
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Synthesis, X-ray characterization and DFT calculations of a series of 3-substituted 4,5-dichloroisothiazoles†
Irina A.
Kolesnik
a,
Vladimir I.
Potkin
a,
Mikhail S.
Grigoriev
b,
Anton P.
Novikov
 b,
Rosa M.
Gomila
c,
Alexandra G.
Podrezova
d,
Vadim V.
Brazhkin
e,
Fedor I.
Zubkov
*d and
Antonio
Frontera
*c
b,
Rosa M.
Gomila
c,
Alexandra G.
Podrezova
d,
Vadim V.
Brazhkin
e,
Fedor I.
Zubkov
*d and
Antonio
Frontera
*c
aInstitute of Physical Organic Chemistry of National Academy of Sciences of Belarus, 13 Surganov Str, 220072 Minsk, Belarus. E-mail: irynakolesnik93@gmail.com
bFrumkin Institute of Physical Chemistry and Electrochemistry, Russian Academy of Sciences, 31 Bldg 4, Leninsky prosp, Moscow, 119071, Russian Federation. E-mail: mickgrig@mail.ru
cDepartment of Chemistry, Universidad de les Islas Baleares, Crta. de Valldemosa km 7.5, 07122 Palma de Mallorca (Baleares), Spain. E-mail: toni.frontera@uib.es
dFaculty of Science, Peoples' Friendship University of Russia (RUDN University), 6 Miklukho-Maklaya St., 117198 Moscow, Russian Federation. E-mail: fzubkov@sci.pfu.edu.ru
eVereshchagin Institute for High Pressure Physics, Russian Academy of Sciences, Kaluzhskoe shosse 14, 108840 Troitsk, Moscow, Russian Federation. E-mail: brazhkin@hppi.troitsk.ru
First published on 28th February 2023
Abstract
This manuscript reports a synthetic protocol to obtain 3-substituted 4,5-dichloroisothiazole derivatives in good yields. This is exemplified by the synthesis of six new compounds, which have been spectroscopically characterized including their solid state structure by single crystal X-ray analysis. As substituents in position 3, both 1,4-dihydropyridinyl and 4H-pyranyl moieties have been used, decorated with several groups like cyano, amino, acetyl, ester and methyl, thus providing a variety of possibilities to establish noncovalent interactions. The assemblies have been analysed theoretically, using DFT calculations, molecular electrostatic potential (MEP) surfaces and QTAIM/NCIplot calculations. The π-stacking between the 4,5-dichloroisothiazole rings is a recurrent motif in the solid state of most compounds, in addition to other less conventional interactions like lone pair (LP)⋯π and halogen bonding contacts. Another interesting motif is the formation of centrosymmetric R22(12) motifs where the cyano groups concurrently establish H-bonds and antiparallel CN⋯CN interactions.
Introduction
The deep understanding of noncovalent interactions is crucial for the development of supramolecular chemistry and crystal engineering.1–3 The analysis of X-ray structures is very useful to keep progressing in the adequate interpretation of the structure directing role of non-covalent interactions.4–6 Moreover, the application of theoretical methods is also convenient to investigate the energetic features of interactions and their geometric requirements, allowing the design and synthesis of new compounds with tailored properties.7,8 Furthermore, the utilization of computational tools like the quantum theory of atoms-in-molecules,9 noncovalent interaction plots10 or molecular electrostatic potentials11 is also very convenient to investigate and disclose the physical nature of interactions, charge transfer effects, etc.Substituted isothiazoles and their fused derivatives are important in the pharmacological industry12 due their analgesic, antipyretic, fungicidal, and herbicidal properties.13 For instance, monocyclic isothiazole skeletons such as sulfasomizole and denotivir display antibacterial and antiviral properties.14 Fused isothiazole derivatives also exhibit interesting effects like cytotoxicity15 and prostaglandin release regulation from human synovial sarcoma cells. Moreover, the antipsychotic drug zipracidone possessing a [d]fused isothiazole ring was proposed to treat schizophrenia and bipolar disorder.16
It has been also demonstrated that isothiazole scaffolds have emissive properties when they are attached to purine or pyridine nucleobases. Polyfunctional isothiazole derivatives have been also used for the construction of metal complexes of different complexities, like organometallic frameworks and functional materials.17 It is documented that Co(II) and Cu(II) complexes of 4,5-dichloroisothiazole-3-carboxylic acid and 1,10-phenanthroline as a coligand present interesting cytotoxicity effects.18,19
4H-pyrans and 1,4-dihydropyridines are also valuable scaffolds for pharmaceutical chemistry due to their wide spectrum of biological activities.20,21 Functionally substituted 4H-pyrans were found among natural products, compounds with antitumor, antibacterial, antiviral, fungicidal, antispasmodic and diuretic activity.20,22 1,4-Dihydropyridines are one of the most important groups of calcium channel modulators widely used in the treatment of cardiovascular diseases.21 These include an extensive group of antihypertensive “dipine” drugs like amlodipine, benidipine, etc.
