
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceNovel high-temperature phase and crystal structure evolution of CsCuBr3 halide identified by neutron powder diffraction†
Carmen
Abia
ab,
Carlos A.
López
 ac,
Javier
Gainza
a,
João Elias F. S.
Rodrigues
d,
María T.
Fernández-Díaz
b,
Eva
Céspedes
a,
José Luis
Martínez
a and
José Antonio
Alonso
*a
ac,
Javier
Gainza
a,
João Elias F. S.
Rodrigues
d,
María T.
Fernández-Díaz
b,
Eva
Céspedes
a,
José Luis
Martínez
a and
José Antonio
Alonso
*a
aInstituto de Ciencia de Materiales de Madrid, CSIC, Cantoblanco, 28049 Madrid, Spain. E-mail: ja.alonso@icmm.csic.es
bInstitut Laue Langevin, 38000 Grenoble Cedex, France
cINTEQUI, (UNSL-CONICET) and, Facultad de Química, Bioquímica y Farmacia, UNSL, Almirante Brown 1455, 5700, San Luis, Argentina
dEuropean Synchrotron Radiation Facility, 38000 Grenoble Cedex, France
First published on 29th May 2023
Abstract
The search for novel lead-free materials with potential optoelectronic applications is a main research topic nowadays for an environment-friendly energy transition. Based on promising all-inorganic CsPbBr3 perovskite, we tested here the substitution of toxic Pb2+ with Cu2+ and explored the understudied CsCuBr3 halide. We present a mechanically-synthesized CsCuBr3 specimen obtained by ball milling, consisting of a well-crystallized and pure sample. Calorimetric measurements revealed a thermal event suggesting a structural phase transition around 422 K, immediately below the decomposition of the sample due to Br loss. A detailed structural analysis was carried out using neutron powder diffraction data from 20 to 420 K, focusing on the evolution of the orthorhombic phase (space-group: C2221) up to 400 K; this crystal arrangement consists of dimer units of face-sharing [CuBr6] octahedra containing Cu–Cu dimers with conspicuously short distances that account for the magnetic coupling between Cu2+ spins described before. Additionally, at 420 K, we identified a novel high-temperature phase as described in the hexagonal P6522 space group, where infinite chains of [CuBr4] square-planar units were observed. An additional study considering the Debye model was carried out, providing information on the relative Cs–Br and Cu–Br chemical bonds. From diffuse reflectance UV-Vis measurements, an optical gap of ∼1.8 eV was estimated for the orthorhombic phase at room temperature. Magnetic measurements indicate that the effective magnetization behavior versus temperature appears close to linear and seems to extrapolate to zero (smaller than 10−5 μB/Cu atom) at temperatures above 420 K, coinciding with onset of phase transition.
Introduction
Hybrid halide organic–inorganic perovskites, typified by methylammonium lead triiodide (MAPbI3 or MAPI) and formamidinium lead triiodide (FAPbI3), have attracted significant attention in recent years due to their promising potential in photovoltaic applications, with over 22% power conversion efficiency.1 The bromine phase, MAPbBr3, has also been studied particularly for their enhanced environmental stability.2,3 Totally inorganic perovskite derivatives are also promising materials for solar cells, because of their remarkable optical properties. With a bandgap of 1.73 eV,4 the cubic CsPbI3 is one of the best choices. This compound has a high fluorescence quantum yield and is more resistant to degradation when exposed to ambient air and humidity as compared to its organic–inorganic counterparts.Unfortunately, CsPbI3 has a cubic (α) to orthorhombic (δ) structural phase transition near room temperature,5 which shifts away from a stable phase and prevents the commercialization of this material inserted in a final device. This limitation can be overcome by replacing I with Br, partially or completely, as in the case of the orthorhombic CsPbBr3 perovskite.6–14 Such modifications improve the optoelectronic and photoresponse properties, besides better stability under ambient conditions, making it a suitable material for a variety of optoelectrical systems.15 On the other hand, the substitution of I for Br raises the bandgap to 2.3 eV, which is considerably large for solar cell applications. The toxicity of Pb is also a major drawback and a big restriction for its commercialization. For this reason, great efforts have been put to develop new lead-free alternatives with convenient optical properties for optoelectronic applications.16
With this framework in mind, we focus on cesium tribromocuprate(II), or CsCuBr3. This material presents an orthorhombic 3D structure with face-sharing [CuBr6] octahedra, forming dimer units. This halide is rather understudied; only Inoue et al. briefly discussed the paramagnetism in 1967,17 possibly due to coupled unpaired electrons within the dimers; Li et al. studied in more detail the room temperature crystal structure of the compound back in 1973.18 The magnetic susceptibility was reported as rather weak (slight paramagnetism)18 in the temperature range from 80 up to 300 K, with an effective magnetic moment on the copper atoms of 0.1 μB, close to zero, due to the experimental sensitivity. This low value seems to indicate that the unpaired electrons of Cu2+ are coupled, forming dimers. Also, due to the distance between Cu ions and the general rules of Goodenough–Kanamori, the prevalent interaction should be antiferromagnetic.18 It was also observed in this early work that the magnetic exchange, described by Anderson et al.,19 through Br ions would need an excited electronic configuration, which will result in a charge transfer from Br− (p orbital) to an empty dx2−y2 orbital of the Cu2+ ion, which in consequence will produce a residual magnetic moment of the copper site. However, no long-term interactions were described between the different Cu dimers.
In this work, a well-crystallized powder of CsCuBr3 was synthesized by a mechano-chemical process using a ball milling device. This method enables solvent-free preparation, reliability, and less energy consumption. For structural characterization, the neutron powder diffraction (NPD) patterns were collected within a wide temperature range to probe the evolution of the crystallographic structure. We revised the C2221 orthorhombic symmetry, maintained in the temperature range of 20–400 K, and at 420 K, a novel P6522 hexagonal phase was identified, just below the onset of decomposition by Br loss. The NPD data allowed determining accurate values of the mean square displacement (MSD) factors for Cs, Cu, and Br atoms. An additional study of the thermal evolution of these atomic MSDs and the consideration of the Debye model revealed the contrasting bonding stiffness of the crystal. The description of the crystallographic structure is complemented by calorimetric and optical measurements. The ball milling prepared CsCuBr3 showed a suitable optical gap of ∼1.8 eV for potential applications in photovoltaic devices.
