
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Sodium tantalates: monitoring crystallization via in situ total X-ray scattering†
Ezgi
Onur Şahin
 a,
Siyuan
Zhang
b,
Christina
Scheu
b and
Claudia
Weidenthaler
*a
a,
Siyuan
Zhang
b,
Christina
Scheu
b and
Claudia
Weidenthaler
*a
aMax-Planck-Institut für Kohlenforschung, Diffraction and Spectroscopy, Kaiser-Wilhelm-Platz 1, 45470 Mülheim an der Ruhr, Germany. E-mail: weidenthaler@mpi-muelheim.mpg.de
bMax-Planck-Institut für Eisenforschung GmbH, Nanoanalytics and Interfaces, Max-Planck-Straße 1, 40237 Düsseldorf, Germany
First published on 14th March 2023
Abstract
Sodium tantalates, which have potential applications in photocatalysis, microelectronics and energy harvesting, are known to crystallize into several polymorphs on which the electrical and optical properties are dependent. As a photocatalyst, amorphous sodium tantalum oxide prepared by a sol–gel method has previously been shown to provide superior activity compared to crystalline counterparts. Intending to understand the structural basis of their activity in catalysis, this work provides insights into the crystallization behavior of sodium tantalates. The structural evolution of amorphous sodium tantalate was monitored during heat-induced crystallization. The local structure was investigated by pair distribution function analysis based on in situ total X-ray scattering experiments. Further information on the spatial arrangements was obtained from complementary scanning transmission electron microscopy and spectroscopy studies. It was found that a composite of natrotantite (R![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) cH) and distorted perovskite (Cmcm or Pbnm) together with a considerable amount of amorphous solid is obtained during heating the amorphous starting material up to 700 °C. The findings of the present work are expected to shine further light on the alterations of the band gap energy and subsequent changes in the photocatalytic performance of this large band gap semiconductor material.
cH) and distorted perovskite (Cmcm or Pbnm) together with a considerable amount of amorphous solid is obtained during heating the amorphous starting material up to 700 °C. The findings of the present work are expected to shine further light on the alterations of the band gap energy and subsequent changes in the photocatalytic performance of this large band gap semiconductor material.
Introduction
Sodium tantalum oxides show tunable electrical and optical properties and have attracted significant attention as promising photocatalysts besides being used for other applications in microelectronics (dielectrics, ferroelectrics, electro-optic waveguides) and in energy harvesting applications (piezoelectrics, thermoelectrics).1,2 Among many other transition-metal oxide photocatalysts investigated for photon-driven catalysis of water into H2 and O2, both amorphous/nanocrystalline and crystalline sodium tantalates have been intensively studied, and they showed promising activity for hydrogen generation.3–9Among studies on the structure of sodium tantalate, the ones involving high-temperature neutron diffraction to determine phase transitions10 and phase coexistence,11 as well as investigation of the structural relationships in synthetic natrotantite12 attracted attention. Reports on Rietveld analysis of X-ray diffraction data carried out on nanocrystalline sodium tantalate2 discussed the effects of the sodium precursor on the crystalline structure and morphology13 and the effects of the preparation method.14
Sodium tantalates crystallize in three main types, namely perovskites (NaTaO3), layered natrotantite (Na2Ta4O11),15,16 and pyrochlore (Na2Ta2O6).17 Perovskite NaTaO3 is orthorhombic with the space group Pbnm at room temperature.14 Upon heating, it undergoes successive structural phase transitions first into the orthorhombic Cmcm (at ∼440 °C) structure, further to tetragonal P4/mbm (at ∼560 °C), and finally to cubic Pmm (at ∼620 °C).10,18,19 These structures consist of corner-sharing TaO6 octahedra with sodium ions at dodecahedral interspaces. With tilting of the TaO6 octahedra, perovskite unit cells with different symmetries are obtained. NaTaO3 frameworks are flexible enough to tolerate this tilting, giving rise to the rich polymorphism of the system.2 On the other hand, natrotantite (Na2Ta4O11) is composed of layers of edge-sharing TaO7 pentagonal bipyramids and TaO6 octahedra sharing corners acting as connectors surrounded by sodium ions.20 The discussed structure models are shown in Fig. 1. Additionally, phases such as Na2Ta8O21 and L-Ta2O5 are also reported for the composite systems of NaTaO3–Ta2O5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 12 where the Na content plays an important role in the formation of these phases.
12 where the Na content plays an important role in the formation of these phases.
