
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Hydrogen-bonded liquid crystals formed from 4-alkoxystilbazoles and chlorophenols†
Oliver D.
Johnson
,
Stephen G.
Wainwright
 ,
Adrian C.
Whitwood
and
Duncan W.
Bruce
*
,
Adrian C.
Whitwood
and
Duncan W.
Bruce
*
Department of Chemistry, University of York, Heslington, YORK YO10 5DD, UK. E-mail: duncan.bruce@york.ac.uk; Tel: +44 (0)1904 324085
First published on 14th April 2023
Abstract
Hydrogen-bonded complexes are formed between two chain lengths of 4-alkoxystilbazoles and six different chlorophenols. Single crystal structure determinations were possible for four of the complexes, all of which showed a similar dimeric motif in which two chlorophenols formed a loose back-to-back dimer with a stilbazole hydrogen-bonded at either side. Two of the complexes showed similarities in their three-dimensional packing. Liquid crystal properties were found for all complexes except those containing pentachlorophenol. The phase behaviour of the longer-chain dodecyloxystilbazole complexes was dominated by the observation of the smectic A phase, while both nematic and smectic A phases were found for the octyloxy homologues. The mesophase stability was appreciably lower in these new complexes when compared with fluorophenol analogues reported previously, attributed to a reduction in anisotropy on account of the effects of the greater size of chlorine compared with fluorine.
Introduction
Hydrogen bonding has been used extensively as a non-covalent linkage in the realisation of new liquid crystals.1,2 From early work on the dimeric 4-alkoxybenzoic acids by Jones3–5 and studies of carbohydrate mesogens,6–8 the subject developed to incorporate some of the self-assembly approaches of the 1980s through to more designed systems, arguably initiated by the seminal studies of Kato and Fréchet.9 Thus, examples exists where a hydrogen-bonded complex is formed from one or more fragment that is itself liquid crystalline as well as many where mesomorphism results only in the newly formed complex, with neither component having any liquid crystal phase behaviour. More recently, hydrogen-bonding in liquid crystal systems has begun to be exploited beyond simply establishing their mesomorphism.10–13 Analogous work has also been carried out in the field of halogen bonding.14,15In both hydrogen- and halogen-bonded liquid crystals, one area of discussion has been the extent to which it can be shown (or not) that the clearing point (the temperature at which the liquid crystallinity is lost and an isotropic liquid is formed) is driven either by the natural clearing of the new supramolecular complex or by the rupture of the supramolecular linkage (hydrogen or halogen bond) to form non-mesomorphic components. Aspects of this have recently been reviewed where it was concluded that at least for halogen-bonded complexes, both possibilities likely exist, whereas for hydrogen-bonded mesogens, there is good experimental evidence that the hydrogen bond is preserved through clearing.15
One of the factors that may be at play in thinking about whether a complex may rupture on heating is the strength of the halogen or hydrogen bond. In the former case, this will depend upon the degree of electrophilicity of the iodine16 (halogen-bonded liquid crystals are almost invariably formed using electron-poor iodines and the basicity of the (invariably) pyridine donor appears to have little effect17), while in the latter case it will relate to the acidity of the hydrogen donor – in this case a phenol.
In a previous systematic study of halogen-bonded systems,16 we prepared and characterised crystallographically a series of complexes formed between 4-(N,N-dimethylamino)pyridine (DMAP) and eight fluoro-substituted iodobenzenes. The relative electrophilicity of the iodine was assigned using the pKa of the related fluorophenol18 as a proxy and in this way a linear relationship was found between pKa and N⋯I distance in the halogen-bonded complex. However, attempts to extend this study to the investigation of what ought to have been a series of halogen-bonded liquid crystals formed between 4-alkoxystilbazoles and the same fluoroiodobenzenes could not proceed as only the pentafluoroiodobenzene complex was mesomorphic and replacement of even one of the ring fluorines by hydrogen suppressed liquid crystal properties entirely.19
However, a related study using hydrogen bonding was somewhat more successful, with alkoxystilbazoles being complexed by a series of fluorophenols.20 Complexes were therefore formed between butyloxy-, octyloxy- and dodecyloxy-stilbazole with sixteen of the nineteen possible isomers of phenols of the formula HOC6H6−xFx (1 ≤ x ≤ 5), with single crystal structures obtained for octyloxystilbazole complexes of ten of them. From this study it was possible to show conclusively that the clearing point was in no way related to the strength of the hydrogen bond so that the clearing behaviour was of the intact complex.
It is very well known that, particularly in calamitic liquid crystals, mesophase identity and stability can be affected strongly by the size, nature and steric influence of substituents.21 As such and following the knowledge gained in the study of the fluorophenols, an analogous and more focused study has been undertaken with a selection of chlorophenols, the results of which are now reported.