Materials and methods
Synthesis
The polycrystalline CsCuBr3 powder was synthesized by mechano-chemical synthesis using ball milling apparatus. Stoichiometric amounts of CsBr and CuBr2, with a total mass of 1 g, were weighed and mixed with 20 zirconia balls (5 mm of diameter) in a N2-filled glove box. The reaction was carried out in a Retsch PM100 mill, within a sealed zirconia-lined jar with a N2 environment, for 3 hours at 400 rpm. A laboratory X ray powder diffraction (XRD) pattern at room temperature was collected on a Bruker D5 diffractometer with Cu-Kα (λ = 1.5418 Å) radiation.Neutron diffraction and analysis
The evolution of the crystallographic structure with temperature was studied with neutron powder diffraction (NPD) using the D20 instrument, a high intensity 2-axis diffractometer at the Institut Laue Langevin (ILL) in Grenoble, France. The NPD patterns were collected at 20, 100, 200, 300, 400, and 420 K with a wavelength of λ = 1.540 Å and a take-off angle of 90°. The sample was contained in a cylindrical vanadium holder of 6 mm diameter. NPD data analysis was performed by the Rietveld method with the FullProf program.20,21 The refined parameters were the following: zero-point error, background coefficients, scale factor, asymmetry factors, lattice parameters, atomic fractional coordinates (x, y, z), and isotropic thermal displacements (Uiso).Thermal and optical properties
Differential scanning calorimetry (DSC) and thermogravimetric (TG) measurements were carried out in the temperature range of 300–480 K in a Q600 SDT system from TA Instruments, with a heating rate of 10 K min−1 and 10 mg of sample. A UV-Vis spectrophotometer Varian Cary 5000 was employed to measure the optical diffuse reflectance spectrum at room temperature, from which the optical bandgap was estimated. A diffuse reflectance cell was considered with an integrating sphere for estimating the absorption coefficient (α). This coefficient can be determined by the Kubelka–Munk function F(R). The bandgap energy was estimated with the spectrophotometer used for the diffuse reflectance data fitted by the equation [hνF(R)]1/n = A(hν − EG) using the Tauc plot. The exponent n depends on the nature of the electron transition: 1/2 and 2 for direct and indirect transition gaps, respectively.Magnetic measurements
The magnetic characterization was performed by means of SQUID magnetometers from Quantum Design (San Diego, USA) in a temperature range from 1.8 up to 400 K and applied magnetic fields up to 7 T. A specific oven system was used to reach the 300–430 K temperature interval. The furnace included an Al2O3 ceramic sample holder, with a resistive heating element and a thermocouple embedded on the backside of the sample holder. To retain heat and reduce thermal gradients between the sample and heater stick, the platinum heater region is wrapped securely with a thin copper-foil radiation shield. We limited the sample temperature to 430 K to avoid the sublimation of bromine above such temperature, with the corresponding destruction of the sample and the contamination of the magnetometer cryostat.Results and discussion
Initial characterization