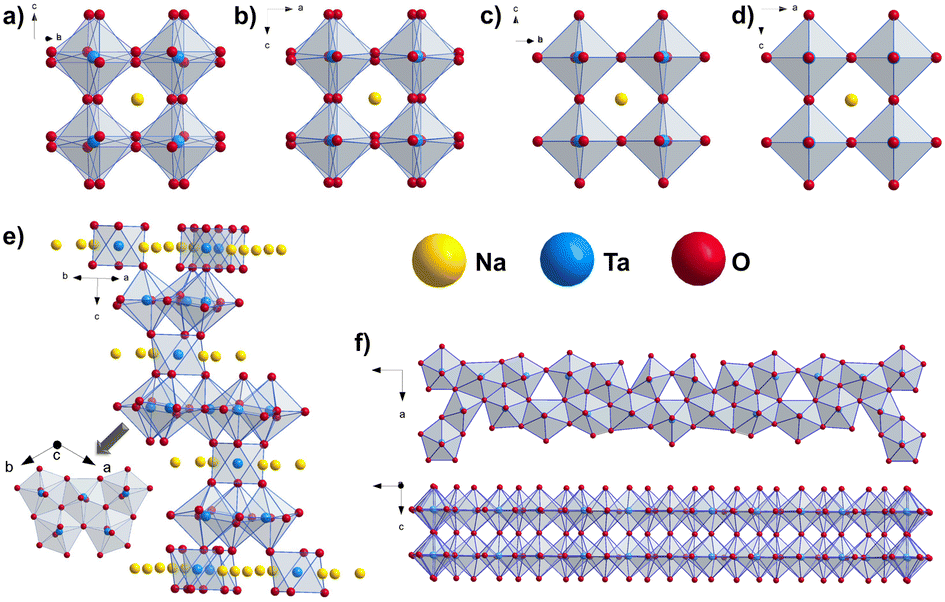 | ||
| Fig. 1 Structure motifs in perovskites pointing out the ordering of the TaO6 octahedra in the structure models for NaTaO3 (a) Pbnm, (b) Cmcm, (c) P4/mbm and (d) Pmm space groups10 and in (e) Na2Ta4O1112 and (f) L-Ta2O5.21 | ||
Both NaTaO3 and Na2Ta2O6 are wide-bandgap semiconductors with the Egap of 4.1 eV22 and 4.6 eV,23 respectively. For the Na2Ta4O11 structure, values between 3.6–4.3 eV were reported.16,20,24 Owing to their large band gap, sodium tantalates give photocatalytic response to the ultraviolet (UV) light of the electromagnetic spectrum. However, their band gaps can be engineered to enhance the performance by e.g. N, Cr or Bi doping as well as Ta4+ self-doping.22
The structure–electronic and optical property relationship for alkali metal tantalates has been investigated before and it was found that the degree of the crystallinity of the tantalates highly depends on the alkali metal and its amount.25 This had also an effect on the photoluminescence intensity and the photocatalytic activity of the solid material or water splitting. The increased activities in the presence of excess alkali were attributed to the absences of grain boundaries and alkali defects that can act as recombination centers for the photogenerated electrons and holes. In line with these findings, the effect of distortion of TaO6 connections and Ta–O–Ta bond angles in the corner sharing TaO6 octahedra on the optical properties was also examined.25–27 The results showed that, as the bond angles get closer to 180°, the band gap is reduced and electrons excited by the incoming light emigrated easier in the crystal.
In addition to the structural complexity already mentioned in the Na–Ta–O system at the solid state, the intricacies brought by the crystallization route starting from the amorphous sodium tantalum oxide precursor make this material an interesting candidate for crystallization studies. Herein, the structural evolution of an amorphous sodium tantalate that was prepared by a sol–gel method was monitored during heat-induced crystallization. To investigate the changes in the structural motifs during the amorphous to crystalline transition, in situ total scattering experiments were performed. The local structure and chemical composition were analyzed using scanning transmission electron microscopy (STEM) imaging and energy-dispersive X-ray spectroscopy (EDS). For imaging, either a high-angle annular dark field (HAADF) or an annular bright field (ABF) detector was used. The results of our work provide detailed information on the atomic arrangement and composition of as-synthesized and calcined Na–Ta–O, which directly affect the electronic and optical properties, and extend existing work on structural studies of sodium tantalates.
Results and discussion
Sodium tantalum oxide (in the following NaxTayOz) was prepared by a previously reported7 sol–gel route described in the ESI.† The as-prepared NaxTayOz powder is semi-amorphous as can be seen from the powder X-ray diffraction (XRD) pattern given in Fig. 2a. The X-ray pair distribution function (PDF) obtained from data collected at a synchrotron facility at 30 °C only shows short-range order with a coherently scattering domain size of around 8 Å (Fig. 2b). The strong pair correlations up to 4.5 Å are followed by weaker ones. These weaker atom pairs can be caused by a few weak interactions and/or by the interaction between the domains. The experimental PDF obtained for NaxTayOz is very similar to that of pure Ta oxide (Fig. 2b). Previous work on tantalum oxide prepared via the same preparation route using the same dimeric tantalum(V) ethoxide as tantalum source, except for the addition of the sodium precursor, showed similar pair correlations.28 The pair correlations observed for NaxTayOz can be explained using a disordered pentagonal model originating from the L-Ta2O5 structure.21 Although this model is composed solely of Ta and O atoms, a similar model can be used to explain the current data for Na–Ta oxide as well. This is due to the dominancy of the interactions between the strongly scattering Ta atom-containing pairs compared to the weakly scattering sodium atoms.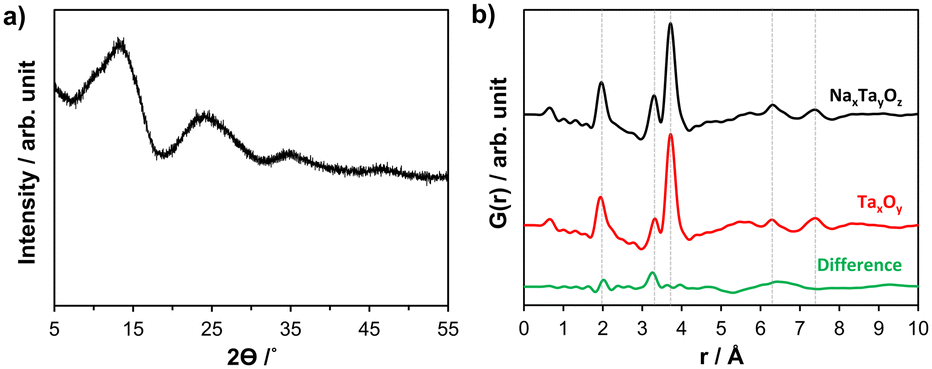 | ||
| Fig. 2 Ex situ powder XRD pattern of as-prepared non-crystalline NaxTayOz powder measured with MoKα1 radiation (a). Experimental PDFs (λ = 0.20723 Å) obtained for NaxTayOz and TaxOy together with the difference PDF (calculated by subtracting the intensities of the PDF of TaxOy from those of NaxTayOz) (b). | ||
The first peak centred at ∼1.97 Å represents the distribution of Ta–O distances within the TaO6 octahedron. The next pair correlation centred at ∼3.32 Å is assigned to Ta–Ta distances between edge-sharing octahedra. This peak is followed by a peak corresponding to Ta–Ta distances between corner-sharing octahedra and gives a maximum at 3.73 Å. The differences between the PDFs obtained for NaxTayOz and TaxOy samples correspond to atom pairs between Ta–O (∼2.0 Å) and Na–Ta (∼3.3 Å). This indicates that the material prepared in the presence of the sodium precursor contains more Ta–O and expectedly Na–Ta interactions, which are limited to the range up to 3.5 Å.
