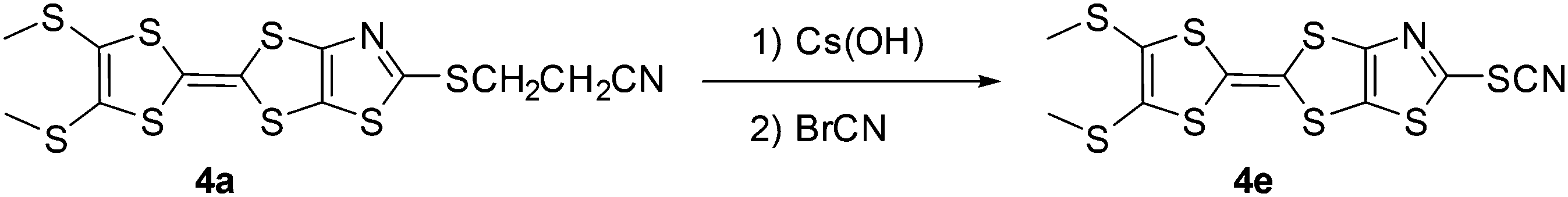DOI:
10.1039/D3CE00257H
(Paper)
CrystEngComm, 2023,
25, 2946-2958
Non-covalent interactions in neutral and oxidized tetrathiafulvalenes†
Received
15th March 2023
, Accepted 24th April 2023
First published on 26th April 2023
Abstract
Tetrathiafulvalenes (TTFs) functionalized with groups able to increase non-covalent intermolecular interactions through either hydrogen, halogen or chalcogen bonding were investigated. For that purpose, we designed several appropriate precursors, fused dithiole-2-one/thiazoles, which under homocoupling led to symmetrically substituted TTFs or through heterocoupling to dissymmetrically substituted TTFs. The electrochemical investigations allowed the assessment of the electronic effect of these substituents on the overall donating ability of the TTFs. In addition two charge transfer salts have been prepared involving two of these TTFs with TCNQF4 as the electron acceptor, (4c)2(TCNQF4) and (4b)(TCNQF4). Furthermore the crystal structures of the neutral TTFs and of the charge transfer salts allowed us to study the stacking mode and the different non-covalent interactions taking place in the crystals, in connection with the calculated electrostatic surface potential (ESP) of the investigated molecules. All the data provide information about non-covalent bonding interactions in the solid state and about their potential contribution to direct to some extent crystallographic intermolecular organization.
Introduction
For five decades now, tetrathiafulvalenes (TTF), a fascinating family of electron donor molecules, have been the focus of a great deal of interest as precursors of conducting molecular materials especially after the discovery of the first organic metal (TTF)(TCNQ).1,2 In addition, remarkable redox properties make them valuable building blocks for a broad range of applications.3–6 The properties of molecular materials are essentially linked to the organization and the interactions of the molecules in the solid state and it is somehow a big challenge to increase the dimensionality of these materials.7 Indeed it is always difficult to predict how the molecules will be organized and interact within the material. Thereby, with the aim of gaining control over the intermolecular architecture, the use of non-covalent intermolecular interactions can be envisioned. For instance, hydrogen bonding interactions (HB),8 first used by Bryce in 1995,9 led to a catechol-fused ethylenedithio-TTF semiconducting material at ambient pressure, while upon application of pressure a metallic behavior was observed.10 Among the various possible non-covalent interactions, halogen bonding (XB)11 has also been investigated in order to favor the organization of the TTFs in the solid state.12,13 More recently, chalcogen bonding (ChB)14 have focused also attention in this research area.15 An interesting characteristic of these non-covalent interactions is that when the hydrogen, halogen or chalcogen atoms are located on an electroactive moiety such as TTF, the lower electronic density called σ-hole, located on the extension of the covalent bond,16 is enhanced upon oxidation. Indeed neutral TTF, substituted by a halogen or chalcogen atom, is an electron donor molecule, but once oxidized, the TTF exerts an electron withdrawing effect that increases the magnitude of the σ-hole and thus favor such intermolecular interactions. Herein we investigate the synthesis of novel substituted TTFs able to form non-covalent intermolecular interactions, such as hydrogen, halogen, or chalcogen bonding. We studied the influence of these non-covalent interactions in the solid state either on the precursors or on the neutral and oxidized TTFs.
Results and discussion
In order to prepare the appropriate precursors of the targeted TTFs, we used N-tBu-1,3-thiazoline-2-thione 1 as starting materials (Scheme 1).17 Indeed, we have shown previously that compound 1 in the presence of an electrophile such as an halogenated alkyl can be converted into the 2-alkylthio-1,3-thiazole derivative.17,18 Moreover, it is also possible to generate the 2-iodo-substituted thiazole core, by reacting 1 with iodine in DMSO forming the non-substituted thiazole core as a side product.19 Thus we decided to explore the reactivity of 1 towards other reagents with the aim of preparing functionalized moieties which could bring the TTF core with a sizeable potential for non-covalent intermolecular interactions. We first focused on the cyanoethylthio functionalization. Indeed, the cyanoethyl group is an excellent protecting group of the thiolate.20 Therefore, this protecting group can be withdrawn at will and replaced by other functional groups. For that purpose, the transformation of the N-tBu-1,3-thiazoline-2-thione 1 into 2-cyanoethylthio-1,3-thiazole derivative 2a was first studied in DMF. The reaction of 1 in DMF in the presence of an excess of bromopropionitrile led after 48 hours under inert atmosphere at 70 °C to the desired 2-cyanoethylthio-1,3-thiazole derivatives 2a in 63% yield. We also attempted the same reaction using DMSO instead of DMF. Unexpectedly, using DMSO in the presence of 1 and an excess of bromopropionitrile, we isolated the non-substituted thiazole ring 2b in 55% yield. We also prepared the iodo substituted precursor 2c according to the procedure described earlier by simply mixing 1 with an excess of iodine in DMSO at 55 °C for four hours under inert atmosphere.19
 |
| | Scheme 1 Reactivity of N-tBu-1,3-thiazoline-2-thione derivative 1 towards various reagents. | |
The 2-ethylthio derivative 2d was prepared by simply refluxing 1 for 2 hours with an excess of iodoethane in dichloromethane as previously reported.17 The same procedure with cyanogen bromide (BrCN) as alkylating agent was not successful in dichloromethane while when the reaction was performed in DMSO at 55 °C for 16 h, the thiocyanate substituted precursor 2e was isolated in 50% yield.
Dithiole-2-ones are known to be excellent precursors for the synthesis of TTF through their homocoupling, at 120 °C under inert atmosphere, either in the presence of a phosphite derivative only, such as triethylphosphite or trimethylphosphite, or by mixing the dithiol-2-one in toluene in the presence of the trialkylphosphite.21 Thus we investigated the reactivity of the different dithiol-2-one 1 and 2a–e in the presence of trialkylphosphite. The first attempt concerns the coupling of the dithiole-2-one 1 in the presence of trimethylphosphite P(OMe)3, acting as solvent and reagent (Scheme 1). After several hours we did not observe the formation of the desired TTF but instead we isolated the 2-methylthio-1,3-thiazole derivative 2f (Scheme 1). Therefore, in the presence of P(OMe)3 alkylation of the exocyclic sulfur occurs followed by the loss of the tBu substituent. It is worth noting that the same compound 2f was previously obtained by simply refluxing 1 with an excess of iodomethane in dichloromethane.17
Then we studied the reactivity of the dithiole-2-one 2a–d with different substituents on position 2 of the thiazole ring toward homocoupling. These building blocks heated at 120 °C for 5 h in triethylphosphite P(OEt)3 afforded the desired symmetric TTFs, 3(a–d), in relatively low yield after purification by column chromatography on silica gel (22–27% yield), Scheme 2.
 |
| | Scheme 2 Synthesis of symmetrical TTF(R)2. | |
We also investigated the formation of dissymmetrically substituted TTF and for that purpose we chose the 4,5-bis(methylthio)-1,3-dithiole-2-thione 3 as the second building block in order to gain some solubility compared to the symmetrically substituted TTFs. Moreover, this dithiole-2-thione motif is known to have a low reactivity towards itself in the conditions used to form a TTF core and thus, the cross-coupling reaction toward unsymmetrical TTFs should be favored.22 We carried out the cross-coupling reaction between the dithiole-2-one 3 and each of the four thiazole derivatives 2a–c and 2e in the same conditions, i.e. 2 hours at 120 °C in P(OEt)3 (Scheme 3). Besides for 2e, all the expected dissymmetrical TTF, 4a–c, are formed (19–38% yield). Using this approach no symmetrical TTF, 3a–c, was isolated. In the case of 2e the only TTF which could be isolated is the dissymmetrical TTF, 4d.