The laboratory powder XRD measurement at room temperature (RT) allowed initial structural characterization, assessing the high purity of the final product. The CsCuBr3 sample presents an orthorhombic symmetry in the space group C2221 (#20), in agreement with unique previous results of this phase reported by Li et al. in 1972.18 The Rietveld refinement of the laboratory XRD data provided a preliminary crystallographic structural model with the unit-cell parameters a = 12.776(2) Å, b = 7.657(2) Å, and c = 12.641(3) Å. Fig. S1 (ESI†) displays the Rietveld plot. Fig. 1 shows the TG and DSC curves as a function of temperature; the sample is stable up to 440 K, where the chemical decomposition starts by Br loss. In the DSC curve, an endothermal event, starting at 410 K and peaking at around 422 K, suggests a structural phase transition, immediately below the decomposition onset of the sample.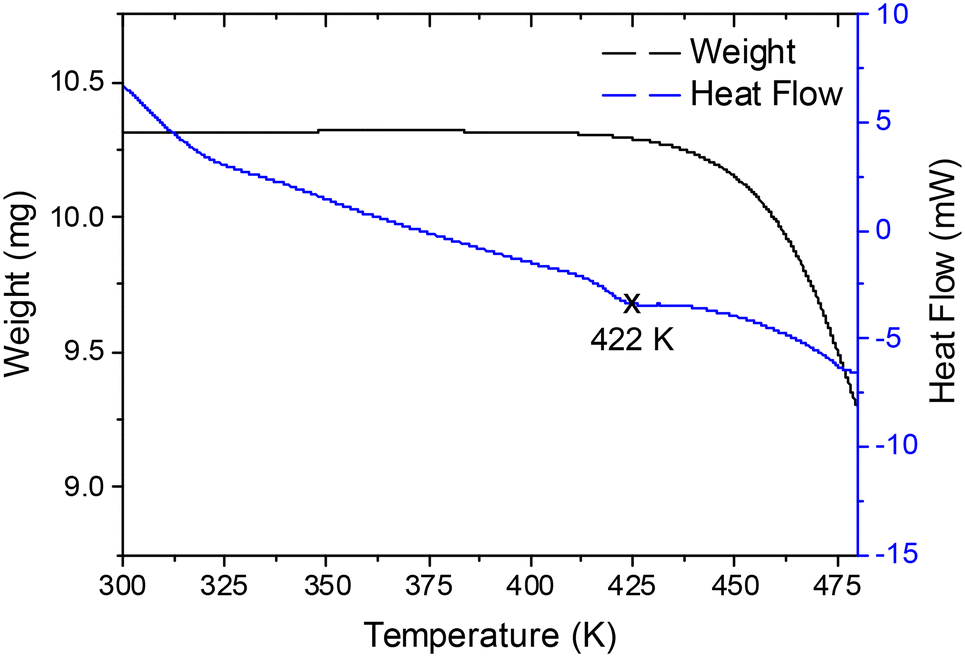 | ||
| Fig. 1 TG and DSC curves of the CsCuBr3 sample, showing a possible phase transition around 422 K before the beginning of the decomposition process at 440 K. | ||
Orthorhombic phase at 300 K
The analysis of the NPD data allowed an updated revision of the crystal structure of CsCuBr3. At 300 K, the compound crystallizes in the orthorhombic C2221 space group (#20), as it was already determined from the laboratory XRD data. The Rietveld refinement, shown in Fig. 2a, yielded the following values of the unit-cell parameters and volume: a = 12.6532(4) Å, b = 7.5718(3) Å, c = 12.5151(5) Å and V = 1199.04(7) Å3. The crystallographic information of the CsCuBr3 sample is displayed in Table 1.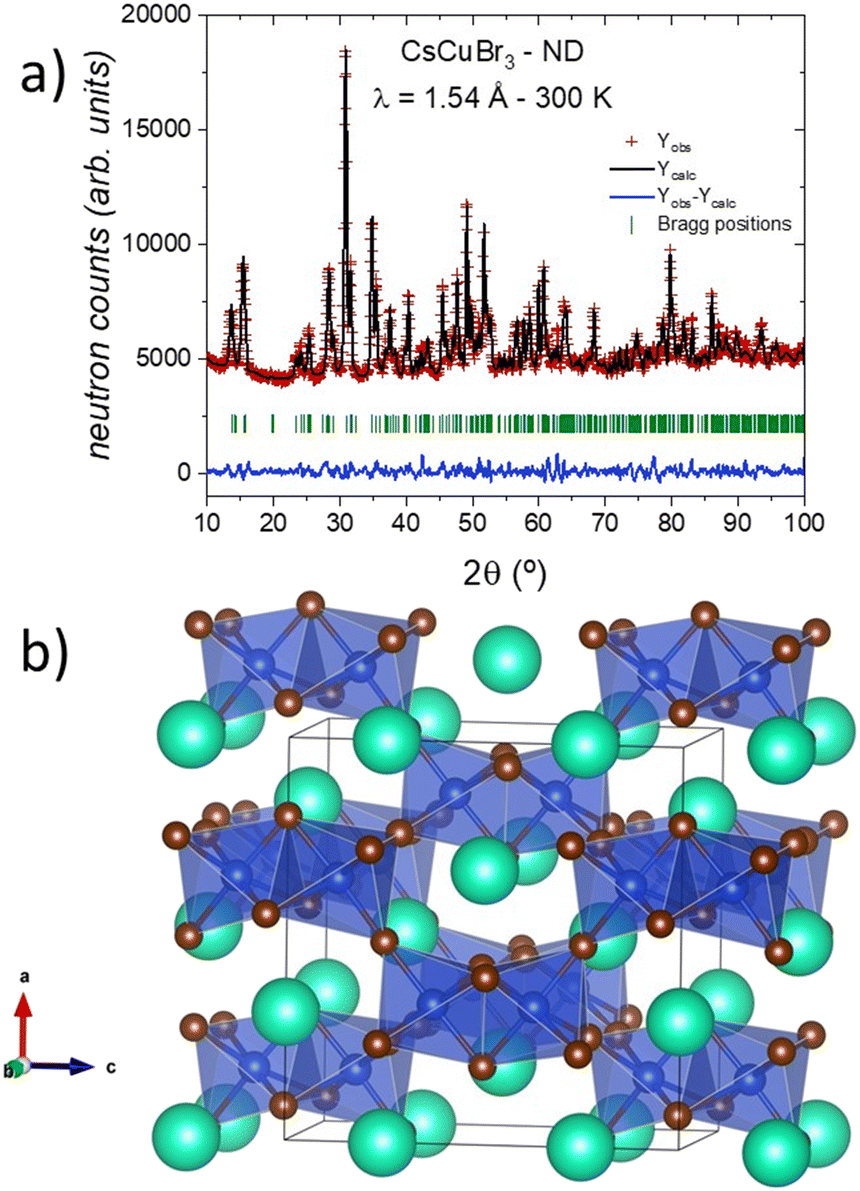 | ||
| Fig. 2 (a) Observed (red crosses), calculated (full black line), and difference (blue line) Rietveld profiles for CsCuBr3 at 300 K from NPD data. (b) View of the 3D arrangement of CsCuBr3 as dimers of face-sharing [CuBr6] octahedra, linked to another six dimers through common corners. | ||
| Atom | Site | x | y | z | U iso (Å2) | f occ |
|---|---|---|---|---|---|---|
| R p = 2.42%, Rwp = 3.16%, χ2 = 5.98, RBragg = 7.30%. | ||||||
| Cs1 | 4a | 0.318(1) | 0 | 0 | 0.031(3) | 1 |
| Cs2 | 4b | 0 | 0.020(2) | 1/4 | 0.031(3) | 1 |
| Cu | 8c | 0.1544(5) | 0.498(2) | 0.1290(5) | 0.228(5) | 1 |
| Br1 | 4a | 0.3053(7) | 1/2 | 0 | 0.228(5) | 1 |
| Br2 | 8c | 0.0739(5) | 0.258(1) | 0.0286(6) | 0.228(5) | 1 |
| Br3 | 8c | 0.2585(7) | 0.2971(9) | 0.2612(6) | 0.228(5) | 1 |
| Br4 | 4b | 0 | 0.515(2) | 1/4 | 0.228(5) | 1 |
The crystal structure consists of a three-dimensional framework, where the Cs atoms are located in two different Wyckoff sites, at 4a (x, 0, 0) and 4b (0, y, 1/4). The Cu atoms, located in 8c (x, y, z), are arranged in a network of [Cu2Br9]5− dimeric units, which are formed by two face-shared distorted [CuBr6] octahedra with copper at the centers and bromine at the corners. There are four types of Br atoms: Br1 at 4a (x, 1/2, 0), Br4 at 4b (0, y, 1/4), and two inequivalent atoms (Br2 and Br3) at 8c (x, y, z) sites. In short, the crystal structure can be described as dimers of face-sharing octahedra, linked to another six dimers through common corners in a 3D arrangement (in Fig. 2b). Within the dimers, there are conspicuously short Cu–Cu distances of 3.260(9) Å, which account for the magnetic coupling between Cu2+ spins described before.18 It is important to notice that although the chemical formula corresponds to the perovskite structure ABX3, this phase does not strictly exhibit a perovskite-like framework, as it could be suggested by the crystallochemistry.