Additionally, the material shows a specific surface area of 40 m2 g−1 determined from the desorption branch of the gas adsorption isotherm (Fig. S3†). The size distribution obtained for NaxTayOz from dynamic light scattering (Fig. S4†) indicates a hydrodynamic diameter accumulated around 29 nm considering the number of particles.
The NaxTayOz sample was subjected to thermal analysis to collect preliminary information on the crystallization behavior of the semi-amorphous material. Combined thermogravimetry-differential scanning calorimetry (TG-DSC) measurements were performed with different heating rates. Above 630 °C, an exothermic reaction is observed, which is represented by broad peaks in the DSC curves. This reaction can be interpreted as the beginning of the reorganization of the structure on an atomic level. The broad peak is followed by a sharp exothermic DSC signal starting at temperatures above 700 °C, which could be attributed to crystallization phenomenon or phase transformations (Fig. S5†). Based on the thermal analysis using three different heating rates, an activation energy of Ea = 418 kJ mol−1 could be determined for the crystallization process (Fig. S6†). This value is higher than the value previously obtained for pure tantalum oxide crystallization (397 kJ mol−1).28
In the following, the local structure of sample NaxTayOz (non-heat treated material) will be discussed based on the evaluation of ex situ STEM and total scattering data. The sample was further calcined to 700 °C under airflow. After the calcination the sample was cooled to room temperature (NaxTayOz-700), and the temperature-induced structural changes were investigated by electron microscopy and PDF analysis. After the description of the ex situ results, the in situ studies will be discussed.
Ex situ investigations of the local structure
 | ||
| Fig. 3 (a) HAADF-STEM image of amorphous NaxTayOz. (b) Respective ABF image and corresponding FFT pattern (inset). Spatial distribution of (c) Na and (d) Ta elements and (e) composition obtained by STEM-EDS spectrum.29 | ||
Fourier transform infrared spectroscopy (FT-IR) performed on the non-heat-treated material (Fig. S8†) exhibits vibrations that could be assigned to OH groups and organic residues, which were formed during the hydrolysis and condensation reactions. These remained partly in the amorphous network, as previously reported.30
cH) (Fig. 4a) and orthorhombic NaTaO3 (Fig. 5a). The reflections of the orthorhombic perovskite can be indexed in both space groups Cmcm and Pbnm. The crystal structures of both perovskites are similar and show differences only in the tilting of the octahedra (Fig. 1a–d). However, the NaTaO3 in space group Pbnm is reported to be the stable form at room temperature.19 The analysis of the electron micrographs reveals that in addition to the regions showing long-range order, short-range ordered regions are still visible in the rim of some particles. Some particles are either completely amorphous (Fig. 6) while some others are crystalline (Fig. 4a and 5a).
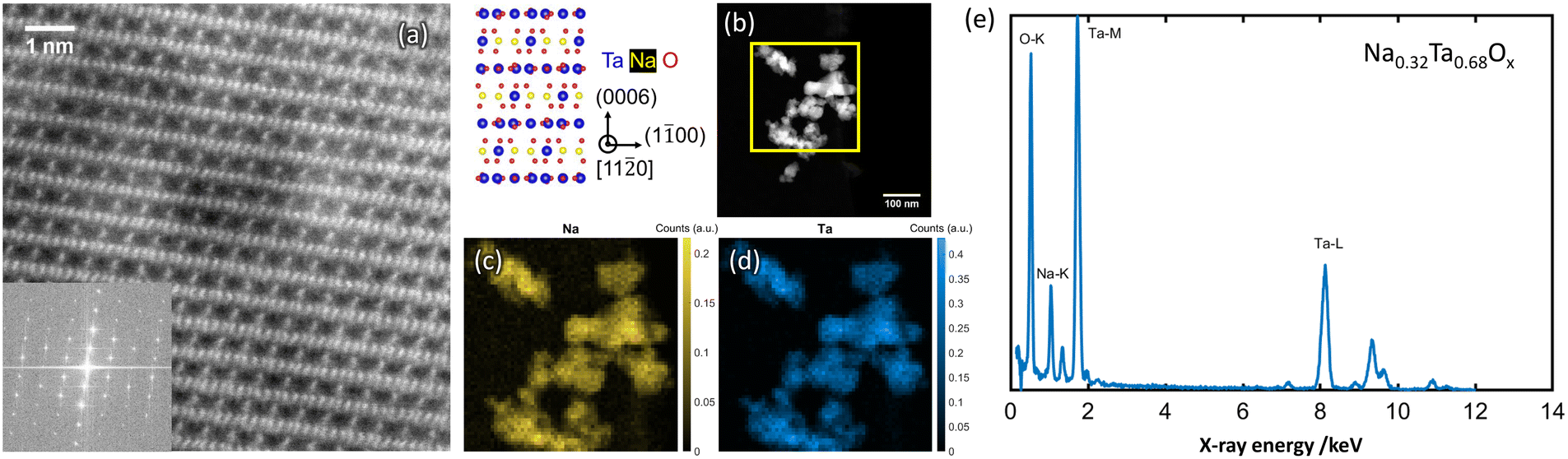 | ||
| Fig. 4 Atomic resolution HAADF-STEM image (a) obtained from NaxTayOz-700 with the corresponding FFT pattern (inset) of the regions showing Na2Ta4O11 (RcH) arrangement in the crystallized powder. Spatial distribution of Na element obtained from the respective regions (b) by STEM-EDS is given in (c) and (d). EDS data processing according to ref. 29 results in Na0.32Ta0.68Ox as average composition (e). | ||
 | ||
| Fig. 5 Atomic resolution HAADF-STEM image (a) obtained from NaxTayOz-700 with the corresponding FFT pattern (inset) of the regions showing NaTaO3 (Cmcm or Pbnm) arrangement in the crystallized powder. Spatial distribution of Na element obtained from the respective regions (b) by STEM-EDS is given in (c) and (d). EDS data processing according to ref. 29 results in Na0.48Ta0.52Ox as average composition (e). | ||
 | ||