1,4-Dihydropyridine is the main heterocyclic core of compounds displaying antitumor, antioxidant, and gero-, hepato- and neuroprotective effects as well as of derivatives effective in the treatment of diabetes, ischemia, and Alzheimer's disease.23
The relevance of 4H-pyran and 1,4-dihydropyridine derivatives is emphasized by the abundance of studies on optimizing their synthesis.24 These biologically active 4H-pyrans and 1,4-dihydropyridines often bear an aromatic substituent in the 4 position. However, there are only a few examples with a heterocyclic core in this position.23–25 Isothiazole containing derivatives of this type are almost unknown.26
In this work, a convenient synthetic protocol to obtain 4H-pyrans and 1,4-dihydropyridines containing a 4,5-dichloroisothiazole fragment in the 4 position is reported, as exemplified by the generation of compounds 1–6 shown in Scheme 1. The target compounds are very rich in functional groups that allow the generation of supramolecular synthons in the solid state that are described in this work and analysed theoretically using a variety of computational tools.
 | ||
| Scheme 1 Chemical diagram of compounds 1–6 reported in this work. | ||
Methods
Synthesis and characterization
Synthesis of heterocycles 1–6 was performed according to known procedures for preparation of 4H-pyrans and 1,4-dihydropyridines using 4,5-dichloroisothiazole-3-carbaldehyde27 and commercially available methylene-active components (acetylacetone, malononitrile, ethyl 2-cyanoacetate) as starting materials. Single crystals of 1–6 for XRD analysis were obtained by slow evaporation of their saturated solutions in methanol.In addition to XRD, all obtained compounds were characterized using standard LCMS, FTIR and NMR spectroscopy. The presence of the 4,5-dichloroisothiazole fragment was elucidated from the characteristic position of quaternary carbon atom signals in 13C NMR spectra (δ 101.8–115.5, 118.1–123.3 and 146.2–149.1 ppm). In 1H NMR spectra, chemical shifts of the protons at the tertiary carbon atoms of the pyran and pyridine systems are observed in the area of δ 4.91–5.50 ppm. Dihydropyridine NH group signals are observed at δ 6.24–6.49 ppm. A slight W-splitting of the proton signals at the tertiary carbon atom (H-4) and one of the methyl groups (5J4,Me ∼ 0.7 Hz) was observed in the cases of 5 and 6.
X-ray analysis
The crystal structure of heterocycles 2, 3, 5 and 6 was determined by X-ray structural analysis using an automatic four-circle area-detector diffractometer Bruker KAPPA APEX II with MoKα radiation. The crystal structure 1 and 4 was determined by X-ray structural analysis using an XtaLAB Synergy, Dualflex, HyPix with CuKα radiation. The cell parameters of 2, 3, 5 and 6 were refined over the entire data set, together with data reduction using SAINT-Plus software.28 Absorption corrections of 2, 3, 5 and 6 were introduced using the SADABS program.29 The structures were solved using the SHELXT-2018/2 program30 and refined by full-matrix least squares on F2 in the anisotropic approximation for all non-hydrogen atoms (SHELXL-2018/3).31 The C–H bonded hydrogen atoms were placed in geometrically calculated positions and refined in an idealized geometry with isotropic temperature factors equal to 1.2Ueq(C) for CH and CH2-groups, and 1.5Ueq(C) for CH3-groups. The N–H bonded atoms were objectively located from difference Fourier synthesis and refined with isotropic temperature factors equal to 1.2Ueq(N) for NH and NH2 groups. The orientation of CH3-groups was refined. Structures 3 and 6 were refined as inversion twins. Tables and figures for the structures were generated using Olex2.32 Crystal data, data collection, and structure refinement details are summarized in Table 1. All other crystallographic parameters of the structures are indicated in Tables S1–S24 (see the ESI†). The atomic coordinates were deposited at the Cambridge Crystallographic Data Centre (CCDC).33 The CCDC numbers are 2213501–2213504 for 2, 3, 5, and 6, correspondingly, and 2243955 and 2243956 for 1 and 4.| Identification code | 1 | 2 | 3 | 4 | 5 | 6 |
|---|---|---|---|---|---|---|
| CCDC number | 2243955 | 2236983 | 2236984 | 2243956 | 2236985 | 2236986 |
| Empirical formula | C16H18N2O4SCl2 | C15H16N2O3SCl2 | C14H14N2O2SCl2 | C12H9N3O2SCl2 | C13H11N3O3SCl2 | C14H14N2O4SCl2 |
| Formula weight | 405.28 | 375.26 | 345.23 | 330.18 | 360.21 | 377.23 |
| Temperature/K | 100(2) | 100(2) | 100(2) | 100(2) | 100(2) | 100(2) |
| Crystal system | Triclinic | Triclinic | Monoclinic | Triclinic | Triclinic | Monoclinic |
| Space group |
P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) |
P |
P21 |
P |
P |
P21 |
| a/Å | 8.7279(3) | 7.3058(11) | 7.2276(5) | 8.1335(2) | 7.5420(14) | 12.2812(6) |
