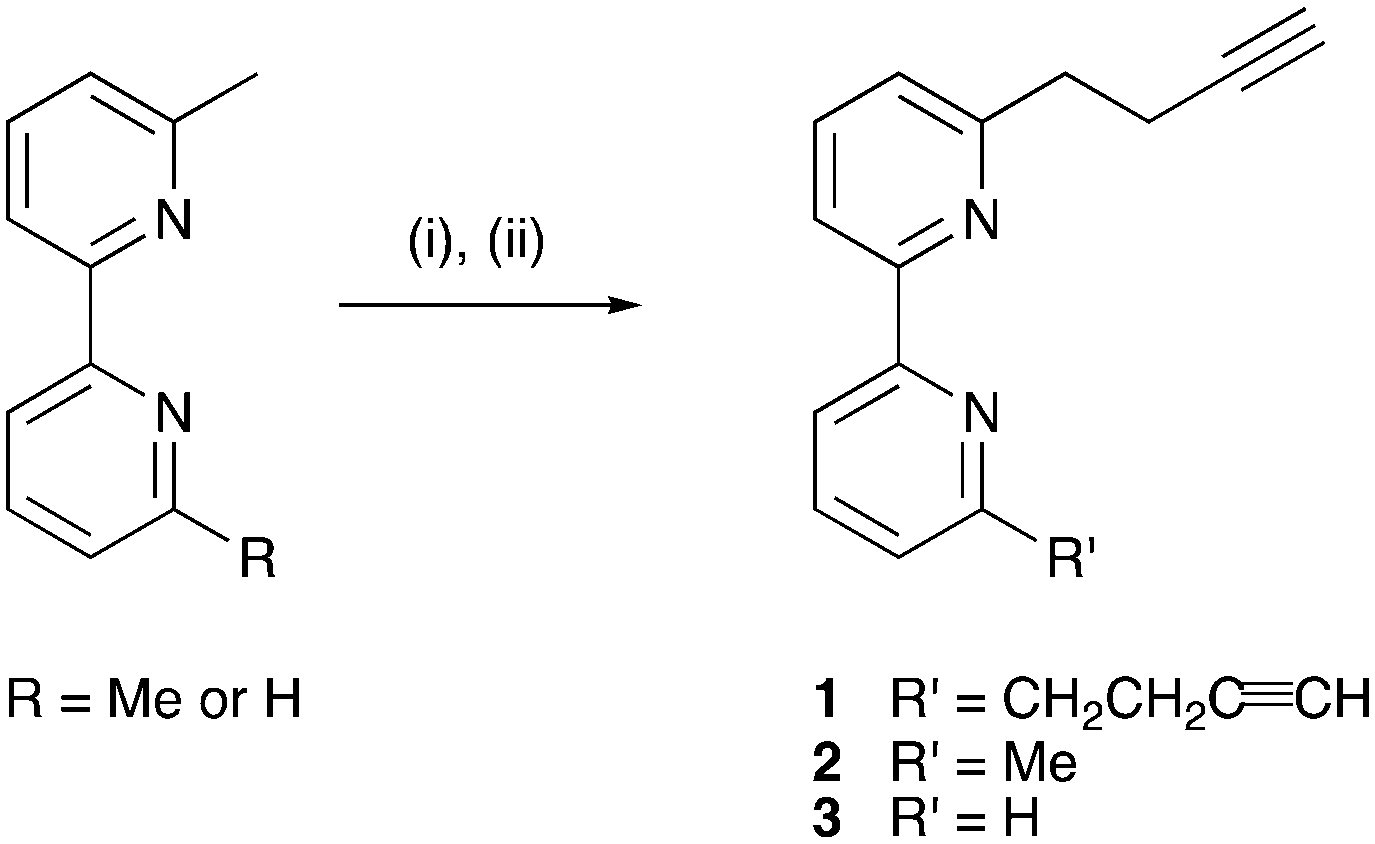
DOI:
10.1039/D3CE00355H
(Paper)
CrystEngComm, 2023,
25, 3000-3012
Introducing intramolecular, interligand arene–alkynyl π-interactions into heteroleptic [Cu(N^N)(P^P)]+ complexes†
Received
12th April 2023
, Accepted 1st May 2023
First published on 1st May 2023
Abstract
The synthesis and characterization of six new heteroleptic copper(I) compounds incorporating wide bite-angle bisphosphanes (POP = [oxydi(2,1-phenylene)]bis(diphenylphosphane), xantphos = (9,9-dimethyl-9H-xanthene-4,5-diyl)bis(diphenylphosphane)) combined with 6,6′-di(but-3-yn-1-yl)-2,2′-bipyridine (1), 6-(but-3-yn-1-yl)-6′-methyl-2,2′-bipyridine (2) or 6-(but-3-yn-1-yl)-2,2′-bipyridine (3) are reported. The single-crystal structures of [Cu(1)(POP)][PF6], [Cu(1)(xantphos)][PF6], [Cu(3)(POP)][PF6] and [Cu(3)(xantphos)][PF6] have been determined and confirm distorted tetrahedral copper(I) centres. In the solid state, the compounds are yellow or green emitters (λemmax = 530–573 nm). The highest solid-state photoluminescence quantum yields (PLQYs) were observed for [Cu(1)(xantphos)][PF6] (46%) and [Cu(1)(POP)][PF6] (41%). A combination of face-to-face arene⋯arene π-stacking interactions and C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C⋯πarene interactions protects the Cu atom in each structurally characterized complex. The CC⋯πarene interactions are characterized by a near parallel alignment of the CCH unit over an arene ring, and Calkyne⋯arenecentroid distances lie in the range of 3.74 to 4.16 Å, and CCcentroid⋯arenecentroid distances lie between 3.86 and 4.09 Å. These distances fall well within the ranges for related interactions for compounds in the Cambridge Structural Database, and data for these interactions are presented.
C⋯πarene interactions protects the Cu atom in each structurally characterized complex. The CC⋯πarene interactions are characterized by a near parallel alignment of the CCH unit over an arene ring, and Calkyne⋯arenecentroid distances lie in the range of 3.74 to 4.16 Å, and CCcentroid⋯arenecentroid distances lie between 3.86 and 4.09 Å. These distances fall well within the ranges for related interactions for compounds in the Cambridge Structural Database, and data for these interactions are presented.
Introduction
Some heteroleptic copper(I) coordination exhibit enhanced photoluminescence quantum yields (PLQYs) as a result of thermally activated delayed fluorescence (TADF).1–5 Neutral compounds are of interest for application in organic light-emitting diodes (OLEDs),6 while cationic complexes are appropriate to light-emitting electrochemical cells (LECs).4 McMillin and co-workers first demonstrated that MLCT excitation of [Cu(bpy)(PPh3)2]+ (bpy = 2,2′-bipyridine) produced photoluminescence (PL) from low-lying charge transfer excited states,7 and subsequent studies of [Cu(N^N)2]+ and [Cu(N^N)(P^P)]+ species (N^N = aromatic diimine; P^P = chelating bis(phosphane)).8–13 Sterically-demanding substituents at the 6,6′-(bpy) or 2,9-positions (1,10-phenanthroline, phen) enhance the PL of [Cu(N^N)(P^P)]+ complexes.4,11,14,15 Photoexcitation causes a flattening of the tetrahedral coordination geometry towards square-planar and the bulky substituents limit solvent attack and prevent the formation of 5-coordinate exciplexes with concomitant quenching of emission.
For [Cu(N^N)(P^P)]+ emitters,3,4 the most efficient LECs contain [Cu(6,6′-Me2bpy)(P^P)][PF6],16 [Cu(4,5,6-Me3bpy)(P^P)][PF6],17 or [Cu(4,4′-(CF3)2-6,6′-Me2bpy)(P^P)][PF6]18 (6,6′-Me2bpy = 6,6′-dimethyl-2,2′-bipyridine, 4,5,6-Me3bpy = 4,5,6-trimethyl-2,2′-bipyridine, 4,4′-(CF3)2-6,6′-Me2bpy = 4,4′-bis(trifluoromethyl)-6,6′-dimethyl-2,2′-bipyridine, P^P = xantphos, Scheme 1) in the active layer. Optimal photophysical properties of [Cu(N^N)(P^P)]+ complexes are achieved when the P^P ligand is a wide-bite angle bis(phosphane),19 such as xantphos or POP (Scheme 1). In [Cu(N^N)(P^P)]+ cations, these ligands can give rise to intramolecular π-stacking interactions which enhance emission,20 and we recently reviewed solid-state structural features that influence the PLQY.4 However, it is difficult to find combinations of N^N and P^P ligands that ensure both high PLQY and optimal LEC performance, and developing guidelines to assist the synthetic chemist is critical. Here, we investigate the effects of introducing bpy ligands with CH2CH2CCH substituents in the bpy 6- and 6,6′-positions; we evaluate the structural data in terms of CC⋯πarene interactions found in compounds in the Cambridge Structural Database.
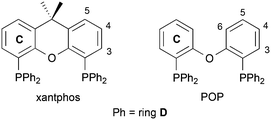 |
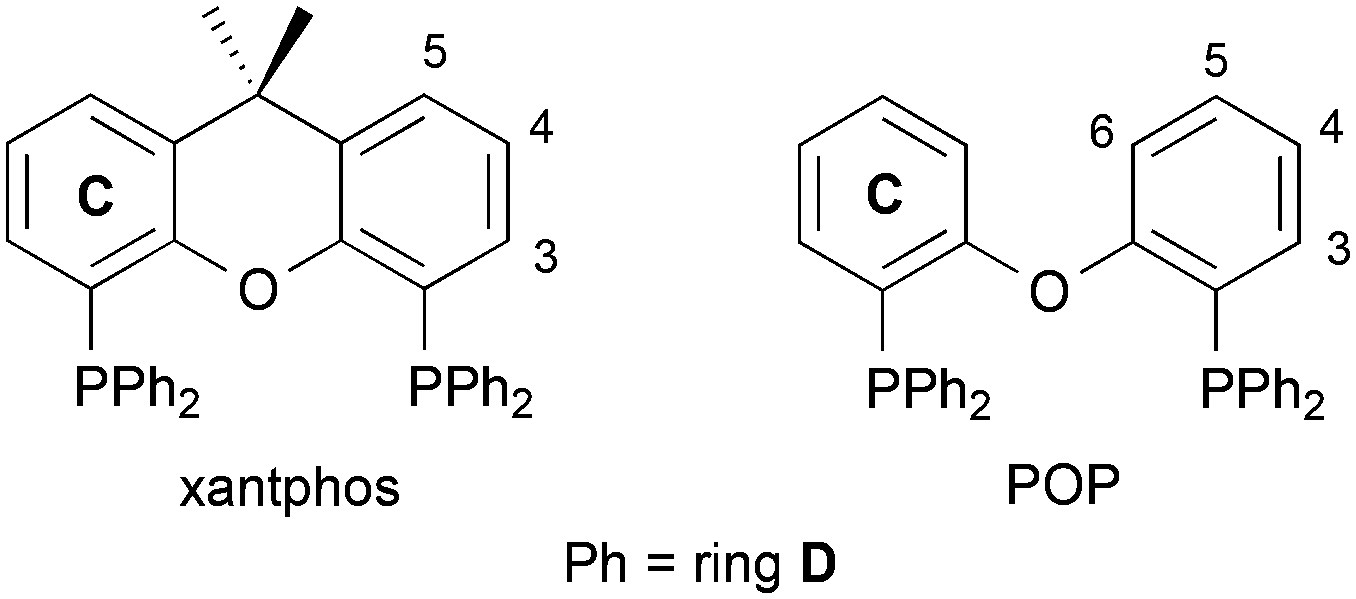
| | Scheme 1 Structures of the wide-bit angle bis(phosphane) ligands xantphos and POP. Ring and atom numbering for NMR assignments is given. | |
Experimental
1H, 13C{1H} and 31P{1H} NMR spectra were recorded at room temperature using a Bruker Avance III-500 NMR spectrometer. 1H and 13C chemical shifts were referenced to residual solvent peaks. 31P{1H} NMR spectra were referenced using δ = 0 ppm (85% aqueous H3PO4).
Column chromatography for purification was performed using a Biotage Selekt equipped with Biotage Sfär silica HC D – Duo 60 μm (10–100 g) cartridges.
Electrospray ionization mass spectra (ESI-MS) were recorded using a Shimadzu LCMS-2020 instrument. Infrared spectra were measured on a Perkin Elmer UATR Two spectrometer. The PLQY for powder samples were measured on a Hamamatsu absolute photoluminescence quantum yield spectrometer C11347 Quantaurus-QY equipped with a Xe lamp (λexc = 365 nm). Lifetimes and the emission spectra for powder samples were measured on a Hamamatsu Compact Fluorescence Lifetime Spectrometer C11367 Quantaurus-Tau equipped with a LED light source (λexc = 365 nm). The lifetime data was fitted to an exponential decay with MATLAB® R2017b and a biexponential fit was used because a single exponential decay curve give a poor fit.
Starting materials were purchased from Acros Organics, Alfa Aesar or Sigma-Aldrich. For reactions carried out under an inert atmosphere, dry solvents with AcroSeal® or crown cap were used and bought from Sigma-Aldrich or Acros Organics. HPLC grade solvents were used for analytical measurements. The compounds xantphos and POP were purchased from Acros Organics, and [Cu(MeCN)4][PF6] was prepared by a literature procedure.21 Abbreviation: 6-Mebpy = 6-methyl-2,2′-bipyridine.