Results and discussion
Given the lessons learned from the study with fluorophenols, the two longer stilbazole chain lengths were chosen for study along with three dichlorophenols, two trichlorophenols and pentachlorophenol (Fig. 1), the choice driven to some degree by their availability. The stilbazoles were prepared as described elsewhere (see ESI†). The complexes were then prepared by dissolving the stilbazole and the phenol separately in the same solvent (pentane for octyloxystilbazole and hexane for dodecyloxystilbazole), mixing the two together and then allowing slow evaporation of the solvent to yield the crystalline complexes, which were recovered by filtration. The colour of the complexes broadly reflected the pKa of the phenol.22 Thus, pure alkoxystilbazoles are colourless (λmax ≈ 320 nm), whereas on full protonation the absorption is red-shifted to 390 nm and the cation is bright yellow.23 In these complexes, the colour varied from a pale yellow (complexes of 3,5-dichlorophenol) to a bright and vibrant yellow (complexes of 2,3,4,5,6-pentachlorophenol) as a function of the pKa of the phenol and hence, by implication, the degree of proton transfer (see also below).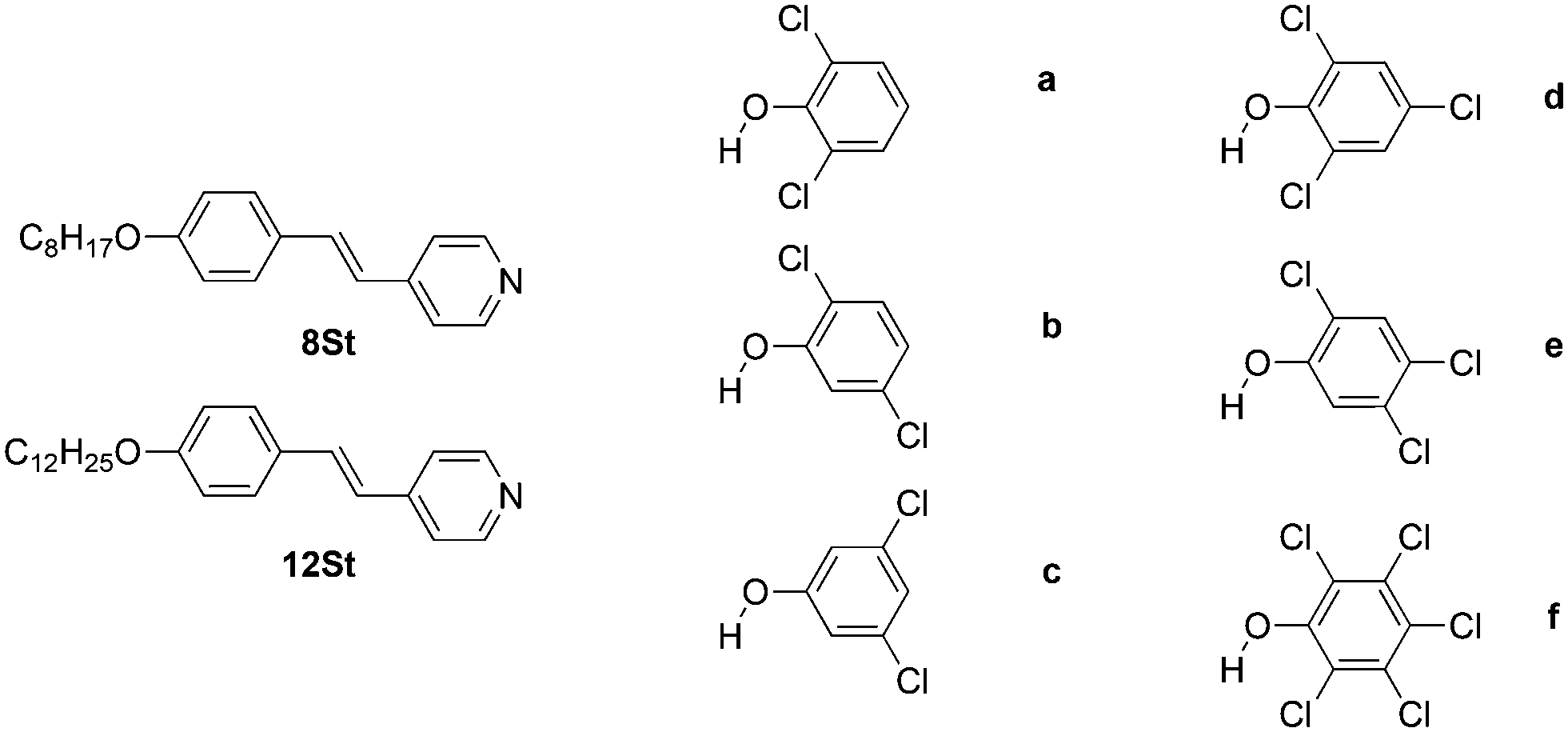 | ||
| Fig. 1 Structure of the two stilbazoles (8St and 12St) and the six chlorophenols (a–f) used in this study. | ||
Crystal structure determinations
Four complexes of octyloxystilbazole formed single crystals and structure determinations were completed for each of them. This stilbazole was chosen in preference to dodecyloxystilbazole as experience shows that shorter chains tend to crystallise more easily and, in addition, use of the octyloxy chain allows comparison with the previous study of fluorophenol complexes. All of complexes crystallised in the P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) space group and essential crystallographic data are found in Table 1. In analysing the structures, we have used a cut-off of the sum of the van der Waals radii minus 0.1 Å to identify meaningful non-covalent interactions.
space group and essential crystallographic data are found in Table 1. In analysing the structures, we have used a cut-off of the sum of the van der Waals radii minus 0.1 Å to identify meaningful non-covalent interactions.
| 8St-b | 8St-c | 8St-e | 8St-f | |
|---|---|---|---|---|
| CCDC number | 2245333 | 2245334 | 2245335 | 2245336 |
| Empirical formula | C27H31Cl2NO2 | C27H31Cl2NO2 | C27H30Cl3NO2 | C27H28Cl5NO2 |
| Formula weight/g mol−1 | 472.43 | 472.43 | 506.87 | 575.75 |
| Temperature/K | 110.15 | 110.15 | 110.15 | 110.15 |
| Crystal system | Triclinic | Triclinic | Triclinic | Triclinic |
| Space group |
P |
P |
P |
P |
| a/Å | a = 7.7755(10) | a = 7.8919(15) | a = 7.8166(16) | a = 7.938(9) |
| b/Å | b = 9.6671(12) | b = 11.242(2) | b = 9.981(2) | b = 11.115(13) |
| c/Å | c = 16.403(2) | c = 14.800(3) | c = 16.223(3) | c = 15.689(18) |
| α/° | α = 91.184(3) | α = 76.592(4) | α = 87.546(4) | α = 74.87(2) |
| β/° | β = 91.699(3) | β = 81.917(4) | β = 89.183(4) | β = 87.11(2) |
| γ/° | γ = 92.084(2) | γ = 76.998(4) | γ = 88.641(4) | γ = 83.09(2) |
| Volume/Å3 | 1231.3(3) | 1239.1(4) | 1264.0(4) | 1326(3) |
| Z | 2 | 2 | 2 | 2 |
| ρ calc g cm−3 | 1.274 | 1.266 | 1.332 | 1.442 |
| μ/mm−1 | 0.288 | 0.286 | 0.387 | 0.573 |
| F(000) | 500 | 500 | 532 | 596 |
| Crystal size/mm3 | 0.25 × 0.21 × 0.19 | 0.26 × 0.06 × 0.05 | 0.26 × 0.22 × 0.11 | 0.21 × 0.16 × 0.02 |
| Radiation/nm | 0.71073 | 0.71073 | 0.71073 | 0.71073 |
| 2θ range for data collection/° | 4.218 to 59.976 | 3.802 to 56.764 | 4.086 to 56.726 | 3.82 to 50.404 |
| Index ranges | −10 ≤ h ≤ 10, −13 ≤ k ≤ 13, −23 ≤ l ≤ 23 | −10 ≤ h ≤ 10, −14 ≤ k ≤ 14, −19 ≤ l ≤ 19 | −10 ≤ h ≤ 10, −13 ≤ k ≤ 13, −21 ≤ l ≤ 21 | −8 ≤ h ≤ 9, −12 ≤ k ≤ 13, −18 ≤ l ≤ 18 |
| Reflections collected | 14![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 040 040 |
12899 |
13040 |
7464 |
| Independent reflections | 6920 [R(int) = 0.0196] | 6116 [R(int) = 0.0238] | 6216 [R(int) = 0.0195] | 4519 [R(int) = 0.0437] |
| Data/restraints/parameters | 6920/0/294 | 6116/0/294 | 6216/0/299 | 4519/0/317 |
| Goodness-of-fit on F2 | 1.033 | 1.015 | 1.036 | 1.023 |
| Final R indexes [I ≥ 2σ(I)] | R 1 = 0.0409, wR2 = 0.1043 | R 1 = 0.0427, wR2 = 0.0978 | R 1 = 0.0357, wR2 = 0.0952 | R 1 = 0.0541, wR2 = 0.1418 |
| Final R indexes [all data] | R 1 = 0.0523, wR2 = 0.1127 | R 1 = 0.0602, wR2 = 0.1073 | R 1 = 0.0423, wR2 = 0.1005 | R 1 = 0.0735, wR2 = 0.15878 |
| Largest diff. peak/hole/e Å−3 | 0.39 and −0.22 | 0.32 and −0.31 | 0.46 and −0.21 | 0.58 and −0.42 |
There are structural similarities across the motifs of the four complexes, yet despite this, only two of them (8St-b and 8St-e) are close to isostructural. While the phenolic hydrogen atoms are notionally located through the refinement, because they are hydrogen bonded the electron density around them is not high and so the accuracy of the positions is debatable. As such, in the descriptions below the N(H)ÔC angle (the hydrogen is shown for its position but is not included in the measurement) is used to describe the hydrogen bond.
