
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Novel cyclam multicomponent crystal forms: synthesis, characterization and antimicrobial activity†
Rajaa
Saied
a,
Paula C.
Alves
ab,
Patrícia
Rijo
cd,
Vânia
André
 *ab and
Luis G.
Alves
*ab
*ab and
Luis G.
Alves
*ab
aCentro de Química Estrutural, Institute of Molecular Sciences, Instituto Superior Técnico, Universidade de Lisboa, Av. Rovisco Pais 1, 1049-001 Lisboa, Portugal. E-mail: vaniandre@tecnico.ulisboa.pt; luis.g.alves@tecnico.ulisboa.pt
bAssociação do Instituto Superior Técnico para a Investigação e Desenvolvimento, Avenida António José de Almeida, no. 12, 1000-043 Lisboa, Portugal
cUniversidade Lusófona's Research Center for Biosciences and Health Technologies (CBIOS), Campo Grande 376, 1749-024 Lisboa, Portugal
dResearch Institute for Medicines (iMed. ULisboa), Faculty of Pharmacy, Universidade de Lisboa (UL), Av. Prof. Gama Pinto, 1649-003 Lisboa, Portugal
First published on 18th July 2023
Abstract
Novel multicomponent crystal forms were obtained from reactions of trans-disubstituted cyclam derivatives with flufenamic and salicylic acids. The reactions of H2(4-CF3PhCH2)2cyclam with those acids led to the formation of salts of the formulae [H2{H2(4-CF3PhCH2)2cyclam}](3-CF3PhNHC6H4COO)2 and [H2{H2(4-CF3PhCH2)2cyclam}](HOC6H4COO)2, respectively. The reaction of H2(PhCH2)2cyclam with flufenamic acid led to the formation of the salt [H2{H2(PhCH2)2cyclam}](3-CF3PhNHC6H4COO)2, whereas its reaction with salicylic acid afforded the ionic cocrystal [H2(PhCH2)2cyclam](HOC6H4COOH)2·[H2{H2(PhCH2)2cyclam}](HOC6H4COO)2. The compounds obtained were fully characterized and tested against several yeasts as well as Gram-positive and Gram-negative bacterial strains. The results have shown that the new multicomponent forms display relevant antimicrobial activity when compared to their parent cyclam derivatives, highlighting the importance of exploring synergistic effects to unveil new and efficient antimicrobial agents.
Introduction
Infections are still responsible for significant mortality rates worldwide due to multidrug resistant bacteria that drastically reduce the options for effective antimicrobial treatments, having a massive negative social and economic global impact.1 For several years, pharmaceutical companies diminished the investment in the research and development of novel antimicrobials, whose financial return is currently lower than that for any other drug or vaccine.2 More recently, pharmaceutical companies are being encouraged to improve the antibiotic pipeline.3,4Considering the variety of bacterial resistance mechanisms, it is necessary to establish a multifaceted response.5 Two possible approaches may be undertaken: (i) the development of entirely new compounds, a time-consuming and risky approach or (ii) the modification of compounds already approved as antimicrobials, the best approach when it comes to short-term solutions for the current shortage of antimicrobials.6
Macrocyclic polyamines can play a role in the quest for alternative antimicrobial agents,7 and thus they have received a great interest over the past years because of their chemical and biological properties. For example, cyclam derivatives, which were previously studied as antiviral drugs against HIV,8 are now being explored as a novel class of antimicrobials.9–12 Recently, Alves et al. have shown that trans-disubstituted cyclam derivatives and their Fe(III) and Cu(II) metal complexes have antibacterial13,14 and antifungal15 properties. These results, together with the fact that cyclams display a highly versatile backbone easily modifiable by the introduction of new chemical substituents at the amino groups, highlight these molecules as a potential new family of antimicrobial drugs.
The efficiency of these tetrazamacrocycles can be further increased by applying crystal engineering and supramolecular chemistry principles for the design of new multicomponent compounds towards improved physicochemical properties that strongly influence the bioavailability, manufacturability, stability and other performance characteristics of the drugs16–20 as well as their activity. Indeed, a multicomponent crystal form enclosing cyclam and paracetamol has already been disclosed.21
According to the nature of the co-former, multicomponent crystal forms may be classified as solvates, salts, molecular cocrystals (MCCs) or ionic cocrystals (ICCs).22 Multicomponent crystal forms of two cyclam derivatives with known antimicrobial properties with flufenamic and salicylic acids envisaging synergistic effects are reported herein. Flufenamic acid has analgesic, anti-inflammatory, and antipyretic properties,23 while salicylic acid displays topical antibacterial properties.24 The design, synthesis, and characterization of novel multicomponent crystal forms by different techniques are presented as well as the assessment of the antimicrobial activity of the final compounds.
Experimental section
Synthesis and characterization
H2(PhCH2)2cyclam, 1,25 and H2(4-CF3PhCH2)2cyclam, 2,26 were prepared according to previously described procedures. All other reagents were purchased from Sigma and used without further purification.![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O), 144.5 (CAr), 143.3 (CAr), 136.6 (CAr), 132.2 (CAr), 131.7 (CAr), 130.6 (CAr), 130.3 (q, 2JC–F = 32 Hz, CAr) 129.9 (CAr), 128.3 (CAr), 127.4 (CAr), 124.3 (q, 1JC–F = 232 Hz, CF3), 122.1 (CAr), 120.8 (CAr), 118.7 (CAr), 117.1 (q, 3JC–F = 4 Hz, CAr), 114.6 (CAr), 114.4 (q, 3JC–F = 4 Hz, CAr), 57.2 (PhCH2N), 49.8 ([C2]CH2N), 48.6 ([C3]CH2N), 46.5 ([C3]CH2N), 45.6 ([C2]CH2N), 23.5 (CH2CH2CH2). 19F NMR (D2O/(CD3)2SO, 282.4 MHz, 296 K): δ (ppm) −61.3 (CF3). Anal. calcd for C52H56F6N6O4: C, 66.23; H, 5.99; N, 8.91. Found: C, 65.91; H, 5.92; N, 8.87.