Compound 1
A hexane solution of nBuLi (2.5 M, 5.20 mL, 13.0 mmol) was added to a deaerated solution of iPr2NH (1.8 mL, 12.7 mmol) in THF (5 mL). The reaction mixture was stirred at −78 °C for 1 h. A solution of 6,6′-Me2bpy (598 mg, 3.25 mmol, 1.0 eq.) in THF (10 mL) was added and the resulting mixture was stirred for 3 h. Afterwards, a solution of propargyl bromide (97%, 500 μL, 5.80 mmol, 1.8 eq.) in THF (30 mL) was added dropwise. The solution was stirred at −78 °C and allowed to warm to room temperature (ca. 22 °C); stirring was continued overnight (ca. 15 h). The reaction mixture was quenched with saturated aqueous NH4Cl (30 mL) and extracted with CH2Cl2 (3 × 20 mL). The combined organic phases were dried over MgSO4 and the solvent was evaporated. The crude material was purified by column chromatography (SiO2, cyclohexane![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :EtOAc, Rf = 0.29), followed by a Kugelrohr distillation (1 mbar, 200 °C) and compound 1 was isolated as an off-white solid (308 mg, 1.18 mmol, 36%). 1H NMR (500 MHz, 298 K, acetone-d6) δ/ppm: 8.36 (d, J = 7.6 Hz, 2H, HB3), 7.84 (dd, J = 7.7, 7.7 Hz, 2H, HB4), 7.34 (m, 2H, HB5), 3.06 (t, J = 7.5 Hz, 4H, Ha), 2.73 (dt, J = 7.5, 2.7 Hz, 4H, Hb), 2.33 (t, J = 2.7 Hz, 2H, Hd). 13C{1H} NMR (126 MHz, 298 K, acetone-d6) δ/ppm: 160.1 (CB2), 156.5 (CB6), 138.0 (CB4), 123.9 (CB5), 119.3 (CB3), 84.5 (Cc), 70.2 (Cd), 37.7 (Ca), 18.7 (Cb). ESI(+)-MS (MeOH, formic acid) m/z 261.11 [M + H]+ (base peak, calc. 261.13), 283.08 [M + Na]+ (calc. 283.12). FTIR (ν/cm−1): 3283s, 3062w, 2955w, 2114w, 1570s, 1439s, 1393w, 1105w, 1082m, 906m, 838w, 791s, 759m, 641s, 419m. Found C 82.90, H 6.20, N 10.79; C18H16N2 requires C 83.04, H 6.19, N 10.76%.
:EtOAc, Rf = 0.29), followed by a Kugelrohr distillation (1 mbar, 200 °C) and compound 1 was isolated as an off-white solid (308 mg, 1.18 mmol, 36%). 1H NMR (500 MHz, 298 K, acetone-d6) δ/ppm: 8.36 (d, J = 7.6 Hz, 2H, HB3), 7.84 (dd, J = 7.7, 7.7 Hz, 2H, HB4), 7.34 (m, 2H, HB5), 3.06 (t, J = 7.5 Hz, 4H, Ha), 2.73 (dt, J = 7.5, 2.7 Hz, 4H, Hb), 2.33 (t, J = 2.7 Hz, 2H, Hd). 13C{1H} NMR (126 MHz, 298 K, acetone-d6) δ/ppm: 160.1 (CB2), 156.5 (CB6), 138.0 (CB4), 123.9 (CB5), 119.3 (CB3), 84.5 (Cc), 70.2 (Cd), 37.7 (Ca), 18.7 (Cb). ESI(+)-MS (MeOH, formic acid) m/z 261.11 [M + H]+ (base peak, calc. 261.13), 283.08 [M + Na]+ (calc. 283.12). FTIR (ν/cm−1): 3283s, 3062w, 2955w, 2114w, 1570s, 1439s, 1393w, 1105w, 1082m, 906m, 838w, 791s, 759m, 641s, 419m. Found C 82.90, H 6.20, N 10.79; C18H16N2 requires C 83.04, H 6.19, N 10.76%.
Compound 2
A hexane solution of nBuLi (2.5 M, 4.3 mL, 9.10 mmol) was added to a deaerated solution of iPr2NH (1.5 mL, 10.7 mmol) in THF (5 mL). The reaction mixture was stirred at −78 °C for 2 h. A solution of 6,6′-Me2bpy (1.02 g, 5.54 mmol) in THF (10 mL) was added, and the resulting mixture was stirred for 3 h. Then, a solution of propargyl bromide (97%, 400 μL, 4.51 mmol) in THF (10 mL) was added dropwise. The solution was stirred at −78 °C and was allowed to warm up to room temperature; stirring was continued for ca. 15 h. The reaction mixture was quenched with saturated aqueous NH4Cl (30 mL) and extracted with CH2Cl2 (3 × 20 mL). The combined organic phases were dried over MgSO4 and the solvent was evaporated. The crude material was purified by a Kugelrohr distillation (1 mbar, 180 °C) and column chromatography (SiO2, cyclohexane:EtOAc, Rf = 0.21). Compound 2 was isolated as a brown oil (584 mg, 2.63 mmol, 48%). 1H NMR (500 MHz, 298 K, acetone-d6) δ/ppm: 8.33 (d, J = 6.8 Hz, 1H, HA3), 8.32 (d, J = 6.8 Hz, 1H, HB3), 7.82 (dd, J = 7.7, 7.7 Hz, 1H, HB4), 7.78 (dd, J = 7.7, 7.7 Hz, 1H, HA4), 7.32 (m, 1H, HB5), 7.26 (dd, J = 7.7, 0.6 Hz, 1H, HA5), 3.06 (t, J = 7.5 Hz, 2H, Ha), 2.73 (dt, J = 7.5, 2.7 Hz, 2H, Hb), 2.57 (s, 3H, HMe), 2.33 (t, J = 2.7 Hz, 1H, Hd). 13C{1H} NMR (126 MHz, 298 K, acetone-d6) δ/ppm: 160.0 (CB2), 158.5 (CA2), 156.5 (CB6), 156.3 (CA6), 138.0 (CA4), 137.9 (CB4), 124.0 (CA5), 123.8 (CB5), 119.1 (CB3), 118.5 (CA3), 84.5 (Cc), 70.2 (Cd), 37.7 (Ca), 24.6 (CMe), 18.6 (Cb). ESI(+)-MS (MeOH, formic acid) m/z 223.12 [M + H]+ (base peak, calc. 223.12), 245.11 [M + Na]+ (calc. 245.11). FTIR (ν/cm−1): 3297m, 3060w, 2919w, 2117w, 1572s, 1438s, 1256w, 1149w, 1082m, 993w, 785s, 635s. Found C 80.41, H 6.28, N 12.45; C15H14N2 requires C 81.15, H 6.35, N 12.60%.
Compound 3
A hexane solution of nBuLi (2.5 M, 4.5 mL, 11.3 mmol) was added to a deaerated solution of iPr2NH (1.52 mL, 10.8 mmol) in THF (5 mL), and the reaction mixture was stirred at −78 °C for 1.5 h. A solution of 6-Mebpy (850 μL, 5.44 mmol) in THF (10 mL) was added and the resulting mixture was stirred for 3 h at −78 °C. A solution of propargyl bromide (97%, 410 μL, 4.75 mmol) in THF (10 mL) was added dropwise. The stirred mixture was allowed to warm from −78 °C to room temperature (ca. 22 °C) was left stirring overnight (ca. 15 h). The reaction mixture was then quenched with saturated aqueous NH4Cl (30 mL) and was extracted with CH2Cl2 (3 × 20 mL). The combined organic phases were dried over MgSO4 and solvent was removed. The crude material was purified by a Kugelrohr distillation (1 mbar, 140 °C) and column chromatography (SiO2, cyclohexane:EtOAc, Rf = 0.34) and compound 3 was isolated as a white solid (323 mg, 1.55 mmol, 29%). 1H NMR (500 MHz, 298 K, acetone-d6) δ/ppm: 8.66 (ddd, J = 4.7, 1.8, 0.9 Hz, 1H, HA6), 8.52 (dd, J = 8.0, 1.1 Hz, 1H, HA3), 8.33 (dd, J = 7.8, 1.0 Hz, 1H, HB3), 7.91 (ddd, J = 7.8, 7.4, 1.8 Hz, 1H, HA4), 7.84 (dd, J = 7.7, 7.7 Hz, 1H, HB4), 7.39 (ddd, J = 7.5, 4.7, 1.2 Hz, 1H, HA5), 7.35 (m, 1H, HB5), 3.06 (t, J = 7.5 Hz, 2H, Ha), 2.73 (dt, J = 7.5, 2.7 Hz, 2H, Hb), 2.34 (t, J = 2.7 Hz, 1H, Hd). 13C{1H} NMR (126 MHz, 298 K, acetone-d6) δ/ppm: 160.1 (CB6), 157.0 (CA2), 156.3 (CB2), 150.0 (CA6), 138.1 (CB4), 137.7 (CA4), 124.7 (CA5), 124.0 (CB5), 121.5 (CA3), 119.2 (CB3), 84.5 (Cc), 70.2 (Cd), 37.6 (Ca), 18.6 (Cb). ESI(+)-MS (MeOH, formic acid) m/z 209.19 [M + H]+ (base peak, calc. 209.10). FTIR (ν/cm−1): 3220s, 1581m, 1563s, 1479m, 1429s, 1259w, 1147w, 1080w, 992w, 779s, 758s, 686s, 621m, 594w, 505w, 409w. Found C 80.36, H 5.79, N 13.40; C14H12N2 requires C 80.74, H 5.81, N 13.45%.
[Cu(1)(POP)][PF6]
[Cu(MeCN)4][PF6] (93.9 mg, 252 μmol) and POP (135.3 mg, 251 μmol) were dissolved in CH2Cl2 (30 mL) and the mixture was stirred for 2 h at room temperature. Then compound 1 (65.1 mg, 250 μmol) was added and the solution immediately turned yellow. The reaction mixture was stirred overnight and was then filtered before the solvent evaporated under reduced pressure. The crude material was redissolved in CH2Cl2 (5 mL) and precipitated by adding Et2O (30 mL). After centrifuging (7000 rpm, 10 min), the supernatant liquid was removed by decantation. The dissolution/precipitation steps were repeated twice. The precipitate was crystallized from a CH2Cl2 solution (5 mL) by layer diffusion with Et2O (30 mL). [Cu(1)(POP)][PF6] was isolated as a yellow solid (207 mg, 205 μmol, 81%). 1H NMR (500 MHz, 298 K, CD2Cl2) δ/ppm: 8.04 (d, J = 7.9 Hz, 2H, HB3), 7.97 (dd, J = 7.8, 7.8 Hz, 2H, HB4), 7.42 (d, J = 7.8 Hz, 2H, HB5), 7.31 (t, J = 7.3 Hz, 4H, HD4), 7.27 (dd, J = 7.9, 1.7 Hz, 2H, HC5), 7.17 (m, 8H, HD3), 7.12 (m, 2H, HC4), 7.03 (m, 8H, HD2), 6.91 (m, 2H, HC6), 6.77 (m, 2H, HC3), 2.85 (t, J = 6.9 Hz, 4H, Ha), 1.85 (m, 4H, Hb), 1.83 (m, 2H, Hd). 13C{1H} NMR (126 MHz, 298 K, CD2Cl2) δ/ppm: 160.9 (CB6), 157.6 (pseudo-t, JPC = 6 Hz, CC1), 153.1 (CB2), 139.4 (CB4), 134.1 (CC3), 133.7 (t, JPC = 8 Hz, CD2), 132.6 (CC5), 131.2 (pseudo-t, JPC = 17 Hz, CD1), 130.5 (CD4), 129.3 (pseudo-t, JPC = 5 Hz, CD3), 125.7 (pseudo-t, JPC = 2 Hz, CC4), 125.5 (pseudo-t, JPC = 15 Hz, CC2), 125.0 (CB5), 121.2 (CB3), 120.2 (pseudo-t, JPC = 2 Hz, CC6), 82.3 (Cc), 70.3 (Cd), 38.5 (Ca), 17.2 (Cb). 31P{1H} NMR (202 MHz, 298 K, CD2Cl2) δ/ppm: −13.6 (broad, FWHM ≈ 150 Hz, POP), −142.7 (septet, JPF = 708 Hz, PF6). ESI(+)-MS (MeOH) m/z 601.11 [Cu(POP)]+ (calc. 601.09), 861.24 [M–PF6]+ (base peak, calc. 861.22). ESI(−)-MS (MeOH) m/z 145.07 [PF6]− (calc. 144.96). UV-vis (CH2Cl2, 5.0 × 10−5 mol dm−3) λ/nm (ε/dm3 mol−1 cm−1): 248 (24700), 292 (18800), 318 (11100), 372 (1500). FTIR (ν/cm−1): 3292m, 1574w, 1462m, 1434s, 1262w, 1220w, 1094w, 877w, 835s, 745w, 697w, 574w, 556m, 505w. Found C 64.18, H 4.42, N 2.83; C54H44CuF6N2OP3 requires C 64.38, H 4.40, N 2.78%.