| b/Å | 9.6603(3) | 10.1615(14) | 26.5464(19) | 9.2637(2) | 8.8365(16) | 7.3500(4) |
| c/Å | 11.4482(4) | 12.4124(18) | 8.5613(6) | 10.0924(3) | 11.384(2) | 17.4340(9) |
| α/° | 86.524(3) | 105.388(6) | 90 | 72.817(2) | 83.566(6) | 90 |
| β/° | 77.743(3) | 106.574(6) | 112.990(3) | 72.328(3) | 83.963(6) | 97.212(2) |
| γ/° | 71.031(3) | 100.321(6) | 90 | 76.935(2) | 88.682(6) | 90 |
| Volume/Å3 | 891.98(6) | 818.5(2) | 1512.16(19) | 684.39(3) | 749.7(2) | 1561.26(14) |
| Z | 2 | 2 | 4 | 2 | 2 | 4 |
| ρ calc g cm−3 | 1.509 | 1.523 | 1.516 | 1.602 | 1.596 | 1.605 |
| μ/mm−1 | 4.588 | 0.539 | 0.572 | 5.746 | 0.587 | 0.571 |
| F(000) | 420.0 | 388.0 | 712.0 | 336.0 | 368.0 | 776.0 |
| Crystal size/mm3 | 0.25 × 0.2 × 0.18 | 0.4 × 0.05 × 0.04 | 0.32 × 0.2 × 0.16 | 0.24 × 0.21 × 0.15 | 0.26 × 0.14 × 0.06 | 0.5 × 0.4 × 0.32 |
| Radiation | CuKα (λ = 1.54184) | MoKα (λ = 0.71073) | MoKα (λ = 0.71073) | CuKα (λ = 1.54184) | MoKα (λ = 0.71073) | MoKα (λ = 0.71073) |
| 2Θ range for data collection/° | 7.904 to 158.912 | 8.186 to 59.998 | 8.674 to 59.998 | 9.484 to 159.122 | 8.162 to 59.986 | 8.192 to 59.998 |
| Index ranges | −10 ≤ h ≤ 11, −12 ≤ k ≤ 12, −14 ≤ l ≤ 14 | −8 ≤ h ≤ 10, −14 ≤ k ≤ 14, −17 ≤ l ≤ 17 | −10 ≤ h ≤ 10, −37 ≤ k ≤ 37, −12 ≤ l ≤ 11 | −9 ≤ h ≤ 10, −11 ≤ k ≤ 11, −12 ≤ l ≤ 12 | −10 ≤ h ≤ 10, −12 ≤ k ≤ 12, −15 ≤ l ≤ 16 | −17 ≤ h ≤ 17, −10 ≤ k ≤ 9, −24 ≤ l ≤ 24 |
| Reflections collected | 19![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 809 809 |
18356 |
25913 |
14966 |
13476 |
26683 |
| Independent reflections | 3836 [Rint = 0.0591, Rsigma = 0.0317] | 4650 [Rint = 0.1086, Rsigma = 0.1363] | 8579 [Rint = 0.0655, Rsigma = 0.0936] | 2945 [Rint = 0.0511, Rsigma = 0.0293] | 4251 [Rint = 0.0823, Rsigma = 0.1054] | 8322 [Rint = 0.0379, Rsigma = 0.0527] |
| Data/restraints/parameters | 3836/0/234 | 4650/0/215 | 8579/2/393 | 2945/0/190 | 4251/0/207 | 8322/1/434 |
| Goodness-of-fit on F2 | 1.061 | 0.965 | 1.016 | 1.031 | 0.993 | 1.037 |
| Final R indices [I ≥ 2σ(I)] | R 1 = 0.0415, wR2 = 0.1139 | R 1 = 0.0633, wR2 = 0.1243 | R 1 = 0.0507, wR2 = 0.0867 | R 1 = 0.0358, wR2 = 0.0985 | R 1 = 0.0527, wR2 = 0.1013 | R 1 = 0.0362, wR2 = 0.0697 |
| Final R indices [all data] | R 1 = 0.0421, wR2 = 0.1147 | R 1 = 0.1396, wR2 = 0.1520 | R 1 = 0.0840, wR2 = 0.0982 | R 1 = 0.0363, wR2 = 0.0992 | R 1 = 0.1077, wR2 = 0.1207 | R 1 = 0.0446, wR2 = 0.0727 |
| Largest diff. peak/hole/e Å−3 | 0.54/−0.61 | 0.47/−0.68 | 0.45/−0.42 | 0.64/−0.50 | 0.58/−0.64 | 0.38/−0.28 |
| Flack parameter | — | — | 0.04(4) | — | — | 0.30(5) |
DFT calculations
The calculations reported herein were performed at the PBE0 (ref. 34)-D3 (ref. 35)/def2-TZVP (ref. 36) level of theory using the Turbomole 7.0 program.37 The binding energies were computed as the difference between the energy of the assembly and the sum of the isolated monomers. The energies have been corrected for the basis set superposition error.38 The MEP surfaces were generated using a 0.001 isosurface to emulate the van der Waals envelope. The QTAIM (ref. 9) and NCIPlot (ref. 39) analyses were performed at the same level using the MultiWFN program40 and represented using the VMD software.41 The NCIplot method is convenient to reveal interactions in real space. It uses reduced density gradient isosurfaces and a colour code (based on the sign of the second eigenvalue of ρ, λ2) to identify the attractive or repulsive nature of the interactions. The following settings were used in this work: RDG = 0.5, density cut-off = 0.04 a.u., and colour scale −0.03 a.u. ≤ signλ2(ρ) ≤ 0.03 a.u. Blue and green colours are used here to identify strongly and moderately attractive interactions, respectively.Results and discussion
Synthesis
Since isothiazole derivatives of 1,4-dihydropyridines and 4H-pyrans were not previously described, their synthesis was carried out by known methods,42 including ones used for the preparation of other heterocyclic derivatives of this type. 4,5-Dichloroisothiazole-3-carbaldehyde 7 (ref. 27) and its condensation products with acetoacetic ester, acetylacetone, and cyanoacetic ester 8–10 were chosen to be starting compounds (Scheme 2). | ||
| Scheme 2 Synthesis of 1,4-dihydropyridines 1–3 and 4H-pyrans 4–6. | ||
Dihydropyridines 1 and 2 were obtained by the interaction of ethyl 3-aminocrotonate with α,β-unsaturated heterocyclic derivatives 8 and 9 (Scheme 2). Ethanol is a common solvent for such processes; however, the reaction in ethanol was not selective. The possible reason for this is side processes involving the nucleophilic substitution of the active chlorine atom in the 5th position of the isothiazole cycle. The same reactions in toluene proceeded smoothly; therefore, compounds 1 and 2 were isolated in 56% and 59% yields, respectively.