| Fig. 6 ABF-STEM image and corresponding FFT pattern (inset) obtained from the heat-treated material NaxTayOz-700 showing the region with only short-range order. | ||
Elemental mapping reveals different amounts of Na for the regions with different crystal structures. Depending on the Na content, different NaxTayOz phases are formed. Fig. 4c and 5c show the spatial distribution of Na in the regions where natrotantite and perovskite are observed. Fig. 4e displays an average Na concentration of 32 at% for the region with Na2Ta4O11 structure, while Na concentration is 48 at% for the region with NaTaO3 as shown in Fig. 5e. While some parts are transformed into the relatively Na-rich NaTaO3 phase, the rest of the particles either remain without long-range order or transform into Na2Ta4O11. This phase contains layers of TaO7 polyhedra and has less Na. During their investigations on natrotantite at 850 °C, Ercit et al. found that all three phases NaTaO3, Na2Ta4O11, and Ta2O5 are present in different proportions depending on the initial compositions.12 Moreover, according to their chemical analyses, NaTa3O8 or Na2Ta8O21 are considered possible compositions for natrotantite.
Based on the information that the heat-treated material NaxTayOz-700 still contains an amorphous solid, it is assumed that also pair correlations present in the amorphous non-heat-treated material NaxTayOz will be observed. As the fraction of the amorphous amount in NaxTayOz-700 is not known, the PDFs of NaxTayOz and NaxTayOz-700 were normalized to each other and a simple subtraction was performed. One needs to keep in mind that short-range pair correlations might get lost by this procedure. The difference PDF (dPDF) (Fig. S9†) was then compared to the PDFs simulated for different Na–Ta–O crystal structure models (Fig. S10†). The comparison with the experimental PDF shows that the perovskite models match. The refinement of the difference PDF in the region 5–20 Å indicates a better fit with NaTaO3 in space group Pbnm rather than Cmcm (Fig. S11†). This is in accordance with the fact that the structure in Pbnm is reported as the stable phase at ambient temperatures.19
The difference at around 6.2 and 9.5 Å can be improved by adding the Na2Ta4O11 structure as a second phase (Fig. S12a and b†). The presence of NaTaO3 and Na2Ta4O11 in the PDFs confirms the findings of the electron microscopy study. Based on the electron microscopy results, sample NaxTayOz-700 still contains a non-crystalline component. The model that best describes the PDF obtained from the non-crystalline material was based on the L-Ta2O5 structure consisting of TaO6 and TaO7 layers. Therefore the L-Ta2O5 structure model was included in the refinements which results in 44.4 wt% NaTaO3-Pbnm, 42.1 wt% Na2Ta4O11, and 13.5 wt% L-Ta2O5. The fit is shown in Fig. 7 and results are tabulated in Table S1.† It can be summarized at this point that, after calcination to 700 °C, the sample still contains some X-ray amorphous portions with an L-Ta2O5-like local atom arrangement with sizes in the subnanometer range. These semi-amorphous regions are the regions with the lowest Na content.
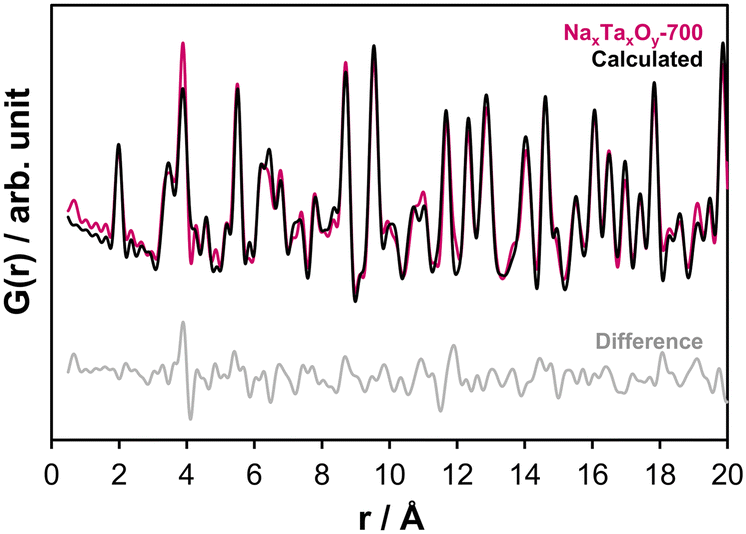 | ||
| Fig. 7 Refinement fit to the experimental PDF (Rw-value: 0.24) obtained at ambient temperature for sample NaxTayOz-700 using Na2Ta4O11,12 L-Ta2O521 and NaTaO3-Pbnm10 structure models. | ||
Following the thermal analysis and ex situ PDF studies, the samples are further subjected to in situ total scattering experiments to determine the temperature interval of interest for the local structure analysis. In the first experiment, the X-ray total scattering data were collected between 30–600 °C in temperature intervals of 50 °C (Fig. S13†). The long-range PDFs (Fig. S14†) showed that the changes in the local structure related to crystallization start between 550 and 600 °C. Accordingly, a second temperature-dependent experiment was performed between 550 and 700 °C in temperature intervals of 10 °C (Fig. S15 and S16†).