[Cu(1)(xantphos)][PF6]
[Cu(MeCN)4][PF6] (93.8 mg, 252 μmol) was dissolved in CH2Cl2 (15 mL). A solution of xantphos (146 mg, 252 μmol, 1.0 eq.) and 1 (65.6 mg, 252 μmol) in CH2Cl2 (15 mL) was added causing an immediate colour change to yellow. The mixture was stirred at room temperature for ca. 15 h, after which it was filtered and the solvent was removed under reduced pressure. The crude material was redissolved in CH2Cl2 (5 mL), precipitated by adding Et2O (30 mL) and centrifuged (7000 rpm, 10 min). The supernatant was removed by decantation. This washing step was repeated and then again with tBuOMe (30 mL) in place of Et2O. The precipitate was crystallized from CH2Cl2 (5 mL) by layer diffusion using Et2O (30 mL). [Cu(1)(xantphos)][PF6] was isolated as a pale yellow solid (212 mg, 202 μmol, 84%). 1H NMR (500 MHz, 298 K, CD2Cl2) δ/ppm: 7.90 (dd, J = 7.8, 7.8 Hz, 2H, HB4), 7.84 (dd, J = 8.1, 8.1 Hz, 2H, HB3), 7.68 (dd, J = 7.8, 1.4 Hz, 2H, HC5), 7.39 (m, 2H, HB5), 7.35 (t, J = 7.7 Hz, 4H, HD4), 7.21 (t, J = 7.7 Hz, 2H, HC4), 7.15 (m, 8H, HD3), 7.07 (m, 8H, HD2), 6.92 (m, 2H, HC3), 2.63 (t, J = 6.6 Hz, 4H, Ha), 1.90 (dt, J = 6.6, 2.7 Hz, 4H, Hb), 1.74 (s, 6H, Hxantphos-Me), 1.61 (t, J = 2.6 Hz, 2H, Hd). 13C{1H} NMR (126 MHz, 298 K, CD2Cl2) δ/ppm: 160.0 (CB6), 155.2 (pseudo-t, JPC = 7 Hz, CC1), 152.5 (CB2), 139.2 (CB4), 134.2 (CC6), 133.6 (pseudo-t, JPC = 8 Hz, CD2), 131.5 (pseudo-t, JPC = 16 Hz, CD1), 131.1 (CC3), 130.5 (CD4), 129.2 (pseudo-t, JPC = 5 Hz, CD3), 128.0 (CC5), 125.9 (pseudo-t, JPC = 3 Hz, CC4), 124.4 (CB5), 121.9 (pseudo-t, JPC = 14 Hz, CC2), 121.0 (CB3), 81.8 (Cc), 70.5 (Cd), 38.9 (Ca), 36.5 (Cxantphos-bridge), 28.5 (Cxantphos-Me), 17.2 (Cb). 31P{1H} NMR (202 MHz, 298 K, CD2Cl2) δ/ppm: −12.9 (broad, FWHM ≈ 155 Hz, xantphos), −142.7 (septet, JPF = 709 Hz, PF6). ESI(+)-MS (MeOH) m/z 641.12 [Cu(xantphos)]+ (calc. 641.12), 901.26 [M–PF6]+ (base peak, calc. 901.25). ESI(−)-MS (MeOH) m/z 145.07 [PF6]− (calc. 144.96). UV-vis (CH2Cl2, 5.0 × 10−5 mol dm−3) λ/nm (ε/dm3 mol−1 cm−1): 248 (27350), 281 (23200), 318 (9700), 371 (2010). FTIR (ν/cm−1): 3294m, 1599w, 1434m, 1402s, 1226m, 1095w, 836s, 740m, 695m, 556s, 502s, 456m. Found C 65.28, H 4.56, N 2.83; C57H48CuF6N2OP3 requires C 65.36, H 4.62, N 2.67%.
[Cu(2)(POP)][PF6]
The method was as for [Cu(1)(POP)][PF6] starting with [Cu(MeCN)4][PF6] (93.9 mg, 252 μmol), POP (135.2 mg, 251 μmol) and 2 (70.1 mg, 315 μmol). The purification steps (dissolution/precipitation) were repeated four times. [Cu(2)(POP)][PF6] was isolated as an orange solid (185 mg, 190 μmol, 75%). 1H NMR (500 MHz, 298 K, acetone-d6) δ/ppm: 8.32 (d, J = 7.9 Hz, 1H, HB3), 8.27 (d, J = 8.0 Hz, 1H, HA3), 8.13 (dd, J = 7.9, 7.9 Hz, 1H, HB4), 8.03 (dd, J = 7.9, 7.8 Hz, 1H, HA4), 7.61 (m, 1H, HB5), 7.43 (m, 2H, HC5), 7.42 (m, 1H, HA5), 7.36 (t, J = 7.4 Hz, 4H, HD4), 7.28 (dd, J = 7.5, 1.1 Hz, 2H, HC4), 7.22 (m, 8H, HD3), 7.21 (m, 2H, HC6), 7.13 (m, 8H, HD2), 7.06 (m, 2H, HC3), 2.96 (t, J = 7.3 Hz, 2H, Ha), 2.32 (s, 3H, HMe), 2.30 (t, J = 2.5 Hz, 1H, Hd), 2.02 (dt, J = 7.3, 2.5 Hz, 2H, Hb). 13C{1H} NMR (126 MHz, 298 K, acetone-d6) δ/ppm: 161.3 (CB6), 159.8 (CA6), 158.8 (pseudo-t, JPC = 6 Hz, CC1), 153.7 (CA2), 140.1 (CB4), 140.0 (CA4), 134.5 (CC3), 134.0 (pseudo-t, JPC = 8 Hz, CD2), 133.3 (CC5), 132.5 (pseudo-t, JPC = 16 Hz, CD1), 130.9 (CD4), 129.7 (pseudo-t, JPC = 6 Hz, CD3), 127.3 (CA5), 126.2 (pseudo-t, JPC = 2 Hz, CC4), 125.4 (CB5), 121.8 (CA3), 121.2 (CB3), 121.0 (pseudo-t, JPC = 2 Hz, CC6), 82.9 (Cc), 71.5 (Cd), 39.4 (Ca), 26.8 (CMe), 17.3 (Cb). Signals for CB2 and CC2 were not resolved in 1D or 2D spectra. 31P{1H} NMR (202 MHz, 298 K, acetone-d6) δ/ppm: −13.6 (broad, FWHM ≈ 215 Hz, POP), −144.2 (septet, JPF = 707 Hz, PF6). ESI(+)-MS (MeOH) m/z 601.16 [Cu(POP)]+ (calc. 601.09), 823.22 [M–PF6]+ (base peak, calc. 823.21). ESI(−)-MS (MeOH) m/z 145.06 [PF6]− (calc. 144.96). UV-vis (CH2Cl2, 5.0 × 10−5 mol dm−3) λ/nm (ε/dm3 mol−1 cm−1): 252 (25340), 289 (20930), 318 (13020), 379 (2850). FTIR (ν/cm−1): 3287m, 3060w, 1600w, 1462m, 1435s, 1258m, 1222m, 1008w, 832s, 744s, 694s, 556s, 511m, 417m. Found C 61.48, H 4.41, N 2.91; C51H42CuF6N2OP3·0.5CH2Cl2 requires C 61.13, H 4.28, N 2.77%.
[Cu(2)(xantphos)][PF6]
The method was as for [Cu(1)(xantphos)][PF6] starting with [Cu(MeCN)4][PF6] (94.3 mg, 253 μmol), xantphos (145.3 mg, 251 μmol) and 2 (60.1 mg, 270 μmol). [Cu(2)(xantphos)][PF6] was isolated as a dark orange solid (197 mg, 194 μmol, 77%). 1H NMR (500 MHz, 298 K, acetone-d6) δ/ppm: 8.21 (d, J = 8.0 Hz, 1H, HB3), 8.18 (d, J = 8.0 Hz, 1H, HA3), 8.07 (dd, J = 7.9, 7.9 Hz, 1H, HB4), 7.98 (dd, J = 7.8, 7.9 Hz, 1H, HA4), 7.85 (dd, J = 7.8, 1.4 Hz, 2H, HC5), 7.57 (m, 1H, HB5), 7.42 (m, 1H, HA5), 7.39 (m, 4H, HD4), 7.32 (t, J = 7.7 Hz, 2H, HC4), 7.23 (m, 8H, HD3), 7.18 (m, 8H, HD2), 6.96 (m, 2H, HC3), 2.71 (t, J = 6.7 Hz, 2H, Ha), 2.18 (s, 3H, HA6-Me), 2.03 (t, J = 2.6 Hz, 1H, Hd), 1.99 (dt, J = 6.7, 2.7 Hz, 2H, Hb), 1.80 (s, 3H, Hxantphos-Me), 1.74 (s, 3H, Hxantphos-Me). 13C{1H} NMR (126 MHz, 298 K, acetone-d6) δ/ppm: 160.7 (CB6), 159.2 (CA6), 155.8 (pseudo-t, JPC = 7 Hz, CC1), 153.2 (CA2), 139.9 (CB4), 139.5 (CA4), 134.8 (pseudo-t, JPC = 3 Hz, CC6), 134.1 (pseudo-t, JPC = 8 Hz, CD2), 132.6 (pseudo-t, JPC = 16 Hz, CD1), 131.3 (CC3), 131.0 (CA5), 129.8 (pseudo-t, JPC = 5 Hz, CD3), 128.7 (CC5), 126.8 (CD4), 126.4 (pseudo-t, JPC = 3 Hz, CC4), 125.1 (CB5), 122.6 (pseudo-t, JPC = 13 Hz, CC2), 121.8 (CB3), 121.1 (CA3), 82.5 (Cc), 71.6 (Cd), 39.4 (Ca), 36.9 (Cxantphos-bridge), 29.1 (Cxantphos-Me), 28.3 (Cxantphos-Me), 27.2 (CA6-Me), 17.2 (Cb). Signal for CB2 was not observed in 1D or 2D spectra. 31P{1H} NMR (202 MHz, 298 K, acetone-d6) δ/ppm: −13.5 (broad, FWHM ≈ 215 Hz, xantphos), −144.2 (septet, JPF = 707 Hz, PF6). ESI(+)-MS (MeOH) m/z 641.18 [Cu(xantphos)]+ (calc. 641.12), 863.29 [M–PF6]+ (base peak, calc. 863.24). ESI(−)-MS (MeOH) m/z 145.06 [PF6]− (calc. 144.96). UV-vis (CH2Cl2, 5.0 × 10−5 mol dm−3) λ/nm (ε/dm3 mol−1 cm−1): 246 (30380), 281 (24900), 318 (11270), 370 (2370). FTIR (ν/cm−1): 3305w, 1574w, 1599w, 1433m, 1403m, 1222m, 1095w, 832s, 749m, 693m, 533w, 556s, 506s, 464m. Found C 62.86, H 4.53, N 2.92; C54H46CuF6N2OP3·0.5CH2Cl2 requires C 62.23, H 4.50, N 2.66%.
[Cu(3)(POP)][PF6]
The method was as for [Cu(1)(POP)][PF6] starting with [Cu(MeCN)4][PF6] (93.6 mg, 251 μmol), POP (137 mg, 254 μmol) and 3 (53 mg, 254 μmol). Purification was by crystallization from a CH2Cl2 solution (5 mL) of the crude compound by layer diffusion using Et2O (30 mL). [Cu(3)(POP)][PF6] was isolated as a pale yellow solid (196 mg, 205 μmol, 82%). 1H NMR (500 MHz, 298 K, acetone-d6) δ/ppm: 8.58 (d, J = 5.2 Hz, 1H, HA6), 8.55 (d, J = 8.1 Hz, 1H, HA3), 8.49 (d, J = 7.9 Hz, 1H, HB3), 8.18 (dd, J = 7.9, 7.9 Hz, 1H, HB4), 8.09 (dd, J = 7.9, 7.9 Hz, 1H, HA4), 7.60 (m, 1H, HB5), 7.45 (d, J = 7.8 Hz, 2H, HC5), 7.37 (m, 1H, HA5), 7.35 (m, 4H, HD4), 7.31 (m, 4H, HD3′), 7.25 (m, 4H, HD3), 7.24 (m, 2H, HC6), 7.15 (m, 4H, HD2), 7.14 (m, 2H, HC4), 7.06 (m, 4H, HD2′), 6.84 (m, 2H, HC3), 3.07 (t, J = 7.5 Hz, 2H, Ha), 2.39 (t, J = 2.7 Hz, 1H, Hd), 2.08 (dt, J = 7.5, 2.7 Hz, 2H, Hb). 13C{1H} NMR (126 MHz, 298 K, acetone-d6) δ/ppm: 161.4 (CB6), 158.7 (pseudo-t, JPC = 6 Hz, CC1), 153.5 (CA2), 152.7 (CB2), 150.2 (CA6), 140.2 (CB4), 139.7 (CA4), 135.0 (CC3), 134.0 (pseudo-t, JPC = 8 Hz, CD2+D2′), 133.3 (CC5), 131.9 (pseudo-t, JPC = 16 Hz, CD1), 131.1 (CD4), 129.8 (pseudo-t, JPC = 7.1 Hz, CD3+D3′), 126.7 (CA5), 126.1 (pseudo-t, JPC = 2 Hz, CC4), 125.6 (CB5), 124.8 (pseudo-t, JPC = 15 Hz, CC2), 123.7 (CA3), 121.8 (CB3), 121.4 (pseudo-t, JPC = 2 Hz, CC6), 83.0 (Cc), 71.6 (Cd), 39.7 (Ca), 17.6 (Cb). 31P{1H} NMR (202 MHz, 298 K, acetone-d6) δ/ppm: −13.1 (broad, FWHM ≈ 205 Hz, POP), −144.2 (septet, JPF = 708 Hz, PF6). ESI(+)-MS (MeOH/CH2Cl2) m/z 601.12 [Cu(POP)]+ (calc. 601.09), 809.22 [M–PF6]+ (base peak, calc. 809.19). ESI(−)-MS (MeOH/CH2Cl2) m/z 145.05 [PF6]− (calc. 144.96). UV-vis (CH2Cl2, 5.0 × 10−5 mol dm−3) λ/nm (ε/dm3 mol−1 cm−1): 247 (27570), 291 (22950), 312 (14940), 383 (2950). FTIR (ν/cm−1): 1597w, 1565w, 1454w, 1435m, 1259w, 1223m, 1069w, 877w, 836s, 745s, 693s, 556s, 510m, 495w. Found C 62.01, H 4.25, N 2.92; C50H40CuF6N2OP3 requires C 62.86, H 4.22, N 2.93%.