The classical Hantzsch synthesis was chosen to obtain symmetric 4-isothiazole-1,4-dihydropyridine 3 from aldehyde 7 (Scheme 2). The yield of product 3 (58%) was similar to those of compounds 1 and 2. This way seems to be more efficient, since it does not require the preliminary synthesis of the intermediate α,β-unsaturated isothiazole from the corresponding aldehyde 7 and acetylacetone, which proceeds with a high but not quantitative yield.
Finally, isothiazole-containing 4H-pyrans 4 and 5 were prepared by the condensation of α,β-unsaturated compounds 8 and 9 with malononitrile; compound 6 was prepared from ethyl 2-cyanocrotonate 10 and acetylacetone. These reactions were catalyzed by piperidine as a base catalyst and yielded the target products with acceptable to good yields (56–76%) even in ethanol.
Structural description of the X-ray structures
The molecular structures of the obtained compounds 1–6 are presented in Fig. 1. Structures 2 and 4 contain one independent molecule of the complex; structures 3 and 6 contain two crystallographically independent molecules. At the same time, 3 contains two molecules with the same conformation (RMSD without inversion is 0.408 Å), and 6 contains right and left independent molecules (Fig. 2), which, however, practically coincide upon inversion (RMSD with inversion is 0.654 Å).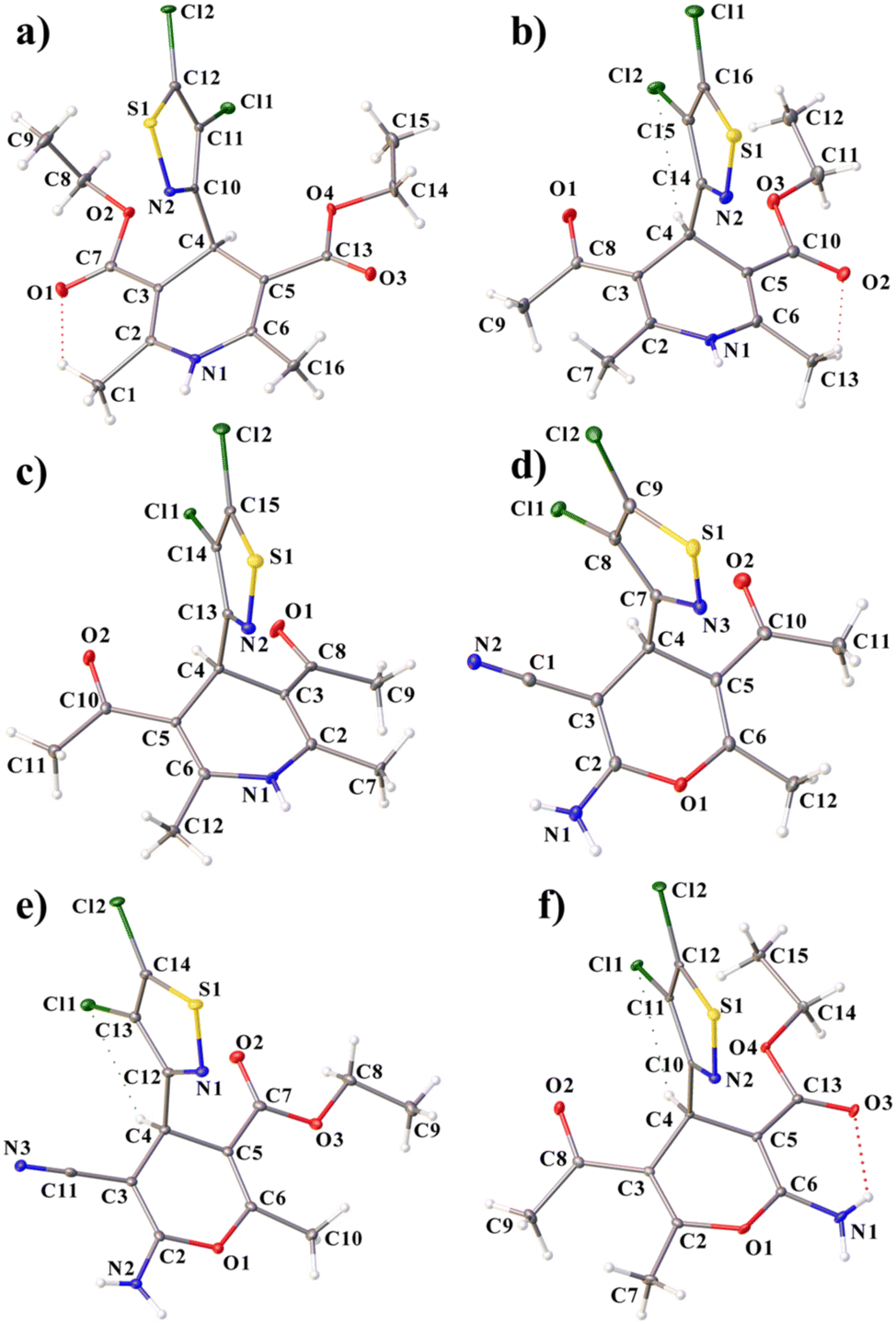 | ||
| Fig. 1 Molecular structure of 1–6 (a–f), including atom labelling. Only one independent molecule is shown in 3 and 6. Displacement ellipsoids are drawn at the 50% probability level. | ||
 | ||
| Fig. 2 Superposition of two crystallographically independent molecules in 3 (a) and 6 (b). | ||
The chlorine atoms are in the plane of the isothiazole rings (the maximum deviation from the plane is 0.02 Å for one of the molecules in 6). The S–N distances in all compounds are close and vary from 1.65 to 1.66 Å. It is worth noting that the isothiazole fragments are turned perpendicular to the plane of the central six-membered ring (the angles between the isothiazole and pyridinyl/pyranyl fragments are close to 90°). In all compounds except 5, the central six-membered rings containing a nitrogen or oxygen atom have a slightly distorted conformation. Most of all, the C4 carbon atom deviates from the plane of the six-membered ring. In 5, distortion of the ring conformation is practically not observed (RMSD is 0.025 Å).