O), 144.6 (CAr), 143.1 (CAr), 141.9 (CAr), 132.1 (2xCAr), 130.6 (CAr), 130.4 (CAr), 130.3 (q, 2JC–F = 33 Hz, CAr) 127.9 (q, 2JC–F = 32 Hz, CAr), 125.1 (q, 3JC–F = 4 Hz, 2xCAr), 124.4 (q, 1JC–F = 270 Hz, CF3), 124.2 (q, 1JC–F = 284 Hz, CF3), 122.3 (CAr), 120.0 (q, 3JC–F = 4 Hz, CAr), 118.7 (CAr), 117.3 (q, 3JC–F = 4 Hz, CAr), 114.6 (CAr), 56.9 (PhCH2N), 50.0 ([C2]CH2N), 48.4 ([C3]CH2N), 46.5 ([C3]CH2N), 45.7 ([C2]CH2N), 23.6 (CH2CH2CH2). 19F NMR (D2O/(CD3)2SO), 282.4 MHz, 296 K): δ (ppm) −60.9 (CF3), −61.4 (CF3). Anal. calcd. for C54H54F12N6O4·H2O: C, 59.12; H, 5.15; N, 7.66. Found: C, 59.75; H, 4.40; N, 6.93.
O), 161.5 (CAr), 135.4 (CAr), 135.2 (CAr), 131.9 (2xCAr), 130.4 (CAr), 129.8 (CAr), 120.6 (CAr), 119.4 (CAr), s117.7 (CAr), 56.6 (PhCH2N), 53.2 ([C3]CH2N), 51.8 ([C2]CH2N), 49.9 ([C3]CH2N), 46.3 ([C2]CH2N), 22.9 (CH2CH2CH2). Anal. calcd for C76H96N8O12·H2O: C, 68.55; H, 7.42; N, 8.41. Found: C, 68.44; H, 7.36; N, 8.39.
O), 161.5 (CAr), 140.3 (CAr), 135.2 (CAr), 132.2 (CAr), 131.7 (CAr), 126.9 (CAr), 126.8 (q, 3JC–F = 3 Hz, CAr), 120.2 (CAr), 118.8 (CAr), 117.6 (CAr), 55.9 (PhCH2N), 52.0 ([C3]CH2N), 50.6 ([C2]CH2N), 48.0 ([C2]CH2N or [C3]CH2N), 45.3 ([C2]CH2N or [C3]CH2N), 23.2 (CH2CH2CH2). CF3 could not be identified due to the low intensity of the signal. 19F NMR (D2O/(CD3)2SO), 376.5 MHz, 296 K): δ (ppm) −61.3 (s, CF3). Anal. calcd for C40H46F6N4O6·H2O: C, 59.25; H, 5.97; N, 6.91. Found: C, 59.50; H, 5.65; N, 7.01.
O), 144.5 (CAr), 143.3 (CAr), 136.6 (CAr), 132.2 (CAr), 131.7 (CAr), 130.6 (CAr), 130.3 (q, 2JC–F = 32 Hz, CAr) 129.9 (CAr), 128.3 (CAr), 127.4 (CAr), 124.3 (q, 1JC–F = 232 Hz, CF3), 122.1 (CAr), 120.8 (CAr), 118.7 (CAr), 117.1 (q, 3JC–F = 4 Hz, CAr), 114.6 (CAr), 114.4 (q, 3JC–F = 4 Hz, CAr), 57.2 (PhCH2N), 49.8 ([C2]CH2N), 48.6 ([C3]CH2N), 46.5 ([C3]CH2N), 45.6 ([C2]CH2N), 23.5 (CH2CH2CH2). 19F NMR (D2O/(CD3)2SO, 282.4 MHz, 296 K): δ (ppm) −61.3 (CF3). Anal. calcd for C52H56F6N6O4: C, 66.23; H, 5.99; N, 8.91. Found: C, 65.91; H, 5.92; N, 8.87.
O), 144.6 (CAr), 143.1 (CAr), 141.9 (CAr), 132.1 (2xCAr), 130.6 (CAr), 130.4 (CAr), 130.3 (q, 2JC–F = 33 Hz, CAr) 127.9 (q, 2JC–F = 32 Hz, CAr), 125.1 (q, 3JC–F = 4 Hz, 2xCAr), 124.4 (q, 1JC–F = 270 Hz, CF3), 124.2 (q, 1JC–F = 284 Hz, CF3), 122.3 (CAr), 120.0 (q, 3JC–F = 4 Hz, CAr), 118.7 (CAr), 117.3 (q, 3JC–F = 4 Hz, CAr), 114.6 (CAr), 56.9 (PhCH2N), 50.0 ([C2]CH2N), 48.4 ([C3]CH2N), 46.5 ([C3]CH2N), 45.7 ([C2]CH2N), 23.6 (CH2CH2CH2). 19F NMR (D2O/(CD3)2SO), 282.4 MHz, 296 K): δ (ppm) −60.9 (CF3), −61.4 (CF3). Anal. calcd. for C54H54F12N6O4·H2O: C, 59.12; H, 5.15; N, 7.66. Found: C, 59.75; H, 4.40; N, 6.93.
O), 161.5 (CAr), 135.4 (CAr), 135.2 (CAr), 131.9 (2xCAr), 130.4 (CAr), 129.8 (CAr), 120.6 (CAr), 119.4 (CAr), s117.7 (CAr), 56.6 (PhCH2N), 53.2 ([C3]CH2N), 51.8 ([C2]CH2N), 49.9 ([C3]CH2N), 46.3 ([C2]CH2N), 22.9 (CH2CH2CH2). Anal. calcd for C76H96N8O12·H2O: C, 68.55; H, 7.42; N, 8.41. Found: C, 68.44; H, 7.36; N, 8.39.