Despite what has just been written, it is interesting to note that according to the locations found through the refinement, the N⋯H distances are statistically identical in all of the structures (Table 2). The more accurate structures (smaller R-factor) for 8St-b, 8St-c and 8St-e allow for a better comparison, showing a shorter N⋯H distance in 8St-b and 8St-c compared to 8St-e (Table 2). A directly analogous comparison for the O–H bond lengths shows the inverse position, namely a longer bond in 8St-b and 8St-c compared to 8St-e. However, these distances do not track the pKa of the phenols and it is only when the O⋯N distance is considered that meaningful correlations emerge. Thus, considering these distances and the attendant esd values, the O⋯N distance increases as 8St-f < 8St-e ≈ 8St-b < 8St-c, which follows the pKa values (4.74 < 7.03 ≈ 7.3 < 8.25, respectively) and hence the expected strength of the hydrogen bond. Finally, it is noted that in 8St-b and 8St-d, there is a ‘contact’ between the ortho-hydrogen of the phenol and the pyridine nitrogen (2.60 Å and 2.62 Å, respectively corresponding to 91 and 91.5%, respectively, of the sum of the corresponding van der Waals radii). These are highlighted in Fig. S1† and, given the likely poor orbital overlap, it is assumed that these are electrostatic in nature.
| 8St-b | 8St-c | 8St-e | 8St-f | |
|---|---|---|---|---|
| O–H/Å | 0.93(2) | 0.96(2) | 0.73(2) | 0.82(5) |
| N⋯H/Å | 1.74(2) | 1.72(2) | 1.93(2) | 1.78(4) |
| O⋯N/Å | 2.660(1) | 2.674(2) | 2.656(2) | 2.561(4) |
In then making a comparison to the structures of the fluorophenol analogues,20 only two are possible, namely 8St-c and 8St-f. In both cases, the structure of the dimeric unit is rather similar, but this similarity does not extend to the packing.
Complex 8St-b
The hydrogen-bonded unit is shown in Fig. 2. The NÔC angle at the phenolic oxygen is 109.52(8)°, which gives a hydrogen bond that is close to linear. The plane of the pyridyl ring of the stilbazole subtends an angle of 60.67(4)° to the phenyl ring of the chlorophenol (better evident in Fig. 3a). | ||
| Fig. 2 The hydrogen-bonded complex of 8St-b. | ||
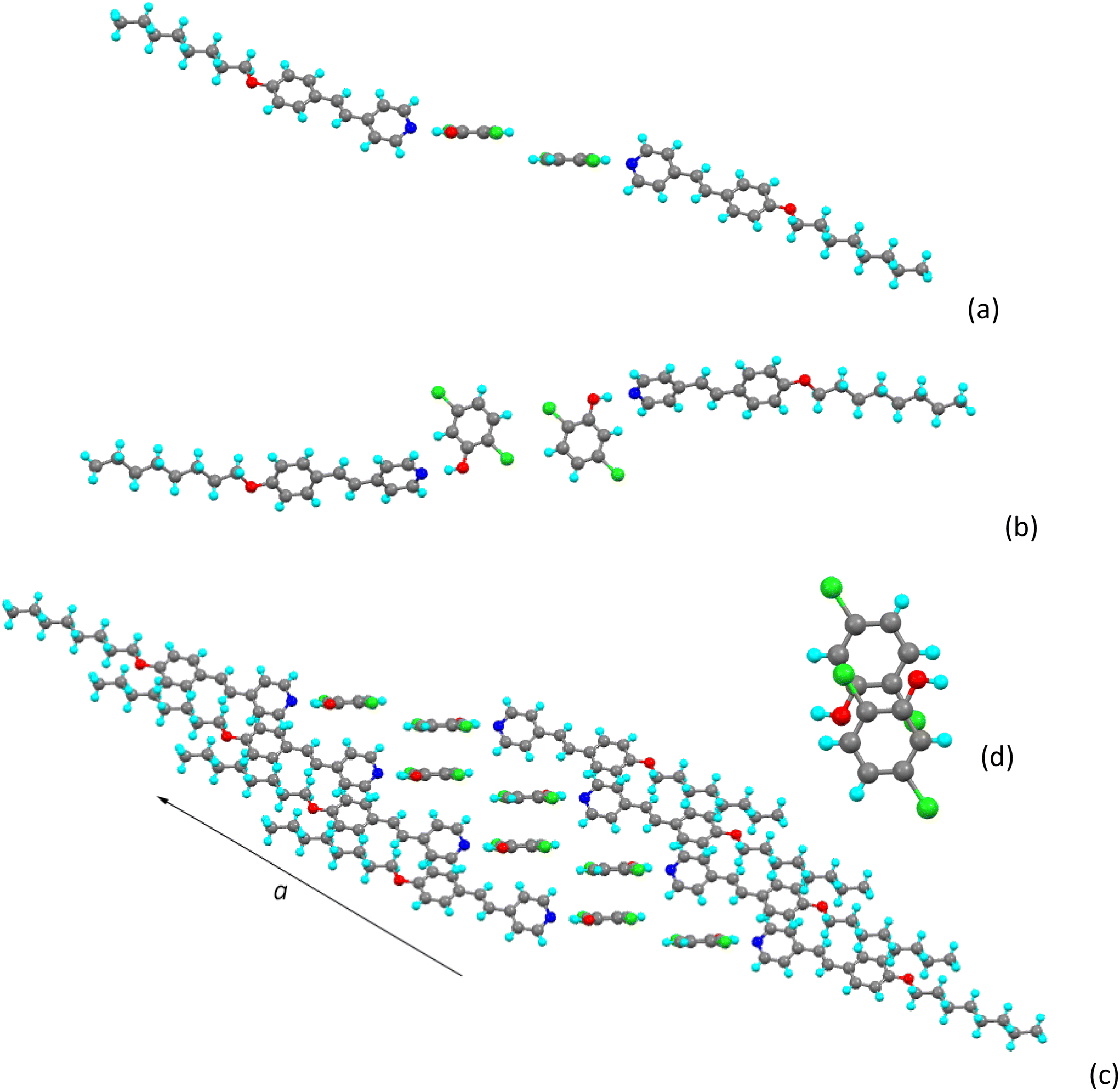 | ||
| Fig. 3 View of the ‘dimeric’ propagating unit of 8St-b (a) from the side and (b) from above; (c) propagation along the a axis and (d) view from above of the stacked pair of antiparallel phenols. | ||
It can be considered that the packing propagates through a dimeric unit (Fig. 3a and b) in which two stilbazoles are disposed either side of a slipped, back-to-back arrangement of two dichlorophenols. The separation between the planes of the two phenols is just 1.670(4) Å and the intermolecular Cl⋯H distances are 2.9037(5) Å, which is just over 0.11 Å less than the sum of the two van der Waals radii (3.02 Å). This dimeric pair then propagates along the a-axis as shown in Fig. 3c with the ‘left-hand’ phenolic ring of one pair sitting under the ‘right-hand’ phenolic ring of the next. The planes of the phenolic rings are separated by 3.475(2) Å, the view from above (Fig. 3d) shows no overlap of the π-systems of the rings themselves.