When searching in the CCDC, only two compounds were found containing isothiazole and pyridinyl fragments,43 but with other bulky substituents in the five-membered ring; therefore, it is difficult to compare them with those obtained in this work. However, it should be noted that in the described analogues, the pyridinyl ring was not substituted and had a planar geometry, while the isothiazole ring was rotated differently.
In all the structures, intramolecular hydrogen bonds of the C–H⋯Cl type are present. In 2, an additional weak hydrogen bond of the C–H⋯O type appears. In 3, a stronger N–H⋯O bond compared to the rest is present.
In compounds 2, 3, and 6, the distances between the center of the six-membered ring and the nitrogen atom of the isothiazole fragment vary from 2.71 Å to 2.78 Å, which are shorter than the sum of the van der Waals radii of C and N, and the angle α is close to 70° (Fig. 3). There is an increase in this growth to 2.93–3.11 Å and a decrease in the angle α to 53–61° in 1, 4 and 5. All this allows us to assume the presence of an intramolecular N⋯π interaction in the molecules.44
 | ||
| Fig. 3 View showing N⋯π interactions in structures 1–6 (a–f). Distances in Å. H-atoms are omitted for clarity. Only one independent molecule for 3 and 6 is shown. | ||
All the compounds present good H-bond donor and acceptor groups and, consequently, exhibit H-bonded assemblies in the solid state as shown in Fig. 4. In most of them, the H-bonds propagate the molecules into 1D supramolecular polymers, as observed in compounds 2, 3, 4 and 6, where either the NH of the 1,4-dihydropyridinyl ring (2 and 3) or the exocyclic NH2 group of the 4H-pyranyl ring (4 and 6) acts as a H-bond donor and the O-atom of the carbonyl group (from either keto or ester groups) acts as a H-bond acceptor. It is interesting to comment on compound 6 (Fig. 4f) where two different and alternate types of interactions connect the monomers. On the one hand, there is a directional NH⋯O H-bond with a typical distance (2.22 Å) and on the other hand, there is a longer and less directional NH⋯O contact (2.75 Å) in combination with a C–Cl⋯O halogen bond (2.98 Å) involving one of the Cl-atoms of the 4,5-dichloroisothiazole moiety and the carbonyl O-atom of the ester group. Compounds 1 and 5 (see Fig. 4a and b) form discrete self-assembled dimers instead of 1D polymers, where the H-bond acceptor is the N-atom of the 4,5-dichloroisothiazole moiety. The NH⋯O distances range from 2.05 to 2.22 Å (apart from the special XB/HB assembly in 6), which are similar to the NH⋯H distances in 1 and 5 (2.09 Å and 2.19 Å, respectively). Further analysis of these H-bonds is provided in the theoretical section below.