To obtain a complete picture of the thermal evolution of the crystal structure, NPD patterns were collected and analyzed at the following temperature points: 20, 100, 200, 300, 400, and 420 K. A first inspection of these neutron data revealed that the orthorhombic symmetry is observed from 20 up to 400 K, without any major change.
In contrast, the pattern at 420 K is completely different from the ones collected at lower temperatures, suggesting a structural phase transition taking place at this temperature. Fig. 3 displays some selected angular 2θ ranges, which illustrate the changes along the diffraction patterns. Therefore, the crystal structure was successfully refined in the C2221 space group from NPD data in the temperature range of 20–400 K.
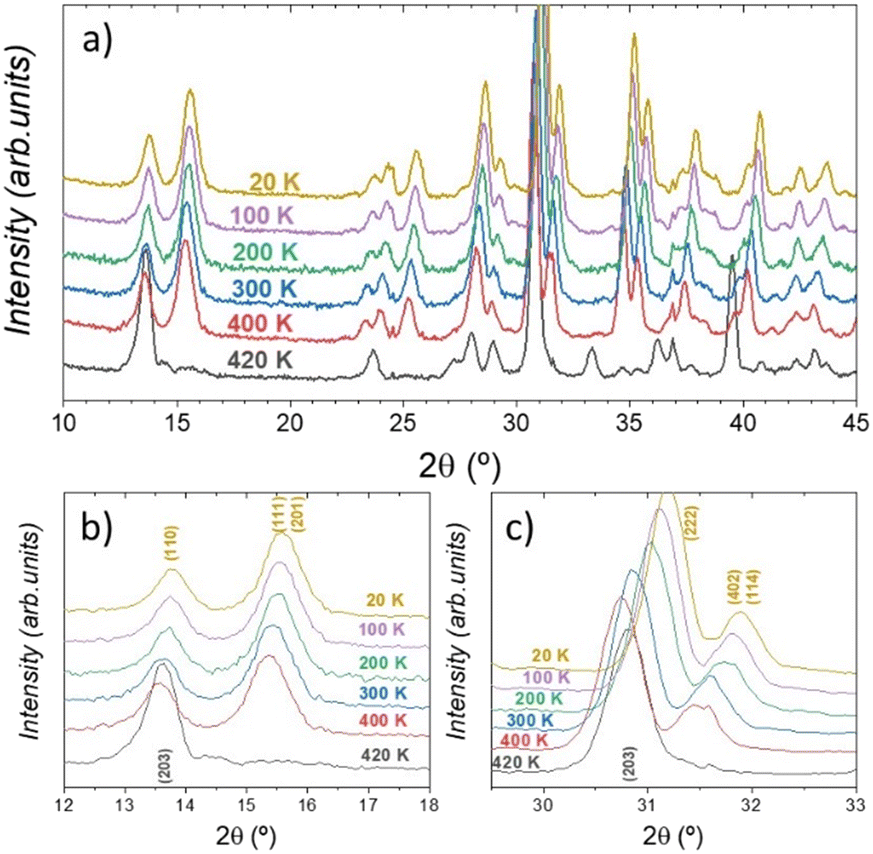 | ||
| Fig. 3 Temperature-dependent neutron diffraction patterns of CsCuBr3 halide: angular 2θ range: (a) 10–45°, (b) 12–18°, and (c) 30–33°. The 420 K NPD pattern has a different peak distribution as compared with the lower-temperature ones. | ||
Novel high temperature phase at 420 K
The first attempts to index the NPD pattern at 420 K revealed a hexagonal symmetry; however, it was not possible to suitably determine the space group. Nevertheless, the comparison with the chlorine counterpart phase, CsCuCl3, was enlightening. This compound exhibits a hexagonal symmetry at room temperature, described in the chiral P6522 space group (#179). By using this reported model as a reference, it was finally possible to properly fit the diffraction data at 420 K, as illustrated in Fig. 4a. In this crystallographic structural model, the cesium and copper cations are located in the 6b (x, 2x, 1/4) and 6a (x, 0, 0) Wyckoff sites, respectively. Moreover, the bromide anions are distributed between the 12c (x, y, z) and 6b (x, y, 3/4) sites. This structure is substantially different from the previously described C2221 orthorhombic phase, mainly due to the geometry of the copper environment with square planar [CuBr4] units. The copper arrangement changed from a 3D connectivity to a 1D connection, in Br vertex-sharing chains along the c-axis, conforming a chiral helix on a screw axis (65), see Fig. 4b for a crystallographic view. The main crystallographic information of the CsCuBr3 sample at 420 K is displayed in Table 2.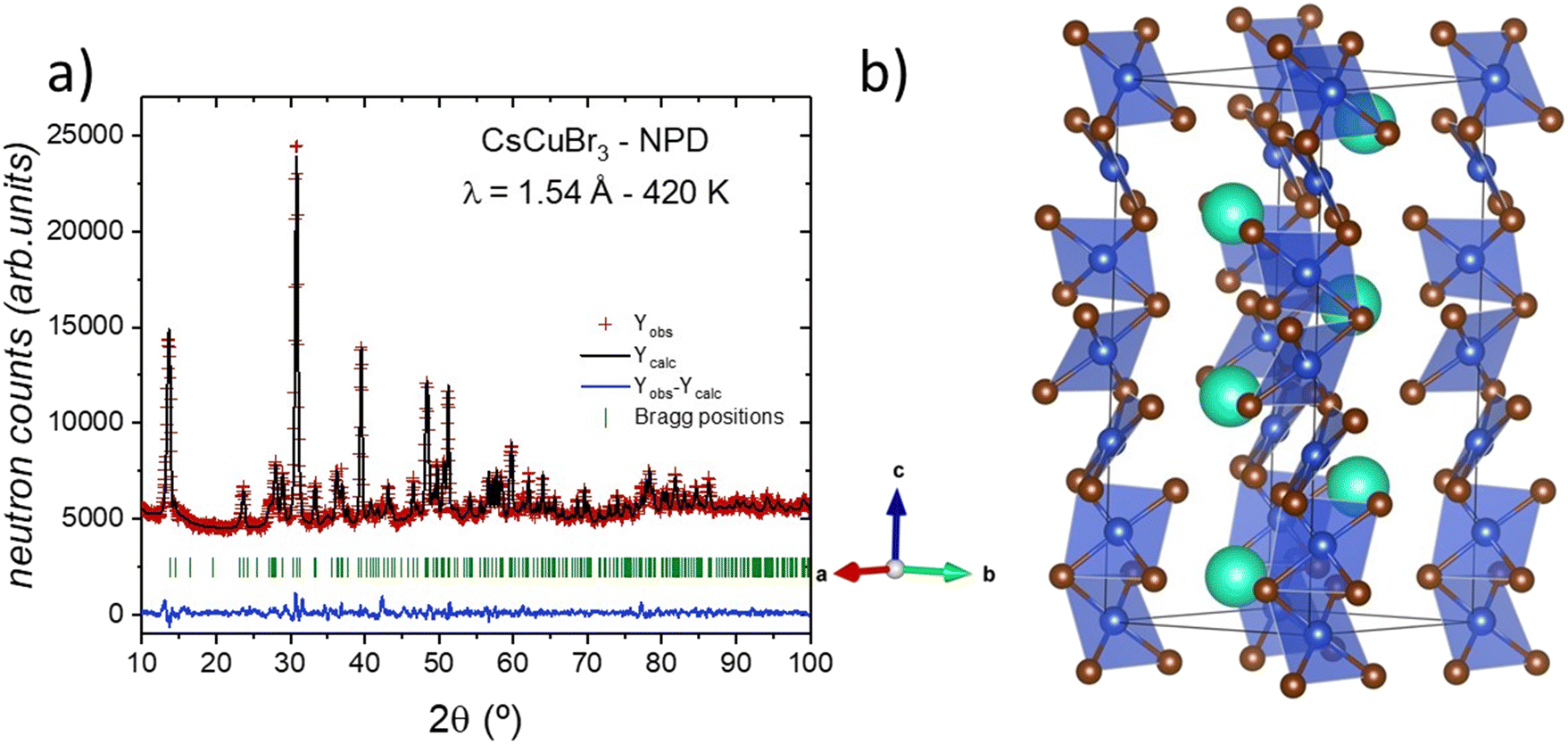 | ||
| Fig. 4 (a) Rietveld refinement of the crystal structure from the NPD data at 420 K and (b) view of the high-temperature hexagonal crystal structure with the chiral P6522 space group. | ||
| Atom | x | y | z | U eq (Å2) | f occ |
|---|---|---|---|---|---|
| Cs | 0.354(2) | 0.708(2) | 0.25 | 0.056(7) | 1 |
| Cu | 0.9451(8) | 0 | 0 | 0.044(4) | 1 |
| Br1 | 0.842(1) | 0.2037(8) | 0.2424(3) | 0.044(4) | 1 |
| Br2 | 0.1198(9) | 0.2397(9) | 0.75 | 0.037(5) | 1 |
| Atomic displacement parameters (Å2) | ||||||
|---|---|---|---|---|---|---|
| U 11 | U 22 | U 33 | U 12 | U 13 | U 23 | |