[Cu(3)(xantphos)][PF6]
The method was as for [Cu(1)(xantphos)][PF6] starting with [Cu(MeCN)4][PF6] (93.9 mg, 252 μmol), xantphos (147 mg, 254 μmol) and 3 (52.7 mg, 253 μmol). Purification was by crystallization from a CH2Cl2 solution (5 mL) by layer diffusion using Et2O (30 mL). [Cu(3)(xantphos)][PF6] was isolated as a dark yellow solid (210 mg, 210 μmol, 83%). 1H NMR (500 MHz, 298 K, acetone-d6) δ/ppm: 8.54 (d, J = 8.1 Hz, 1H, HA3), 8.49 (d, J = 7.9 Hz, 1H, HB3), 8.37 (m, 1H, HA6), 8.23 (dd, J = 7.9, 7.9 Hz, 1H, HB4), 8.10 (dd, J = 7.8, 7.8 Hz, 1H, HA4), 7.87 (dd, J = 7.8, 1.4 Hz, 2H, HC5), 7.67 (m, 1H, HB5), 7.45 (m, 1H, HA5), 7.38 (t, J = 7.4 Hz, 2H, HD4′), 7.34 (t, J = 7.5 Hz, 2H, HD4), 7.29 (t, J = 7.7 Hz, 2H, HC4), 7.24 (t, J = 7.7 Hz, 4H, HD3), 7.17 (m, 4H, HD3′), 7.13 (m, 4H, HD2), 6.98 (m, 4H, HD2′), 6.67 (m, 2H, HC3), 2.77 (t, J = 7.1 Hz, 2H, Ha), 2.18 (t, J = 2.6 Hz, 1H, Hd), 1.95 (dt, J = 7.1, 2.6 Hz, 2H, Hb), 1.89 (s, 3H, Hxantphos-Me), 1.74 (s, 3H, Hxantphos-Me). 13C{1H} NMR (126 MHz, 298 K, acetone-d6) δ/ppm: 160.9 (CB6), 155.8 (pseudo-t, JPC = 6 Hz, CC1), 153.4 (CA2), 152.5 (CB2), 149.8 (CA6), 140.3 (CB4), 140.0 (CA4), 135.0 (pseudo-t, JPC = 3 Hz, CC6), 134.0 (pseudo-t, JPC = 8 Hz, CD2), 133.6 (pseudo-t, JPC = 8 Hz, CD2′), 132.4 (pseudo-t, JPC = 17 Hz, CD1), 131.7 (CC3), 131.1 (CD4′), 131.0 (CD4), 129.9 (pseudo-t, JPC = 5 Hz, CD3), 129.7 (pseudo-t, JPC = 5 Hz, CD3′), 128.6 (CC5), 127.0 (CA5), 126.4 (pseudo-t, JPC = 2 Hz, CC4), 125.9 (CB5), 123.9 (CA3), 122.0 (CB3), 121.3 (pseudo-t, JPC = 13 Hz, CC2), 82.5 (Cc), 71.7 (Cd), 39.8 (Ca), 37.0 (Cxantphos-bridge), 29.7 (Cxantphos-Me), 27.1 (Cxantphos-Me), 17.3 (Cb). 31P{1H} NMR (202 MHz, 298 K, acetone-d6) δ/ppm: −12.6 (broad, FWHM ≈ 210 Hz, xantphos), −144.2 (septet, JPF = 709 Hz, PF6). ESI(+)-MS (MeOH/CH2Cl2) m/z 641.20 [Cu(xantphos)]+ (calc. 641.12), 849.25 [M–PF6]+ (base peak, calc. 849.22). ESI(−)-MS (MeOH/CH2Cl2) m/z 145.04 [PF6]− (calc. 144.96). UV-vis (CH2Cl2, 5.0 × 10−5 mol dm−3) λ/nm (ε/dm3 mol−1 cm−1): 247 (32450), 282 (32050), 312 (12100), 377 (2790). FTIR (ν/cm−1): 3306w, 3052w, 1596w, 1564w, 1451w, 1435m, 1403s, 1229m, 1095w, 999w, 877w, 832s, 747m, 693m, 556s, 505s, 464m, 432w. Found C 63.61, H 4.43, N 3.01; C53H44CuF6N2OP requires C 63.95, H 4.46, N 2.81%.
X-ray crystallography
Single crystal data were collected using a Eulerian 4-circle STOE STADIVARI Cu diffractometer, and data reduction and solution used the programs ShelXT 2018/222 and Olex223 as the graphical interface. The model was refined with ShelXL 2018/324 using full matrix least squares minimization on F2. All non-hydrogen atoms were refined anisotropically. Hydrogen atom positions were calculated geometrically and refined using a riding model. Structure analysis used Mercury CSD v. 2022.3.0.25 A solvent mask had to be used in [Cu(3)(xantphos)][PF6]·1.5CH2Cl2 because of disorder; the electron density removed was accounted for in terms of added solvent molecules, and these were added to the formulae and all appropriate numbers.
[Cu(1)(POP)][PF6].
C54H44CuF6N2OP3, Mr = 1007.36, yellow block, monoclinic, space group P21/c, a = 10.8682(2), b = 32.7516(8), c = 13.6339(2) Å, β = 98.5070(10)°, V = 4799.61(16) Å3, Dc = 1.394 g cm−3, T = 150 K, Z = 4, μ(CuKα) = 2.136 mm−1. Total 56154 reflections, 9442 unique (Rint = 0.0427). Refinement of 8033 reflections (605 parameters) with I > 2σ(I) converged at final R1 = 0.0871 (R1 all data = 0.1035), wR2 = 0.1739 (wR2 all data = 0.1900), gof = 1.128. CCDC 2247205.
[Cu(1)(xantphos)][PF6].
C57H48CuF6N2OP3, Mr = 1047.42, yellow plate, monoclinic, space group P21/c, a = 17.0919(3), b = 14.2952(3), c = 20.6426(3) Å, β = 103.5210(10)°, V = 4903.86(15) Å3, Dc = 1.419 g cm−3, T = 150 K, Z = 4, μ(CuKα) = 2.113 mm−1. Total 97842 reflections, 9704 unique (Rint = 0.0419). Refinement of 8338 reflections (634 parameters) with I > 2σ(I) converged at final R1 = 0.0829 (R1 all data = 0.0936), wR2 = 0.1745 (wR2 all data = 0.1846), gof = 1.013. CCDC 2247208.
[Cu(3)(POP)][PF6].
C50H40CuF6N2OP3, Mr = 955.29, yellow plate, triclinic, space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) , a = 10.8302(4), b = 11.9965(4), c = 17.2894(6) Å, α = 98.862(3), β = 95.289(3), γ = 102.775(3)°, V = 2145.90(13) Å3, Dc = 1.478 g cm−3, T = 150 K, Z = 2, μ(CuKα) = 2.355 mm−1. Total 42991 reflections, 8373 unique (Rint = 0.0525). Refinement of 6386 reflections (569 parameters) with I > 2σ(I) converged at final R1 = 0.0885 (R1 all data = 0.1150), wR2 = 0.1749 (wR2 all data = 0.1998), gof = 0.950. CCDC 2247206.
, a = 10.8302(4), b = 11.9965(4), c = 17.2894(6) Å, α = 98.862(3), β = 95.289(3), γ = 102.775(3)°, V = 2145.90(13) Å3, Dc = 1.478 g cm−3, T = 150 K, Z = 2, μ(CuKα) = 2.355 mm−1. Total 42991 reflections, 8373 unique (Rint = 0.0525). Refinement of 6386 reflections (569 parameters) with I > 2σ(I) converged at final R1 = 0.0885 (R1 all data = 0.1150), wR2 = 0.1749 (wR2 all data = 0.1998), gof = 0.950. CCDC 2247206.
[Cu(3)(xantphos)][PF6]·1.5CH2Cl2.
C54.5H47Cl3CuF6N2OP3, Mr = 1122.74, yellow block, triclinic, space group P, a = 10.9817(8), b = 15.1190(8), c = 18.4780(12) Å, α = 112.459(3), β = 93.961(3), γ = 108.337(5)°, V = 2627.9(3) Å3, Dc = 1.419 g cm−3, T = 150 K, Z = 2, μ(CuKα) = 3.379 mm−1. Total 55621 reflections, 10279 unique (Rint = 0.0567). Refinement of 7323 reflections (598 parameters) with I > 2σ(I) converged at final R1 = 0.0889 (R1 all data = 0.1179), wR2 = 0.1724 (wR2 all data = 0.1935), gof = 0.995. CCDC 2247207.
Results and discussion
Rationale for N^N ligand design
In a tetrahedral [Cu(N^N)(P^P)]+ cation, the CuN2 and CuP2 planes lie approximately orthogonal to each other. Coordinated xantphos ligands exhibit a bowl-shaped xanthene cavity which can host the 6-substituent of the bpy ligand in [Cu(6-Rbpy)(xantphos)]+ or [Cu(6,6′-R2bpy)(xantphos)]+ cations. In a [Cu(6-Rbpy)(xantphos)]+ cation, the 6-Rbpy could adopt one of two orientations (Scheme 2). Significantly, a search of the Cambridge Structural Database (CSD)26 using Conquest (version 2022.3.0 including November 2022 updates)27 for salts of [Cu(6-Rbpy)(xantphos)]+ containing a range of R groups demonstrates a preference for the R group to point towards the xanthene cavity (Table 1, Scheme 2A). Methyl, methoxy and ethyl groups sit neatly in the cavity,16,28,29 and the intramolecular, interligand interaction should protect the copper(I) centre in the excited state. Heteroleptic copper(I) complexes containing larger 6-substituents are relatively rare (Table 1). Still, it is noteworthy that even sterically demanding substituents, other than large aromatic units,30 may be partially hosted within the xanthene cavity.
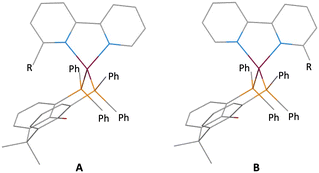 |
| | Scheme 2 Schematic representations of the two possible orientations of the 6-Rbpy ligand in a [Cu(6-Rbpy)(xantphos)]+ cation with the R group facing towards (A) or away from (B) the xanthene unit. | |
Table 1 Orientations of 6-Rbpy ligands in the solid-state structures of salts of [Cu(6-Rbpy)(xantphos)]+. See Scheme 2 for definitions of A and B
| R |
CSD refcode |
Orientation A or B |
Ref. |
|
6-CF3bpy is orientationally disordered; fractional occupancies 0.75:0.25 with the major site in orientation A.
|
| Me |
ZAQFEG |
A
|
28
|
| Me |
ZAQFOQ |
A
|
28
|
| Me |
GABTUB |
A
|
16
|
| Et |
GABWAK |
A
|
16
|
| Ph |
GABMEE |
B
|
16
|
| Br |
MEWZAS |
A
|
31
|
| OMe |
UDOWAO |
A
|
29
|
| OEt |
UDOWIW |
A
|
29
|
| SMe |
UDOXOD |
A
|
29
|
| SEt |
UDOXUJ |
A
|
29
|
| OPh |
UDOXIX |
A
|
29
|
| SPh |
UDOWOC |
A
|
29
|
| CF3 |
VICQUW |
A/B
|
32
|
| CN |
GURDOP |
B
|
33
|
|
ILORUZ |
A
|
34
|
| Naphthalen-2-yl |
VAWDUV |
B
|
30
|
| Pyren-1-yl |
VAWDIJ |
B
|
30
|
In the solid state, [Cu(bpy)(xantphos)]+ cations (here, bpy represents unsubstituted or substituted 2,2′-bipyridine) typically exhibit face-to-face π-stacking between phenyl rings of two PPh2 units of the xantphos ligand. However, additional intramolecular interligand π-stacking is expected to increase complex stability with respect to ligand dissociation. While the single-crystal structure of [Cu(6-Phbpy)(xantphos)][PF6] shows orientation B (Table 1), variable temperature NMR spectra are consistent with there being two conformers in solution, related by inversion of the xanthene unit.16 In the solid state, there are no significant face-to-face or edge-to-face π-contacts between phenyl rings within the [Cu(6-Phbpy)(xantphos)]+ cation.16 Kaeser et al. have reported that using bulky (notably phenyl) substituents in heteroleptic copper(I) complexes may be detrimental to dynamic ligand exchange.35 In contrast, we have recently demonstrated that [Cu(N^N)(POP)][PF6] and [Cu(N^N)(xantphos)][PF6] in which N^N = 6,6′-bis(but-3-en-1-yl)-2,2′-bipyridine, 6-(but-3-en-1-yl)-6′-methyl-2,2′-bipyridine, 6,6′-bis(pent-4-en-1-yl)-2,2′-bipyridine or 6-(pent-4-en-1-yl)-6′-methyl-2,2′-bipyridine (Scheme S1†) are both stable to ligand dissociation in solution and exhibit high PLQYs (28.5 to 62.3% in the solid state).36 These results and the beneficial effects of intramolecular π-stacking interactions motivated us to investigate copper(I) complexes incorporating ligands 1–3 (Scheme 3). Although analysis by Moore and co-workers in 1996 of structures in the CSD concluded that “π–π stacking between triple bonds and aromatic rings is not a particularly significant noncovalent interaction”,37 recent work from Karki et al. suggests that intermolecular π–π interactions between alkynes and fluoroarenes are significant in influencing solid-state packing.38 We were, therefore, interested to see whether the incorporation of the terminal alkyne moieties in ligands 1–3 would lead to intramolecular interactions involving the CCH unit and arene domains in [Cu(N^N)(P^P)][PF6] complexes (N^N = 1–3, P^P = POP or xantphos).