Complex 8St-c
The hydrogen-bonded unit is shown in Fig. 4. In this complex the NÔC angle at the phenolic oxygen is 118.5(1)° and, as such, at least in the solid state there is deviation from a linear hydrogen bond. The plane of the pyridyl ring of the stilbazole subtends an angle of 74.98(6)° to the phenyl ring of the chlorophenol, which is more evident in Fig. 5a. | ||
| Fig. 4 The hydrogen bonded complex 8St-c. | ||
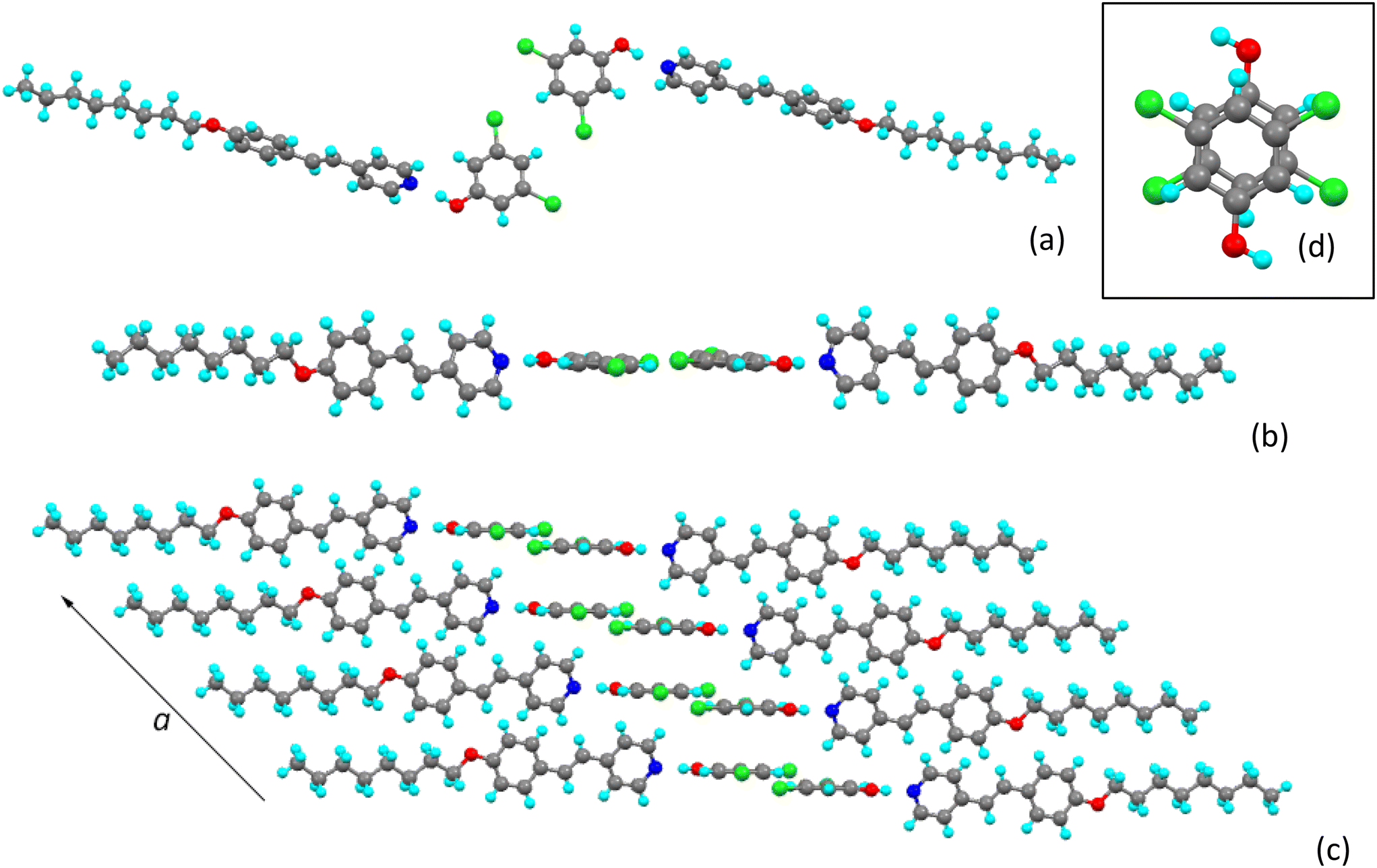 | ||
| Fig. 5 View of the ‘dimeric’ propagating unit of 8St-c (a) from above and (b) from the side; (c) propagation along the a axis and (d) view from above of the stacked pair of antiparallel phenols. | ||
Although once more there is a dimeric propagating unit (Fig. 5a), it is distinct from that of 8St-b. Thus, there is a different relative orientation of the core dichlorophenol units on account of the different chlorine substitution, leading to a different relative disposition of the two OH groups. Furthermore, while in 8St-b the planes of dichlorophenol rings were separated by 1.670 Å, here they are much closer at 0.621(5) Å, although this is not readily seen in Fig. 5b. The intermolecular H⋯Cl contacts that appear to stabilise the dimeric arrangement are found to be 2.9634(7) Å, which is rather close to the sum of the van der Waals radii (98% of the sum) and do not show up if close contacts are defined as the sum of the van der Waals radii less 0.1 Å. As such, it is unclear if these contacts are actively structure-directing.
The structure propagates along the a-axis in a manner not dissimilar to that found in 8St-b (Fig. 5c), except that now pairs of dichlorophenol rings are more directly superposed (Fig. 5d) in an antiparallel manner with a separation of 3.531(2) Å.