In situ investigations of the local structure
In situ total scattering experiments and subsequent PDF analysis of the NaxTayOz enables the assignment of structural order during heating on an atomic scale. The experimental PDFs obtained between 550 and 700 °C are displayed in Fig. 8. The first atom pair corresponding to the first neighbor Ta–O pair correlation remains stable during heating. The major changes in the short-range are observed in the region around 3.3 Å. The main component of this peak is represented by the distance distribution of Ta–Ta pairs between edge-sharing octahedra. This peak becomes broader with temperature which points to a variation in the bond length. Additionally, the Na–Ta pair correlations, which are expected to get more frequent by the formation of the perovskite structure, are expected in this range. The Na–Ta distances vary in the range of 3.35–3.40 Å and get even larger for the natrotantite structure. Therefore, the change of this peak might be attributed to the formation of crystalline Na–Ta–O structures, keeping in mind that most probably there is still a contribution from the edge-sharing polyhedra in the semi-amorphous component represented by an L-Ta2O5-type local arrangement. | ||
| Fig. 8 Overview of the short-range PDFs obtained from the scattering data collected between 550–700 °C with 10 °C increments together with the PDF obtained at 30 °C after cooling for comparison. | ||
With the increase of temperature up to 600 °C, the distribution of the Ta–Ta distances between corner-sharing octahedra also extends to larger distances (3.72 Å at 30 °C, 3.74 Å at 550 °C and 3.79 Å at 600 °C). Besides the thermal expansion effects, this can originate from the reorganization of the structure as the absolute intensity also changes. With the increase of the temperature from 600 to 700 °C, this peak shifts from 3.79 Å at to 3.90 Å. This increment is expected to happen during the formation of the perovskite phases, in which the Ta–Ta distances are slightly larger than 3.95 Å.
Above 600 °C, the weak pair correlations in the experimental PDFs above 5 Å get more intense. To monitor the evolution of the phases and changes in their fractions, the PDFs obtained in the temperature interval between 600 and 700 °C were fitted for the range 0.5–20 Å. Different structure models were used based on the qualitative comparison of the pair correlations calculated for the different structures (Fig. 1). At this point, the phase compositions obtained from the ex situ data of NaxTayOz-700 discussed in the previous section were used as a guide for the refinement of the PDF measured in situ at 700 °C. According to the results of the ex situ examination, the sample still contains an amorphous fraction after calcination at 700 °C. Based on this information, the in situ PDF obtained for the amorphous material was subtracted from the PDF obtained during the heat treatment at 700 °C (dPDF).
As a first attempt, the dPDF was refined against the high-temperature polymorph of NaTaO3 in space group Pmm. The refinement of the data in the range 5–20 Å using a single phase Pmm (Rw: 0.57, Fig. S17†) was significantly improved by the inclusion of Na2Ta4O11 (Rw: 0.36, Fig. S18†). However, a more disordered perovskite structure (P4/mbm) (stable over 560 °C) together with Na2Ta4O11 gave the best goodness of fit value for the dPDF refinement (Rw: 0.33, Fig. S19†).
Based on the refinements of the dPDF obtained in situ at 700 °C, the experimental PDFs were refined starting from 700 °C to 550 °C using NaTaO3-P4/mbm and Na2Ta4O11 structures with the addition of L-Ta2O5 to compensate for the amorphous/nanocrystalline content. The results of the refinement (Rw: 0.29, Fig. S20†) were improved by introducing the low temperature perovskite polymorph (stable above 440 °C) in Cmcm symmetry (Rw: 0.26, Fig. S21†). The results and the details of the refinements are given in Fig. S22† and tabulated as wt% in Table 1 (for at% see Table S2 and Fig. S23†). The phase fractions in terms of wt% are displayed in Fig. 9.