O), 161.5 (CAr), 140.3 (CAr), 135.2 (CAr), 132.2 (CAr), 131.7 (CAr), 126.9 (CAr), 126.8 (q, 3JC–F = 3 Hz, CAr), 120.2 (CAr), 118.8 (CAr), 117.6 (CAr), 55.9 (PhCH2N), 52.0 ([C3]CH2N), 50.6 ([C2]CH2N), 48.0 ([C2]CH2N or [C3]CH2N), 45.3 ([C2]CH2N or [C3]CH2N), 23.2 (CH2CH2CH2). CF3 could not be identified due to the low intensity of the signal. 19F NMR (D2O/(CD3)2SO), 376.5 MHz, 296 K): δ (ppm) −61.3 (s, CF3). Anal. calcd for C40H46F6N4O6·H2O: C, 59.25; H, 5.97; N, 6.91. Found: C, 59.50; H, 5.65; N, 7.01.
Single-crystal X-ray diffraction (SCXRD) studies
Crystals of compounds 3–6 suitable for single-crystal X-ray diffraction studies were mounted on a loop with Fomblin© protective oil. Data were collected on a Bruker AXS-KAPPA D8 QUEST with graphite-monochromated radiation (Mo Kα, λ = 0.71073 Å) at 293 K. An X-ray generator was operated at 50 kV and 30 mA and the APEX3 program monitored the data collection. Data were corrected for Lorentzian polarization and absorption effects using SAINT27 and SADABS28 programs. SHELXT 2014/429 was used for structure solution and SHELXL 2014/730 was used for full matrix least-squares refinement on F2. These two programs are included in the WINGX-Version 2014.1 program package.31 A full-matrix least-squares refinement was used for the non-hydrogen atoms with anisotropic thermal parameters. The hydrogens of carbons were inserted in idealized positions and allowed to refine in the parent carbon atom. The hydrogen atoms of OH, NH, NH2+ and COOH moieties were located in the electron density map and the distances were restrained. Disorder in CF3 moieties in 3, 4 and 6 has been modelled. Data quality of 5 precluded better refinement. MERCURY 2022.3.032 was used for packing diagrams. PLATON33 was used for determination of hydrogen bond interactions. Table 1 summarizes data collection and refinement details. Crystallographic data of compounds 3–6 were deposited at the Cambridge Crystallographic Data Centre (CCDC 2243135–2243138).| 3 | 4 | 5 | 6 | |
|---|---|---|---|---|
| a R 1 = Σ‖Fo| − |Fc‖/Σ|Fo|. b wR2 = [Σ[w(Fo2 − Fc2)2]/Σ[w(Fo2)2]]1/2. | ||||
| Empirical formula | C24H38N4·2(C14H9F3NO2) | C26H36F6N4·2(C14H9F3NO2) | C24H36N4·C24H38N4·2(C7H6O3)·2(C7H5O3) | C26H36F6N4·2(C7H5O3) |
| Formula weight | 943.03 | 1079.03 | 1313.61 | 792.81 |
| Crystal form, colour | Block, colourless | Block, colourless | Block, colourless | Block, colourless |
| Crystal size (mm) | 0.20 × 0.20 × 0.20 | 0.20 × 0.20 × 0.10 | 0.20 × 0.20 × 0.20 | 0.40 × 0.40 × 0.16 |
| Crystal system | Monoclinic | Monoclinic | Triclinic | Monoclinic |
| Space group | P21/n | C2/c |
P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) |
P21/n |
| a, Å | 15.790(1) | 19.785(4) | 10.881(2) | 10.352(2) |
| b, Å | 16.494(1) | 18.079(4) | 11.187(2) | 18.345(4) |
| c, Å | 19.737(2) | 16.343(3) | 16.516(3) | 20.704(5) |
| α, ° | 90 | 90 | 70.972(6) | 90 |
| β, ° | 110.881(3) | 115.629(7) | 71.325(6) | 93.095(9) |
| γ, ° | 90 | 90 | 85.357(7) | 90 |
| Z | 4 | 4 | 1 | 4 |
| V, Å3 | 4802.7(7) | 5270.9(19) | 1799.9(5) | 3926.4(15) |
| T, K | 293 | 293 | 293 | 293 |
| D c, g cm−3 | 1.304 | 1.360 | 1.212 | 1.341 |
| μ (Mo Kα), mm−1 | 0.099 | 0.115 | 0.082 | 0.109 |
| θ range (°) | 2.052–24.999 | 2.253–24.994 | 1.978–24.999 | 2.929–25.000 |
| Refl. collected | 137596 | 49510 | 50632 | 114517 |
| Independent refl. | 8460 | 4621 | 6323 | 6805 |
| R int | 0.1213 | 0.1440 | 0.1052 | 0.1180 |
| R 1,awR2b [I ≥ 2σ(I)] | 0.0609, 0.1453 | 0.0887, 0.2464 | 0.0857, 0.2495 | 0.0605, 0.1438 |
| GOF on F2 | 1.055 | 1.064 | 1.088 | 1.055 |
General characterization
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2) were performed and 10 μL of microbial inoculum was added to each well. The microtiter plates were incubated at 37 °C for 24 h and 48 h for bacteria and yeasts, respectively, and their growth was assessed through analysis of the optical density of cultures at 620 nm using a Thermo Scientific Multiskan FC (Loughborough, UK) microplate reader.
:2) were performed and 10 μL of microbial inoculum was added to each well. The microtiter plates were incubated at 37 °C for 24 h and 48 h for bacteria and yeasts, respectively, and their growth was assessed through analysis of the optical density of cultures at 620 nm using a Thermo Scientific Multiskan FC (Loughborough, UK) microplate reader.