Complex 8St-e
The hydrogen-bonded unit is shown in Fig. 6 and there are many similarities between this structure and that of 8St-b. Thus, the NÔC angle at the phenolic oxygen is 109.09(8)° leading to a hydrogen bond that is close to linear, while the plane of the pyridyl ring of the stilbazole subtends an angle of 59.35(5)° to the phenyl ring of the chlorophenol (60.67(4)° in 8St-b). | ||
| Fig. 6 The hydrogen-bonded complex of 8St-e. | ||
The basic dimeric unit (Fig. 7a) is once more scaffolded on a back-to-back arrangement of two chlorophenols, which interact in a manner that is almost identical to that in 8St-c on account of the m-dichloro substitution pattern on the ring and with very similar intramolecular H⋯Cl contacts (2.9601(6) Å as against 2.9634(7) Å in 8St-c). However, in this complex the m-dichloro ‘unit’ is part of a 2,4,5-trisubstitution arrangement, which has the effect of leaving the OH groups disposed as found in 8St-b rather than in 8St-c, resulting in a propagation motif along a (Fig. 7b) resembling that of 8St-b (Fig. S2†). Thus, within each dimer the phenol planes are separated by 1.505(4) Å (1.670(4) Å in 8St-b), while between the dimers the interplane distance between phenols is 3.496(2) Å (3.475(2) Å in 8St-b). However, there is slightly greater superposition of the two rings in this complex (Fig. 7c).
 | ||
| Fig. 7 (a) View of the ‘dimeric’ propagating unit of 8St-e from above; (b) propagation along the a axis and (c) view from above of the stacked pair of antiparallel phenols. | ||
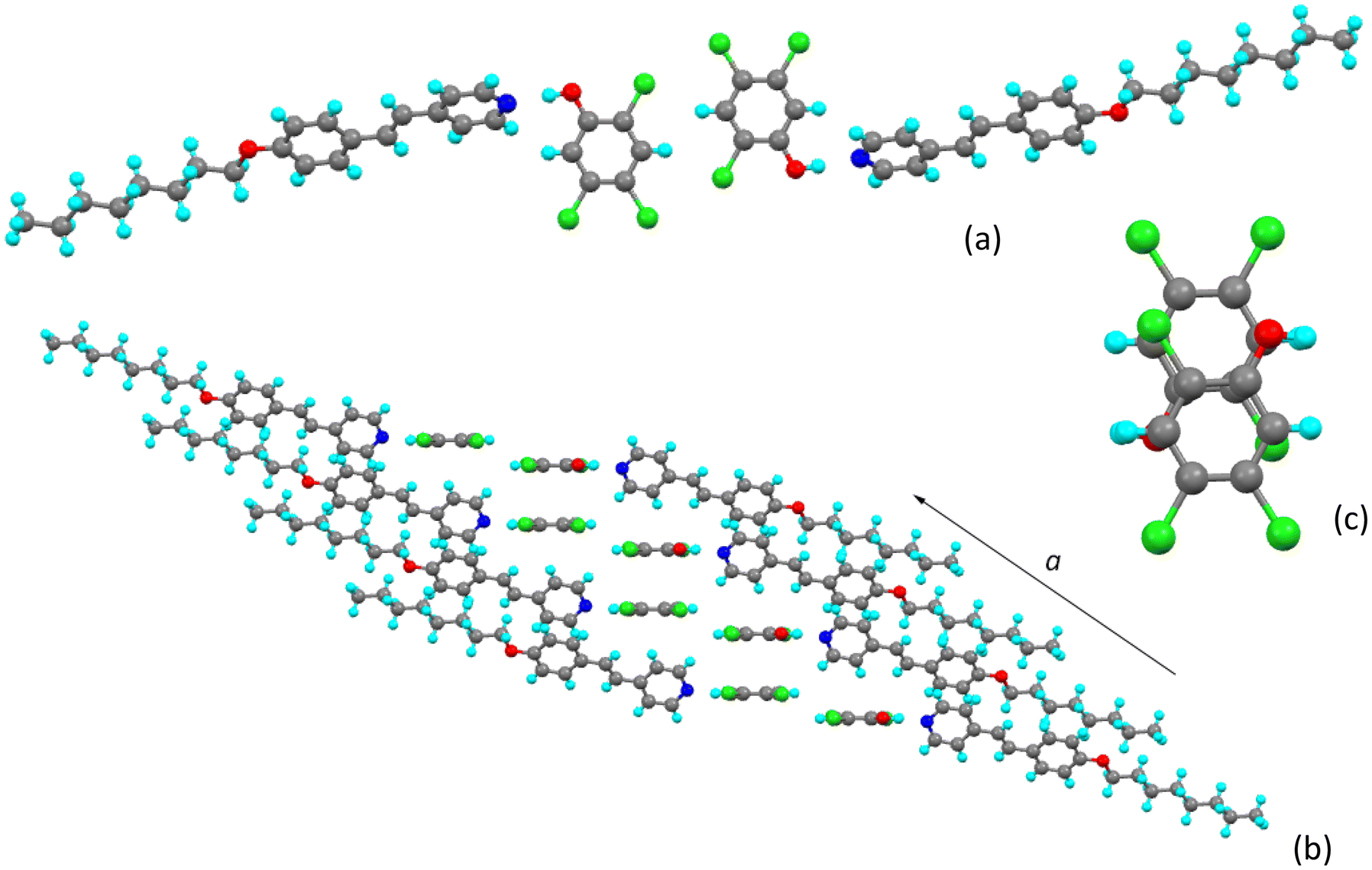
8St-f
Although with crystal parameters not dissimilar to those of 8St-c, the solid-state arrangement of 8St-f is different to the other three complexes described here, although retaining some similarities. The basic complex is shown in Fig. 8 and has the largest CÔN angle found in these materials at 137.0(2)°, giving quite a non-linear hydrogen bond. Although the most acidic of the phenols used in this study with pKa = 4.74, pentachlorophenol is less acidic than benzoic acid (pKa = 4.20) and so by analogy with many hydrogen-bonded liquid crystals involving alkoxystilbazoles and benzoic acids, there is no proton transfer at ambient temperature. Indeed, comparison of the C–O distances in all four phenols shows them to be statistically identical (ca. 1.34 Å, cf. 1.35(2) Å in phenol itself). Previous studies of mixed hydrogen- and halogen-bonded co-crystals, where the phenol was unequivocally deprotonated (in the presence of the much stronger base, 4-(N,N-dimethylamino)pyridine), revealed C–O distances as short as 1.240(2) Å, consistent with the presence of a phenate anion in a delocalised, Meissenheimer-like structure.20 This is not the case here. | ||
| Fig. 8 The hydrogen-bonded complex unit in 8St-f. | ||
In common with the other complexes, there is a dimeric repeat unit formed from two stilbazoles hydrogen bonded to two back-to-back phenols (Fig. 9a), with a Cl⋯Cl distance of 3.575(4) Å, which is all but twice the van der Waals radius of chlorine (1.82 Å). However, when viewed from the side (Fig. 9b), while the planes of the two phenols are extremely close with a separation of 0.336(13) Å (cf. 0.621(5) Å in 8St-c), in this complex the planes of the pyridyl ring of the stilbazole and of the phenol to which it is hydrogen bonded are almost co-planar, subtending an angle of just 3.61(11)°.