| T/°C | NaTaO3 (Pbnm) | NaTaO3 (Cmcm) | NaTaO3 (P4/mbm) | Na2Ta4O11 (RcH) |
L-Ta2O5 (P2mm) |
|---|---|---|---|---|---|
| 550 | 0.0 | 5.7 | 4.7 | 82.5 | 7.1 |
| 560 | 0.0 | 6.1 | 4.3 | 82.2 | 7.4 |
| 570 | 0.0 | 7.1 | 2.6 | 83.1 | 7.2 |
| 580 | 0.0 | 9.8 | 2.5 | 80.5 | 7.2 |
| 590 | 0.0 | 13.0 | 3.0 | 76.6 | 7.4 |
| 600 | 0.0 | 22.7 | 3.0 | 66.0 | 8.3 |
| 610 | 0.0 | 24.4 | 3.1 | 64.0 | 8.5 |
| 620 | 0.0 | 26.9 | 3.4 | 61.0 | 8.7 |
| 630 | 0.0 | 29.7 | 4.1 | 57.5 | 8.7 |
| 640 | 0.0 | 31.7 | 5.1 | 54.3 | 8.9 |
| 650 | 0.0 | 34.6 | 6.6 | 50.4 | 8.4 |
| 660 | 0.0 | 37.8 | 8.2 | 46.3 | 7.7 |
| 670 | 0.0 | 44.5 | 9.5 | 37.0 | 9.0 |
| 680 | 0.0 | 26.5 | 24.2 | 40.2 | 9.1 |
| 690 | 0.0 | 23.6 | 24.4 | 43.3 | 8.7 |
| 700 | 0.0 | 12.3 | 32.4 | 44.7 | 10.6 |
| 30 | 44.4 | 0.0 | 0.0 | 42.1 | 13.5 |
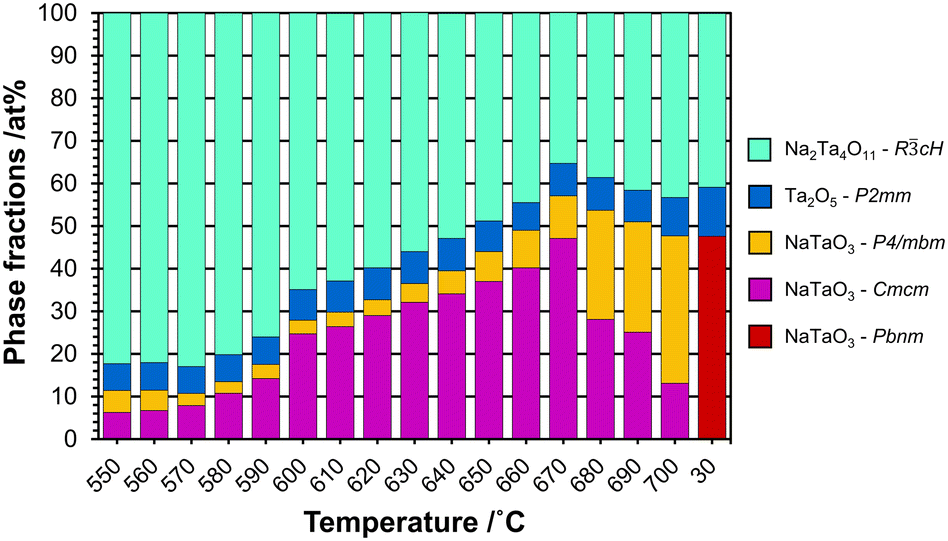 | ||
| Fig. 9 Plot showing the phase fractions in wt% as a function of temperature in the regime from 550 to 700 °C, together with the ex situ results after cooling to 30 °C (sample NaxTayOz-700). | ||
At 550 °C, the dominant phase is Na2Ta4O11 with over 82 wt%. The high content of this phase in the disordered material at this temperature can be explained by the Ta–O layers present in the structure resembling the L-Ta2O5-like local atomic arrangement with sizes in the subnanometer range proposed for the starting X-ray amorphous material. The amount of Na2Ta4O11 reaches 66 wt% at 600 °C. The material also contains 8 wt% L-Ta2O5 and ∼26 wt% perovskite with Cmcm symmetry. With increasing temperature, the amount of Na2Ta4O11 decreases, giving rise to an increase in the total amount of perovskite. While in the beginning, the low-temperature polymorph (Cmcm) was the dominant perovskite structure, the tetragonal polymorph (P4/mbm) becomes the dominant one above 680 °C. On the other hand, the amount of L-Ta2O5 stays almost the same.
Additional to the in situ X-ray total scattering experiments, in situ XRD experiments were performed on the as-prepared powder. The resultant powder diffraction patterns are given in Fig. 10, together with the results of the Rietveld refinements in Fig. S24a and b.†
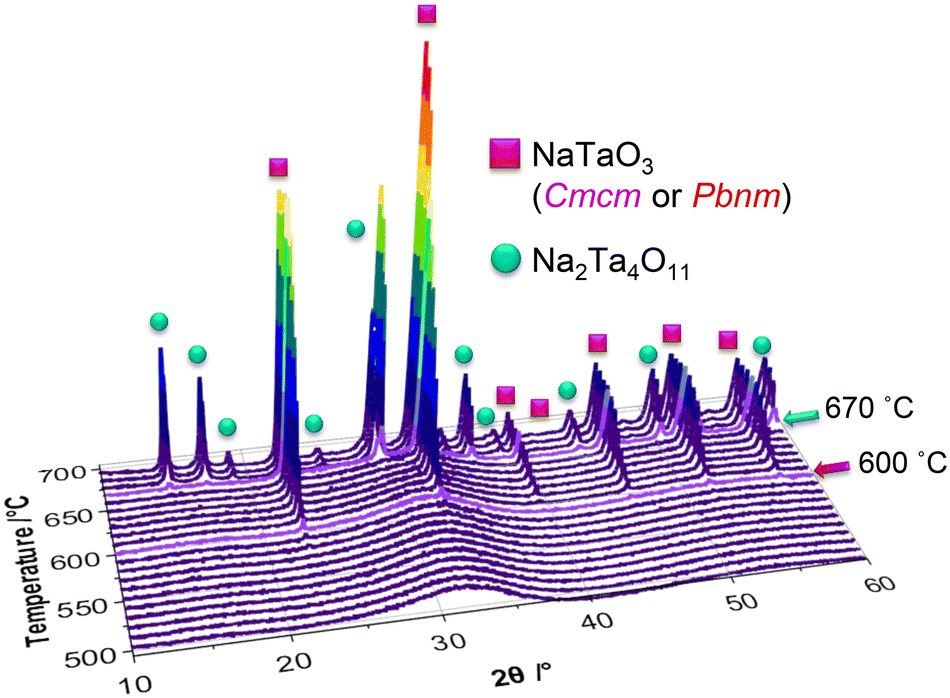 | ||
| Fig. 10 Overview of the in situ XRD patterns collected between 550–700 °C with 10 °C increments. | ||
Phase analysis of the in situ collected XRD patterns indicate that the first reflections forming at 590 °C belong to the perovskite phase in space group Cmcm, which are followed by natrotantite reflections emerging at 670 °C. While the Rietveld refinements performed on the in situ XRD patterns show the increase of natrotantite with temperature, the local structure analysis performed on the X-ray total scattering data and subsequent PDFs displays that the local structure shows a better match to natrotantite phase starting from 550 °C on, decreasing in amount as the temperature increases. This discrepancy between the results of the average and local structure analysis can be explained by the nanostructure of the composite material. According to the deviation of the cell parameters of the major phase, natrotantite, obtained from the PDF analysis at 550 °C, it is clear that the refined structure of this phase deviates from the averaged crystal structure model. The distorted structure of natrotantite at relatively low temperatures results in broad scattering contributions rather than sharp Bragg reflections.