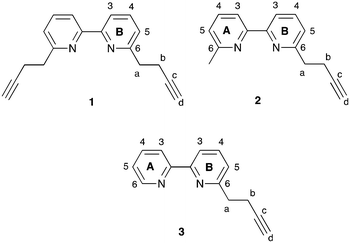 |
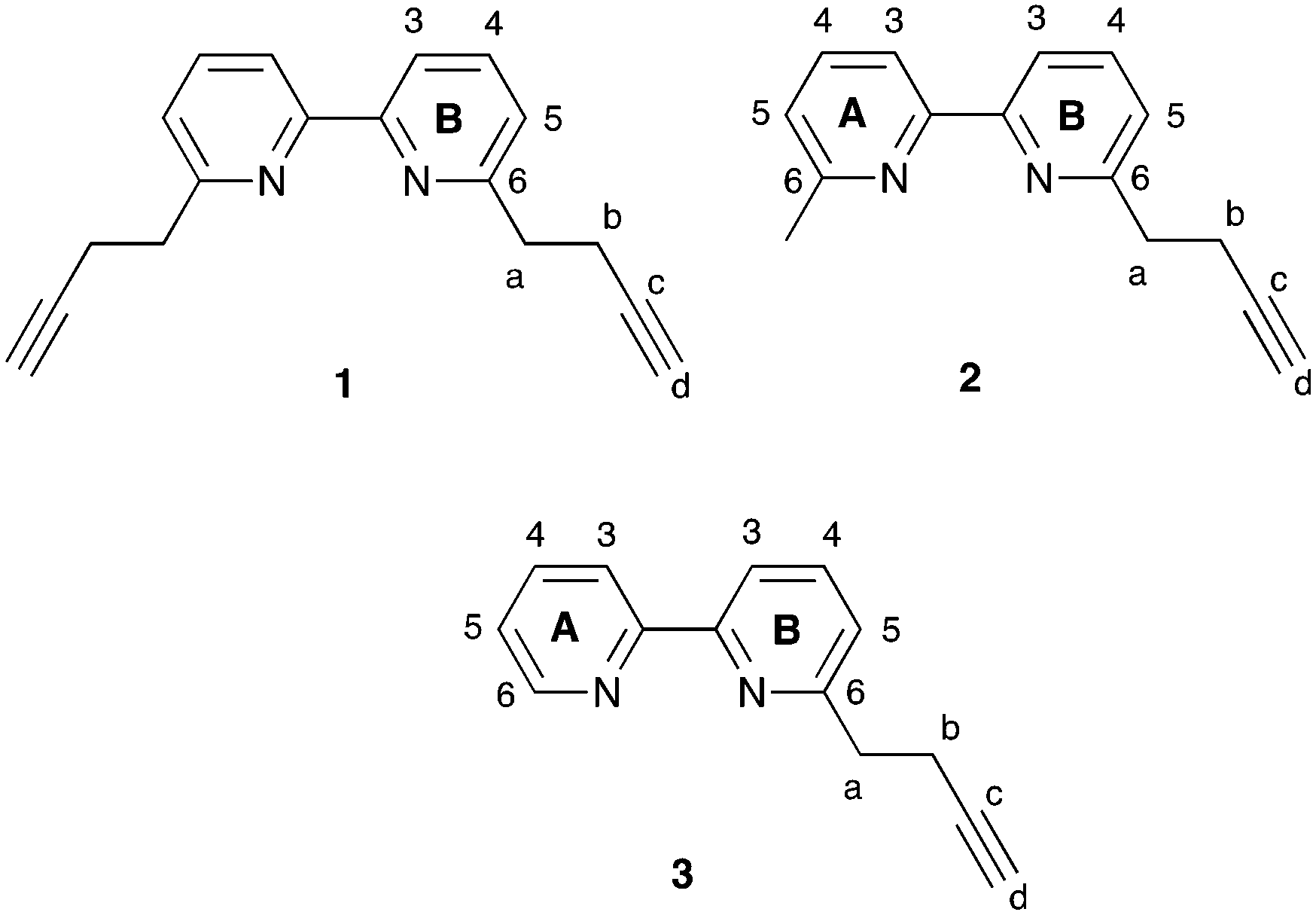
| | Scheme 3 Structures of N^N ligands 1–3 with ring and atom numbering for NMR assignments. | |
Ligand synthesis and characterization
Compounds 1–3 were prepared according to Scheme 4, the strategy being adapted from a literature reaction.39 The reactions were conducted under Schlenk conditions in THF at −78 °C. The first step is the in situ formation of lithium diisopropylamide (LDA) from nBuLi and diisopropylamine, followed by lithiation of the methyl substituents of 6-methyl-2,2′-bipyridine or 6,6′-dimethyl-2,2′-bipyridine. Subsequent addition of propargyl bromide in appropriate molar ratios (see Experimental section) yielded 1, 2 or 3. After purification, the compounds were isolated in 36, 48 and 29% yields, respectively.
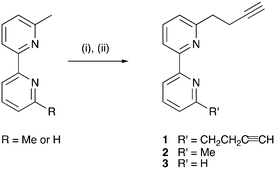 |
| | Scheme 4 Syntheses of compounds 1–3. Conditions: (i) LDA, THF, −78 °C; (ii) propargyl bromide (1.8 molar equivalents for 1, 0.8 equivalents for 2 and 3), THF, −78 °C warm to 22 °C, ca. 15 h. | |
In the electrospray mass spectrum of each of 1–3, the base peak corresponded to the [M + H]+ ion (m/z = 261.11 for 1, 223.12 for 2, and 209.19 for 3). The 1H and 13C{1H} NMR spectra were assigned using 2D methods and were in accord with the structures shown in Scheme 4. The 1D spectra, along with the HMQC and HMBC spectra, are displayed in Fig. S1–S12.† The terminal CCH unit showed a characteristic triplet (J = 2.7 Hz) at δ 2.33 ppm in 1 and 2 and δ 2.34 ppm in 3, while the CH2 protons appeared as a triplet at δ 3.06 ppm, and doublet of triplets at δ 2.73 ppm, respectively, in all three compounds. The solid-state IR spectra of 1, 2 and 3 exhibited a strong or medium intensity absorption at 3283, 3297 and 3220 cm−1, respectively, assigned to the CC stretching mode.
Preparation and characterization of the copper(I) complexes
The heteroleptic complexes [Cu(N^N)(POP)][PF6] and [Cu(N^N)(xantphos)][PF6] with N^N = 1, 2 and 3 were prepared using one of two methods depending on the bisphospane ligands.16,40 For the POP-containing compounds, POP and [Cu(MeCN)4][PF6] were first combined, and the mixture was stirred for two hours, after which the N^N ligand was added. The xantphos ligand is less conformationally flexible than POP and, as has been detailed previously,16,40 [Cu(MeCN)4][PF6], xantphos and the N^N ligand could be combined in one step. The [Cu(N^N)(P^P)][PF6] compounds were isolated as yellow solids in yields ranging from 75 to 84%. The base peak in each compound's electrospray mass spectrum (positive mode) corresponded to the [M–PF6]+ ion. In addition, the [Cu(POP)]+ or [Cu(xantphos)]+ fragment gave rise to a dominant peak in the mass spectrum. 1H, 13C{1H} and 31P{1H} NMR spectra were recorded, and the 1H and 13C{1H} NMR spectra were assigned with the aid of COSY, NOESY, HMQC and HMBC experiments. 1H NMR, HMQC and HMBC spectra for all the copper(I) complexes are shown in Fig. S13–S30,† and include assignments of the 1H signals. The change from a symmetrical to asymmetrical N^N ligand upon going from [Cu(1)(POP)][PF6] and [Cu(1)(xantphos)][PF6] to the corresponding compounds with ligands 2 and 3 is supported by the appearance of additional signals in the aromatic region (e.g. compare Fig. S13† for [Cu(POP)(1)][PF6] with Fig. S19† for [Cu(POP)(2)][PF6]). The 1H NMR spectra reveal that the aliphatic protons for the CH2CH2CCH substituent are typically shifted to a lower frequency upon complexation. The chemical shifts for the bpy protons are largely unaffected on going from the free ligands 1–3 to the coordinated ligands. We have previously discussed in detail the solution dynamic behaviour of several [Cu(6-Rbpy)(P^P)]+ cations (R = alkyl)16,40 and, in this regard, the differences in the 1H NMR spectra of [Cu(N^N)(POP)][PF6] and [Cu(N^N)(xantphos)][PF6] as N^N changes from 1 and 2 to 3 are worthy of note. Upon going from [Cu(1)(xantphos)][PF6] to [Cu(3)(xantphos)][PF6], the symmetry of the cation is lowered and the phenyl rings D (labelled in Scheme 1) split into two sets, those adjacent to the CH2CH2CCH substituent of 3 (rings D) and those facing the unsubstituted pyridine ring (rings D′) as depicted in Scheme 5. The chemical shift separation of the phenyl D and D′-ring protons is smaller for POP-containing complexes compared to the xantphos-containing complexes (compare Fig. S25 and S28†). The 31P{1H} NMR spectrum of each compound showed a broadened signal close to δ −13 ppm (see Experimental section) which was assigned to the POP or xantphos ligand, in addition to a septet at δ −144 ppm arising from the [PF6]− anion.
 |
| | Scheme 5 Schematic representation of [Cu(3)(xantphos)]+ showing the distinction between phenyl rings D and D′. | |
Emission behaviour
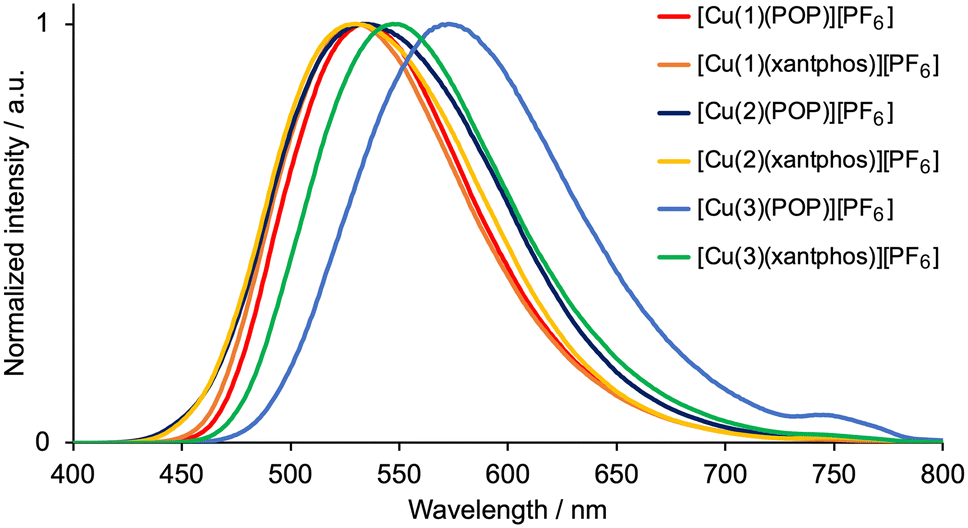
The emission behaviour of the copper(I) complexes was investigated in the solid state. The visible region of the CH2Cl2 solution absorption spectra of [Cu(1)(POP)][PF6], [Cu(1)(xantphos)][PF6], [Cu(2)(POP)][PF6], [Cu(2)(xantphos)][PF6], [Cu(3)(POP)][PF6] and [Cu(3)(xantphos)][PF6] exhibits a broad band in the range λmax = 370–383 nm (Table 2) assigned to MLCT. This is typical for heteroleptic [Cu(N^N)(P^P)][PF6] compounds.28 Excitation into the MLCT bands (λexc = 365 nm) of powdered samples resulted in a yellow or green emission with maxima in the range 530–573 nm (Fig. 1 and Table 2). The emission spectra are broad and structureless, and the maxima compare to values of 565, 550, 549 and 535 nm for powdered samples of [Cu(6-Mebpy)(POP)][PF6], [Cu(6-Mebpy)(xantphos)][PF6], [Cu(6,6′-Me2bpy)(POP)][PF6] and [Cu(xantphos)(6,6′-Me2bpy)][PF6], respectively (all with λexc = 365 nm).28 The PLQY values of 41 and 46%, respectively, for solid [Cu(1)(POP)][PF6] and [Cu(1)(xantphos)][PF6] are relatively high for [Cu(N^N)(P^P)][PF6] complexes in which N^N is a bpy derivative,28,36,41 and the excited-state lifetimes (Table 2) are similar to those of [Cu(POP)(6,6′-Me2bpy)][PF6] (8.7 μs), [Cu(xantphos)(6-Mebpy)][PF6] (10.5 μs) and [Cu(xantphos)(6,6′-Me2bpy)][PF6] (14.7 μs).28 [Cu(3)(POP)][PF6] exhibits low values of the PLQY and excited-state lifetime (Table 2). Factors that have been discussed which may impact on solid-state PLQY are intra-cation π-stacking4,20 (see earlier) and the non-bonded Cu⋯OPOP or Cu⋯Oxantphos distance.41 A survey of literature data indicates that PL is commonly weak when the Cu⋯O distance is less than 3.10 Å,41 and we return to this in the structural discussion below.
Table 2 Photophysical data for the heteroleptic copper(I) compounds
| Compound |
Absorptiona |
Solid-state emissionb |
| MLCT λmax/nm |
λ
max/nm |
PLQY/% |
<τ>/μsc |
|
Solution, CH2Cl2, 5 × 10−5 mol dm−3.
λ
exc = 365 nm.