Results and discussion
Cyclam derivatives/flufenamic acid
Cyclam derivatives of the general formula H2(RPhCH2)2cyclam, where R = H (1) and R = 4-CF3 (2), reacted with two equivalents of flufenamic acid in DMF to yield salts of the formula [H2{H2(RPhCH2)2cyclam}](3-CF3PhNHC6H4COO)2 (R = H, 3, and R = 4-CF3, 4), as shown in Scheme 1. | ||
| Scheme 1 Synthetic route for the preparation of 3 and 4. | ||
Compounds 3 and 4 are dicationic salts that resulted from the protonation of the nitrogen atoms of the cyclam ring by the carboxylic acid moieties of flufenamic acid. The 1H NMR spectra of both 3 and 4 reveal the presence of five signals integrating to four protons each that correspond to the CH2 groups of the cyclam ring. The methylene protons of the pendant arms of the macrocycle show up as singlets at 3.69 ppm in 3 and at 3.75 ppm in 4. In addition, the resonances due to the benzylic groups of cyclam and flufenamate anions appear in the aromatic region of the spectra. The NH2+ protons are not observed due to proton exchange in D2O. The 13C{1H} NMR spectra display five different resonances for the macrocycle backbone and one set of resonances that correspond to the benzylic moieties. The proton and carbon NMR spectra of 3 and 4 are in agreement with a C2 symmetry in solution similarly to those obtained for other trans-disubstituted cyclam salts.36 The 1H and 13C{1H} NMR spectra of compounds 3 and 4 are presented in Fig. S1 and S2,† respectively.
The IR spectra of 3 and 4 (Fig. S3 and S4,† respectively) reveal stretching vibrational bands assigned to the C–F bond stretching of –CF3 groups between 1162 and 1065 cm−1. The bands at 1326 cm−1 in 3 and at 1327 cm−1 in 4 assigned to the combination of νC–C, νC–N and νN–H modes are within the characteristic stretching vibrational bands of C–C and C–N modes (1400–1000 cm−1).37 The νN–H vibrational modes are also observed at higher frequencies but could be overlapped with the νC–H vibrational modes (1450–1300 cm−1).37 Hence, the bands ranging from 1453 to 1367 cm−1 are assigned to the combination of νN–H and νC–H modes. Nevertheless, these bands are overlapped by the strong absorption stretching vibration bands at 1581 to 1367 cm−1 assigned to νCO and νC–O, respectively, of the flufenamate anions. The νC–C stretching (in-ring) bands observed at 1630 cm−1 in 3 and at 1653 cm−1 in 4 are assigned to the aromatic rings.
The structural elucidation of compounds 3 and 4 was confirmed from single-crystal X-ray diffraction data that allowed the structure determination of both compounds. The purity of both bulk samples was checked by powder X-ray diffraction data (Fig. S5 and S6,† respectively).
The asymmetric unit of 3 consists of two deprotonated flufenamic acid moieties and two halves of crystallographically independent protonated cyclam moieties residing on inversion centres (see Fig. 1). The determination of the salt nature of 3 arose from the confirmation of the deprotonation of the carboxylic acid group of flufenamic acid by the C–O distances (1.250(3), 1.258(4), 1.263(3) and 1.249(3) Å) and the location of hydrogen atoms from the electron density maps revealing the protonation of the cyclam secondary amines. Thus, compound 3 is a dicationic salt with the presence of two NH2+ groups in the cyclam ring.
 | ||
| Fig. 1 Asymmetric unit of 3, representing two crystallographically independent flufenamate moieties and two independent protonated cyclam moieties residing on inversion centres (represented by the yellow dots). The full protonated cyclams are represented for better understanding and hydrogen atoms have been omitted for clarity. | ||
Flufenamate moieties assume antiparallel orientations in the overall packing, interacting almost perpendicularly with cyclam. Charge-assisted hydrogen bonds are established between the cyclam ring and flufenamate anions via several N+–H⋯OCOO– interactions (2.853(3), 3.151(3), 2.766(3), 2.752(3), and 2.759(3) Å), with each cyclam interacting only with a single type of crystallographically independent flufenamate, as shown in Fig. 2a. Apart from these intermolecular hydrogen bonds, there are also intramolecular bonds found in flufenamate between amines and carboxylate groups (2.652(3) and 2.617(4) Å) (see Fig. 2a). The list of the main hydrogen bonds found in compound 3 is presented in Table S1.† The combination of these interactions results in a supramolecular arrangement where it is possible to see that in a view along the b axis, the flufenamate–cyclam–flufenamate groups align along the ac diagonal (Fig. 2b) with alternate lines being formed by crystallographically independent entities.
 | ||
| Fig. 2 (a) Detailed view of the hydrogen bonds established in 3 between each crystallographically independent cyclam and two crystallographically dependent flufenamic acid molecules, with intramolecular interactions represented in dark blue and non-contact hydrogen atoms omitted for clarity. (b) Supramolecular arrangement of 3 in a view along the b axis, with hydrogen atoms omitted for clarity. | ||
The asymmetric unit of compound 4 is formed by half of a protonated cyclam molecule residing on an inversion center and one deprotonated flufenamic acid moiety (see Fig. 3). The C–O bond distances in the flufenamate (1.264(4) and 1.251(6) Å) indicate that these molecules have transferred the carboxylic protons to the secondary amines of cyclam, giving rise to flufenamate anions, as observed in the formation of 3. Thus, compound 4 is also a dicationic salt with the presence of two NH2+ groups in the cyclam ring, confirmed by the electron density maps.