 |
| | Scheme 3 Synthesis pathways of dissymmetrical TTF 4a–d. | |
Here again we attempted the cross-coupling reaction between the fused dithiole-2-one thiazoline 1 with 3 in P(OEt)3 for 2 hours at 120 °C (Scheme 4). In these conditions, we isolated TTF 4d in 46% yield. According to what we observed previously, a plausible explanation would be that thiazoline 1 in the presence of P(OEt)3 is first transformed into the thiazole 2d and then the heterocoupling occurs between 2d and 3 to afford TTF 4d.
 |
| | Scheme 4 Reactivity of 1 in the presence of dithiole-2-thione 3 and P(OEt3). | |
Thus, in order to achieve the synthesis of the thiocyanate TTF 4e, we used another approach that relies on the use of the protected TTF thiolate namely 4a. The cyanoethyl group can indeed be easily removed in basic medium by adding caesium hydroxide to a DMF solution of TTF 4a and after stirring for 30 min at room temperature cyanogen bromide (BrCN) was added. TTF 4e was obtained in 70% yield after purification by chromatography on silica gel (Scheme 5). Thus we successfully synthetized various TTFs substituted either with hydrogen atoms, 3b and 4b, or with iodine atoms, 3c and 4c, susceptible to participate respectively into hydrogen or halogen bonding interactions. On the other hand, the nitrile on the sulfur atom of 4e should increase the charge depletion on one σ-hole of this atom, in the prolongation of the NC–S bond, and potentially promote the formation of chalcogen bonding.
 |
| | Scheme 5 Synthetic route towards TTF 4e. | |
Electrochemical investigation
The redox properties of the TTF derivatives were investigated by cyclic voltammetry (CV) and the redox potentials are listed in Table 1 together with those of the tetramethylthio-TTF (MeS)4-TTF for comparison. All these TTFs exhibit a typical cyclic voltammogram for a TTF, namely two reversible oxidation waves corresponding to the successive oxidation of the neutral TTF into the radical cation and then to the dication. The redox potentials of all the investigated TTFs are shifted to higher oxidation potentials than those observed for (MeS)4-TTF (Table 1). This anodic shift is more pronounced for the symmetrically substituted TTFs than for the dissymetrically one indicating the electron withdrawing effect of the fused thiazole ring on the electron donating ability of the TTFs. Depending on the substituent on the thiazole ring, the same order from the easier to oxidize to the most difficult one is observed, either in the symmetrical series or in the dissymmetrical one, namely: SEt, SCH2CH2CN, H, I (d, a, b, c).
Table 1 Redox potentials of the investigated TTFs in CH2Cl2, 0.1 M TBAPF6 E in V vs. SCE
| TTF |
E
1/2
1 (ΔEp mV) |
E
1/2
2 (ΔEp mV) |
| (MeS)4TTF |
0.49 (60) |
0.83 (60) |
|
3a
|
0.63 (60) |
0.97 (70) |
|
3b
|
0.68/0.46 |
1.08/0.75 |
|
3c
|
0.74 (60) |
1.12 (70) |
|
3d
|
0.61 (60) |
0.97 (60) |
|
4a
|
0.56 (70) |
0.91 (70) |
|
4b
|
0.57 (80) |
0.94 (80) |
|
4c
|
0.59 (70) |
0.94 (90) |
|
4d
|
0.53 (60) |
0.87 (60) |
|
4e
|
0.58 (80) |
0.89 (70) |
Solid state investigations
Crystals suitable for X-ray diffraction studies were obtained by slow evaporation of concentrated dichloromethane solution of the three dithiole-2-one 2a, 2e and 2f. The molecular structure of 2a, 2e and 2f are presented in Fig. 1. In all cases the fused heterocycles (dithiol-2-one and the thiazole ring) are coplanar.
 |
| | Fig. 1 Molecular structures of 2a (left), 2e (middle) and 2f (right). | |
For 2a, with the cyanoethylthio substituent on the thiazole ring, the molecule crystallizes in the triclinic system, space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) . There is no relevant interatomic contacts shorter than 95% of the sum of the van der Waals radii between neighboring molecules indicating none or weak non-covalent intermolecular interactions. Contrariwise, for 2e the presence of the thiocyanate group induces some intermolecular interactions. The crystal structure of 2e is solved in the monoclinic system, space group C2/c, with the molecule in general position. The molecules form “dimers” in the ac plane, stacking along the b axis. The “dimers” are related by an inversion center, and are held by chalcogen bonding between the nitrogen of the thiocyanate group (N2) as ChB acceptor of one molecule, and two opposite sulfur atoms (S3, S2) as ChB donors from the other molecule (Fig. 2 and Table 2). The C–S⋯N angles are in the range of 170°, and the S⋯N contacts are short with a 90% reduction ratio relative to the sum of the vdW radii (Fig. 2). There also exists strong S⋯O interactions, between the oxygen of dithiol-2-one and the sulfur of the thiocyanate group along the c axis, with a S⋯O distance of 2.911 Å indicating a reduction ratio of 87.6% compared to the sum of the vdW radii (3.32 Å). Weaker S⋯O interactions (3.168 Å, RR: 95%) are observed along the a axis between the oxygen atom of the dithiol-2-one and the sulfur atom of the neighboring molecule.
. There is no relevant interatomic contacts shorter than 95% of the sum of the van der Waals radii between neighboring molecules indicating none or weak non-covalent intermolecular interactions. Contrariwise, for 2e the presence of the thiocyanate group induces some intermolecular interactions. The crystal structure of 2e is solved in the monoclinic system, space group C2/c, with the molecule in general position. The molecules form “dimers” in the ac plane, stacking along the b axis. The “dimers” are related by an inversion center, and are held by chalcogen bonding between the nitrogen of the thiocyanate group (N2) as ChB acceptor of one molecule, and two opposite sulfur atoms (S3, S2) as ChB donors from the other molecule (Fig. 2 and Table 2). The C–S⋯N angles are in the range of 170°, and the S⋯N contacts are short with a 90% reduction ratio relative to the sum of the vdW radii (Fig. 2). There also exists strong S⋯O interactions, between the oxygen of dithiol-2-one and the sulfur of the thiocyanate group along the c axis, with a S⋯O distance of 2.911 Å indicating a reduction ratio of 87.6% compared to the sum of the vdW radii (3.32 Å). Weaker S⋯O interactions (3.168 Å, RR: 95%) are observed along the a axis between the oxygen atom of the dithiol-2-one and the sulfur atom of the neighboring molecule.
 |
| | Fig. 2 Intermolecular short contacts with the indicated reduction ratio relative to the sum of the vdW radii of the atoms for 2e (left) and 2f with hydrogen atoms omitted for clarity (right). | |
Table 2 Relevant interatomic distances in Å and angles (°) chalcogen bonding interactions
| Distances (Å) |
Type |
RR% |
Angles (°) |
| For 2e |
| S2⋯N2 3.051(2) |
ChB |
91.0 |
C1–S2⋯N2 172.8(6) |
C5–N2⋯S2 162.9(7) |
| S3⋯N2 3.037(2) |
ChB |
90.6 |
C4–S3⋯N2 168.6(9) |
C5–N2⋯S3 132.3(1) |
| S1⋯O1 3.168(2) |
ChB |
95.4 |
C1–S1⋯O1 136.1(3) |
C1–O1⋯S1 101.4(6) |
| S4⋯O1 2.911(0) |
ChB |
87.6 |
C4–S4⋯O1 162.8(8) |
C1–O1⋯S4 163.7(5) |
| For 2f |
| S2⋯O1 2.862(2) |
ChB |
86.1 |
C2–S2⋯O1 177.4(2) |
C2–O1⋯S2 172.2(2) |
| S3⋯O1 3.178(3) |
ChB |
95.4 |
C6–S3⋯O1 166.3(1) |
C2–O1⋯S3 110.6(2) |
| S1⋯S4 3.395(2) |
ChB |
94.3 |
C2–S1⋯S4 175.3(1) |
C6–S4⋯S1 129.4(1) |
For 2f, which crystallizes in the monoclinic system, space group P21/c, a closer look at the structure shows the existence of chalcogen bonding interactions, involving the sulfur atoms of both heterocycles as ChB donors toward the O atom and the S-Me substituent as ChB acceptors, with a short S4⋯O1 interatomic distance of 2.862 Å which corresponds to a reduction ration of 86.1% relative to the van der Waals contact distance (3.32 Å). This chalcogen bonding interaction is reminiscent of what was previously observed with the iodo analogue 2c where a short S⋯O interatomic distance of 2.940 Å corresponding to a reduction ratio of 88.5% was observed.19,23
Concerning the neutral TTFs, crystals were obtained by slow evaporation of concentrated dichloromethane solution of the symmetrical TTF, 3d, and the three dissymmetrical TTF-R, 4a, 4b, 4e (R = SCH2CH2CN, H, and SCN respectively). The molecular structures of these TTFs are presented in Fig. 3. The crystal structure of 3d is solved in the monoclinic system, space group P21/a, with the molecule in general position. The donor core is essentially planar with the two thioethyl substituents in the plane of the molecule. One TTF moiety is disordered on two positions (89![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :11) compatible with the presence of the two configurations, Z and E. The molecules organize into face-to-face inversion-centered dimers with a plane-to-plane distance of 3.50 Å, slightly shorter that the S⋯S van der Waals contact distance (Fig. S1†). The dimers adopt a herringbone organization (Fig. S1b†), reminiscent of the kappa-type phases observed in BEDT-TTF salts.24 No other short intermolecular contacts are identified.