| R p = 1.87%, Rwp = 2.53%, χ2 = 4.13, RBragg = 4.71%. | ||||||
| Cs | 0.040(6) | 0.040(6) | 0.089(8) | 0.020(6) | 0.021(5) | 0 |
| Cu | 0.055(4) | 0.048(5) | 0.029(3) | 0.024(5) | 0.004(2) | 0.008(2) |
| Br1 | 0.041(5) | 0.046(4) | 0.047(3) | 0.024(4) | −0.003(4) | 0.001(4) |
| Br2 | 0.038(5) | 0.020(4) | 0.053(6) | 0.010(4) | −0.007(5) | 0 |
Thermal evolution of the CsCuBr3 crystal structure
The thermal evolution of the crystal structure was analyzed, initially, through the unit-cell parameters. Fig. S2† plots the thermal evolution of a, b and c unit-cell parameters and Fig. 5 plots the volume normalized by Z (number of chemical formulas per unit-cell), where an abrupt increase is observed between 400 and 420 K due to the orthorhombic–hexagonal phase transition under heating. This abrupt volume variation agrees with the nature of the structural change involving a large atomic reorganization. Besides, two main differences between the C2221 and P6522 space groups can be highlighted: the first one is the change in the Br coordination of the copper, which goes from octahedral [CuBr6] to square planar [CuBr4] units, as mentioned before; the second one concerns the connection between copper polyhedra, which evolve from a 3D framework of octahedral dimers to a 1D helical chain of square planes. Both cases are illustrated in Fig. 5 as insets. | ||
| Fig. 5 Thermal evolution of the unit-cell volume, showing the crystallographic views of the 3D octahedral dimers and the 1D helical chain of square planes for the two detected phases. | ||
Additionally, there is a peculiar behavior along the unit-cell volume evolution within the temperature range of the orthorhombic phase. Naturally, the expected thermal expansion is observed, but this expansion is not gradual, and it presents an inflexion at about 200 K, suggesting subtle changes in the crystalline structure, as exhibited in Fig. 5 and S2.†
To further elaborate on the above observations, the thermal behavior of Cu polyhedra was analyzed from Cu–Br distances. To carry out such analysis, two additional parameters that describe the octahedral distortion were calculated considering distances and angles: the distortion index (DI)22,23 and the quadratic elongation (QE).24 Both parameters are plotted in Fig. 6b. Fig. 6c and d illustrate a detailed view of the Cu polyhedral arrangement in C2221 and in P6522 space groups, with all the Br atoms suitably labelled. The octahedral change is also observed in the DI and QE parameters, as they abruptly increase from 400 to 420 K, indicating a distortion event along both distances and angles. This abrupt change is also seen in the Cu–Br distances, which are 2.479(9) Å, 2.516(7) Å, and 2.951(8) Å for Cu–Br1, Cu–Br2, and Cu–Br1*, respectively (see Fig. 6d). This sharp variation does not occur in the orthorhombic phase about 200 K, since they present an octahedral arrangement with hardly distorted Cu–Br distances, see Fig. 6a.