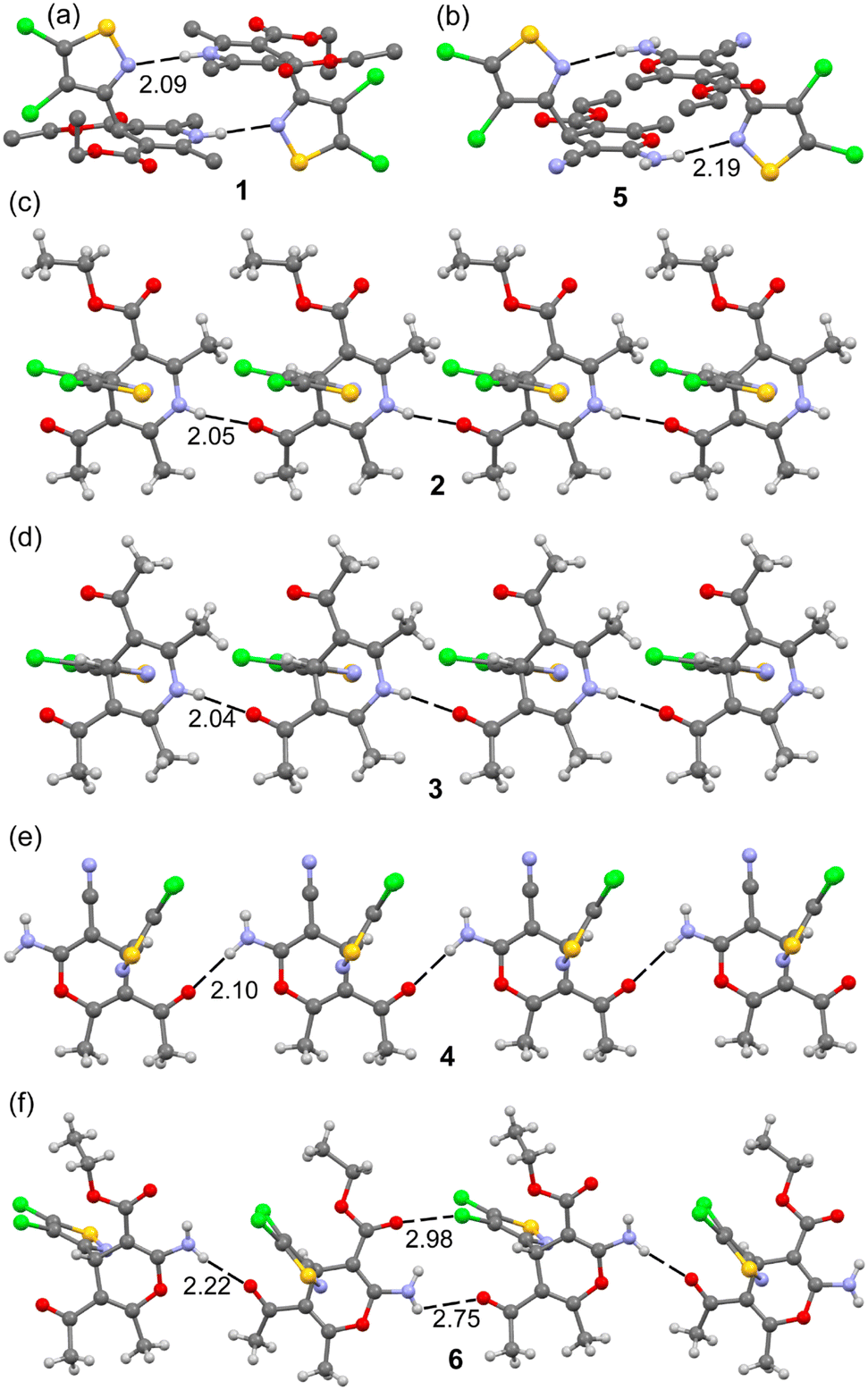 | ||
| Fig. 4 H-bonded assemblies observed in the solid state X-ray structures of compounds 1 (a), 5 (b), 2 (c), 3 (d), 4 (e) and 6 (f). Distances in Å. | ||
Additional H-bonded assemblies were also observed in the solid state of compounds 4 and 5 that contain the cyano and amino substituents attached to the 4H-pyranyl ring. Actually, the exocyclic amino group has two protons suitable to form H-bonds, the ones shown in Fig. 4 and another one with the N-atom of the CN group as an acceptor. The ability of the cyano group to establish noncovalent interactions including HBs has been analyzed before in the literature.45 Both compounds form centrosymmetric R22(12) supramolecular rings in the solid state, as highlighted in Fig. 5. The NH⋯N distances are 2.28 and 2.22 Å for 4 and 5, respectively. The conjugation of the electron lone pair of the amino group to the cyano group likely increases the electron acceptor ability of the sp-hybridized N-atom, as studied below.
 | ||
| Fig. 5 H-bonded centrosymmetric dimer observed in the solid state X-ray structures of compounds 4 (a) and 5 (b). Distances in Å. | ||
It is also worthy to comment on the different π-stacked assemblies observed in some of the compounds reported herein. That is, three different binding modes are observed for the π–π interactions between the 4,5-dichloroisothiazole rings, which are detailed in Fig. 6. In the first one, only one carbon atom of each ring are in contact (van der Waals distance), as observed in compound 1 (see Fig. 6a).
 | ||
| Fig. 6 π-Stacked assemblies observed in the solid state X-ray structures of compounds 1 (a), 2 (b), 4 (c) and 5 (d). Distances in Å. | ||
This binding mode leaves one of the Cl-atoms over the centre of the aromatic ring of the adjacent molecule and vice versa (see blue dashed lines in Fig. 6a), thus establishing LP⋯π interactions. In the second binding mode, two atoms of each ring are in contact, one C-atom of one ring interacts with the S-atom of the other ring and vice versa. This binding mode is observed in compounds 2 and 5 with distances of 3.53 Å and 3.63 Å, respectively. Finally, in the third binding mode, the N-atom of one ring interacts with the S-atom of the other ring and vice versa. This binding mode is observed in compound 4 with a distance of 3.52 Å. The energetic differences between these three binding modes are small, as discussed in the following section.