:11) compatible with the presence of the two configurations, Z and E. The molecules organize into face-to-face inversion-centered dimers with a plane-to-plane distance of 3.50 Å, slightly shorter that the S⋯S van der Waals contact distance (Fig. S1†). The dimers adopt a herringbone organization (Fig. S1b†), reminiscent of the kappa-type phases observed in BEDT-TTF salts.24 No other short intermolecular contacts are identified.
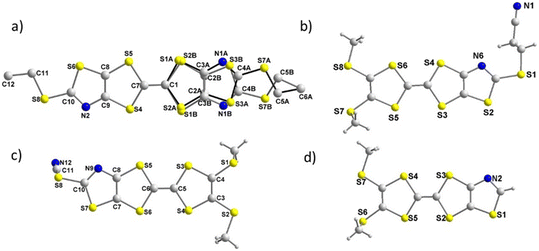 |
| | Fig. 3 Molecular structures of symmetrical TTF 3d (a) and dissymmetrical TTFs 4a (b) 4e (c) and 4b (d). | |
The crystal structure of the dissymmetrically substituted TTFs are solved in the triclinic system, space group P. Analysis of the structure reveals some similarities for the three investigated TTFs, such as a planar TTF core with one thiomethyl group in the plane of the TTF while the other one points above the plane. Concerning the TTF 4a, the cyanoethyl group is located below the plane (Fig. 3). A closer look at the interatomic distances in 4a indicates that no contact shorter than the sum of the van der Waals radii can be observed between adjacent molecules. The molecules organize through π–π stacking into uniform stacks running along a axis with a plane-to-plane distance of 3.60 Å, i.e. at van der Waals contact distance (Fig. S2†).
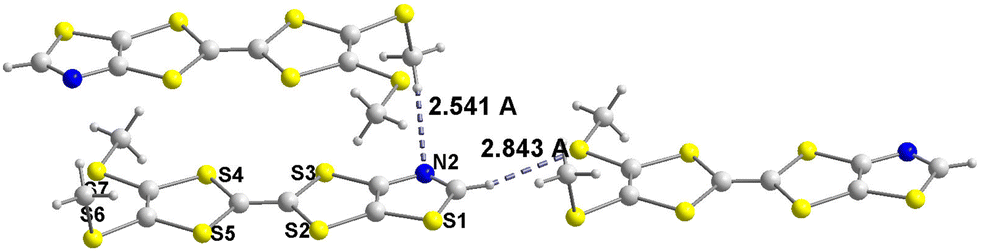
Contrariwise, for 4b, as shown in Fig. 4, two main short interatomic distances can be observed between adjacent TTFs. These short contacts involve the hydrogen of the methyl group of one TTF as HB donor with the nitrogen atom (N2) of the second TTF as HB acceptor, with a H⋯N distance of 2.541 Å, and the hydrogen atom of the thiazole ring on one TTF with the sulfur (S7) atom of another TTF, with a H⋯S distance of 2.843 Å (Fig. 4). Both distances are shorter than the van der Waals contact distances which amount to 2.75 Å for H⋯N (1.2 + 1.55) and to 3 Å for H⋯S (1.2 + 1.8) which leads to a reduction ratio of 92.4% for H⋯N and 94.7% for S⋯H. Moreover the C–H⋯N and C–H⋯S angles amount to 176.3(2)° and 160.4(1)° respectively indicating the existence of hydrogen bonding interaction (HB) between neighboring TTFs. Concerning TTF 4e, no intermolecular interactions through chalcogen bonding involving the sulfur atom of the thiocyanate group is observed, in contrast to the strong ones observed for 2e. Molecules are organized into strongly dimerized stacks, with a plane-to-plane distance of 3.50 Å within face-to-face dimers and a longer 4.00 Å plane-to-plane-distance between dimers (Fig. S3†). No other characteristics intermolecular short contacts are identified.
 |
| | Fig. 4 Hydrogen bonding interactions in TTF 4b. | |
Charge transfer complexes
The oxidation potentials of all the obtained TTFs are too high for using the TCNQ as oxidizing agent. Indeed, TCNQ is reduced at 0.18 V vs. SCE and all the TTFs 3 and 4 are oxidized above 0.53 V vs. SCE and with such potential difference no redox reaction is expected between the TCNQ and the TTFs. In order to generate a charge transfer complex, we decided to use a stronger electron acceptor such as TCNQF4 which is reduced at 0.53 V vs. SCE. Crystals were obtained when mixing 4b and 4c with TCNQF4. For the complex obtained with 4c, it crystallizes in the triclinic system, space group P, with one TTF in general position in the unit cell and the TCNQF4 on an inversion center. The crystal structure determination indicates a stoichiometry of two donors for one TCNQF4, (4c)2(TCNQF4). Within this complex, dyads of TTFs alternate with TCNQF4 in a –(D2–A)x– motif.25 Within the donor dyad the TTF are organized head to tail with a bond over ring overlap (Fig. 5). Several short intermolecular contacts can be observed within the ac plane: i) I⋯S2 between the iodo atom of one TTF and the sulfur atom of the thiomethyl group of a neighboring TTF (3.404 Å) and ii) F⋯S between a fluorine atom of one TCNQF4 and the sulfur atom within a dithiole ring of the neighboring TTF (3.122 Å). The bond distances, reduction ratio and angles are collected in Table 3. Chalcogen interactions are also found: i) short S⋯S distances between the sulfur atom of one TTF and the sulfur atom of the neighboring TTF (3.279 Å) and ii) S⋯N between the nitrogen atom of one TCNQF4 and the sulfur atom of the neighboring TTF (3.019 Å) (Fig. 5).
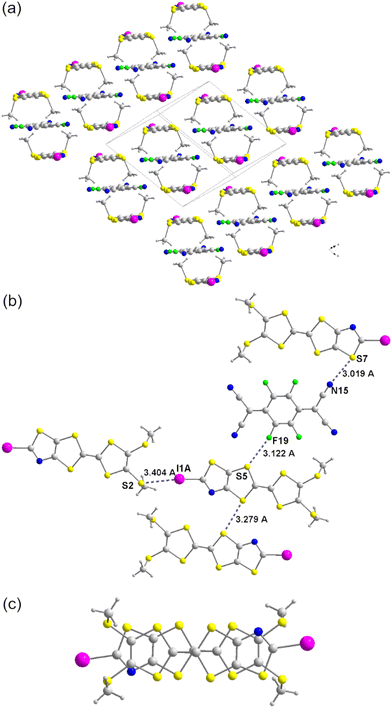 |
| | Fig. 5 Projection view of (4c)2(TCNQF4) complex along the long axis of TTF and TCNQF4 showing the –(D2–A)x– motif (a). View of the short contacts observed in the organization of (4c)2(TCNQF4) along the ac plane (b) and view of the TTF dyad overlap (c). | |
Table 3 Relevant interatomic distances (Å), reduction ratio (RR) and angles (°) for the halogen bonding and chalcogen interactions in (4c)2(TCNQF4). The van der Waals contact distances amount to S⋯I: 3.78 Å, S⋯S: 3.60 Å, S⋯F: 3.27 Å, S⋯N: 3.35 Å
|
|
Distances (Å) |
RR |
Angles (°) |
| (4c)2(TCNQF4) |
S6⋯S6 3.279 |
91% |
C–S6⋯S6 164.7 |
| S7⋯N15 3.019 |
95% |
C–S7⋯N15 163.4 |
| S2⋯I1 3.404 |
90% |
C–I1⋯S2 172.7 |
| S5⋯F19 3.122 |
95% |
C–F19⋯S5 166.5 |
The charge transfer complex obtained with 4b crystallizes in the triclinic system, space group P. Crystal structure determination indicates a stoichiometry of one donor for one TCNQF4, (4b)(TCNQF4), with 4b and TCNQF4 organized in segregated stacks (Fig. 6). The thiazole ring of the TTF is disordered on two positions (86:14). Within the donor stacks, the dissymmetrical TTFs are organized head to tail with the thiazole ring of one TTF over the bisthiomethyl substituents of the dithiole ring of the neighboring TTF. The stacks are not uniform as shown by the two distances measured between the planes of the TTF, 3.575 Å and 3.355 Å indicating that the 4b are actually dimerized along the stacking axis. Similarly the TCNQF4 form dimerized stacks with a short interplanar distance within the dimer (3.086 Å) and an almost eclipsed overlap. A lateral slip exist between neighboring dimers with a longer interplanar distance of 3.399 Å. No specific hydrogen, halogen or chalcogen interactions can be observed within this complex.