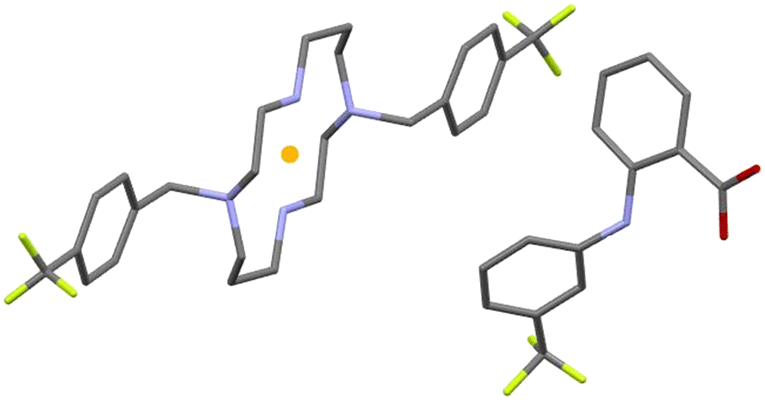 | ||
| Fig. 3 Asymmetric unit of 4, with representation of the full protonated cyclam residing on an inversion center (represented by the yellow dot) for better understanding; hydrogen atoms have been omitted for clarity. | ||
The interactions between the cyclam ring and flufenamate are established by N+–H⋯OCOO– charge-assisted hydrogen bonds (2.766(5), 2.754(5) and 3.223(5) Å), as shown in Fig. 4a. Apart from these intermolecular hydrogen bonds, there are also intramolecular bonds found in flufenamate between amines and carboxylate groups (2.653(5) Å) as well as within cyclam via a N+–H⋯N (3.013(4) Å) hydrogen bond. A list of the main hydrogen bonds found in compound 4 is presented in Table S2.†
 | ||
| Fig. 4 (a) Detailed view of the hydrogen bonds established in 4, with intramolecular interactions represented in dark blue and non-contact hydrogen atoms omitted for clarity. (b) Supramolecular arrangement of 4 in a view along the a axis, with hydrogen atoms omitted for clarity. | ||
Regarding the supramolecular arrangement, it is possible to see that in a view along the a axis, each of the flufenamate–cyclam–flufenamate groups shown in Fig. 4b aligns along the ac diagonal. Alternate lines are formed by crystallographically independent groups.
Cyclam derivatives/salicylic acid
The cyclam derivative H2(PhCH2)2cyclam, 1, reacted with two equivalents of salicylic acid in DMF to yield an ionic cocrystal of the formula [H2(PhCH2)2cyclam](HOC6H4COOH)2·[H2{H2(PhCH2)2cyclam}](HOC6H4COO)2, 5. On the other hand, the reaction of H2(4-CF3PhCH2)2cyclam, 2, with two equivalents of salicylic acid in DMF afforded the salt [H2{H2(3-CF3PhCH2)2cyclam}](HOC6H4COO)2, 6. The synthetic route to obtain compounds 5 and 6 is shown in Scheme 2. | ||
| Scheme 2 Synthetic route for the preparation of 5 and 6. | ||
Compounds 5 and 6 can also be obtained using acetylsalicylic acid instead of salicylic acid as starting material. This result is attributed to the hydrolysis of acetylsalicylic acid into salicylic acid in solution, as a similar behaviour has already been reported for similar systems.38 The proton and carbon NMR spectra of 5 and 6 are similar to those obtained for compounds 3 and 4. The similarity of the NMR spectra of the ionic cocrystal 5 with those obtained for the salts 3, 4 and 6 might be due to the fact that in a D2O/(CD3)2SO solution, hydrogen bonds between cations and anions are broken. Furthermore, the COOH, NH and NH2+ protons cannot be distinguished due to proton/deuterium exchange with D2O. The 1H and 13C{1H} NMR spectra of compounds 5 and 6 are shown in Fig. S7 and S8,† respectively.
The IR spectra of 5 and 6 (Fig. S9 and S10,† respectively) show bands at 1326 and 1324 cm−1, respectively, which are assigned to the combination of νC–C, νC–N and νN–H modes. The νN–H vibrational modes are also observed at higher frequencies but could be overlapped with the νC–H vibrational modes (1450–1300 cm−1).37 Hence, the bands ranging from 1457 to 1381 cm−1 are assigned to the combination of νN–H and νC–H modes. Nevertheless, these bands are overlapped by the strong absorption stretching vibration bands at 1595 to 1381 cm−1 assigned to νCO and νC–O, respectively, of the salicylate anions. The νC–C stretching (in-ring) bands observed at 1643 cm−1 in both compounds are assigned to the aromatic rings. In the IR spectrum of 6, stretching vibrational bands assigned to the C–F bond stretching of –CF3 groups are observed between 1162 and 1066 cm−1.
Crystals of 5 and 6 suitable for single-crystal X-ray diffraction were obtained from slow evaporation of a DMF solution after one week. Crystallographic and experimental details of data collection and crystal structure determination are presented in Table 1. The asymmetric unit of compound 5 is formed by one half of neutral cyclam with one salicylic acid moiety, and one half of protonated cyclam with a salicylate moiety. Both cyclams are placed on inversion centres as shown in Fig. 5. The C–O bond distances in salicylic acid are 1.185(9) and 1.282(8) Å and the C–O bond distances in the salicylate moiety are 1.264(5) and 1.246(7) Å, revealing the presence of both neutral and protonated parts, and thus the formation of an ionic cocrystal.
 | ||
| Fig. 5 Asymmetric unit of compound 5, representing the neutral [H2(PhCH2)2cyclam](HOC6H4COOH)2 and the [H2{H2(PhCH2)2cyclam}]2+(HOC6H4COO)−2 pairs in the asymmetric unit, depicting the relevant hydrogen atoms to depict the ICC character of form 5, with both cyclams residing on inversion centers (represented by the yellow dots) being fully represented for better understanding. Hydrogen atoms have been omitted for clarity, except the ones determining the formation of an ICC. | ||
Interactions within the neutral salicylic acid and cyclam are established via N–H⋯OCOOH and O–HCOOH⋯N hydrogen bonds (2.741(5) and 2.799(6) Å). These are replaced by two N+–H⋯OCOO– charge-assisted hydrogen bonds (2.807(6) and 2.746(6) Å) in the ionic part.