DFT analysis
The molecular electrostatic potential surfaces of compounds 2, as a representative example for the 1,4-dihydropyridinyl series, and 4 for the 4H-pyranyl series are presented in Fig. 7. It can be observed that the MEP maximum is located at the NH group in 2 (51.5 kcal mol−1) and at the NH2 group in 4. The MEP minimum in 2 is located at the keto O-atom (−42.7 kcal mol−1) which is a better H-bond acceptor than the carbonyl O-atom of the ester group (−35.1 kcal mol−1). In compound 4, it is interesting to highlight that the MEP minimum is located at the sp-hybridized N atom of the cyano group, confirming that the conjugated LPs of the N and O-atoms bonded at the other end of the double bond increase the electron density at the N-atom of the cyano group. The MEP at the O-atom of the keto group is also large and negative (−35.7 kcal mol−1); thus it is also adequate to participate in H-bonds. The MEP also reveals the existence of a σ-hole opposite to the C–Cl bond that is more intense in the Cl-atom bonded at position 5. A detail of the MEP surface around these Cl-atoms is presented at the bottom part of the figure for both compounds, revealing the anisotropy and a modest MEP value at the σ-hole (+12.0 and +13.2 kcal mol−1 for 2 and 4, respectively). | ||
| Fig. 7 MEP surfaces of compounds 2 (a) and 4 (b). The values at selected points are given in kcal mol−1. At the bottom, a detail of the MEP surface around the Cl-atom in position 5 is given for both compounds using a reduced scale (±14 kcal mol−1). | ||
H-bonded dimers extracted from the 1D assemblies described above (see Fig. 4) have been further analysed energetically and characterized using the NCIplot and QTAIM methods. The results are gathered in Fig. 8, evidencing that in all dimers, the NH⋯O interaction is characterized by a bond critical point (CP, small red sphere) and bond path (orange line) and a blue reduced density gradient (RDG) isosurface, thus evidencing the strong nature of these contacts. In the dimers of compounds 2 and 3, the QTAIM analysis evidences the existence of a weaker halogen bond (XB) contact established between one of the Cl-atoms of the 4,5-dichloroisothiazole ring of one monomer and the N-atom of the isothiazole ring of the other monomer, also characterized by the corresponding bond CPs, bond paths and blue RDG isosurfaces. Moreover, additional CH⋯Cl and CH⋯O contacts are revealed in compounds 2 and 3, respectively, which are weaker according to the green colour of the RDG isosurfaces.
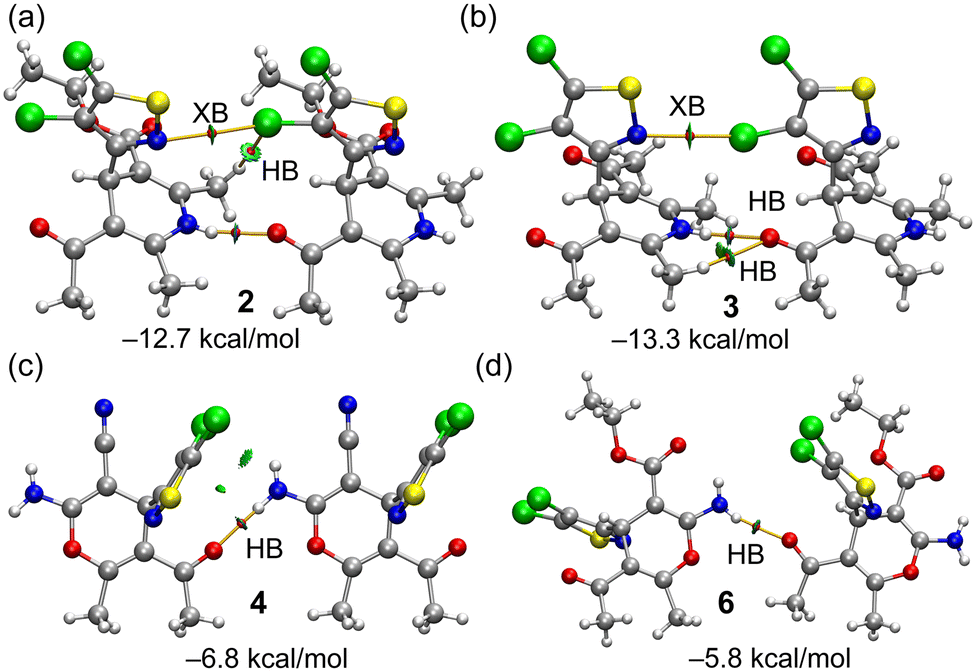 | ||
| Fig. 8 QTAIM (bond CPs in red and bond paths as orange lines) and RDG isosurfaces (see computational methods for NCIPlot settings) of compounds 2 (a), 3 (b), 4 (c) and 6 (d). Only intermolecular interactions are presented. The dimerization energies are also given. | ||
In the dimers of compounds 4 and 6, only one bond CP and bond path are observed corresponding to the strong NH⋯O contact. As expected, taking into consideration the QTAIM/NCIplot analysis, the dimerization energies are larger in the dimers 2 and 3 than in 4 and 6, due to the presence of the additional CH⋯Cl,O contacts, XBs and shorter NH⋯O distances (−12.7 kcal mol−1 and −13.3 kcal mol−1, respectively). The dimerization energies of 4 (−6.8 kcal mol−1) and 6 (−5.8 kcal mol−1) are moderately strong, and basically correspond to the H-bond strength. The contribution of the XBs has been estimated by using the potential energy density (Vr) value at the bond CP that characterizes the XB. This QTAIM parameter can be used as an energy predictor via the equation proposed by Bartashevich & Tsirelson (i.e., E = 0.49 × Vr).46 The Cl⋯N XB energies are very small in compounds 2 and 3 (−0.6 and −0.8 kcal mol−1, respectively). Such small energies disclose that these dimers are dominated by the NH⋯O HBs in line with the dark blue colour of the RDG isosurfaces. Finally, we have also estimated the Cl⋯O XB energy of the XB/HB dimer of compound 6 presented at the bottom of Fig. 4 (middle) and commented above. The XB energy is −1.9 kcal mol−1, thus evidencing that the Cl⋯O XB in compound 6 is stronger than the Cl⋯N XBs in compounds 2 and 3.