Components for the biexponential fit are given in Table S1.†
|
| [Cu(1)(POP)][PF6] |
372 |
535 |
41 |
11.1 |
| [Cu(1)(xantphos)][PF6] |
371 |
530 |
46 |
12.8 |
| [Cu(2)(POP)][PF6] |
379 |
533 |
26 |
17.8 |
| [Cu(2)(xantphos)][PF6] |
370 |
530 |
18 |
9.5 |
| [Cu(3)(POP)][PF6] |
383 |
573 |
8 |
3.5 |
| [Cu(3)(xantphos)][PF6] |
377 |
547 |
27 |
9.6 |
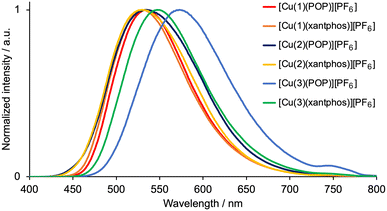 |
| | Fig. 1 Solid-state emission spectra of [Cu(N^N)(POP)][PF6] and [Cu(N^N)(xantphos)][PF6] with N^N = 1–3. λexc = 365 nm. | |
Single-crystal structures
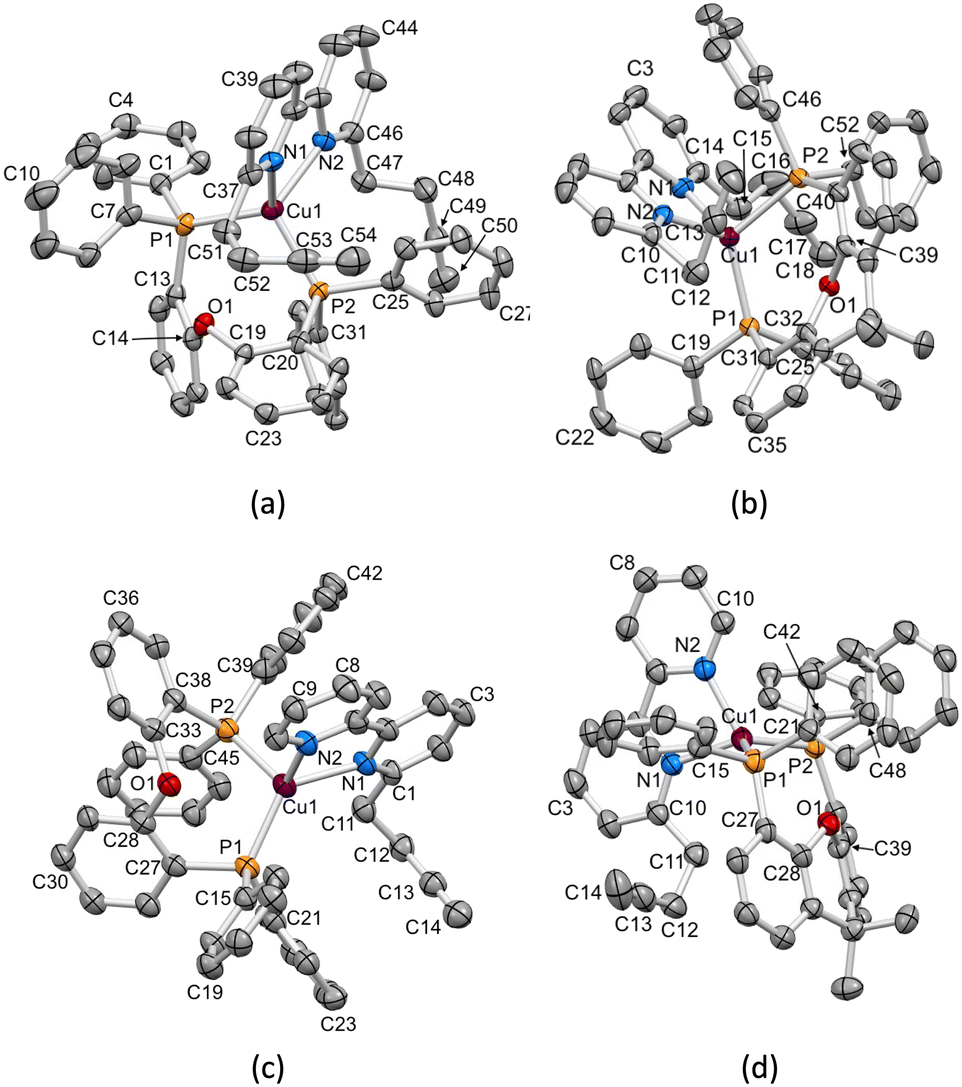
X-ray quality single crystals of [Cu(1)(POP)][PF6], [Cu(1)(xantphos)][PF6], [Cu(3)(POP)][PF6] and [Cu(3)(xantphos)][PF6]·1.5CH2Cl2 were obtained by slow diffusion of Et2O into CH2Cl2 solutions containing the complex. The compounds containing 1 crystallize in the monoclinic space group P21/c, while those containing 3 crystallize in the triclinic space group P (see the Experimental section for crystallographic details). The structures of the [Cu(1)(POP)]+, [Cu(1)(xantphos)]+, [Cu(3)(POP)]+ and [Cu(3)(xantphos)]+ cations are displayed in Fig. 2. Atom Cu1 in each cation is in a distorted tetrahedral environment, and Cu–N and Cu–P bond lengths, along with P–Cu–P and N–Cu–N bond angles are given in Table 3. In keeping with the greater flexibility of the coordinated POP ligand with respect to xantphos, the P–Cu–P bond angles in the POP-containing complexes are smaller than those in the corresponding xantphos-containing complex cations. The N–Cu–P bond angles lie in the ranges 111.81(12)–123.83(11)°, 100.16(11)–125.64(11)°, 106.00(14)–130.47(14)° and 111.95(13)–117.73(12)° in [Cu(1)(POP)]+, [Cu(1)(xantphos)]+, [Cu(3)(POP)]+ and [Cu(3)(xantphos)]+, respectively. The C–O–C bond angles in the P^P ligands are similar, irrespective of P^P being POP or xantphos (Table 4).
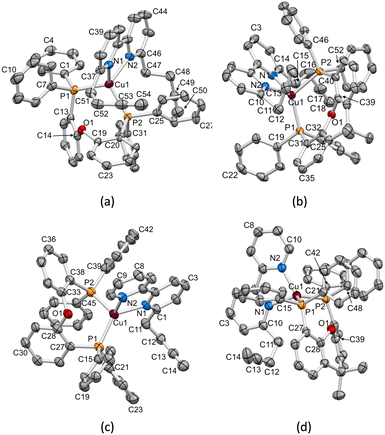 |
| | Fig. 2 ORTEP-style representations of the structures of the cations (a) [Cu(1)(POP)]+, (b) [Cu(1)(xantphos)]+, (c) [Cu(3)(POP)]+ and (d) [Cu(3)(xantphos)]+. Ellipsoids are plotted at a 40% probability level; H atoms are omitted. | |
Table 3 Bond lengths and angles in the copper(I) coordination sphere of each [Cu(N^N)(P^P)]+ cations
| Cation |
Cu–N/Å |
Cu–P/Å |
N–Cu–N/° |
P–Cu–P/° |
| [Cu(1)(POP)]+ |
2.115(4), 2.156(4) |
2.2860(13), 2.2916(14) |
78.99(15) |
108.08(5) |
| [Cu(1)(xantphos)]+ |
2.114(4), 2.127(3) |
2.2699(12), 2.2982(12) |
79.47(15) |
118.47(5) |
| [Cu(3)(POP)]+ |
2.111(4), 2.073(5) |
2.2306(16), 2.2879(17) |
79.81(18) |
109.95(6) |
| [Cu(3)(xantphos)]+ |
2.092(4), 2.057(4) |
2.2710(16), 2.2579(14) |
80.27(17) |
113.40(6) |
Table 4 Additional bond parameters in the [Cu(N^N)(P^P)]+ cations
| Cation |
C–O–C/° |
Angle between ring planes in bpy/° |
C–N–N–C torsion angle in bpy/° |
Cu⋯O/Å |
| [Cu(1)(POP)]+ |
119.0(4) |
13.7 |
−10.6(7) |
3.313(3) |
| [Cu(1)(xantphos)]+ |
116.0(3) |
19.7 |
18.1(6) |
3.120(3) |
| [Cu(3)(POP)]+ |
115.8(4) |
9.8 |
−9.8(8) |
3.063(4) |
| [Cu(3)(xantphos)]+ |
114.3(4) |
4.0 |
0.4(7) |
3.218(3) |
The distortion away from planarity of the bpy domain is described by the angles between the least squares planes of the two pyridine rings and the N–C–C–N torsion angles (Table 4). The larger inter-plane angles in the complex cations containing 1 are most probably associated with the accommodation of two CH2CH2CCH substituents versus only a single CH2CH2CCH group in [Cu(3)(POP)]+ and [Cu(3)(xantphos)]+.
We noted above that the intramolecular Cu⋯O distance may influence solid-state PLQY values of the heteroleptic copper(I) compounds. The values of Cu⋯O are given in Table 4, and, significantly, the smallest non-bonded separation of 3.063(4) Å is for [Cu(3)(POP)][PF6], which exhibits the lowest solid-state PLQY value (8%, Table 2).
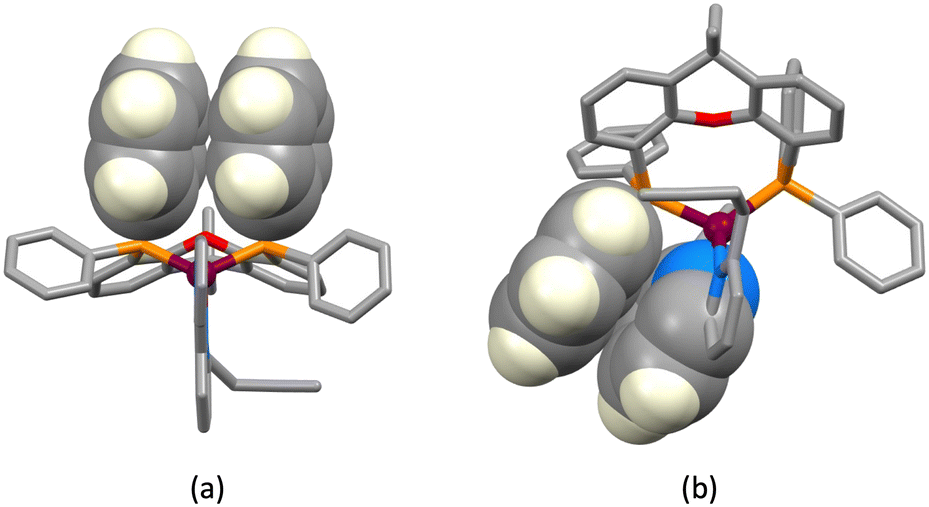
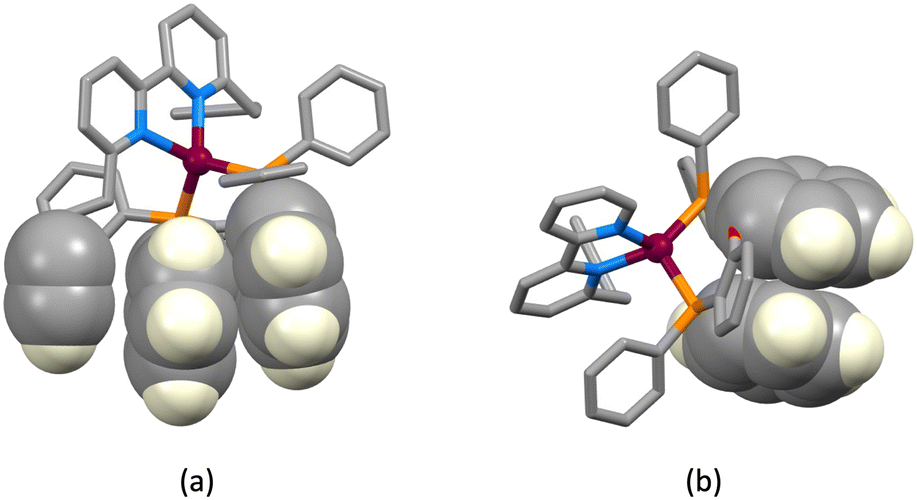
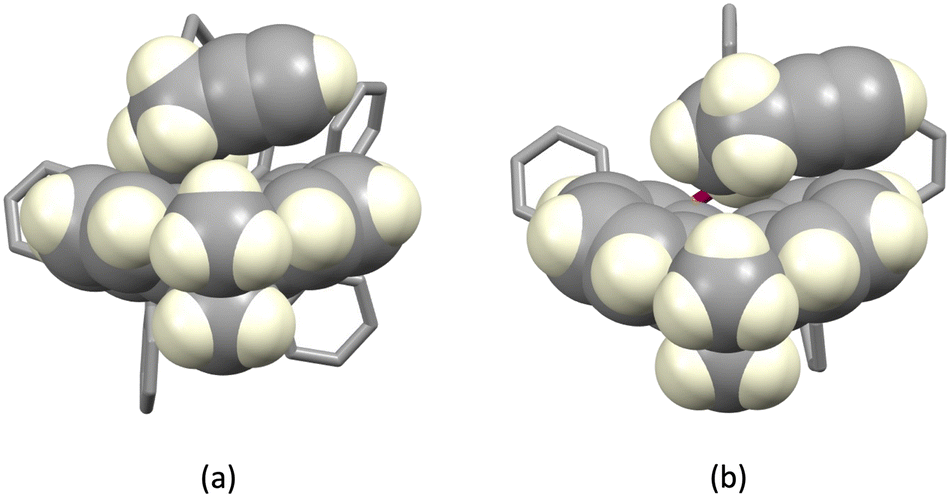
The structures of the hexafluoridophosphate salts of [Cu(1)(POP)]+, [Cu(1)(xantphos)]+, [Cu(3)(POP)]+ and [Cu(3)(xantphos)]+ reveal some interesting differences in intramolecular π-stacking interactions. Typically, [Cu(N^N)(xantphos)]+ complexes exhibit face-to-face π-stacking between two phenyl rings of different PPh2 units of the xantphos ligand.4 This is indeed observed in [Cu(3)(xantphos)]+ (Fig. 3a); the interaction is characterized by a ring centroid⋯centroid distance of 3.75 Å, and an interplane angle of 4.4°. In contrast, in [Cu(1)(xantphos)]+, one phenyl ring of a PPh2 unit is involved in a weak π-stacking interaction with one pyridine ring of ligand 1 (interplane angle = 13.2°, centroid⋯centroid distance = 3.94 Å) as shown in Fig. 4b. In the POP-containing complexes, an intraligand face-to-face π-stacking interaction is present in [Cu(1)(POP)]+ and [Cu(3)(POP)]+, and this is typical of [Cu(N^N)(POP)]+ complexes.4 However, the metrics of the interactions differ and are consistent with an efficient interaction only in [Cu(1)(POP)]+. In the latter, the centroid⋯centroid distance = 3.51 Å and the angle between the ring planes = 10.3° (Fig. 4a), while in [Cu(3)(POP)]+, the corresponding metrics are 3.91 Å and 24.2° (Fig. 4b). Significantly, the face-to-face contact is extended in [Cu(1)(POP)]+ to incorporate an alkyne⋯arene π-interaction in which the angle between the CC vector and the arene ring-plane is 0.4° (Fig. 4a). The distances from the alkyne atoms C50 and C49 to the centroid of the phenyl ring containing C31 are 3.99 and 4.28 Å, respectively. The second CH2CH2CCH unit is also positioned over an arene ring (ring with C19), with an angle between the CC vector and the ring-plane of 2.0°; the alkyne atoms Calkyne⋯ringcentroid distances are 3.74 and 4.15 Å. Despite these rather long separations, the interactions appear to be significant, since the CH2CH2CCH substituent in [Cu(3)(POP)]+ is also positioned over a phenyl ring (ring with C21) with similar distances between the alkyne C atoms and the ring-centroid (3.82 and 4.01 Å for C13 and C14, respectively) and an angle between the CC vector and the arene ring-plane of 0.1°. In both xantphos-containing complexes, the xanthene unit hosts a CH2CH2CCH substituent such that the alkyne unit resides over one arene ring (Fig. 5). In [Cu(1)(xantphos)]+, the distances from the alkyne atoms C13 and C14 to the centroid of the ring containing atom C40 (4.16 and 4.07 Å) are longer than the corresponding distances in [Cu(3)(xantphos)]+ (3.75 and 4.05 Å). The angles between the CC vector and the xantphos–arene ring-plane in [Cu(1)(xantphos)]+ and [Cu(3)(xantphos)]+ are 4.8 and 13.5°, respectively. In [Cu(1)(xantphos)]+, the second CH2CH2CCH substituent also lies over an arene ring (the PPh2 phenyl ring containing C52) but with long Calkyne⋯centroidphenyl distances (4.26 and 4.85 Å). For the CC⋯arene contacts, the distances between the centroids of the CC bond and the aromatic ring lie within the range 3.86–4.09 Å. None of the complexes exhibits intramolecular CCH⋯πarene interactions of the type previously described in the literature.42–47