 | ||
| Fig. 6 Thermal evolution of (a) Cu–Br distances and (b) distortion parameters (distortion index and quadratic elongation) of [CuO6] octahedra. Schematic views of [CuO6] polyhedra in (c) orthorhombic C2221 and (d) hexagonal P6522 space groups. | ||
In spite of this, this structural distortion is not concomitant with the tetragonal-octahedral distortion observed in Cu2+ ions. As can be seen in Fig. 6c, the elongated bonds are Cu–Br2* and Cu–Br3*, which are not opposite in the octahedron. This situation is probably due to the absence of Jahn–Teller effects or to a disorder of this distortion in Cu2+. Furthermore, the distance and angle distortion revealed subtle changes, showing that the [CuBr6] octahedron is not monotonically affected by the temperature but the octahedral distortion increases as temperature decreases. A non-cooperative Jahn–Teller effect in the orthorhombic phase may be assumed to occur through the structural differences observed between high (300 and 400 K) and low (20 and 100 K) temperatures. It is possible to assume that the non-cooperative Jahn–Teller effect is more dynamic as temperature increases, probably reducing the distortion of the [CuO6] octahedron until the structural phase transition is completed.
Mean-square displacements within the orthorhombic phase
An alternative way to study the temperature effect on the crystal structure is analyzing the mean-square displacements (MSDs). Thus, based on the NPD data in the 20–400 K range, we have extracted the mean-square displacements (MSDs) of the atomic species Cs, Cu, and Br within the CsCuBr3 compound. However, few approximations were considered: Cs1 and Cs2 have the same MSD values; all the bromides (Br1, Br2, Br3, and Br4) are constrained to exhibit equal displacement factors. Therefore, those MSD coefficients indeed represent average displacements for Cs 〈Cs〉 and Br 〈Br〉. We have used the Debye model25–27 to describe the temperature-dependence of the aforementioned MSDs (or isotropic displacement Uiso, in units of Å2), i.e. | (1) |
In Fig. 7, the temperature-dependence of the MSDs for 〈Cs〉, Cu, and 〈Br〉 with their respective best fittings to the Debye equation are shown. The Debye temperatures were obtained, as follows: 〈Cs〉 (101.6 K), Cu (180.4 K), and 〈Br〉 (159.7 K). The bonding stiffness evaluation for 〈Cs〉–〈Br〉 and Cu–〈Br〉 can be done by the harmonic one-particle potential model (h-OPP),28,29 such that a force constant is defined from the Debye temperature (θD) and atomic mass (m):
 | (2) |
 | ||
| Fig. 7 Temperature dependence of Uiso (MSDs) for 〈Cs〉, Cu, and 〈Br〉 atoms. Sphere symbols denote the MSD values extracted from the Rietveld refinement of the NPD data. The dotted lines are the best fittings to the experimental data using the Debye model (eqn (1)). Standard deviations are smaller than the size of the symbols. | ||
Optical properties
Diffuse reflectance UV-vis spectroscopy was performed to probe the absorption capacity of the CsCuBr3 powder sample. The optical absorption coefficient related to the Kubelka–Munk function [F(R) = a = (1 − R)2/2R, where R is the reflectance] versus wavelength in units of eV is displayed in Fig. 8 (Tauc plot). By extrapolating the linear region to the abscissa, it is possible to estimate a bandgap value of ∼1.8 eV, denoting that it would be promising for lead-free solar cell devices. This bandgap estimation method (Tauc) only provides a lower bound for the actual value;30 however, any comparison with other reported values can be done directly, since most of the reported bandgaps were determined using the Tauc plot methodology (see Materials and methods). The CsCuBr3 bandgap value is close to the one reported for the chlorine counterpart (∼1.92 eV),31 but far from the one reported for the iodine phase (∼0.29 eV).32 These differences may be related to the structural features that the bromine and chlorine phases present with respect to CsCuI3, such that the former exhibits a standard perovskite structure.32 Nonetheless, the bandgap of CsCuBr3 is within the range of other all-inorganic halide phases of interest in the optoelectronic field, such as CsPbBr3 (∼2.22 eV)14 and CsSnBr3 (∼1.78 eV).33 In the Materials Project database, a preliminary DFT calculation provided the electronic band structure of CsCuBr3, attesting for an indirect transition (Γ → X).34 In such a calculation, the spin configuration was not properly set, and therefore the bandgap is said to be 0.0 eV, which is in contrast with our experimental result. This difference is mainly concerned with the spin configuration that was absent in this calculation. | ||
| Fig. 8 Kubelka–Munk (KM) transformed diffuse reflectance spectrum of CsCuBr3 halide. The extrapolation by the linear curve provides a bandgap of ∼1.8 eV. | ||
Magnetic properties
The magnetic susceptibility of CsCuBr3 is presented in the inset of Fig. 9a. The magnetic susceptibility is relatively low at room temperature, exhibiting a significant increase at low temperatures, as a Curie tail. Fig. 9a represents the temperature dependence of χ × T in a wide temperature range. If the system follows a Curie–Weiss law in the full temperature range, the term (χ × T)1/2 is proportional to an effective magnetic moment (μeff). In Fig. 9a, an increase of χ × T is observed, with a maximum effective moment as low as μeff = 0.06 at 120 K. Above this temperature, the effective moment monotonously decreases, at 400 K with a value close to μeff = 0.02. This result is coherent with the suggestion of Anderson et al.,19 although the obtained experimental effective magnetic moment is very low. | ||
| Fig. 9 a) Temperature dependence of χ × T for CsCuBr3. The inset shows the temperature dependence of the magnetic susceptibility measured with an applied magnetic field of 0.7 T. b) Magnetic field dependence of the magnetization for CsCuBr3 at different temperatures in the temperature range of 1.8–400 K. | ||
To shed some light on the magnetic behavior, we measured the field dependence of the magnetization for CsCuBr3 at different temperatures in the 1.8–400 K range (see Fig. 9b). The variation of the magnetization is very large between 1.8 and 400 K. Moreover, for temperatures around 300 K and high magnetic fields (above 1.5 T, up to our maximum field of 7 T) the total magnetic signal becomes diamagnetic, dominated by Br and Cs ion contribution. The field dependence of the magnetization indicates a tendency to saturation (see the log scale in Fig. 9b). The extrapolation of the magnetization from large magnetic fields to zero (some type of effective localized magnetization) is rather low (10−2 μB/Cu atom) at 1.8 K, and it decreases to even lower values (after correcting the diamagnetic signal), as the temperature increases.
