chai, Y. Q. Sun, R. Lei, Y. P. Chen, S. Zhang, Y. N. Cao and H. H. Zhang, Cryst. Growth Des., 2010, 10, 658 CrossRef CAS;
(d) G. A. Farnum, C. Y. Wang, R. M. Supkowski and R. L. LaDuca, Inorg. Chim. Acta, 2011, 375, 280 CrossRef CAS;
(e) K. M. Blake, J. S. Lucas and R. L. LaDuca, Cryst. Growth Des., 2011, 11, 1287 CrossRef CAS;
(f) C. Y. Wang, Z. M. Wilseck, R. M. Supkowski and R. L. LaDuca, CrystEngComm, 2011, 13, 1391 RSC;
(g) C. H. Ke, G. R. Lin, B. C. Kuo and H. M. Lee, Cryst. Growth Des., 2012, 12, 3758 CrossRef CAS;
(h) X. Q. Wang, M. X. Li, X. He, M. Shao and Z. X. Wang, Inorg. Chim. Acta, 2015, 427, 273 CrossRef CAS;
(i) M. S. Deshmukh, A. Chaudhary, P. N. Zolotarev and R. Boomishankar, Inorg. Chem., 2017, 56, 11762 CrossRef CAS PubMed;
(j) N. Y. Li and D. Liu, Acta Crystallogr., Sect. C: Struct. Chem., 2018, 74, 1581 CrossRef CAS PubMed;
(k) K. Chainok, N. Ponjan, C. Theppitak, P. Khemthong, F. Kielar, W. Dungkaew, Y. Zhou and S. R. Batten, CrystEngComm, 2018, 20, 7446 RSC.
-
(a) C. R. Groom, I. J. Bruno, M. P. Lightfoot and S. C. Ward, Acta Crystallogr., Sect. B: Struct. Sci., Cryst. Eng. Mater., 2016, 72, 171 CrossRef CAS;
(b) R. Taylor and P. A. Wood, Chem. Rev., 2019, 119, 9427 CrossRef CAS PubMed.
- A. T. Royappa, A. L. Rheingold, W. C. Teuchtler and N. L. Auld, J. Mol. Struct., 2020, 1202, 127268 CrossRef CAS.
-
(a) V. A. Blatov, M. O'Keeffe and D. M. Proserpio, CrystEngComm, 2010, 12, 44 RSC;
(b) L. Öhrström, Crystals, 2015, 5, 154 CrossRef.
-
(a) N. S. Bobbitt, M. L. Mendonca, A. J. Howarth, T. Islamoglu, J. T. Hupp, O. K. Farha and R. Q. Snurr, Chem. Soc. Rev., 2017, 46, 3357 RSC;
(b) R. Medishetty, J. K. Zareba, D. Mayer, M. Samoc and R. A. Fischer, Chem. Soc. Rev., 2017, 46, 4976 RSC;
(c) X. Zhang, P. Li, M. Krzyaniak, J. Knapp, M. R. Wasielewski and O. K. Farha, Inorg. Chem., 2020, 59, 16795 CrossRef CAS PubMed;
(d) S. Rojas and P. Horcajada, Chem. Rev., 2020, 120, 8378 CrossRef CAS PubMed;
(e) E. R. Engel and J. L. Scott, Green Chem., 2020, 22, 3693 RSC;
(f) Q. Wang, Q. Gao, A. M. Al-Enizi, A. Nafady and S. Ma, Inorg. Chem. Front., 2020, 7, 300 RSC;
(g) C. Castillo-Blas, C. Montoro, A. E. Platero-Plats, P. Ares, P. Amo-Ochoa, J. Conesa and F. Zamora, Adv. Inorg. Chem., 2020, 76, 73 CrossRef;
(h) D. Sud and G. Kaur, Polyhedron, 2021, 193, 114897 CrossRef CAS;
(i) K. Suresh, D. Aulakh, J. Purewal, D. J. Siegel, M. Veelstra and A. J. Matzger, J. Am. Chem. Soc., 2021, 143, 10727 CrossRef CAS PubMed;
(j) M. Guo, M. Zhang, R. Liu, X. Zhang and G. Li, Adv. Sci., 2022, 9, 2103361 CrossRef CAS PubMed;
(k) K. Berijani, L. M. Chang and Z. G. Gu, Coord. Chem. Rev., 2023, 474, 214852 CrossRef CAS.