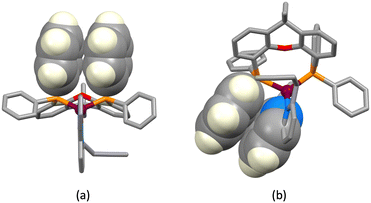 |
| | Fig. 3 Face-to-face π-stacking interactions involving (a) PPh2 phenyl rings in [Cu(3)(xantphos)]+ and (b) one PPh2 phenyl ring and a pyridine ring of the bpy domain [Cu(1)(xantphos)]+. | |
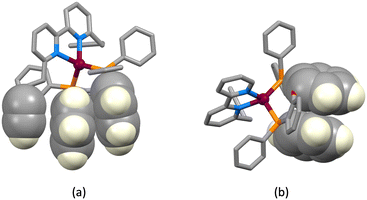 |
| | Fig. 4 Intraligand face-to-face π-stacking within the POP ligand in (a) [Cu(1)(POP)]+ and (b) [Cu(3)(POP)]+. Diagram (a) also shows the extension of the stacking to incorporate an alkyne⋯arene π-interaction in [Cu(1)(POP)]+. | |
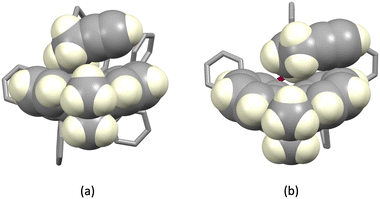 |
| | Fig. 5 Hosting of a CH2CH2CCH substituent in the xanthene bowl-shaped cavity in (a) [Cu(1)(xantphos)]+ and (b) [Cu(3)(xantphos)]+. | |
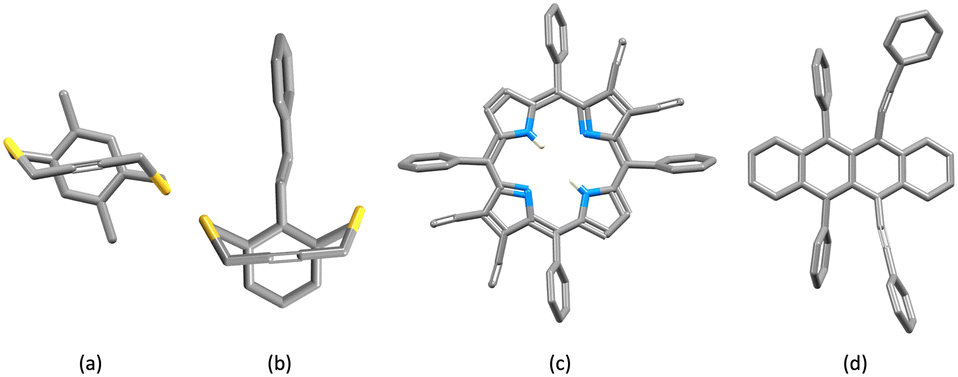
Comments on C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/h3_char_e002.gif) C⋯arene contacts in compounds in the CSD
C⋯arene contacts in compounds in the CSD
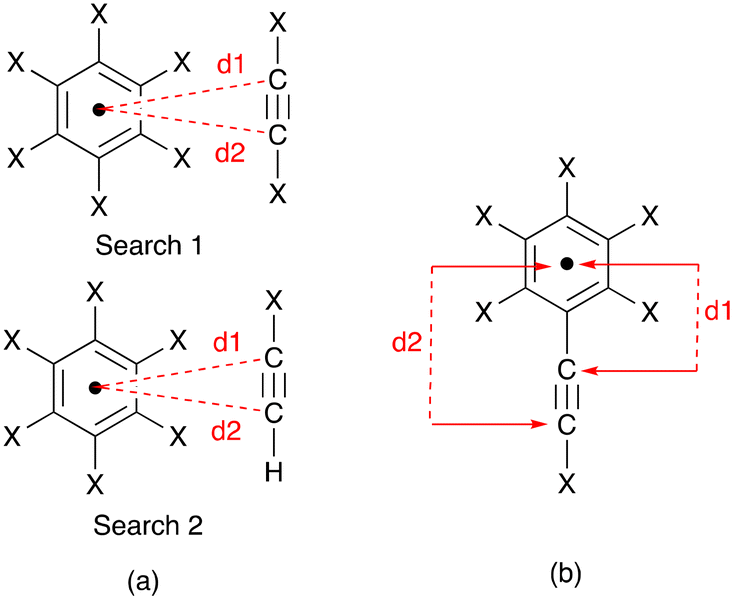
Each of the complex cations [Cu(1)(POP)]+, [Cu(1)(xantphos)]+, [Cu(3)(POP)]+ and [Cu(3)(xantphos)]+ exhibits a near-parallel alignment of one or two CCH units over an arene ring. We consider that these alkyne⋯arene π-interactions are significant and contribute to the protection of the copper(I) centre in the solid state. To the best of our knowledge, there has been little detailed discussion of such alkyne⋯arene π-interactions (intra- or intermolecular) in the literature.37,38 We therefore decided to search the CSD using Conquest (version 2022.3.0 including November 2022 updates)27 with a focus on intramolecular CC⋯arene contacts; ‘arene’ was defined as an aromatic C6 ring with no constraints on substituents, and an initial search had no restriction on the groups attached to the alkyne unit. The CC unit was defined with a terminal hydrogen atom in a second search. Distances, d1 and d2, between the alkyne C atoms and the arene ring centroid (Scheme 6a) were determined using Conquest with a defined range of 3.0 ≤ d ≤ 4.5 Å. Values less than 3.0 Å corresponded to the through-bond arenecentroid–C distance, d1, shown in Scheme 6b, and several hits with values of d1 ≥ 3.0 Å, which also corresponded to Scheme 6b were discarded. For the first and second searches, 4029 and 324 hits were found, respectively; for these, 7464 and 454 intramolecular CC⋯arene contacts were identified.
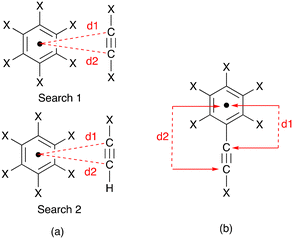 |
| | Scheme 6 (a) Definitions for the two searches of distances, d1 and d2, between the alkyne C atoms and the arene ring centroid; X = any atom. (b) Definitions of occurrences of d1 and d2 that were discarded. | |
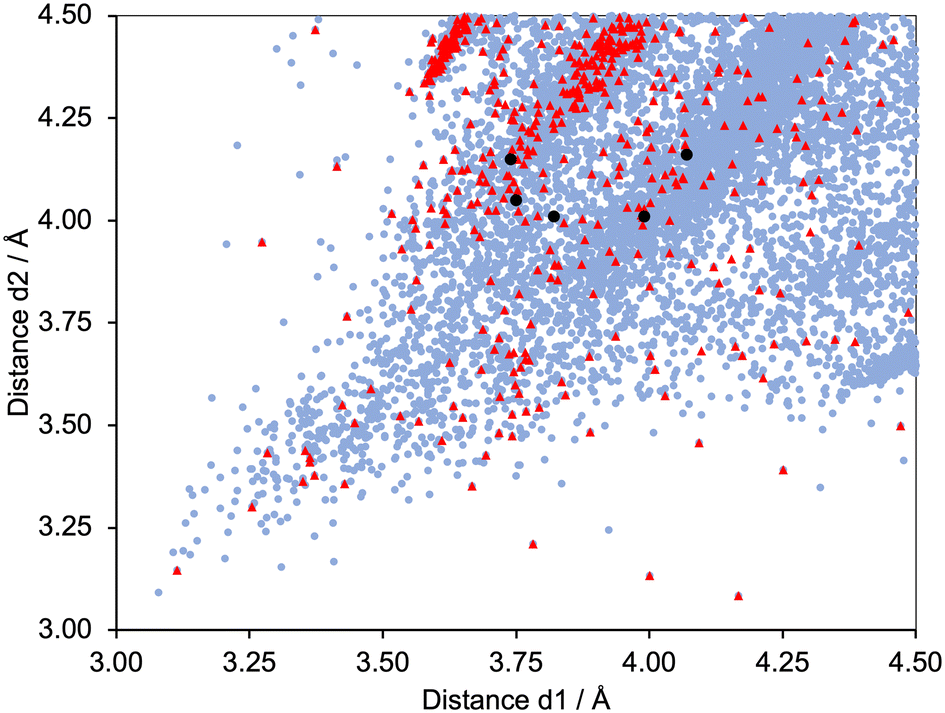
Fig. 6 shows a scatter plot of Calkyne⋯arenecentroid distances d1 against d2. Points lying along the diagonal of the plot correspond to CC units symmetrically positioned with respect to the centroid of the arene ring. A primary factor contributing to this arrangement is the structural constraint of the arene and alkyne, as illustrated by the structures shown in Fig. 7. The shortest contacts are observed for the structure displayed in Fig. 7a (CSD refcode TOSLOC, d1 = 3.079 Å, d2 = 3.092 Å),48 and a similar motif is found in the molecule shown in Fig. 7b (CSD refcode FINXIJ, d1 = 3.24 Å, d2 = 3.28 Å).49 The porphyrin derivative in Fig. 7c was designed to undergo Bergman cyclization,50 and the close CC⋯arene π-contacts are characterized by values of d1 = 3.114 Å and d2 = 3.146 Å, and d1 = 3.363 Å and d2 = 3.411 Å. The forced close proximity of phenyl and alkynyl groups appended to the porphyrin framework leads to CC⋯arene π-contacts in several structures, for example, CSD refcodes AKICIG,51 BOVDIB,52 DOKRIG53 and FEKYOK.54Fig. 6 shows some outliers; for example, the points with 3.085/4.167 Å, 3.134/4.001 Å, and 3.227/4.184 Å, correspond to CSD refcodes GIGBAZ,55 VIYBIR56 and INDMOB,57 respectively, containing metal π-bonded η2-alkynes or η6-arenes. The black circles in Fig. 6 correspond to the four structurally characterized copper(I) compounds [Cu(1)(POP)][PF6] (d1/d2 = 3.99/4.01 and 3.74/4.15 Å), [Cu(1)(xantphos)][PF6] (d1/d2 = 3.82/4.01 Å), [Cu(3)(POP)][PF6] (d1/d2 = 4.07/4.16 Å) and [Cu(3)(xantphos)][PF6] (d1/d2 = 3.75/4.05 Å). The points fall within the central part of the scatter plot and represent meaningful CC⋯πarene interactions.
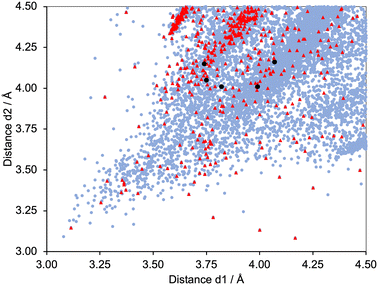 |
| | Fig. 6 Scatter plot of the hits found in the CSD using Conquest (version 2022.3.0 including November 2022 updates) corresponding to the searches defined in Scheme 6a.27 All data, pale blue points; CCH units, red triangles. Values of d1 and d2 for the copper(I) complexes from this work are shown as black circles – two interactions in [Cu(1)(POP)][PF6], and one in each of [Cu(1)(xantphos)][PF6], [Cu(3)(POP)][PF6] and [Cu(3)(xantphos)]. Points falling on the diagonal line correspond to CC units symmetrically positioned with respect to the centroid of the arene ring. | |
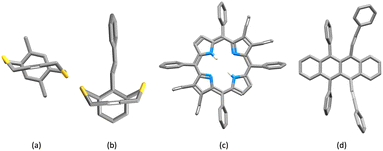 |
| | Fig. 7 Structurally constrained short CC⋯arene π-contacts: CSD refcodes (a) TOSLOC (d1/d2 = 3.079, 3.092 Å),48 (b) FINXIJ (d1/d2 = 3.24/3.28 Å),49 (c) NAPHUJ (d1/d2 = 3.114/3.146 Å, and 3.363/3.411 Å),50 and (d) NOYXOR (d1/d2 = 3.303/3.320 Å, and 3.295/3.381 Å).58 | |
The distance between the centroids of the CC bond and the arene ring was also considered, and Fig. 8 displays a histogram with the values of the CCcentroid⋯arenecentroid distances for all of the compounds presented in Fig. 6. The shortest value (3.028 Å) corresponds to the structure shown in Fig. 7a (CSD refcode TOSLOC) and the majority of the hits with values <3.35 Å involve structures similar to those in Fig. 7. The mean and median values for Fig. 8 are 4.01 and 4.04 Å, respectively, and we note that the CCcentroid⋯arenecentroid distances for the copper(I) compounds reported in this work fall in the range 3.86 to 4.09 Å (Fig. 8, white circles).