According to the literature, the transformation from tetragonal P4/mbm to cubic Pmm, which is the highest symmetry polymorph, takes place around 620 °C.10,17 However, our refinements of the high-temperature data do not yield good fits with this type of perovskite structure. This indicates that the TaO6 octahedra present at high temperatures are still not as linearly aligned as in the case of NaTaO3-Pmm. Interestingly, the highest symmetry perovskite cannot be obtained at temperatures even above 690 °C, which might be due to the high degree of disorder of the amorphous starting material which is used as the precursor for the crystallization experiments. The amount of energy needed for the alignment of the polyhedra to reach higher symmetries might be higher than can be supplied by the experimental conditions. Instead of NaTaO3-Pmm, the refinements show perovskites with P4/mbm and Cmcm symmetries.
For Na2Ta4O11, the Ta–O–Ta bond angles formed between corner-sharing octahedra are ∼133°.25 For Na–Ta–O perovskites, different Ta–O–Ta bond angles for the corner-sharing octahedra are reported. They increase from 158–160° for the Pbnm structure, to 164–169° for the orthorhombic Cmcm structure, to 169–180° for tetragonal P4/mbm, and to about 180° for cubic Pmm. It is expected to observe a reduction in the band gap as the bond angles get closer to 180° during transformation to high-symmetry perovskites.25–27 According to the results of our PDF refinements of the range 0.5–20 Å, while 71% of the Ta–O–Ta bond angles of the perovskites formed at 700 °C are between 164–169°, the rest falls between 169–180°. After the heat treatment, the angles for perovskite in Pbnm symmetry decrease to values between 158 and 160°.
One of the factors playing the most important role in the changes of the phase fractions is the change of the Na content. Due to the inhomogeneous sodium distribution in the non-crystalline starting material, the formation of phases with different Na content was expected. For bulk ceramics, evaporation of sodium during temperature increase is also a previously reported phenomenon taking place at temperatures around 1550 °C.31–33 Due to sodium evaporation, the formation of the Na-poor phase Na2Ta4O11 besides NaTaO3 was reported. The results show that the Na2Ta4O11 content decreases while NaTaO3 content increases with increasing temperature. This means that the mobility of the sodium atoms in the amorphous phase increases during heating. These atoms might migrate to regions with low sodium content and lead to the formation of perovskites with a nearly perfect stoichiometry.32
The PDF analyses show that natrotantite is the dominant phase at the beginning of the crystallization. At 700 °C, it reaches almost the same amount as perovskite. As perovskite becomes the dominant phase, the crystallite size was changed. The results show that this phase grows in size from 14 Å at 550 °C to ∼70 Å after heating to 700 °C. The coexistence of natrotantite and perovskite phases is reported to provide superior photocatalytic performance advancing from the interfaces of the two distinct structures. Similarly, composites of amorphous and crystalline samples or mixtures of different crystalline phases have proven to enhance the photocatalytic activity of samples toward water splitting, possibly benefiting from the formation of the heterojunctions influencing the band gap and charge separation13 as well as increasing the efficiency and activity.7,34
Conclusions
PDFs obtained from total scattering experiments provide information on the local structure of non-crystalline materials. NaxTayOz prepared by hydrolysis of NaOEt and TaOEt5 is nanocrystalline. The local structure resembles that of pure tantalum oxide with L-Ta2O5 entities of a domain size of less than 10 Å. Scanning transmission electron microscopy combined with EDS provides insights into composition-related structural information. The examinations indicate an inhomogeneous sodium distribution. Additionally, short-range-ordered regions observed from the PDF do not show a high amount of sodium-containing pairs. It is concluded that the sodium atoms are enriched in a highly disordered environment. It was observed that the regions with a low Na content show a natrotantite-like arrangement, whereas sodium-rich regions transform into perovskite structures as expected from the stoichiometry. Additionally, regions with the lowest sodium content among those examined, remain amorphous during heating to 700 °C.In situ temperature-dependent crystallization experiments starting from the non-crystalline material provide detailed information on the evolution of the local structure. Upon heating, the material transformed into natrotantite and perovskite phases with residual pure tantalum oxide. At 700 °C, the formed perovskite phases are still not in the highest symmetry. The reason could be that the energy supplied at the applied temperature is not sufficient to transform the disordered domains into fully ordered structures.
With our semi-amorphous starting material, it was possible to obtain crystalline domains in the local structure that are different from the ones reported to be stable in the bulk at a given temperature. In the case of the sodium tantalates, it can be summarized that the compositional and structural variations might be the reason for the deviation of the reported phase transition track. Control over the material at the local atomic level would provide the opportunity to influence the electronic and optical properties.
Both electron microscopy and atomic pair distribution analysis of the X-ray scattering data give valuable insights into the local structure during the crystallizing of sodium tantalates. However, to obtain a spatially resolved distribution of the atomic pairs, one could benefit from the pair distribution functions obtained from electron diffraction data for future studies.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
E. O. acknowledges the International Max Planck Research School for Interface Controlled Materials for Energy Conversion (IMPRS-SurMat) for funding. C. W. thanks the Max Planck Society for basic funding. We acknowledge DESY (Hamburg, Germany), a member of the Helmholtz Association HGF, for the provision of experimental facilities. Parts of this research were carried out at PETRAIII and we would like to thank Dr. Martin Etter in using P02.1. Beamtime was allocated for proposal I-20190175. S. Z. acknowledges funding from the German Research Foundation (DFG) within the framework of SPP 2370 (Project number 502202153). We gratefully acknowledge Priv. Doz. Dr. Harun Tüysüz for scientific discussions and the material preparation method together with Prof. Candace K. Chan and Dr. Tobias Grewe. We would like to express our gratitude to Dr. Hilke Petersen (MPI-KOFO) for scientific discussions and support for synchrotron experiments and to Jan Ternieden (MPI-KOFO) who supported us during our in-house and synchrotron experiments. We thank the mechanical workshop of the MPI-KOFO for the fabrication of the heating cell. Open Access funding provided by the Max Planck Society.Notes and references
- S. Zlotnik, D. M. Tobaldi, P. Seabra, J. A. Labrincha and P. M. Vilarinho, ChemPhysChem, 2016, 17, 3570–3575 CrossRef CAS PubMed
.