-
(a) J. Jiang, M. J. Sarsfield, J. C. Renshaw, F. R. Livens, D. Collison, J. M. Charnock, M. Helliwell and H. Eccles, Inorg. Chem., 2002, 41, 2799 CrossRef CAS PubMed;
(b) D. K. Unruh, A. Libo, L. Streicher and T. Z. Forbes, Polyhedron, 2014, 73, 110 CrossRef CAS;
(c) J. A. Ridenour, M. M. Pyrch, Z. J. Manning, J. A. Bertke and C. L. Cahill, Acta Crystallogr., Sect. C: Struct. Chem., 2017, 73, 588 CrossRef CAS PubMed.
- K. Carter-Fenk and J. M. Herbert, Phys. Chem. Chem. Phys., 2020, 22, 24870 RSC.
-
(a) A. N. Khlobystov, A. J. Blake, M. R. Champness, D. A. Lemonovskii, A. G. Majouga, N. V. Zyk and M. Schröder, Coord. Chem. Rev., 2001, 222, 155 CrossRef CAS;
(b) R. León-Zárate and J. Valdés-Martínez, Cryst. Growth Des., 2021, 21, 3756 CrossRef.
- J. Jiang, H. Furukawa, Y. B. Zhang and O. M. Yaghi, J. Am. Chem. Soc., 2016, 138, 10244 CrossRef CAS PubMed.
- J. J. Liu, S. B. Xia, D. Liu, J. Hou, H. Suo and F. X. Cheng, Dyes Pigm., 2020, 177, 108269 CrossRef CAS.
-
(a) C. Bianchini, C. J. Elsevier, J. M. Emsting, M. Peruzzini and F. Zanobini, Inorg. Chem., 1995, 34, 84 CrossRef CAS;
(b) S. Wörl, D. Hellwinkel, H. Pritzkow, M. Hofmann and R. Krämer, Dalton Trans., 2004, 2750 RSC;
(c) A. J. Plajer, A. L. Colebatch, F. J. Rizzuto, P. Pröhm, A. D. Bond, R. García-Rodríguez and D. S. Wright, Angew. Chem., Int. Ed., 2018, 57, 6648 CrossRef CAS PubMed.
- D. Munz and K. Meyer, Nat. Rev. Chem., 2021, 5, 422 CrossRef CAS PubMed.
- A. W. G. Platt, Coord. Chem. Rev., 2017, 340, 62 CrossRef CAS.
- W. L. Driessen and W. L. Groeneveld, Recl. Trav. Chim. Pays-Bas, 1969, 88, 491 CrossRef CAS.
- J. Harrowfield, J. Chem. Soc., Dalton Trans., 1996, 3165 RSC.
- T. G. Mitina and V. A. Blatov, Cryst. Growth Des., 2013, 13, 1655 CrossRef CAS.
-
(a) H. D. Burrows, S. J. Formosinho, M. da G. Miguel and F. Pinto Coelho, J. Chem. Soc., Faraday Trans. 1, 1976, 72, 163 RSC;
(b) A. T. Kerr and C. L. Cahill, Cryst. Growth Des., 2014, 14, 1914 CrossRef CAS;
(c) A. T. Kerr and C. L. Cahill, Cryst. Growth Des., 2014, 14, 4094 CrossRef CAS;
(d) J. A. Ridenour, M. M. Pyrch, Z. J. Manning, J. A. Bertke and C. L. Cahill, Acta Crystallogr., Sect. C: Struct. Chem., 2017, 73, 588 CrossRef CAS PubMed;
(e) A. T. Kerr, J. A. Ridenour, A. A. Noring and C. L. Cahill, Inorg. Chim. Acta, 2019, 494, 204 CrossRef CAS;
(f) G. E. Gomez, J. A. Ridenour, N. M. Byrne, A. P. Shevchenko and C. L. Cahill, Inorg. Chem., 2019, 58, 7243 CrossRef CAS PubMed.
- P. Thuéry, Y. Atoini and J. Harrowfield, Inorg. Chem., 2020, 59, 2503 CrossRef PubMed.
-
(a) A. Brachmann, G. Geipel, G. Bernhard and H. Nitsche, Radiochim. Acta, 2002, 90, 147 CrossRef CAS;
(b) M. Demnitz, S. Hilpmann, H. Lösch, F. Bok, R. Steudtner, M. Patzschke, T. Stumpf and N. Huittinen, Dalton Trans., 2020, 49, 7109 RSC.
- P. Thuéry and J. Harrowfield, Inorg. Chem., 2017, 56, 13464 CrossRef PubMed.
- L. Baklouti and J. Harrowfield, Dalton Trans., 2023, 52, 7772 RSC.
|
| This journal is © The Royal Society of Chemistry 2023 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *a,
Youssef
Atoini
b and
Jack
Harrowfield
*a,
Youssef
Atoini
b and
Jack
Harrowfield