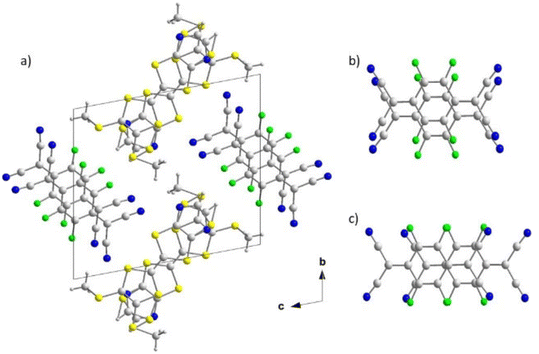 |
| | Fig. 6 View of the unit cell of (4b)(TCNQF4) along the bc plane (a) and view of the TCNQF4 intra-dimer (b) and inter-dimer (c) overlap patterns in the acceptor stacks. | |
In order to determine the degree of charge transfer within (4b)(TCNQF4) and (4c)2(TCNQF4) complexes, we analyzed the bond lengths of the TCNQF4 skeleton. For that purpose we collected in Table 4 the bond lengths of the neutral TCNQF4 and the bond length of the TCNQF4 involved in each complex. As can be seen, the acceptor bond lengths within our complex are different than those reported for the neutral TCNQF4 indicating that these complexes are not neutral. The charge of TCNQF4 can be estimated by the Kistenmacher relationship first established for TCNQ salts by Kistenmacher et al.,26 and adapted to TCNQF4 by Miyasaka et al.27 It is estimated from the bond lengths b, c and d of the acceptor through the equation ρTCNQF4 = A[c/(b + d)] + B, where A = −46.729 and B = 22.308.
Table 4 TCNQF4 bond lengths in Å
|

|
a
|
b
|
C
|
d
|
ρ
TCNQF4 (calc) |
| TCNQF40 |
1.334 |
1.437 |
1.372 |
1.437 |
0 |
| (4c)2(TCNQF4) |
1.352 |
1.419 |
1.409 |
1.432 |
−0.786 |
| (4b)(TCNQF4) |
1.356 |
1.414 |
1.410 |
1.425 |
−0.90 |
| (TBA)(TCNQF4)28 |
1.357 |
1.417 |
1.418 |
1.425 |
−1 |
From this equation we found a charge of −0.78 on the TCNQF4 for (4c)2(TCNQF4) and a charge of −0.90 on the TCNQF4 for (4b)(TCNQF4) confirming that both complexes are charge transfer salts. Based on the nitrile stretching absorption band of TCNQF4, it is also possible to infer the degree of charge transfer in those TCNQF4 complexes. The value obtained for the neutral TCNQF4 is observed at υCN 2228 cm−1 and the value for the radical anion TCNQF4˙− at υCN 2190 cm−1.29 The value obtained for the two complexes amounts to 2189 cm−1 which is close to that observed for the radical anion TCNQF4˙−. This is consistent with the values determined through the bond distances analysis in TCNQF4. Moreover if we compare the bond length of the neutral 4b (see above) with the one of the TTF within (4b)(TCNQF4), we can see that the central C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bond is lengthened in the charge transfer salt in accordance with the presence of an oxidized TTF (1.344 Å in 4b and 1.385 Å in (4b)(TCNQF4)). Thus this complex is a fully ionic complex. Resistivity measurements were performed on (4b)(TCNQF4) and the room temperature resistivity was found to be 1.5 × 104 Ω cm. This high resistivity is essentially a consequence of the strong dimerization within the TTF stacks.
C bond is lengthened in the charge transfer salt in accordance with the presence of an oxidized TTF (1.344 Å in 4b and 1.385 Å in (4b)(TCNQF4)). Thus this complex is a fully ionic complex. Resistivity measurements were performed on (4b)(TCNQF4) and the room temperature resistivity was found to be 1.5 × 104 Ω cm. This high resistivity is essentially a consequence of the strong dimerization within the TTF stacks.
Electrostatic potential surface study
Electrostatic potential (ESP) surface calculations have been carried out on the DFT-optimized geometry of the precursors that have been crystallographically characterized, namely 2e, and 2f as well as on three TTFs, 4b, 4c and 4e (R = H, I, SCN). These calculations were performed in order to rationalize the interactions taking place in the crystal.
As shown in Fig. 7, for 2e and 2f the positive extremum of the ESP surface is found close to the carbon atom connecting the thiazole and the dithiole rings (+40.2 kcal mol−1 for 2e and +32.0 for 2f) and to a lesser extent on the side of the sulfur atom of the S–CN fragment (+33.0 kcal mol−1 for 2e). The location of this positive charge on the sulfur atom, on the opposite side of the thiazole ring, indicates that the thiazole ring exerts a stronger electron withdrawing effect than the cyano group. For 2f interestingly, a non-negligible positive ESP is also found on the hydrogen atoms of the thiomethyl substituent. The most negative extremum is −26.3 kcal mol−1 and is located on the nitrogen atom of the thiocyanate group and to a lesser extent on the oxygen atom of the dithiol-2-one ring (−24.0 kcal per mole) for 2e while for 2f the most negative extremum is −29.2 kcal mol−1 and is located on the oxygen atom. This calculated charge repartition is in good agreement with the organization of the molecules 2e and 2f in the solid state where predominant ChB interactions (S⋯O) are observed together with S⋯N interactions for 2e.
 |
| | Fig. 7 Molecular electrostatic potential surfaces mapped at 0.001 e− au−3 isodensity surface for (left) 2e and (right) 2f. The common color scale ranges from −28 kcal mol−1 (red) to +40 kcal mol−1 (blue). | |
The hydrogen and halogen bond donor abilities of the TTF derivatives can also be estimated via the calculation of the ESP and especially its magnitude at the σ-holes. These calculations have been performed on 4b, 4c and 4e in order to analyze the impact of the R group. As shown in Fig. 8, the most depleted charged area is associated with the σ-hole on the iodine atom of 4c and on the σ-hole of the hydrogen atom of 4b. Both TTF exhibit also a charge depletion on the hydrogen atom of the thiomethyl substituents as shown for 4b in Fig. 8. For 4b and 4c, the most negative extremum, is located on the nitrogen atom of the thiazole ring and to a lesser extent on the sulfur atoms of the thiomethyl substituent (−23.7 kcal mol−1 for 4b). This charge distribution is consistent with the obtained solid-state arrangement, and the preferred HB interactions observed for 4b (Fig. 4) but not for 4e.
 |
| | Fig. 8 Molecular electrostatic potential surfaces mapped at 0.001 e− au−3 isodensity surface for 4b (TTF-H), 4c (TTF-I), and 4e (TTF-SCN). The common color scale ranges from −37 kcal mol−1 (red) to +32.6 kcal mol−1 (blue). | |
The electrostatic surface potential at the iodine atom of 4c is found to logically increase with the charge of molecule from +32.0 to +82.8 kcal mol−1 for the neutral, radical cation respectively (blue dot on iodine in Fig. 8 and 9). Similarly on 4b the charge depletion on the hydrogen atom is increased on the 4b cation radical. Another interesting observation is that the negative charge at the nitrogen atom of the thiocyanate of 4e is reversed (from −36.4 to +13.1 kcal mol−1) as the molecule is oxidized. As a charge of +0.5 is borne out by each TTF in the complex (4c)2(TCNQF4) it is not possible to draw the corresponding ESP surface. However the fact that intermolecular interactions exist between the iodine atom of one 4c+0.5 and the sulfur atom of the neighboring 4c+0.5 molecule is consistent with the charge distribution.