Apart from these intermolecular interactions, there are also intramolecular bonds found in the salicylic acid moieties between the hydroxyl and the carboxylic acid/carboxylate groups (2.531(7) and 2.497(5) Å) (see Fig. 6a). The list of the main hydrogen bonds formed in compound 5 is presented in Table S3.†
 | ||
| Fig. 6 (a) Detailed view of the hydrogen bonds established in 5, with intramolecular interactions represented in dark blue and non-contact hydrogen atoms omitted for clarity. (b) Supramolecular arrangement of 5 in a view along the b axis, with hydrogen atoms omitted for clarity. | ||
Regarding the supramolecular arrangement, it is possible to see that in a view along the b axis, cyclam aligns in zigzag chains of alternate protonated and neutral cyclam 1, leaving cages occupied by two salicylate anions and two neutral salicylic acid molecules (Fig. 6b).
The asymmetric unit of 6 consists of one protonated cyclam and two salicylate moieties, as shown in Fig. 7. The C–O bond distances (1.278(4) and 1.244(4); 1.271(4) and 1.256(4) Å) indicate that the carboxylic acid groups exchanged the proton with cyclam, giving rise to salicylate anions. The salt character of 6 is confirmed by the protonation of both of cyclam's NH groups.
 | ||
| Fig. 7 Asymmetric unit of 6. Hydrogen atoms have been omitted for clarity. | ||
The interactions between cyclam and salicylate moieties are established by N+–H⋯OCOO– charge-assisted hydrogen bonds (2.876(4), 3.179(3), 2.824(4), 2.845(4) and 2.792(4) Å). Apart from these, there are intramolecular bonds in both salicylates established between the hydroxyl and carboxylic groups (2.557(3) and 2.558(3) Å) and in cyclams between one of the protonated NH groups and one of the N-substituted (3.051(3) and 3.071(4) Å) (see Fig. 8a). A list of the main hydrogen bonds found in compound 6 is presented in Table S4.† The overall supramolecular arrangement of 6 is very similar to the packing of 5 with cyclam aligning in zigzag chains, forming cages that are occupied by both crystallographically independent salicylates, in a view along the a axis (Fig. 8b).
 | ||
| Fig. 8 (a) Detailed view of the hydrogen bonds established in 6, with intramolecular interactions represented in dark blue and non-contact hydrogen atoms omitted for clarity. (b) Supramolecular arrangement of 6 in a view along the a axis, with hydrogen atoms omitted for clarity. | ||
Biological assays
The minimum inhibitory concentration (MIC) values of compounds 1–6 were determined against a set of yeasts and Gram-positive and Gram-negative bacteria, revealing that the new multicomponent forms 3–6 display relevant antimicrobial activity when compared to their parent compounds 1 and 2 (Table S5† and Fig. 9). | ||
| Fig. 9 Minimum inhibitory concentration values (MIC, μg mL−1) of compounds 3–6 and the corresponding starting materials for Candida albicans and Saccharomyces cerevisiae (yeasts), Escherichia coli and Pseudomonas aeruginosa (Gram-negative bacteria) and Staphylococcus aureus, Enterococcus faecalis and Mycobacterium smegmatis (Gram-positive bacteria) after 24 h for bacteria and 48 h for yeasts. DMSO was used as negative control, and nystatin, norfloxacin and vancomycin were used as positive controls for yeasts, Gram-negative bacteria and Gram-positive bacteria, respectively. | ||
Compound 3 presents lower MIC values for E. faecalis, P. aeruginosa and S. cerevisiae than compound 1, but both compounds show similar antibacterial activity for S. aureus, E. coli and C. albicans. The highest MIC value detected for 3 is 40.63 μg mL−1 for both M. smegmatis and methicillin-resistant S. aureus (MRSA), which represents a decreased activity compared to 1, but when compared to flufenamic acid, 3 presents augmented antimycobacterial activity to M. smegmatis. Compound 4 displays lower or similar antimicrobial activity when compared to 2. Moreover, 4 seems to be not selective, as it presents the same MIC value (7.81 μg mL−1) for the set of tested bacteria. Comparing 3 and 4, it can be noted that 4 is more active against M. smegmatis and MRSA and that both compounds show a similar impact on cultures of E. faecalis, P. aeruginosa, both S. aureus strains and E. coli. The antimicrobial activity of 5 to P. aeruginosa is higher than that of compound 1 but very similar for M. smegmatis, E. coli and MRSA. Compounds 6 and 2 present similar effects to S. aureus and E. coli. When compared to 5, compound 6 is more active against all tested bacteria except P. aeruginosa, with 5 being more active against both yeasts. In addition, compounds 4 and 6 display MIC values against M. smegmatis very close to the control vancomycin. On the other hand, 3 and 5 are more active to P. aeruginosa and both yeasts. Compound 1 revealed to be a better antimycotic agent, showing lower MIC values for C. albicans and S. cerevisiae, while compound 2 displays higher antimicrobial activity against M. smegmatis, P. aeruginosa, E. faecalis and MRSA. Considering the microorganisms selected for this study, it is clear that the combined effect of the cyclams 1 and 2 with flufenamic acid, increases the antimicrobial activity, except against MRSA.
Conclusions
Novel flufenamic and salicylic acid–cyclam multicomponent crystal forms were synthesized and characterized. Structural characterization allowed concluding the formation of three salts (3, 4 and 6) and an ionic cocrystal (5).The antimicrobial activity of the novel forms 3–6 was compared with the activity of the starting compounds, revealing promising results. In an attempt to establish a trend in the efficiency of the tested compounds, it can be said that flufenamic acid is the most active compound against methicillin-resistant S. aureus (MRSA). Compound 1 is a better antimycotic agent, displaying lower MIC values for C. albicans, while compounds 2, 4 and 6 show higher activity against M. smegmatis. Compound 5 can strongly inhibit the growth of the tested Gram-negative bacteria, with compound 3 a good choice to battle P. aeruginosa.