- V. Shanker, S. L. Samal, G. K. Pradhan, C. Narayana and A. K. Ganguli, Solid State Sci., 2009, 11, 562–569 CrossRef CAS
- H. Onishi, ChemSusChem, 2019, 12, 1825–1834 CrossRef CAS PubMed
- M. Yang, X. Huang, S. Yan, Z. Li, T. Yu and Z. Zou, Mater. Chem. Phys., 2010, 121, 506–510 CrossRef CAS
- H. Tüysüz and C. K. Chan, Nano Energy, 2013, 2, 116–123 CrossRef
- H. Kato and A. Kudo, Catal. Today, 2003, 78, 561–569 CrossRef CAS
- T. Grewe and H. Tüysüz, ACS Appl. Mater. Interfaces, 2015, 7, 23153–23162 CrossRef CAS PubMed
- H. Kato and A. Kudo, Catal. Lett., 1999, 58, 153–155 CrossRef CAS
- W.-J. Yin, B. Weng, J. Ge, Q. Sun, Z. Li and Y. Yan, Energy Environ. Sci., 2019, 12, 442–462 RSC
- B. J. Kennedy, A. K. Prodjosantoso and C. J. Howard, J. Phys.: Condens. Matter, 1999, 11, 6319–6327 CrossRef CAS
- K. S. Knight and B. J. Kennedy, Solid State Sci., 2015, 43, 15–21 CrossRef CAS
- T. S. Ercit, F. C. Hawthorne and P. Černý, Bull. Mineral., 1985, 108, 541–549 CAS
- L. Huang, Q. Chan, B. Zhang, X. Wu, P. Gao, Z. Jiao and Y. Liu, Chin. J. Catal., 2011, 32, 1822–1830 CrossRef CAS
- C.-C. Hu and H. Teng, Appl. Catal., A, 2007, 331, 44–50 CrossRef CAS
- A. Ratnamala, G. Suresh, V. D. Kumari and M. Subrahmanyam, Mater. Chem. Phys., 2008, 110, 176–179 CrossRef CAS
- N. McLamb, P. P. Sahoo, L. Fuoco and P. A. Maggard, Cryst. Growth Des., 2013, 13, 2322–2326 CrossRef CAS
- P. Kanhere, Y. Tang, J. Zheng and Z. Chen, J. Phys. Chem. Solids, 2013, 74, 1708–1713 CrossRef CAS
- S. Kamba, V. Goian, V. Bovtun, D. Nuzhnyy, M. Kempa, M. Spreitzer, J. König and D. Suvorov, Ferroelectrics, 2012, 426, 206–214 CrossRef CAS
- L. E. Cross, Philos. Mag., 1956, 1, 76–92 CrossRef CAS
- O. Palasyuk, A. Palasyuk and P. A. Maggard, Inorg. Chem., 2010, 49, 10571–10578 CrossRef CAS PubMed
- N. C. Stephenson and R. S. Roth, Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem., 1971, 27, 1037–1044 CrossRef CAS
- B. Modak, K. Srinivasu and S. K. Ghosh, Phys. Chem. Chem. Phys., 2014, 16, 17116–17124 RSC
- S. Ikeda, M. Fubuki, Y. K. Takahara and M. Matsumura, Appl. Catal., A, 2006, 300, 186–190 CrossRef CAS
- Y. G. Su, X. Yang, T. T. Wang, B. L. Zhu and X. J. Wang, Adv. Mater. Res., 2014, 1058, 35–39 CAS
- H. Kato and A. Kudo, J. Phys. Chem. B, 2001, 105, 4285–4292 CrossRef CAS
- J. Ye, Int. J. Hydrogen Energy, 2003, 28, 651–655 CrossRef CAS
- C.-C. Hu, C.-C. Tsai and H. Teng, J. Am. Ceram. Soc., 2009, 92, 460–466 CrossRef CAS
- E. Onur Şahin, H. Tüysüz, C. K. Chan, G. H. Moon, Y. Dai, W. Schmidt, J. Lim, C. Scheu and C. Weidenthaler, Nanoscale, 2021, 13, 150–162 RSC
- S. Zhang and C. Scheu, Microscopy, 2018, 67, i133–i141 CrossRef CAS PubMed
- T. Ogihara, T. Ikemoto, N. Mizutani, M. Kato and Y. Mitarai, J. Mater. Sci., 1986, 21, 2771–2774 CrossRef CAS
- A. Tkach, A. Almeida, J. A. Moreira, J. Perez de la Cruz, Y. Romaguera-Barcelay and P. M. Vilarinho, Appl. Phys. Lett., 2012, 100, 192909 CrossRef
- J. König, B. Jančar and D. Suvorov, J. Am. Ceram. Soc., 2007, 90, 3621–3627 CrossRef
- W. van Gool and D. D. Yue, J. Electrochem. Soc., 1974, 121, 154C–155C CrossRef
- R. Marschall, Top. Curr. Chem., 2016, 371, 143–172 CrossRef CAS PubMed
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ce00121k |
| This journal is © The Royal Society of Chemistry 2023 |