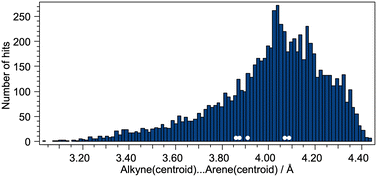 |
| | Fig. 8 Distribution of alkynecentroid⋯arenecentroid distances for the complete set of compounds shown in Fig. 6. The white circles correspond to the alkynecentroid⋯arenecentroid distances in the copper(I) compounds. | |
Conclusions
We have prepared and characterized a series of six heteroleptic copper(I) compounds incorporating wide bite-angle bisphosphanes combined with N^N ligands 1–3 containing CH2CH2CCH substituents in the 6- or 6,6′-positions of the bpy domain. The single crystal structures of [Cu(1)(POP)][PF6], [Cu(1)(xantphos)][PF6], [Cu(3)(POP)][PF6] and [Cu(3)(xantphos)][PF6] were determined. In the solid state, the compounds are yellow or green emitters with λemmax in the 530–573 nm range. The solid-state PLQY values range from 46% for [Cu(1)(xantphos)][PF6] to 8% for [Cu(3)(POP)][PF6], and the lowest PLQY correlates to the shortest Cu⋯O non-bonded distance. The Cu atom in each structurally characterized complex is protected by a combination of face-to-face arene⋯arene π-interactions and CC⋯πarene interactions. The latter shows a near parallel alignment of the CCH unit over an arene ring and Calkyne⋯arenecentroid distances lie between 3.74 and 4.16 Å, and the CCcentroid⋯arenecentroid distances are in the range 3.86 to 4.09 Å. These distances fall well within the ranges for related interactions for compounds (organic and inorganic) in the CSD, and we consider them to be meaningful contacts which contribute to the stability in solution and solid-state photophysical properties of the copper(I) complexes. However, we do not speculate on the exact nature of the CC⋯πarene interactions. The study should provide new structural guidelines to aid in designing heteroleptic copper(I) compounds with high solid-state PLQYs.
Author contributions
Project conceptualization, administration, supervision, and funding acquisition, E. C. C. and C. E. H.; investigation and experimental work, D. G.-S. and M. M.; crystallography, A. P.; CSD data mining and analysis: C. E. H. and E. C. C.; manuscript writing, D. G.-S. and C. E. H.; manuscript editing, all authors. All authors have read and agreed to the published version of the manuscript.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
We thank the University of Basel for financial support.
References
- R. Czerwieniec, M. J. Leitl, H. H. H. Homeier and H. Yersin, Coord. Chem. Rev., 2016, 325, 2 CrossRef CAS.
- H. Yersin, R. Czerwieniec, M. Z. Shafikov and A. F. Suleymanova, ChemPhysChem, 2017, 18, 3508 CrossRef CAS PubMed.
- M. J. Leitl, D. M. Zink, A. Schinabeck, T. Baumann, D. Volz and H. Yersin, Top. Curr. Chem., 2016, 374, 1 CrossRef CAS PubMed.
- C. E. Housecroft and E. C. Constable, J. Mater. Chem. C, 2022, 10, 4456 RSC.
- C. Sandoval-Pauker, M. Santander-Nelli and P. Dreyse, RSC Adv., 2022, 12, 10653 RSC.
- L. P. Ravaro, K. P. S. Zanoni and A. S. S. de Camargo, Energy Rep., 2020, 6, 37 CrossRef.
- M. T. Buckner and D. R. McMillin, J. Chem. Soc., Chem. Commun., 1978, 759 RSC.
- G. Blasse and D. R. McMillin, Chem. Phys. Lett., 1980, 70, 1 CrossRef CAS.
- R. A. Rader, D. R. McMillin, M. T. Buckner, T. G. Matthews, D. J. Casadonte, R. K. Lengel, S. B. Whittaker, L. M. Darmon and F. E. Lytle, J. Am. Chem. Soc., 1981, 103, 5906 CrossRef CAS.
- C. O. Dietrich-Buchecker, P. A. Marnot, J.-P. Sauvage, J. R. Kirchhoff and D. R. McMillin, J. Chem. Soc., Chem. Commun., 1983, 513 RSC.
- J. R. Kirchhoff, R. E. Gamache, M. W. Blaskie, A. A. Del Paggio, R. K. Lengel and D. R. McMillin, Inorg. Chem., 1983, 22, 2380 CrossRef CAS.
- C. E. A. Palmer and D. R. McMillin, Inorg. Chem., 1987, 26, 3837 CrossRef CAS.
- D. J. Casadonte and D. R. McMillin, Inorg. Chem., 1987, 26, 3950 CrossRef CAS.
- N. Armaroli, Chem. Soc. Rev., 2001, 30, 113 RSC.
- M. K. Eggleston, D. R. McMillin, K. S. Koenig and A. J. Pallenberg, Inorg. Chem., 1997, 36, 172 CrossRef CAS.
- S. Keller, A. Pertegás, G. Longo, L. Martínez, J. Cerda, J. M. Junquera-Hernández, A. Prescimone, E. C. Constable, C. E. Housecroft, E. Ortí and H. J. Bolink, J. Mater. Chem. C, 2016, 4, 3857 RSC.
- S. Keller, A. Prescimone, M.-G. La Placa, J. M. Junquera-Hernández, H. J. Bolink, E. C. Constable, M. Sessolo, E. Ortí and C. E. Housecroft, RSC Adv., 2020, 10, 22631 RSC.
- S. Keller, F. Brunner, J. M. Junquera-Hernàndez, A. Pertegàs, M.-G. La-Placa, A. Prescimone, E. C. Constable, H. J. Bolink, E. Ortí and C. E. Housecroft, ChemPlusChem, 2018, 83, 217 CrossRef CAS PubMed.
- P. C. J. Kamer, P. W. N. M. van Leeuwen and J. N. H. Reek, Acc. Chem. Res., 2001, 34, 895 CrossRef CAS PubMed.
- E. Leoni, J. Mohanraj, M. Holler, M. Mohankumar, I. Nierengarten, F. Monti, A. Sournia-Saquet, B. Delavaux-Nicot, J.-F. Nierengarten and N. Armaroli, Inorg. Chem., 2018, 57, 15537 CrossRef CAS PubMed.
- G. J. Kubas, B. Monzyk and A. L. Crumbliss, Inorg. Synth., 1979, 19, 90 CAS.
- G. M. Sheldrick, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3 CrossRef PubMed.
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, J. Appl. Crystallogr., 2009, 42, 339 CrossRef CAS.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Adv., 2015, 71, 3 CrossRef PubMed.
- C. F. Macrae, I. Sovago, S. J. Cottrell, P. T. A. Galek, P. McCabe, E. Pidcock, M. Platings, G. P. Shields, J. S. Stevens, M. Towler and P. A. Wood, J. Appl. Crystallogr., 2020, 53, 226 CrossRef CAS PubMed.
- C. R. Groom, I. J. Bruno, M. P. Lightfoot and S. C. Ward, Acta Crystallogr., Sect. B: Struct. Sci., Cryst. Eng. Mater., 2016, 72, 171 CrossRef CAS PubMed.
- I. J. Bruno, J. C. Cole, P. R. Edgington, M. Kessler, C. F. Macrae, P. McCabe, J. Pearson and R. Taylor, Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem., 2002, 58, 389 CrossRef PubMed.
- M. Meyer, L. Mardegan, D. Tordera, A. Prescimone, M. Sessolo, H. J. Bolink, E. C. Constable and C. E. Housecroft, Dalton Trans., 2021, 50, 17920 RSC.
- M. Alkan-Zambada, S. Keller, L. Martinez-Sarti, A. Prescimone, J. M. Junquera-Hernández, E. C. Constable, H. J. Bolink, M. Sessolo, E. Ortí and C. E. Housecroft, J. Mater. Chem. C, 2018, 6, 8460 RSC.
- F. Brunner, S. Graber, Y. Baumgartner, D. Häussinger, A. Prescimone, E. C. Constable and C. E. Housecroft, Dalton Trans., 2017, 46, 6379 RSC.
- S. Keller, A. Prescimone, H. Bolink, M. Sessolo, G. Longo, L. Martínez-Sarti, J. M. Junquera-Hernández, E. C. Constable, E. Ortí and C. E. Housecroft, Dalton Trans., 2018, 47, 14263 RSC.
- S. Keller, F. Brunner, J. M. Junquera-Hernández, A. Pertegás, M.-G. La-Placa, A. Prescimone, E. C. Constable, H. J. Bolink, E. Ortí and C. E. Housecroft, ChemPlusChem, 2018, 83, 217 CrossRef CAS PubMed.
- W.-M. Wang, P. Ju, M.-H. Jing, P. Yu and Q. Huang, Aust. J. Chem., 2020, 73, 640 CrossRef CAS.
- M. Meyer, F. Brunner, A. Prescimone, E. C. Constable and C. E. Housecroft, Molecules, 2021, 26, 125 CrossRef CAS PubMed.
- A. Kaeser, M. Mohankumar, J. Mohanraj, F. Monti, M. Holler, J.-J. Cid, O. Moudam, I. Nierengarten, L. Karmazin-Brelot, C. Duhayon, B. Delavaux-Nicot, N. Armaroli and J.-F. Nierengarten, Inorg. Chem., 2013, 52, 12140 CrossRef CAS PubMed.
- J. Wöhler, M. Meyer, A. Prescimone, E. C. Constable and C. E. Housecroft, Dalton Trans., 2022, 51, 13094 RSC.
- A. S. Shetty, J. Zhang and J. S. Moore, J. Am. Chem. Soc., 1996, 118, 1019 CrossRef CAS.
- S. Karki, L. J. Karas, X. Wang, J. I. Wu and O. Š. Miljanić, Cryst. Growth Des., 2022, 22, 2076 CrossRef CAS.
- K. Maeyama, C. Okumura and N. Yonezawa, Synth. Commun., 2006, 32, 3159 CrossRef.
- F. Brunner, A. Babaei, A. Pertegás, J. M. Junquera-Hernández, A. Prescimone, E. C. Constable, H. J. Bolink, M. Sessolo, E. Ortí and C. E. Housecroft, Dalton Trans., 2019, 48, 446 RSC.
- S. Keller, M. Alkan-Zambada, A. Prescimone, E. C. Constable and C. E. Housecroft, Crystals, 2020, 10, 255 CrossRef CAS.
- M. Nishio, CrystEngComm, 2004, 6, 130 RSC.
- J. M. A. Robinson, B. M. Kariuki, R. J. Gough, K. D. M. Harris and D. Philp, J. Solid State Chem., 1997, 134, 203 CrossRef CAS.
- K. Dziubek, M. Podsiadło and A. Katrusiak, J. Am. Chem. Soc., 2007, 129, 12620 CrossRef CAS PubMed.
- T. S. Thakur, R. Sathishkumar, A. G. Dikundwar, T. N. Guru Row and G. R. Desiraju, Cryst. Growth Des., 2010, 10, 4246 CrossRef CAS.
- T. Steiner, E. B. Starikov, A. M. Amado and J. J. C. Teixeira-Dias, J. Chem. Soc., Perkin Trans. 2, 1995, 1321 RSC.
- T. Steiner, M. Tamm, B. Lutz and J. van der Maas, Chem. Commun., 1996, 1127 RSC.
- I. Pischel, M. Nieger, A. Archut and F. Vögtle, Tetrahedron, 1996, 52, 10043 CrossRef CAS.
- D. Cao, D. Schollmeyer and H. Meier, Eur. J. Org. Chem., 1999, 1999, 791 CrossRef.
- L. J. K. Boerner, S. Mazumder, M. Pink, M.-H. Baik and J. M. Zaleski, Chem. – Eur. J., 2011, 17, 14539 CrossRef CAS PubMed.
- T. Chandra, B. J. Kraft, J. C. Huffman and J. M. Zaleski, Inorg. Chem., 2003, 42, 5158 CrossRef CAS PubMed.
- P. Bhyrappa, U. K. Sarangi, V. Velkannan and V. Ramkumar, Eur. J. Inorg. Chem., 2014, 5760 CrossRef CAS.
-
R. Kumar and M. Sankar, CSD Commun., 2014, CCDC 1018393 Search PubMed.
- M. Nath, M. Pink and J. M. Zaleski, J. Am. Chem. Soc., 2005, 127, 478 CrossRef CAS PubMed.
- H. G. Alt, H. E. Engelhardt, A. Razavi, M. D. Rausch and R. D. Rogers, Z. Naturforsch., B: J. Chem. Sci., 1988, 43, 438 CrossRef CAS.
- T. Y. Lai, K. L. Gullett, C.-Y. Chen, J. C. Fettinger and P. P. Power, Organometallics, 2019, 38, 1421 CrossRef CAS.
- S. R. Allen, P. K. Baker, S. G. Barnes, M. Green, L. Trollope, L. Manojlovic-Muir and K. W. Muir, J. Chem. Soc., Dalton Trans., 1981, 873 RSC.
- J. Ly, K. Martin, S. Thomas, M. Yamashita, B. Yu, C. A. Pointer, H. Yamada, K. R. Carter, S. Parkin, L. Zhang, J.-L. Bredas, E. R. Young and A. L. Briseno, J. Phys. Chem. A, 2019, 123, 7558 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Scheme 1: Structures of ligands from previous work. Fig. S1–S12, NMR spectra of ligands 1–3; Fig. S13–S30, NMR spectra of the copper(I) coordination compounds. Table S1: Components of biexponential fits to emission decay. CCDC 2247205–2247208. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3ce00355h |
|
| This journal is © The Royal Society of Chemistry 2023 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence ,
Marco
Meyer
,
Alessandro
Prescimone
,
Marco
Meyer
,
Alessandro
Prescimone