 |
| | Fig. 9 Molecular electrostatic potential surfaces mapped at 0.001 e− au−3 isodensity surface for the cation radical state of 4b+˙ (TTF-H+˙), 4c+˙ (TTF-I+˙) and 4e+˙ (TTF-SCN+˙). The common color scale ranges from 13.13 kcal mol−1 (red) to +98.5 kcal mol−1 (blue). | |
Conclusions
Within this work, we have designed TTF precursors with both, hydrogen, halogen and chalcogen bonding, donor and acceptor sites thanks to an original transformation of the N-tBu-1,3-thiazoline-2-thione fused dithiol-2-one. For that purpose, we prepared the precursors with a dithiole-2-one ring that serves as a TTF building block substituted either with cyanoethyl groups, iodine atoms, thiocyanate-groups or simply an H atom. Symmetrically and dissymmetrically substituted TTFs able to form hydrogen, halogen and chalcogen bonds depending on the substitution pattern have been synthesized. Symmetrical TTFs were prepared by a homocoupling of the dithiole-2-one precursors, while the dissymmetrical TTFs were formed, in higher yields, through a heterocoupling. All the synthesized TTFs were studied by cyclic voltammetry, and the effect of the different functional groups on the redox potentials were determined. Structural investigations of the precursors and the TTFs highlight some differences in the intermolecular interactions which could be corroborated with ESP calculations. Charge transfer salts using TCNQF4 as an acceptor were also prepared and X-ray diffraction studies of these salts, (4c)2(TCNQF4) and (4b)(TCNQF4), show different stacking mode. Investigation of the bond length allowed us to determine that (4b)(TCNQF4), is fully ionic while a partial degree of charge transfer was found in (4c)2(TCNQF4). In the latter salt strong non-covalent interactions between the donor molecules themselves and between the donor and acceptor moieties have been evidenced. We have identified and characterized intermolecular non-covalent interactions through analysis of intermolecular angles and distances in the crystal structure that are significantly shorter than the sum of the van der Waals radii. This has been supported by calculations of electrostatic surface potential on the DFT optimized structure of the molecules. To further characterize and quantify non-covalent bonds in single crystals, application of Bader's theory of atoms in molecules (AIM)30 is planned for our subsequent works.
Experimental section
General information
All commercial chemicals were used without further purification. The solvents were purified and dried by standard methods. NMR spectra were obtained in CDCl3 unless indicated otherwise. Chemical shifts are reported in ppm, 1H NMR spectra were referenced to residual CHCl3 (7.26 ppm) and 13C NMR spectra were referenced to CHCl3 (77.2 ppm). Compounds 2b and 2e were prepared in degassed 99% DMSO. Melting points were measured on a Kofler hot-stage apparatus and are uncorrected. Mass spectra were recorded at the Centre Régional de Mesures Physiques de l'Ouest, Rennes. Elemental analyses were performed at the Service de Microanalyse, Gif sur Yvette. Cyclic voltammetry were carried out on a 10−3 M solution in CH2Cl2, containing 0.1 M nBu4NPF6 as supporting electrolyte. Voltammograms were recorded at 0.1 V s−1 on a platinum electrode and the potentials were measured versus the saturated calomel electrode (SCE). The starting derivative 1 (ref. 17) as well as 2c19 and 2d17 were prepared according to the procedure previously described.
3-((2-Oxo-[1,3]dithiolo[4,5-d]thiazol-5-yl)thio)propanenitrile 2a
Under inert atmosphere, to a solution of 1 (600 mg; 2.28 mmol) in 8 mL of degassed DMF, 3-bromopropionitrile (1.52 mL; 18.16 mmol) was added. The reaction mixture was stirred at 70 °C for 48 hours. The reaction was quenched with 200 mL of water and the product was extracted with CH2Cl2. The organic phase was washed 10 times with water and dried over MgSO4. The concentrated solution was purified by flash chromatography using a mixture of petroleum ether and CH2Cl2 starting from 0 to 15% of CH2Cl2 to afford 2a as orange powder in 63% yield (m = 375 mg). Crystals of sufficient quality for X-ray diffraction were obtained by slow evaporation of a concentrated dichloromethane solution. Rf = 0.37, (SiO2, CH2Cl2); mp = 103 °C; 1H NMR (300 MHz) δ 3.50 (t, 3J = 6.9 Hz, 2H), 2.95 (t, 3J = 6.9 Hz, 2H); 13C NMR (75 MHz) δ 190.0 (CO), 164.3 (CN), 142.2 (CC), 117.5 (CC), 117.0 (![[C with combining low line]](https://www.rsc.org/images/entities/char_0043_0332.gif) N), 30.0 (S–CH2), 18.6 (H2CN); HRMS (ASAP) calcd for [M]+˙: 259.9201; found: 259.9199; anal. calcd for C7H4N2OS4: C, 32.29; H, 1.55; N, 10.76; S, 49.25; found: C, 32.17; H, 1.80; N, 10.28; S, 49.26.
N), 30.0 (S–CH2), 18.6 (H2CN); HRMS (ASAP) calcd for [M]+˙: 259.9201; found: 259.9199; anal. calcd for C7H4N2OS4: C, 32.29; H, 1.55; N, 10.76; S, 49.25; found: C, 32.17; H, 1.80; N, 10.28; S, 49.26.
[1,3]Dithiolo[4,5-d]thiazol-2-one 2b
Under inert atmosphere, to a solution of 1 (300 mg; 1.14 mmol) in 4 mL of degassed DMSO, 3-bromopropionitrile (0.76 mL; 9.04 mmol) was added. The reaction mixture was stirred at 55 °C for 48 hours. The reaction was quenched with 200 mL of water, and the product was extracted with CH2Cl2. The organic phase was washed 10 times with water and dried over MgSO4. The concentrated solution was purified by flash chromatography using a mixture of petroleum ether and CH2Cl2 starting from 0 to 15% of CH2Cl2 to afford 2b as white powder in 55% yield (m = 110 mg); Rf = 0.56, (SiO2, CH2Cl2); mp = 120 °C; 1H NMR (300 MHz) δ 8.97 (s, 1H); 13C NMR (75 MHz) δ 190.7 (CO), 153.5 (NC–H), 143.9 (CC), 118.1 (CC); HRMS (ESI) calcd for C4H2NOS3+: 175.9293. Found: 175.9294.19
5-Thiocyanato-[1,3]dithiolo[4,5-d]thiazol-2-one 2e
Under inert atmosphere, to a solution of 1 (300 mg, 1.14 mmol) in 3 mL of DMSO, cyanogen bromide (0.97 g, 9.1 mmol), was added. The reaction mixture was let to stir at 55 °C for 16 hours. The reaction was then quenched with water (200 mL), the product was then extracted with CH2Cl2 and the organic phase was washed with water and then dried over MgSO4. The solvent was evaporated under vacuum and the crude product was purified by flash chromatography using CH2Cl2/petroleum ether (40:60) as eluent giving compound 2e as a white powder. Yield = 50% (133 mg); Rf = 0.52, (SiO2, CH2Cl2); mp = 132 °C. 13C NMR (75 MHz) δ 188.3 (CO), 150.0 (C–S–CN), 144.5 (CC), 123.5 (CN), 106.4 (CC). HRMS (ASAP) calcd for C5HN2OS4+: 232.89662, found: 232.8969; anal. calcd for C5N2OS4: C, 25.85; N, 12.06. Found: C, 25.87; N, 12.34.
TTF 3a
Under inert atmosphere, a solution of 2a (100 mg; 0.38 mmol) in 5 mL of distilled triethylphosphite was stirred at 120 °C for 5 hours. The solvent was evaporated in vacuo and the residue was extracted with CH2Cl2. The organic phase was washed with water and dried over MgSO4. The concentrated solution was purified by chromatography using CH2Cl2 as eluent to afford the expected TTF as an orange powder. Yield = 22% (m = 21 mg); Rf = 0.17 (SiO2, CH2Cl2); mp = 220 °C; 1H NMR (400 MHz) δ 3.46 (t, 3J = 7.1 Hz, 4H), 2.93 (t, 3J = 7.1 Hz, 4H). HRMS (ASAP) calcd for [C14H8N4S8 + H]+: 438.85875; found: 488.8590.