 | ||
| Fig. 9 Dimeric arrangement in 8St-f, (a) viewed from above and (b) viewed from the side. | ||
This co-planarity has consequences for the packing as seen in Fig. 10a showing the two-dimensional arrangement in the ac plane. It is noted that there are short, inter-complex Cl⋯H distances (Fig. S3†) which, from their directionality, could be electrostatic in nature pointing off to the side of the weak σ-hole expected on the chlorine24 and so possibly contributing to the stability of this propagating arrangement. However, there is also propagation out-of-plane – this time in the c-direction – and Fig. 10b shows that in this case pairs of pyridyl and phenol rings stack with appreciable overlap of the two π-systems (Fig. 10c). However, the planes of the two ring systems are not co-parallel and the angle subtended is 3.61(11)° as dictated by the crystal symmetry.
 | ||
| Fig. 10 (a) Packing in 8St-f in the ac plane viewed down the b-axis; (b) propagation out of the ac plane and (c) the superposition of pyridyl and phenol rings. Note that the view in (b) is slightly misleading as it implies an infinite stack of pyridyl and phenol rings, which in fact does not occur (see Fig. S4†). | ||
Liquid crystal properties
The liquid crystal properties of the materials were determined by polarised optical microscopy, which was sufficient given that only the nematic (N) and smectic A (SmA) phases were observed. These were readily identified from their optical textures, examples of which are shown in Fig. 11. With only one complex showing enantiotropic behaviour and with most complexes showing only a clearing transition, DSC data were not recorded as they do not provide any particularly meaningful data. Transition temperatures are found in Table 3 in which monotropic temperatures are recorded on re-heating from the lower phase in order to obtain a thermodynamic temperature. | ||
| Fig. 11 Textures of the nematic (left) and smectic A (right) phase of the complexes. The microscope magnification used was 200×. | ||
| Compound | Transition | T (°C) |
|---|---|---|
| 8St-a | Cr-Iso | 53.0–65.0 |
| (SmA-Iso) | (45.0) | |
| 8St-b | Cr-Iso | 77.0–79.0 |
| (N-Iso) | (62.0) | |
| (SmA-N) | (51.0) | |
| 8St-c | Cr-Iso | 78.0–84.0 |
| (N-Iso) | (81.4) | |
| 8St-d | Cr-Iso | 61.0 |
| (N-Iso) | (58.5) | |
| (SmA-N) | (52.0) | |
| 8St-e | Cr-Iso | 91.5 |
| (N-Iso) | (71.0) | |
| 8St-f | Cr-Iso | 101.0 |
| 12St-a | Cr-Iso | 74.0 |
| (SmA-Iso) | (62.5) | |
| 12St-b | Cr-Iso | 78.0–83.0 |
| (SmA-Iso) | (72.8) | |
| 12St-c | Cr-SmA | 72.0 |
| SmA-N | 74.0 | |
| N-Iso | 83.5 | |
| 12St-d | Cr-Iso | 74.1 |
| (SmA-Iso) | (72.9) | |
| 12St-e | Cr-Iso | 96.9 |
| (SmA-Iso) | (82.8) | |
| 12St-f | Cr-Iso | 87.1 |
All of the complexes showed monotropic phases with three exceptions. Thus, neither of the complexes of pentachlorophenol (f) showed a mesophase, while the phases of 12St-c were enantiotropic.
For the majority of complexes, the crystalline solids melted into the isotropic phase and on cooling showed a nematic and/or a smectic A phase. The SmA phase was seen for all complexes of 12St (with 12St-c also showing a N phase), while for 8St, two complexes (8St-c and 8St-e) showed only a nematic phase, 8St-a showed only SmA, while 8St-b and 8St-d showed both. The predominance of the SmA phase in complexes of 12St mirrors precisely the observations made in the study of the analogous fluorophenol complexes. As shown in Fig. 12, the clearing points were always higher for the complexes of the more anisotropic 12St. The observation of nematic and SmA phases is unsurprising and is indeed typical for small, dipolar mesogens.25–27
 | ||
| Fig. 12 Comparison of the mesophase transition temperatures for the new complexes in this study. Note that melting points are not shown and that, with the exception of the data for 12St-c, all temperatures are monotropic. Melting points have not been indicated in the plot. | ||
Consideration of the data in Table 3 shows that for some complexes, the melting point is recorded as a range. While perhaps at first sight surprising, such behaviour is not uncommon in hydrogen-bonded systems and has been observed before,28,29 including in the previous study of fluorophenol complexes.20 The behaviour reflects the fact that while the hydrogen-bonded complex is the thermodynamically preferred form obtained from solvent crystallisation, at some temperature above ambient (i.e. where the melting is observed to commence), the thermodynamically preferred arrangement can be for the two components to exists separately. Perhaps by coincidence, for 8St-b and 8St-c, the broad melting event coincides approximately with the melting point of the neat stilbazole,30 whereas for 8St-a and 12St-b the broad melting is below the stilbazole melting point. The melting events occur over ranges of between 2 and 12 °C and it is evident that at the high-temperature end of the range the complexes ought to have re-formed as in every case a homogeneous liquid is obtained and the temperature is below the melting point of the phenol. As such, sharp transitions into a mesophase are observed on cooling for the four complexes, also consistent with the presence of a single species.
Discussion
The stability of the crystal phase does not generally lend itself to useful comparisons between liquid-crystalline materials, even despite the fact that here, all four of the complexes that crystallise do so in the P space group. However, the clearing point (in this case TN-Iso or TSmA-Iso) does provide information and lends itself to structure/property considerations. Thus, Fig. S3† shows the data from Fig. 12 re-plotted so that the clearing point decreases from left to right, revealing the ability of the different chlorophenols to stabilise LC phases in these complexes. The order is shown graphically in the upper part of Fig. 13 and reveals that 3,5-dichlorophenol stabilises mesophases most strongly, while 2,6-dichlorophenol is least effective.‡ Interestingly, 3,5-dichlorophenol is the one phenol that stabilises a nematic phase in complexes with 12St, while 2,6-dichlorophenol in the one phenol that does not do so in complexes with 8St. This observation is interesting when compared with the data obtained in the previous study with fluorophenols, where it was found that 3,5-difluorophenol was almost the least effective (of sixteen fluorophenols) at stabilising mesophases.20 How might this be understood?