The same CsCuBr3 powder sample was dismounted and placed on the furnace sample holder, to be measured up to 420 K (assuring that the temperature is lower than 430 K). Due to experimental issues, it is complex to assess the final sample mass at the oven holder, nonetheless being smaller than the initially measured one. For this reason, magnetic units are just expressed in emu, without any normalization, in Fig. 10a. It shows the magnetic field dependence of the magnetization at 420 K, noticing a magnetic signal dominated by the diamagnetic components, including the heater holder ones (negative, opposed to the applied magnetic field). Only at a rather low magnetic field, the obtained signal is positive, evidencing a residual magnetic moment in the order of 10−5 μB/Cu, depending on the estimation of the real sample mass, see lower inset of Fig. 10a.
 | ||
| Fig. 10 a) Magnetic field dependence of the magnetization at 420 K for CsCuBr3. The upper inset shows the magnetic susceptibility in the temperature range from 300 to 430 K, measured with an applied magnetic field of 200 Oe. The lower inset shows the corrected hysteresis loop, after the subtraction of the diamagnetic signal. b) Temperature dependence of the effective magnetization in CsCuBr3 obtained from the extrapolation of the high field magnetization to the zero field from the data in Fig. 9b. The line is just a guide to the eye. | ||
The upper inset of Fig. 10a shows the temperature dependence of the magnetic susceptibility, measured with an applied field of 200 Oe in the 300–420 K range. The observed signal, rather weak, decreases linearly, pointing towards a zero-value slightly above 420 K. The measured data are qualitatively coherent with the lower temperature range (inset Fig. 9a), indicating that around 420 K, in accordance with the structural phase transition, some effective magnetic moment starts to be localized in the Cu atoms.
From the field dependence magnetization shown in Fig. 9b, we could infer the temperature dependence of the effective localized magnetization (see details above). The data are presented in Fig. 10b. The effective magnetization behavior versus temperature appears close to linear and seems to extrapolate to zero (smaller than 10−5 μB/Cu atom) at temperatures above 420 K.
In summary, the magnetic behavior of CsCuBr3 seems to indicate that at 420 K and associated with the structural phase transition, an effective weak magnetic moment appears, probably related to a fraction of localized unpaired electrons of the Cu dimers, due to the exchange interaction mediated by Br. This effective moment is rather weak in that temperature range, but increases from 10−5 μB/Cu-atom at 420 K to 10−2 μB/Cu-atom at very low temperature (1.8 K). Also, it seems that there is no magnetic coupling between the different Cu–Cu dimers of the face-sharing octahedra, which are crystallographically linked to six dimers through common corners (below 420 K).
Conclusions
CsCuBr3 was successfully synthesized by a mechano-chemical method, achieving a well-crystallized and pure powder sample. The evolution of its crystal structure has been studied from NPD data in the temperature range of 20–420 K. Up to 400 K, the compound presents a C2221 orthorhombic symmetry, formed by a three-dimensional infinite network of dimeric units [Cu2Br9]5−. At 420 K, an abrupt phase transition has already occurred, which had never been reported before; at this temperature, CsCuBr3 exhibits a P6522 hexagonal symmetry containing infinite chains of vertex-sharing [CuBr4] square planar units. The combination of the thermal evolution of the atomic mean square displacements and the Debye model allowed the determination of the Debye temperatures of all the atoms within the unit-cell. Considering the harmonic one-particle potential (OPP) model, it was possible to analyze the relative stiffness of the Cs–Br and Cu–Br chemical bonds, resulting in more covalent interactions for this last couple. Finally, an optical bandgap of approximately ∼1.8 eV was measured, comparable to the values of other all-inorganic phases reported in the literature and promising for lead-free solar cell devices. The magnetic measurements in CsCuBr3 indicated that at 420 K, the structural transition C2221 → P6522 takes place, in which an effective weak magnetic moment appears related to a fraction of localized unpaired electrons of the Cu dimers.Author contributions
Conceptualization, C. A., J. L. M., M. T. F. D. and J. A. A.; methodology, J. E. R., J. G., E. C., J. L. M., M. T. F. D. and J. A. A.; formal analysis, C. A., C. A. L., J. E. R., M. T. F. D., and J. G.; resources, J. L. M. and J. A. A.; writing original draft preparation, C. A., C. A. L., J. E. R., J. A. A., and J. G.; funding acquisition, J. L. M. and J. A. A. All authors have read and agreed to the published version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge the financial support from the Spanish Ministry of Science and Innovation (MCIN/AEI/10.13039/501100011033) to the projects PID2021-122477OB-I00, TED2021-129254B-C21 and TED2021-129254B-C22. C. A. thanks financial support from the Institut Laue-Langevin through the DA/SRH/GRI/AS/19-214 contract. J. G. thanks MCIN for granting the contract PRE2018-083398. C. A. L. thanks UNSL (PROICO 2320) and CONICET (PIP 2972) for the financial support. The authors wish to express their gratitude to ILL for making all facilities available for the neutron diffraction experiments.Notes and references
- W. S. Yang, B.-W. Park, E. H. Jung, N. J. Jeon, Y. C. Kim, D. U. Lee, S. S. Shin, J. Seo, E. K. Kim, J. H. Noh and S. Il Seok, Science, 2017, 356, 1376–1379 CrossRef CAS PubMed.
- C. A. López, M. C. Álvarez-Galván, M. V. Martínez-Huerta, M. T. Fernández-Díaz and J. A. Alonso, Chem. – Eur. J., 2019, 25, 4496–4500 CrossRef PubMed.
- A. Liang, J. Gonzalez-Platas, R. Turnbull, C. Popescu, I. Fernandez-Guillen, R. Abargues, P. P. Boix, L.-T. Shi and D. Errandonea, J. Am. Chem. Soc., 2022, 144, 20099–20108 CrossRef CAS PubMed.