TTF 3b
Under inert atmosphere, a solution of 2b (60 mg; 0.34 mmol) in 5 mL of distilled triethylphosphite was stirred at 120 °C for 2 hours. An orange precipitate was observed. The precipitate was filtered off and washed with methanol to afford 3b as a brown powder. Yield = 24% (m = 13 mg); Rf = 0.33 (SiO2, CH2Cl2); mp = >260 °C; 1H NMR (400 MHz, DMSO) δ (ppm) = 9.10 (s, 2H); 13C NMR (75 MHz, DMSO) δ (ppm) = 159.8 (H), 147.5 (CC), 123.7 (CC), 123.4 (CC). HRMS (ASAP) calcd for [M + H] + C8H3N2S6: 318.8615; found: 318.8617; anal. calcd for C8H2N2S6·CH3OH: C, 30.84; H, 1.73; N, 7.99. Found: C, 30.98; H, 1.41; N, 7.39.
TTF 3c
Under inert atmosphere, a solution of 2c (60 mg; 0.19 mmol) in 5 mL of distilled triethylphosphite was stirred at 120 °C for 2 hours. The solvent was evaporated in vacuo and the residue was extracted with CH2Cl2. The organic phase was washed with water and dried over MgSO4. The concentrated solution was purified by chromatography using CH2Cl2 as eluent to afford 3c as an orange powder. Yield = 23% (m = 13 mg); Mp > 240 °C; HRMS (ESI) calcd for [C8N2I2S8+˙]: 569.64698, found: 569.6474.
TTF 3d
In a two necked flask purged with argon, the bicyclic derivative 2d (100 mg, 0.42 mmol) was solubilized in 3 mL of degassed chlorobenzene, 0.5 mL of triethylphosphite was then added to the solution followed by 3 hours at reflux. The solvent was then evaporated and the product was purified using flash chromatography (eluent: CH2Cl2/petroleum ether; 20:80), after evaporation of the solvent the product was obtained as an orange powder which was recrystallized in CH2Cl2 to afford bright orange needles. Yield = 27% (25 mg); Rf = 0.52, (SiO2, CH2Cl2); mp = 205 °C; 1H NMR (300 MHz) δ 3.17 (q, 3J = 7.3 Hz, 4H), 1.41 (t, 3J = 7.3 Hz, 6H); HRMS (ESI) calcd for [A+˙] C12H10N2S8: 437.86042, found: 437.8603; anal. calcd for C12H10N2S8: C, 32.85; H, 2.30; N, 6.39. Found: C, 32.62; H, 2.17; N, 6.32.
General synthesis of TTFs 4a–d through the heterocoupling
Under inert atmosphere, a solution of 2a–c (0.22 mmol, 2a 57 mg, 2b 38 mg, 2c 66 mg) and 3 (50 mg; 0.22 mmol) in 5 mL of distilled triethylphosphite was stirred at 120 °C for 2 hours. The solvent was evaporated in vacuo and the residue was extracted with CH2Cl2. The organic phase was washed with water and dried over MgSO4.
TTF 4a
The reaction mixture was purified by chromatography using CH2Cl2 as eluent to afford the TTF as an orange powder. Crystals of sufficient quality for X-ray diffraction were obtained by slow evaporation of a concentrated dichloromethane solution. Yield = 24% (m = 23 mg); Rf = 0.67 (SiO2, CH2Cl2); mp = 173 °C; 1H NMR (300 MHz, CD2Cl2) δ 3.40 (t, 3J = 6.9 Hz, 2H), 2.89 (t, 3J = 7.0 Hz, 2H), 2.44 (s, SC![[H with combining low line]](https://www.rsc.org/images/entities/char_0048_0332.gif)
![[3 with combining low line]](https://www.rsc.org/images/entities/char_0033_0332.gif) ), 2.43 (s, SC); 13C NMR (75 MHz, CD2Cl2) δ 163.8(CC), 148.1 (CN), 127.7 (CC), 127.2 (CC), 117.4 (CN), 112.4 (CC), 30.5 (SCH2), 18.8 (C–CH2) 18.3 (CH3); HRMS (ASAP) calcd for [C12H10N2S8]+˙: 437.86097. Found: 437.86042; anal. calcd for C12H10N2S8·0.5CH2Cl2: C, 31.20; H, 2.30; N, 5.82. Found: C, 30.82; H, 2.00; N, 6.00.
), 2.43 (s, SC); 13C NMR (75 MHz, CD2Cl2) δ 163.8(CC), 148.1 (CN), 127.7 (CC), 127.2 (CC), 117.4 (CN), 112.4 (CC), 30.5 (SCH2), 18.8 (C–CH2) 18.3 (CH3); HRMS (ASAP) calcd for [C12H10N2S8]+˙: 437.86097. Found: 437.86042; anal. calcd for C12H10N2S8·0.5CH2Cl2: C, 31.20; H, 2.30; N, 5.82. Found: C, 30.82; H, 2.00; N, 6.00.
TTF 4b
The concentrated solution was purified by chromatography using CH2Cl2 as eluent to afford 4b as a red powder. Crystals of sufficient quality for X-ray diffraction were obtained by slow evaporation of a concentrated dichloromethane solution. Yield = 38% (m = 30 mg); Rf = 0.6 (SiO2, CH2Cl2); mp = 125 °C; 1H NMR (400 MHz) δ 8.68 (s, 1H), 2.44 (s, SC), 2.43 (s, SC); 13C NMR (75 MHz) δ 154.8 (C–H), 150.6 (CC), 128.1 (CC), 127.1 (CC), 122.1 (CC), 114.2 (CC), 19.2 (CH3); HRMS (ASAP) calcd for [C9H7NS7 + H]+: 353.86963. Found: 353.8693; anal. calcd for C9H7NS7: C, 30.57; H, 2.00; N, 3.96. Found: C, 30.08; H, 1.82; N, 3.80.
TTF 4c
The concentrated solution was purified by chromatography using CH2Cl2/petroleum ether (60/40) as eluent to afford 4c as a red powder. Yield = 19% (m = 20 mg); Rf = 0.83 (SiO2, CH2Cl2); mp = 147 °C; 1H NMR (300 MHz) δ 2.44 (s, SC), 2.43 (s, SC); 13C NMR (75 MHz) δ 155.2 (C–I), 149.6 (CN), 128.7 (CC), 128.2 (CC), 127.0 (CC), 126.1 (CC), 100.7 (CC), 19.3 (SH3); HRMS (ASAP) calcd for [C9H6NIS7]+: 478.75845; found: 478.7588.
TTF 4d
Under inert atmosphere, a solution of 1 (58 mg; 0.22 mmol) and 3 (50 mg; 0.22 mmol) in 10 mL of distilled triethylphosphite was stirred at 120 °C for 2 hours. The solvent was evaporated in vacuo and the residue was extracted with CH2Cl2. The organic phase was washed with water and dried over MgSO4. The concentrated solution was purified by chromatography using CH2Cl2/petroleum ether (90/10) as eluent to afford 4d as a red powder. Yield = 46% (m = 42 mg); Rf = 0.73 (SiO2, CH2Cl2); mp = 100 °C; 1H NMR (400 MHz) δ 3.17 (q, 3J = 7.3 Hz, C![[2 with combining low line]](https://www.rsc.org/images/entities/char_0032_0332.gif) CH3), 2.44 (s, 6H, SC), 1.40 (t, 3J = 7.3 Hz, CH2C); 13C NMR (75 MHz) δ 166.9 (CC), 148.3 (SEt), 128.1 (CC), 127.3 (CC), 120.4 (CC), 113.7 (CC), 30.2 (H2), 19.4 (SH3), 14.7(CH2H3); HRMS (ESI) calcd for [C11H11NS8Na]+: 435.85494; found: 435.8550; anal. calcd for C11H11NS8: C, 31.94; H, 2.68; N, 3.39. Found: C, 31.66; H, 2.05; N, 3.28.