 | ||
| Fig. 13 Upper line: The chlorophenols used in this study that generate liquid crystal mesophases in complexes with stilbazole, organised in decreasing stability of the induced mesophase. Lower line: Ordering of mesophase stability of the 12St complexes of the equivalent fluorophenols. | ||
The lower part of Fig. 13 shows the relative ability of the different fluorophenols (from the previous study) to stabilise a mesophase in complexes with 12St (12St was chosen as all of its complexes showed a mesophase, unlike complexes of 8St and 4St). The previous study showed that the most stable mesophases were always found in complexes where there was a fluorine ortho to the hydroxyl group of the phenol. The proposition was that this enabled the formation of an extra, intra-complex, hydrogen-bonded ring structure that added rigidity and so enhanced anisotropy (Fig. 14a, X = F). After that, the next most significant feature was the presence of one or more fluorine atoms disposed meta or para to the hydroxyl and conferring a longitudinal dipole, which would also promote mesophase stability. These proposals were consistent across the 48 complexes studied (three series of sixteen). Support for the existence of an intramolecularly hydrogen-bonded structure came from the observation of such an arrangement in two of the solved crystal structures, namely 8St with pentafluorophenol and with 2,3,5,6-tetrafluorophenol (Fig. 14b). Thus, while this motif was found only in these complexes, it is perfectly reasonable to assume that with increased molecular motion as in the fluid liquid crystal phases, the ring forms.
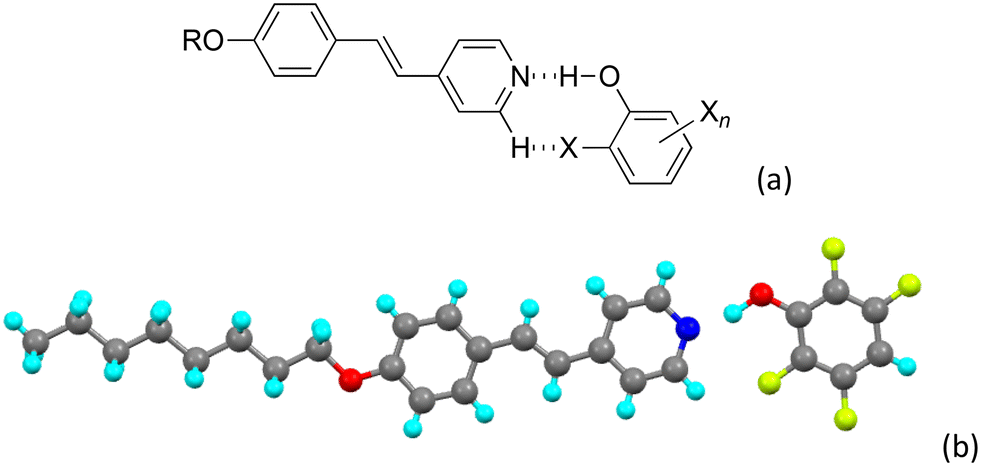 | ||
| Fig. 14 (a) Structure of the possible ring formation using intramolecular hydrogen bonding and (b) an example from the study of the analogous fluorophenols (re-drawn from the cif file). | ||
Of course, this motif is found also in the structure of 8St-f shown above, but there are differences that take account of the greater size of chlorine compared to fluorine. Thus, in the pentafluorophenol complex, the Cipso![[N with combining circumflex]](https://www.rsc.org/images/entities/char_004e_0302.gif) O angle (Cipso is the 4-carbon of the pyridine ring of the stilbazole) is close to linear at 170°, while in the pentachloro analogue it is opened out to 156.56(13)°. This difference is reflected also in the NÔC angle, which is 124.9° in the pentafluorophenol and 137.0(2)° in the pentachlorophenol. Finally, the difference in the o-H⋯X distances is more than twice the difference in the van der Waals radii of the two halogens. Taken together, these variations show both the steric effect of the larger chlorine and the consequence of its lower electronegativity, suggesting that intramolecular ring formation is unlikely to be particularly favoured.
O angle (Cipso is the 4-carbon of the pyridine ring of the stilbazole) is close to linear at 170°, while in the pentachloro analogue it is opened out to 156.56(13)°. This difference is reflected also in the NÔC angle, which is 124.9° in the pentafluorophenol and 137.0(2)° in the pentachlorophenol. Finally, the difference in the o-H⋯X distances is more than twice the difference in the van der Waals radii of the two halogens. Taken together, these variations show both the steric effect of the larger chlorine and the consequence of its lower electronegativity, suggesting that intramolecular ring formation is unlikely to be particularly favoured.
In then considering the generally lower mesophase stability (Fig. 15) of the chlorophenol complexes (between 20–30 °C less stable), this is entirely consistent with the proposal that the absence of a planar, hydrogen-bonded structure would require the ring chlorines to twist out of the plane of the stilbazole hence reducing the anisotropy and destabilising any mesophase that might form. Indeed, recall that in rod-like liquid crystals, even a lateral fluorine atom, small as it is, can destabilise a mesophase when compared to an equivalent compound with a hydrogen in the same position.21 Furthermore, the absence of ortho chlorine atoms in complexes of 3,5-dichlorophenol means that this is the only one with the possibility to be planar across both the stilbazole and the phenol. As such, these complexes have the greatest stability within the chlorophenols and, interestingly, the same mesophase stability as their fluoro counterparts. Finally here, it is appropriate to note that even though for the most part, the crystal phases of the fluorophenol complexes are more stable than those of the new chlorophenol-based materials reported here, the reduction in mesophase stability in the new complexes is sufficient to ensure that, as noted, mesophases are monotropic in all but one case.
 | ||
| Fig. 15 Comparison of the clearing points of the chlorophenol complexes and their fluorophenol analogues plotted in descending order of mesophase stability for the chlorophenols with complexes of 12St on the left of the plot and those of 8St on the right. | ||
In addition to the general observation about more stable mesophases being observed with complexes of fluorophenols, it is worth noting that, despite having a smaller sub-set of phenols to compare, the general order of mesophase stability as a function of substitution pattern is rather similar between the two series (Fig. 13). As discussed, complexes of 3,5-dichlorophenol are the main exception although there is a swop in positions for the 2,6- and 2,4,6-substituted complexes, although it is noted that the different in phase stability in the fluoro complexes of these isomers is minimal (ca. 2 °C).