Overall, these results highlight the benefits of the synergistic effects that can be established between active principal ingredients to produce new and efficient antimicrobial agents able to fight drug resistant bacteria like S. aureus, which are responsible for several nosocomial infections.
Author contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge funding from Fundação para a Ciência e a Tecnologia (projects UIDB/00100/2020, UIDP/00100/2020, LA/P/0056/2020, UIDB/04567/2020 and UIDP/04567/2020 and PTDC/QUI-OUT/30988/2017), contracts under DL No. 57/2016 regulation, and CEECIND/00283/2018; FEDER, Portugal2020 and Lisboa2020 (project LISBOA-01-0145-FEDER-030988).References
- C. J. L. Murray, K. S. Ikuta, F. Sharara, L. Swetschinski, G. R. Aguilar, A. Gray, C. Han, C. Bisignano, P. Rao, E. Wool, S. C. Johnson, A. J. Browne, M. G. Chipeta, F. Fell, S. Hackett, G. Haines-Woodhouse, B. H. K. Hamadani, E. A. P. Kumaran, B. McManigal, R. Agarwal, S. Akech, S. Albertson, J. Amuasi, J. Andrews, A. Araykin, E. Ashley, F. Bailey, S. Baker, B. Basnyat, A. Bekker, R. Bender, A. Bethou, J. Bielicki, S. Boonkasidecha, J. Bukosia, C. Carvalheiro, C. Castañeda-Orjuela, V. Chansamouth, S. Chaurasia, S. Chiurchiù, F. Chowdhury, A. J. Cook, B. Cooper, T. R. Cressey, E. Criollo-Mora, M. Cunningham, S. Barboe, N. P. J. Day, M. D. Luca, K. Dokova, A. Dramowski, S. J. Dunachie, T. Eckmanns, D. Eibach, A. Emami, N. Feasy, N. Fisher-Pearson, K. Forrest, D. Garrett, P. Gastmeier, A. Z. Giref, R. C. Greer, V. Gupta, S. Haller, A. Haselbeck, S. I. Hay, M. Holm, S. Hopkins, K. C. Iregbu, J. Jacobs, D. Jarovsky, F. Javanmardi, M. Khorana, N. Kissoon, E. Kobeissi, T. Kostyanev, F. Krapp, R. Krumkamp, A. Kumar, H. H. Kyu, C. Lim, D. Limmathurotsakul, M. J. Loftus, M. Lunn, J. Ma, T. Munera-Huertas, P. Musicha, M. M. Mussi-Pinhata, T. Nakamura, R. Nanayati, S. Nangia, P. Newton, C. Ngoun, A. Novotney, D. Nwakanma, C. W. Obiero, A. Olivas-Martinez, P. Olliaro, E. Ooko, E. Ortiz-Brizuela, A. Y. Peleg, C. Perrone, N. Plakkal, A. Ponce-de-Leon, M. Raad, T. Ramdin, T. Roberts, J. V. Robotham, A. Roca, K. E. Rudd, N. Russell, J. Schnall, J. A. G. Scott, M. Shivamallappa, J. Sifuentes-Osornio, N. Steenkeste, A. J. Stewardson, T. Stoeva, N. Tasak, A. Thaiprakong, G. Thwaites, C. Turner, P. Turner, H. R. van Doorn, S. Velaphi, A. Vongpradith, H. Vu, T. Walsh, S. Waner, T. Wangrangsimakul, T. Wozniak, P. Zheng, B. Sartorius, A. D. Lopez, A. Stergachis, C. Moore, C. Dolecek and M. Naghavi, Lancet, 2022, 399, 629 CrossRef CAS PubMed.
- B. Plackett, Nature, 2020, 586, S50 CrossRef CAS.
- C. Morel, O. Lindahl, S. Harbarth, M. de Kraker, S. Edwards and A. Hollis, J. Antibiot., 2020, 73, 421 CrossRef CAS PubMed.
- S. A. Hillier and I. A. Dutescu, Infect. Drug Resist., 2021, 14, 415 CrossRef PubMed.
- S. A. Sousa, J. R. Feliciano, T. Pita, C. F. Soeiro, B. L. Mendes, L. G. Alves and J. H. Leitão, Antibiotics, 2021, 10, 942 CrossRef CAS PubMed.
- T. T. Ashburn and K. B. Thor, Nat. Rev. Drug Discovery, 2004, 3, 673 CrossRef CAS PubMed.
- X. Liang and P. J. Sadler, Chem. Soc. Rev., 2004, 22, 246 RSC.
- G. J. Bridger, R. T. Skerlj, D. Thornton, S. Padmanabhan, S. A. Martellucci, G. W. Henson, M. J. Abrams, N. Yamamoto, K. De Vreese, R. Pauwels and E. J. De Clercq, Med. Chem., 1995, 38, 366 CrossRef CAS PubMed.
- M. Spain, J. K.-H. Wong, G. Nagalingam, J. M. Batten, E. Hortle, S. H. Oehlers, X. F. Jiang, H. E. Murage, J. T. Orford, P. Crisologo, J. A. Triccas, P. J. Rutledge and M. H. J. Todd, Med. Chem., 2018, 61, 3595 CrossRef CAS PubMed.
- T. J. Hubin, P. N.-A. Amoyaw, K. D. Roewe, N. C. Simpson, R. D. Maples, T. N. C. Freeman, A. N. Cain, J. G. Le, S. J. Archibald, S. I. Khan, B. L. Tekwani and M. O. F. Khan, Bioorg. Med. Chem., 2014, 22, 3239 CrossRef CAS PubMed.
- M. O. F. Khan, J. Keiser, P. N. A. Amoyaw, M. F. Hossain, M. Vargas, J. G. Le, N. C. Simpson, K. D. Roewe, T. N. C. Freeman, T. R. Hasley, R. D. Maples, S. J. Archibald and T. J. Hubin, Antimicrob. Agents Chemother., 2016, 60, 5331 CrossRef PubMed.