CH3), 2.44 (s, 6H, SC), 1.40 (t, 3J = 7.3 Hz, CH2C); 13C NMR (75 MHz) δ 166.9 (CC), 148.3 (SEt), 128.1 (CC), 127.3 (CC), 120.4 (CC), 113.7 (CC), 30.2 (H2), 19.4 (SH3), 14.7(CH2H3); HRMS (ESI) calcd for [C11H11NS8Na]+: 435.85494; found: 435.8550; anal. calcd for C11H11NS8: C, 31.94; H, 2.68; N, 3.39. Found: C, 31.66; H, 2.05; N, 3.28.
TTF 4e
To a solution of 4a (65 mg; 0.148 mmol) in 4 mL DMF under inert atmosphere was added dropwise a solution of caesium hydroxide (24.4 mg; 0.163 mmol) in 1 mL of degassed methanol. The mixture was stirred 30 min then a solution of cyanogen bromide (23 mg, 0.22 mmol) in 2 mL of degassed DMF was added. The mixture was stirred 1 hour and the reaction was quenched with 100 mL water, the organic phase was extracted with CH2Cl2, and dried over MgSO4. The solvent was evaporated in vacuo and the residue was purified by chromatography using CH2Cl2 as eluent to afford 4e as a red powder. Yield = 70% (m = 43 mg); Rf = 0.80 (SiO2, CH2Cl2); mp = 148 °C; 1H NMR (300 MHz) δ 2.62 (s, 6H). 13C NMR (75 MHz, CD2Cl2) δ (ppm) = 151.1 (CC), 147.8 (CN), 128.9 (CC), 128.2 (CC), 127.2 (CC), 116.6 (CN), 110.8 (CC), 106.9 (CC), 19.3 (CH3), 19.2 (CH3). HRMS (ASAP) calcd for [C10H6N2S8]+˙: 409.82912. Found: 409.8091; anal. calcd for C10H6N2S8: C, 29.25; H, 1.47; N, 6.82. Found: C, 29.73; H, 1.44; N, 6.32.
Charge transfer salt
(4B)(TCNQF4).
A mixture of 4B (10.16 mg, 0.028 mmol) and TCNQF4 (7.66 mg, 0.028 mmol) in 15 mL of acetonitrile and 4 mL of dichloromethane was left at refluxed for 10 min. The solution was filtered and slow concentration of the solution afforded crystals of sufficient quality for X ray diffraction.
(4c)2(TCNQF4).
A mixture of 4c (8.2 mg, 0.017 mmol) and TCNQF4 (4.60 mg, 0.017 mmol) in 15 mL of acetonitrile and 4 mL of dichloromethane was left at refluxed for 10 min. The solution was filtered and slow concentration of the solution afforded crystals of sufficient quality for X ray diffraction.
X-ray crystallographic analysis
Data were collected on a D8 VENTURE Bruker AXS diffractometer, equipped with a (CMOS) Photon 100 detector, for all the crystals except for 3d where data collection was performed on an APEXII Bruker-AXS diffractometer, using Mo Kα radiation (λ = 0.71073 Å, multilayer monochromator). The structures were solved by dual-space algorithm using the SHELXT program,31 and the refined with full-matrix least-square methods based on F2 (SHELXL).32 All non-hydrogen atoms were refined with anisotropic atomic displacement parameters. H atoms were finally included in their calculated positions. Crystallographic data on X-ray data collection and structure refinements are given in Table 5. CCDC 2245494–2245502 contain the supplementary crystallographic data for this paper.
Table 5 Crystallographic data
| Compound |
2a
|
2e
|
2f
|
3d
|
| CCDC |
2245494
|
2245495
|
2245496
|
2245497
|
| Formula |
C7H4N2OS4 |
C5N2OS4 |
C5H3NOS4 |
C12H10N2S8 |
| FW (g mol−1) |
260.36 |
232.31 |
221.32 |
438.7 |
| Crystal system |
Triclinic |
Monoclinic |
Monoclinic |
Monoclinic |
| Space group |
P |
C2/c |
P21/c |
P21/a |
|
a (Å) |
7.0683(15) |
19.606(4) |
9.1900(18) |
14.4980(13) |
|
b (Å) |
8.6879(17) |
3.8452(7) |
10.0186(17) |
7.5276(9) |
|
c (Å) |
9.462(2) |
20.687(4) |
8.6765(17) |
16.6742(17) |
|
α
|
111.186(8) |
90 |
90 |
90 |
|
β
|
108.771(8) |
91.669(5) |
98.049(7) |
111.731(5) |
|
γ
|
97.407(8) |
90 |
90 |
90 |
|
V (Å3) |
492.79(18) |
1558.9(5) |
791.0(3) |
1690.4(3) |
|
T (K) |
150(2) |
150(2) |
150(2) |
296(2) |
|
Z
|
2 |
8 |
4 |
4 |
|
D
calc (g cm−3) |
1.755 |
1.98 |
1.859 |
1.724 |
|
μ (mm−1) |
0.926 |
1.158 |
1.132 |
1.05 |
| Total refls. |
5511 |
5045 |
1775 |
14546 |
| Abs. Corr. |
Multi-scan |
Multi-scan |
Multi-scan |
Multi-scan |
| Uniq. refls. (Rint) |
2195(0.0356) |
1709(0.0567) |
1775 |
3876(0.0406) |
| Unique refls. (I > 2σ(I)) |
2084 |
1399 |
1607 |
2851 |
|
R
1, wR2 |
0.0274, 0.0766 |
0.0571, 0.1197 |
0.0385, 0.0981 |
0.0374, 0.0894 |
|
R
1, wR2 (all data) |
0.0287, 0.0787 |
0.0738, 0.1257 |
0.0455, 0.1036 |
0.060, 0.1013 |
| GoF |
1.029 |
1.09 |
1.167 |
1.049 |
| Compound |
4a
|
4b
|
4e
|
(4b)(TCNQF4) |
(4c)2(TCNQF4) |
| CCDC |
2245498
|
2245499
|
2245500
|
2245501
|
2245502
|
| Formula |
C12H10N2S8 |
C9H7NS7 |
C10H6N2S8 |
C21H7F4N5S7 |
C30H12F4 I2N6S14 |
| FW (g mol−1) |
438.70 |
353.58 |
410.65 |
629.74 |
1235.10 |
| Crystal system |
Triclinic |
Triclinic |
Triclinic |
Triclinic |
Triclinic |
| Space group |
P |
P |
P |
P |
P |
|
a (Å) |
6.0133(3) |
7.906(2) |
7.4506(12) |
7.1063(15) |
7.2339(10) |
|
b (Å) |
10.3719(7) |
8.704(3) |
8.1175(12) |
12.405(3) |
10.7451(15) |
|
c (Å) |
14.1989(9) |
11.633(3) |
13.345(2) |
13.462(4) |
13.7344(19) |
|
α
|
84.691(2) |
100.130(10) |
73.335(5) |
100.797(12) |
77.886(5) |
|
β
|
79.633(2) |
101.742(10) |
87.672(6) |
91.933(13) |
87.003(5) |
|
γ
|
85.606(2) |
113.220(10) |
81.049(6) |
92.390(9) |
72.421(5) |
|
V (Å3) |
865.76(9) |
690.4(3) |
763.8(2) |
1163.7(6) |
995.0(2) |
|
T (K) |
296(2) |
296(2) |
150(2) |
150(2) |
150(2) |
|
Z
|
2 |
2 |
2 |
2 |
1 |
|
D
calc (g cm−3) |
1.683 |
1.701 |
1.786 |
1.797 |
2.061 |
|
μ (mm−1) |
1.026 |
1.116 |
1.156 |
0.735 |
2.370 |
| Total refls. |
17937 |
14575 |
21489 |
24703 |
20058 |
| Abs. Corr. |
Multi-scan |
Multi-scan |
Multi-scan |
Multi-scan |
Multi-scan |
| Uniq. refls. (Rint) |
3918(0.0269) |
3075(0.0303) |
3500(0.0316) |
5303(0.0589) |
4514(0.0450) |
| Unique refls. (I > 2σ(I)) |
3494 |
2738 |
3295 |
4361 |
3913 |
|
R
1, wR2 |
0.0306, 0.0807 |
0.0274, 0.0687 |
0.0274, 0.0785 |
0.0436, 0.0899 |
0.0382, 0.0895 |
|
R
1, wR2 (all data) |
0.0349, 0.0839 |
0.0319, 0.0727 |
0.0291, 0.0806 |
0.0589, 0.0964 |
0.0469, 0.0940 |
| GoF |
1.062 |
1.042 |
1.039 |
1.009 |
1.081 |
Theoretical modeling
Electrostatic Surface Potential calculations were carried out on the optimized geometry of the molecules (with density functional theory using the Gaussian 09 Revision D.01 software, the B3LYP functional and the 6-31+G** basis set for all atoms and the LANLdp basis set for iodine). GaussView 5.0.9 was used to generate the figures.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work was financially supported by Université de Rennes 1 & through a PhD grant (to H. H.). This work was granted access to the HPC resources of CEA-TGCC under the allocation 2023 – AD010814136 awarded by GENCI.