Further, it is also worth reprising the discussion that relates the strength of the hydrogen bond in the complex to its clearing point. Fig. 16 shows a scatter plot of the pKa values for both the chlorophenols and the fluorophenols plotted against the clearing points 8St and 12St complexes of each of them. Once more, there is evidently no correlation between clearing point and pKa, providing yet further examples where clearing of a supramolecular species is of the intact complex and is not driven by thermally induced dissociation into the components.
 | ||
| Fig. 16 Scatter plot of pKavs. clearing point for the isomeric chlorophenol complexes studied here and the analogous fluorophenol complexes studies previously.16 | ||
Conclusion
Hydrogen-bonded complexes form readily between alkoxystilbazoles and chlorophenols in a manner directly analogous to that found in related fluorophenols. There are features in the solid-state organisation of the chlorophenols characterised by X-ray crystallography that are strongly reminiscent of those seen in the fluorophenols, but from the examples to hand there were no examples of isostructural or isomorphous ‘pairs’ of materials. There are however direct analogies in the liquid crystal behaviour inasmuch as 8St complexes of the chlorophenols showed nematic and smectic A phases, while the mesomorphism of complexes of 12St was dominated by the smectic A phase.There was also a broad similarity in the way that different chlorophenols stabilised the mesophases of the complexes they formed, except that it appeared that steric (and perhaps dipolar) influences reduced the propensity for the formation of an extra ring in the complexes via intramolecular hydrogen bonding. As this ring had been shown to promote mesophase stability in the complexes with fluorophenols, then the reduced tendency to planarity coupled with the increased size of chlorine compared with fluorine meant that the clearing point of the chlorophenol complexes were some 20 °C lower than those of the analogous fluorophenol complexes. Perhaps the most striking example of this difference is that the complexes of 3,5-dichlorophenol, being able to planarise owing to the absence of steric encumbrance, shows the most stable mesophases of the complexes studied. However, in the wider study using fluorophenols where most of the complexes could form additional ring structures via intramolecular hydrogen bonding, its complexes were almost the least stable.
Finally, consideration of the clearing points of the chlorophenols as a function of their pKa revealed the absence of any correlation showing once more that it is the intact hydrogen-bonded moieties that clear.
Author contributions
DWB conceived and oversaw the project, verified the liquid crystal assignments and wrote the manuscript. The work was carried out by ODJ with the assistance of SGW, while ACW solved the crystal structures, refined the data and contributed to the crystallographic analysis.Conflicts of interest
The authors report no conflict of interests.Acknowledgements
We thank the EPSRC for support to SGW (EP/E046754/1).References
- T. Kato and Y. Kamikawa, et al., in Handbook of Liquid Crystals, ed. J. W. Goodby, Wiley-VCH, Weinheim, 2nd edn, 2014, ch. 10, vol. 5 Search PubMed.
- C. M. Paleos and D. Tsiourvas, Angew. Chem., Int. Ed. Engl., 1995, 34, 1696–1711 CrossRef CAS.
- E. Bradfield and B. Jones, J. Chem. Soc., 1929, 2660–2661 RSC.
- B. Jones, J. Chem. Soc., 1935, 1874 Search PubMed.
- G. M. Bennett and B. Jones, J. Chem. Soc., 1939, 420–425 RSC.
- G. A. Jeffrey, Acc. Chem. Res., 1986, 19, 168–173 CrossRef CAS.
- H. A. van Doren, E. Smits, J. M. Pestman, J. B. F. N. Engberts and R. M. Kellogg, Chem. Soc. Rev., 2000, 29, 183–199 RSC.
- J. W. Goodby, V. Görtz, S. J. Cowling, G. Mackenzie, P. Martin, D. Plusquellec, T. Benvengu, P. Boullanger, D. Lafont, Y. Queneau, S. Chambert and J. Fitreman, Chem. Soc. Rev., 2007, 36, 1971–2032 RSC.
- T. Kato and J. M. J. Fréchet, J. Am. Chem. Soc., 1989, 111, 8533–8534 CrossRef CAS.
- T. Kato, M. Gupta, D. Yamaguchi, K. P. Gan and M. Nakayama, Bull. Chem. Soc. Jpn., 2021, 94, 357–376 CrossRef CAS.
- S. J. D. Lugger, S. J. A. Houben, Y. Foelen, M. G. Debije, A. P. H. J. Schenning and D. J. Mulder, Chem. Rev., 2022, 122, 4946–4975 CrossRef CAS PubMed.
- J. Uchida, B. Soberats, M. Gupta and T. Kato, Adv. Mater., 2022, 34, 2109063 CrossRef CAS PubMed.
- S. Saha, M. K. Mishra, C. M. Reddy and G. R. Desiraju, Acc. Chem. Res., 2018, 51, 2957–2967 CrossRef CAS PubMed.
- D. Devadiga and T. N. Ahipa, J. Mol. Liq., 2021, 333, 115961 CrossRef CAS.
- C. Präsang and D. W. Bruce, Helv. Chim. Acta, 2023, 106, e202300008 Search PubMed.
- C. Präsang, A. C. Whitwood and D. W. Bruce, Cryst. Growth Des., 2009, 9, 5319–5326 CrossRef.
- A. Wasilewska, M. Gdaniec and T. Połoński, CrystEngComm, 2007, 9, 203–206 RSC.
- J. Han and F.-M. Tao, J. Phys. Chem. A, 2006, 110, 257–263 CrossRef CAS PubMed.
- C. Präsang, L. J. McAllister, A. C. Whitwood and D. W. Bruce, CrystEngComm, 2013, 15, 8947–8958 RSC.
- J. P.-W. Wong, A. C. Whitwood and D. W. Bruce, Chem. – Eur. J., 2012, 18, 16073–16089 CrossRef CAS PubMed.
- See e.g. M. Hird, Chem. Soc. Rev., 2007, 36, 2070–2095 RSC.
- J. Han, R. L. Denning and F.-M. Tao, J. Phys. Chem. A, 2005, 109, 1159–1167 CrossRef CAS PubMed.
- D. J. Price, PhD Thesis, University of Sheffield, 1995 Search PubMed.
- T. Clark, M. Hennemann, J. S. Murray and P. Politzer, J. Mol. Model., 2007, 13, 291–296 CrossRef CAS PubMed.
- G. W. Gray, Philos. Trans. R. Soc., A, 1983, 309, 77–92 CrossRef CAS.
- G. W. Gray, Philos. Trans. R. Soc., A, 1990, 330, 73–94 CAS.
- G. W. Gray, K. J. Harrison and J. A. Nash, Electron. Lett., 1973, 9, 130–131 CrossRef CAS.
- D. J. Price, H. Adams and D. W. Bruce, Mol. Cryst. Liq. Cryst., 1996, 289, 127–140 CrossRef CAS.
- This reference deals with the phenomenon where there is ligand dissociation from a metal complex: B. Donnio, D. W. Bruce, B. Heinrich, D. Guillon, H. Delacroix and T. Gulik-Krzywicki, Chem. Mater., 1997, 9, 2951–2961 CrossRef CAS.
- D. W. Bruce, D. A. Dunmur, E. Lalinde, P. M. Maitlis and P. Styring, Liq. Cryst., 1988, 3, 385–395 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 2245333–2245336. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3ce00266g |
| ‡ Note that mesophase stability refers to the upper temperature at which a particular mesophase is stable and is unrelated to the range over which the phase exists. |
| This journal is © The Royal Society of Chemistry 2023 |