- I. Grabchev, S. Yordanova, E. Vasileva-Tonkova, M. Cangiotti, A. Fattori, R. Alexandrova, S. Stoyanov and M. F. Ottaviani, Dyes Pigm., 2016, 129, 71 CrossRef CAS.
- L. G. Alves, P. F. Pinheiro, J. R. Feliciano, D. P. Dâmaso, J. H. Leitão and A. M. Martins, Int. J. Antimicrob. Agents, 2017, 49, 646 CrossRef CAS PubMed.
- L. G. Alves, J. F. Portel, S. A. Sousa, O. Ferreira, S. Almada, E. R. Silva, A. M. Martins and J. H. Leitão, Antibiotics, 2019, 8, 224 CrossRef CAS PubMed.
- S. Almada, L. B. Maia, J. C. Waerenborgh, B. J. C. Vieira, N. P. Mira, E. R. Silva, F. Cerqueira, E. Pinto and L. G. Alves, New J. Chem., 2022, 46, 16764 RSC.
- A. V. Yadav, A. S. Shete, A. P. Dabke, P. V. Kulkarni and S. S. Sakhare, Indian J. Pharm. Sci., 2009, 71, 359 CrossRef CAS PubMed.
- J. B. Ngilirabanga and H. Samsodien, Wiley Online Library, 2021, vol. 2, p. 512 Search PubMed.
- M. B. Hickey, M. L. Peterson, L. A. Scoppettuolo, S. L. Morrisette, A. Vetter, H. Guzmán, J. F. Remenar, Z. Zhang, M. D. Tawa, S. Haley, M. J. Zaworotko and O. Almarsson, Eur. J. Pharm. Biopharm., 2007, 67, 112 CrossRef CAS PubMed.
- M. Djaló, A. E. S. Cunha, J. P. Luís, S. Quaresma, A. Fernandes, V. André and M. T. Duarte, Cryst. Growth Des., 2021, 21, 995 CrossRef.
- O. Shemchuk, V. André, M. T. Duarte, D. Braga and F. Grepioni, CrystEngComm, 2019, 21, 2949 RSC.
- V. André, M. F. M. Piedade and M. T. Duarte, CrystEngComm, 2012, 14, 5005 RSC.
- E. Grothe, H. Meekes, E. Vlieg, J. H. ter Horst and R. de Gelder, Cryst. Growth Des., 2016, 16, 3237 CrossRef CAS.
- N. K. Evanson, xPharm: The Compreensive Pharmacology Reference, Elsevier, 2007 Search PubMed.
- B. L. Furman, Reference Module in Biomedical Sciences, Elsevier, 2018 Search PubMed.
- G. Royal, V. Dahaoui-Gindrey, S. Dahaoui, A. Tabard, R. Guilard and C. L. Pullumbi, Eur. J. Org. Chem., 1998, 9, 1971 CrossRef.
- L. G. Alves, M. A. Antunes, I. Matos, R. F. Munhá, M. T. Duarte, A. C. Fernandes, M. M. Marques and A. M. Martins, Inorg. Chim. Acta, 2010, 363, 1823 CrossRef CAS.
- Bruker AXS, SAINT+, release 6.22, Bruker Analytical Systems, Madison, WI, USA, 2005 Search PubMed.
- Bruker AXS, SADABS, Bruker Analytical Systems, Madison, WI, USA, 2005 Search PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Adv., 2015, 71, 3 CrossRef PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3 Search PubMed.
- L. J. Farrugia, J. Appl. Crystallogr., 1999, 32, 837 CrossRef CAS.
- C. F. Macrae, I. Sovago, S. J. Cottrell, P. T. A. Galek, P. McCabe, E. Pidcock, M. Platings, G. P. Shields, J. S. Stevens, M. Towler and P. A. Wood, J. Appl. Crystallogr., 2020, 53, 226 CrossRef CAS PubMed.
- A. L. Spek, J. Appl. Crystallogr., 2003, 36, 7 CrossRef CAS.
- F. Siopa, T. Figueiredo, R. F. M. Frade, I. Neto, A. Meirinhos, C. P. Reis, R. G. Sobral, C. A. M. Afonso and P. Rijo, ChemistrySelect, 2016, 1, 5909 CrossRef CAS.
- M. P. Weinstein, J. B. Patel, C.-A. Burnham, S. Campeau, P. S. Conville, C. Doern, G. M. Eliopoulos, M. F. Galas, R. M. Humphries, S. G. Jenkins, S. M. Kircher, J. S. Lewis, B. Limbago, A. J. Mathers, T. Mazzulli, S. D. Munro, M. O. S. de Danies, R. Patel, S. S. Satlin, J. M. Swenson, A. Wong, W. F. Wang and B. L. Zimmer, Methods for Dilution Antimicrobial Susceptibility Tests for Bacteria that Grow Aerobically, Clinical and Laboratory Standards Institute, Wayne, USA, 2018 Search PubMed.
- L. G. Alves, M. T. Duarte and A. M. Martins, J. Mol. Struct., 2015, 1098, 277 CrossRef CAS.
- D. Lin-Vien, N. B. Colthup, W. G. Fateley and J. G. Grasselli, The handbook of infrared and Raman characteristic frequencies of organic molecules, Academic Press, San Diego, USA, 1991 Search PubMed.
- M. S. Fonari, E. V. Ganin, S. S. Basok, K. A. Lyssenko, M. J. Zaworotko and V. C. Kravtsov, Cryst. Growth Des., 2010, 10, 5210 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: NMR, FT-IR, PXRD, hydrogen bonding details and antimicrobial activity data. CCDC 2243135–2243138. Crystallographic data of compounds 3–6 were deposited at the Cambridge Crystallographic Data Centre. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3ce00354j |
| This journal is © The Royal Society of Chemistry 2023 |