- D. B. Straus, S. Guo and R. J. Cava, J. Am. Chem. Soc., 2019, 141, 11435–11439 CrossRef CAS PubMed.
- Z. Chen, L. Dong, H. Tang, Y. Yu, L. Ye and J. Zang, CrystEngComm, 2019, 21, 1389–1396 RSC.
- J. Peng, C. Q. Xia, Y. Xu, R. Li, L. Cui, J. K. Clegg, L. M. Herz, M. B. Johnston and Q. Lin, Nat. Commun., 2021, 12, 1531 CrossRef CAS PubMed.
- X. Li, W. Cai, H. Guan, S. Zhao, S. Cao, C. Chen, M. Liu and Z. Zang, Chem. Eng. J., 2021, 419, 129551 CrossRef CAS.
- L. Peng, A. Dutta, R. Xie, W. Yang and N. Pradhan, ACS Energy Lett., 2018, 3, 2014–2020 CrossRef CAS.
- B. J. Bohn, Y. Tong, M. Gramlich, M. L. Lai, M. Döblinger, K. Wang, R. L. Z. Hoye, P. Müller-Buschbaum, S. D. Stranks, A. S. Urban, L. Polavarapu and J. Feldmann, Nano Lett., 2018, 18, 5231–5238 CrossRef CAS PubMed.
- Y. Gao, L. Zhao, Q. Shang, Y. Zhong, Z. Liu, J. Chen, Z. Zhang, J. Shi, W. Du, Y. Zhang, S. Chen, P. Gao, X. Liu, X. Wang and Q. Zhang, Adv. Mater., 2018, 30, 1801805 CrossRef PubMed.
- J. Song, J. Li, L. Xu, J. Li, F. Zhang, B. Han, Q. Shan and H. Zeng, Adv. Mater., 2018, 30, 1800764 CrossRef PubMed.
- J. Duan, Y. Zhao, X. Yang, Y. Wang, B. He and Q. Tang, Adv. Energy Mater., 2018, 8, 1802346 CrossRef.
- Y. He, L. Matei, H. J. Jung, K. M. McCall, M. Chen, C. C. Stoumpos, Z. Liu, J. A. Peters, D. Y. Chung, B. W. Wessels, M. R. Wasielewski, V. P. Dravid, A. Burger and M. G. Kanatzidis, Nat. Commun., 2018, 9, 1609 CrossRef PubMed.
- C. A. López, C. Abia, M. C. Alvarez-Galván, B.-K. Hong, M. V. Martínez-Huerta, F. Serrano-Sánchez, F. Carrascoso, A. Castellanos-Gómez, M. T. Fernández-Díaz and J. A. Alonso, ACS Omega, 2020, 5, 5931–5938 CrossRef PubMed.
- P. Acharyya, K. Kundu and K. Biswas, Nanoscale, 2020, 12, 21094–21117 RSC.
- M. Aamir, M. Sher, M. A. Malik, N. Revaprasadu and J. Akhtar, Mater. Lett., 2016, 183, 135–138 CrossRef CAS.
- M. Inoue, M. Kishita and M. Kubo, Inorg. Chem., 1967, 6, 900–902 CrossRef CAS.
- T.-I. Li and G. D. Stucky, Inorg. Chem., 1973, 12, 441–445 CrossRef CAS.
- P. W. Anderson, Magnetism, Academic Press, New York, vol. 1, 1963 Search PubMed.
- H. M. Rietveld, J. Appl. Crystallogr., 1969, 2, 65–71 CrossRef CAS.
- J. Rodríguez-Carvajal, Phys. B, 1993, 192, 55–69 CrossRef.
- J. A. Alonso, M. J. Martínez-Lope, M. T. Casais and M. T. Fernández-Díaz, Inorg. Chem., 2000, 39, 917–923 CrossRef CAS PubMed.
- M. W. Lufaso and P. M. Woodward, Acta Crystallogr., Sect. B: Struct. Sci., 2004, 60, 10–20 CrossRef PubMed.
- K. Robinson, G. V. Gibbs and P. H. Ribbe, Science, 1971, 172, 567–570 CrossRef CAS PubMed.
- J. L. Feldman and G. K. Horton, Phys. Rev., 1963, 132, 644–647 CrossRef CAS.
- J.-L. Mi, M. Christensen, E. Nishibori and B. B. Iversen, Phys. Rev. B: Condens. Matter Mater. Phys., 2011, 84, 064114 CrossRef.
- C. Abia, C. A. López, J. Gainza, J. E. F. S. Rodrigues, M. M. Ferrer, G. Dalenogare, N. M. Nemes, O. J. Dura, J. L. Martínez, M. T. Fernández-Díaz, C. Álvarez-Galván and J. A. Alonso, Inorg. Chem., 2022, 61, 5502–5511 CrossRef CAS PubMed.
- A. Nakatsuka, A. Yoshiasa, K. Fujiwara and O. Ohtaka, J. Mineral. Petrol. Sci., 2018, 113, 280–285 CrossRef CAS.
- P. Coppens, Acta Crystallogr., Sect. A: Cryst. Phys., Diffr., Theor. Gen. Crystallogr., 1975, 31, 879 CrossRef.
- A. B. Garg, D. Vie, P. Rodriguez-Hernandez, A. Muñoz, A. Segura and D. Errandonea, J. Phys. Chem. Lett., 2023, 14, 1762–1768 CrossRef CAS PubMed.
- S. Cui, Y. Chen, S. Tao, J. Cui, C. Yuan, N. Yu, H. Zhou, J. Yin and X. Zhang, Eur. J. Inorg. Chem., 2020, 2020, 2165–2169 CrossRef CAS.
- M. N. Islam, J. Podder and M. L. Ali, RSC Adv., 2021, 11, 39553–39563 RSC.
- M. Coduri, T. A. Strobel, M. Szafrański, A. Katrusiak, A. Mahata, F. Cova, S. Bonomi, E. Mosconi, F. De Angelis and L. Malavasi, J. Phys. Chem. Lett., 2019, 10, 7398–7405 CrossRef CAS PubMed.
- Available at Materials Project (ref: CsCuBr3 mp-27336) in https://materialsproject.org/materials/mp-27336.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ce00432e |
| This journal is © The Royal Society of Chemistry 2023 |