References
-
(a) J. Ferraris, D. O. Cowan, V. T. Walatka and J. H. Perlstein, J. Am. Chem. Soc., 1973, 95, 948–949 CrossRef CAS;
(b) L. B. Coleman, M. J. Cohen, D. J. Sandman, F. G. Yamagishi, A. F. Garito and A. J. Heeger, Solid State Commun., 1973, 12, 1125–1132 CrossRef CAS.
- P. Batail, Special issue on Molecular Conductors, Chem. Rev., 2004, 104, 4887–5781 CrossRef CAS.
- N. Martín and J.-L. Segura, Angew. Chem., Int. Ed., 2001, 40, 1372–1409 CrossRef.
- D. Canevet, M. Sallé, G. Zhang, D. Zhang and D. Zhu, Chem. Commun., 2009, 2245–2269 RSC.
- M. B. Nielsen, C. Lomholt and J. Becher, Chem. Soc. Rev., 2000, 29, 153–164 RSC.
- H.-Y. Wang, L. Cui, J.-Z. Xie, C.-F. Leong, D. M. D'Alessandro and J.-L. Zuo, Coord. Chem. Rev., 2017, 345, 342–361 CrossRef CAS.
- H. Jiang, P. Hu, J. Ye, K. K. Zhang, Y. Long, W. Hu and C. Kloc, J. Mater. Chem. C, 2018, 6, 1884–1902 RSC.
- E. Arunan, G. R. Desiraju, R. A. Klein, J. Sadlej, S. Scheiner, I. Alkorta, D. C. Clary, R. H. Crabtree, J. J. Dannenberg, P. Hobza, H. G. Kjaergaard, A. C. Legon, B. Mennucci and D. J. Nesbitt, Pure Appl. Chem., 2011, 83, 1637–1641 CrossRef CAS.
- M. R. Bryce, J. Mater. Chem., 1995, 5, 1481–1496 RSC.
- T. Isono, H. Kamo, A. Ueda, K. Takahashi, A. Nakao, R. Kumai, H. Nakao, K. Kobayashi, Y. Murakami and H. Mori, Nat. Commun., 2013, 4, 1344–1349 CrossRef PubMed.
-
(a) G. Cavallo, P. Metrangolo, R. Milani, T. Pilati, A. Priimagi, G. Resnati and G. Terraneo, Chem. Rev., 2016, 116, 2478–2601 CrossRef CAS PubMed;
(b) L. C. Gilday, S. W. Robinson, T. A. Barendt, M. J. Langton, B. R. Mullaney and P. D. Beer, Chem. Rev., 2015, 115, 7118–7195 CrossRef CAS PubMed.
-
(a) M. Fourmigue and P. Batail, Chem. Rev., 2004, 11, 5379–5418 CrossRef PubMed;
(b) O. Jeannin, E. Canadell, P. Auban-Senzier and M. Fourmigué, Chem. Commun., 2016, 52, 308–311 RSC;
(c) A. Frąckowiak, R. Świetlik, L. Maulana, D. Liu, M. Dressel, O. Jeannin and M. Fourmigué, J. Phys. Chem. C, 2020, 124, 5552–5558 CrossRef.
-
(a) R. Oliveira, S. Groni, C. Fave, M. Branca, F. Mavre, D. Lorcy, M. Fourmigué and B. Schöllhorn, Phys. Chem. Chem. Phys., 2016, 18, 15867–15873 RSC;
(b) R. Oliveira, S. Groni, A. Vacher, F. Barrière, D. Lorcy, M. Fourmigué, E. Maisonhaute, B. Schöllhorn and C. Fave, ChemistrySelect, 2018, 3, 8874–8880 CrossRef CAS;
(c) H. Hijazi, A. Vacher, S. Groni, D. Lorcy, E. Levillain, C. Fave and B. Schöllhorn, Chem. Commun., 2019, 55, 1983–1986 RSC.
- R. Gleiter, G. Haberhauer, D. B. Werz, F. Rominger and C. Bleiholder, Chem. Rev., 2018, 118, 2010–2041 CrossRef CAS PubMed.
-
(a) O. Jeannin, E. W. Reinheimer, P. Foury-Leylekian, J. P. Pouget, P. Auban-Senzier, E. Trzop, E. Collet and M. Fourmigué, IUCrJ, 2018, 5, 361–372 CrossRef CAS PubMed;
(b) M. Beau, O. Jeannin, M. Fourmigué, P. Auban-Senzier, F. Barrière and I.-R. Jeon, CrystEngComm, 2022, 24, 7535–7539 RSC.
- P. Politzer and S. J. Murray, Crystals, 2017, 7, 212–226 CrossRef.
-
(a) A. Filatre-Furcate, P. Auban-Senzier, M. Fourmigué, T. Roisnel, V. Dorcet and D. Lorcy, Dalton Trans., 2015, 44, 15683–15689 RSC;
(b) A. Filatre-Furcate, T. Roisnel and D. Lorcy, J. Organomet. Chem., 2016, 819, 182–188 CrossRef CAS.
-
(a) H. Hachem, Z. Xu, N. Bellec, O. Jeannin, P. Auban-Senzier, T. Guizouarn, M. Fourmigué and D. Lorcy, Dalton Trans., 2018, 47, 6580–6589 RSC;
(b) H. Hachem, N. Bellec, M. Fourmigué and D. Lorcy, Dalton Trans., 2020, 49, 6056–6064 RSC.
- H. Hachem, O. Jeannin, M. Fourmigué, F. Barrière and D. Lorcy, CrystEngComm, 2020, 22, 3579–3587 RSC.
- N. Svenstrup, K. M. Rasmussen, T. K. Hansen and J. Becher, Synthesis, 1994, 809–912 CrossRef CAS.
-
(a) M. Narita and C. U. Jr Pittman, Synthesis, 1976, 489–514 CrossRef CAS;
(b) A. Krief, Tetrahedron, 1986, 42, 1209–1252 CrossRef CAS;
(c) G. Schukat, A. M. Richter and E. Fanghänel, Sulfur Rep., 1987, 7, 155–231 CrossRef CAS.
- T. Konoike, K. Namba, T. Shinida, K. Sakaguchi, G. C. Papavassiliou, K. Murata and Y. Ohfune, Synlett, 2001, 9, 1476–1478 CrossRef.
- Y. Le Gal, A. Colas, F. Barrière, V. Dorcet, T. Roisnel and D. Lorcy, CrystEngComm, 2019, 21, 1934–1939 RSC.
- R. P. Shibaeva and E. B. Yagubskii, Chem. Rev., 2004, 104, 5347–5378 CrossRef CAS PubMed.
- J. Lieffrig, O. Jeannin, A. Frąckowiak, I. Oleniczak, R. Świetlik, S. Dahaoui, E. Aubert, E. Espinosa, P. Auban-Senzier and M. Fourmigué, Chem. – Eur. J., 2013, 19, 14804–14813 CrossRef CAS PubMed.
- T. J. Kistenmacher, T. J. Emge, A. N. Bloch and D. O. Cowan, Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem., 1982, 38, 1193–1199 CrossRef.
- H. Miyasaka, N. Motokawa, S. Matsunaga, M. Yamashita, K. Sugimoto, T. Mori, N. Toyota and K. R. Dunbar, J. Am. Chem. Soc., 2010, 132, 1532–1544 CrossRef CAS PubMed.
- S. A. O'Kane, R. Clérac, H. Zhao, X. Ouyang, J. R. Galan-Mascaros, R. Heintz and K. R. Dunbar, J. Solid State Chem., 2000, 152, 159–173 CrossRef.
- P. Hu, H. Li, Y. Li, H. Jiang and C. Kloc, CrystEngComm, 2017, 19, 618–624 RSC.
- R. F. W. Bader, Chem. Rev., 1991, 91, 893–928 CrossRef CAS.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Adv., 2015, 71, 3–5 CrossRef PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3–5 Search PubMed.
|
| This journal is © The Royal Society of Chemistry 2023 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence ,
Olivier
Jeannin
,
Olivier
Jeannin