The QTAIM/NCIPlot analyses of the H-bonded self-assembled dimers of compounds 1 and 5 are shown in Fig. 9. In both assemblies, the two symmetrically equivalent NH⋯N H-bonds are characterized by bond CPs and bond paths interconnecting the H and N-atoms. An intricate combination of bond CPs and bond paths emerges upon formation of the two strong NH⋯N H-bonds due to the close proximity of the 1,4-dihydropyridinyl rings in 1 and the 4H-pyranyl rings in 5. In particular, five bond CPs and bond paths interconnect the 1,4-dihydropyridinyl rings (including the substituents) in compound 1 and four bond CPs and bond paths interconnect the 4H-pyranyl rings in compound 5. Moreover, extended green RDG isosurfaces are located between the rings, as typical in van der Waals interactions. The dimerization energies are large and negative (−20.9 kcal mol−1 and −17.1 kcal mol−1 in 1 and 5 respectively) due to the contribution of both NH⋯N H-bonds and the van der Waals interactions.
 | ||
| Fig. 9 QTAIM (bond CPs in red and bond paths as orange lines) and RDG isosurfaces (see computational methods for NCIPlot settings) of compounds 1 (a) and 5 (b). Only intermolecular interactions are presented. The dimerization energies are also given. | ||
The energetic features of the centrosymmetric R22(12) dimers have been also studied. Fig. 10 presents the combined QTAIM/NCIPlot analyses of the dimers of compounds 4 and 5, where the NH⋯NC H-bonds are characterized by the corresponding bond CPs, bond paths and blue RDG isosurfaces. It is interesting to highlight the existence of a bond CP interconnecting the CN groups. Moreover, an extended green RDG isosurface is located between the CN groups, thus disclosing the presence of antiparallel CN⋯CN interactions. The dimerization energies are −10.4 kcal mol−1 and −11.9 kcal mol−1 for 4 and 5, respectively, thus confirming the relevance of these R22(12) synthons in the solid state.
 | ||
| Fig. 10 QTAIM (bond CPs in red and bond paths as orange lines) and RDG isosurfaces (see computational methods for NCIPlot settings) of the R22(12) synthons of compounds 4 (a) and 5 (b). Only intermolecular interactions are presented. The dimerization energies are also given. | ||
Finally, the different π-stacking modes between the 4,5-dichloroisothiazole rings observed in several compounds and described in Fig. 6 have been also studied energetically and analysed via QTAIM/NCIplot. The results are summarized in Fig. 11, evidencing that all π-stacked dimers exhibit similar interaction energies, ranging from −9.6 kcal mol−1 in 2 to −10.5 kcal mol−1 in 5, thus explaining the observation of different stacking modes in the solid state. The C⋯C π-stacking is characterized only by one bond CP and bond path (in 1).
 | ||
| Fig. 11 QTAIM (bond CPs in red and bond paths as orange lines) and RDG isosurfaces (see computational methods for NCIPlot settings) of the π-stacked dimers of compounds 1 (a), 2 (b), 4 (c) and 5 (d). Only intermolecular interactions are presented. The dimerization energies are also given. | ||
The double C⋯S stacking is characterized by two bond CPs and bond paths (compounds 2 and 5) and the double S⋯N stacking is characterized by three bond CPs and bond paths (two interconnecting the N and S-atoms and one extra CP connecting both S-atoms (see Fig. 11c)). In compounds 1, 2 and 5, the interaction is further complemented by Cl⋯π interactions, as evidenced by both the QTAIM and NCIplot methods. In all systems, additional van der Waals interactions exist between the 4,5-dichloroisothiazole rings and either the 1,4-dihydropyridinyl (in 1 and 2) or 4H-pyranyl rings (4 and 5), characterized by several bond CPs. bond paths and extended green RDG isosurfaces. It is worthy to emphasize that the interaction energies of the π-stacked assemblies are similar to those of the R22(12) synthons shown in Fig. 11, evidencing that these π-stacked assemblies are energetically very relevant and have an important role in the crystal packing.
Concluding remarks
A convenient synthetic protocol to obtain 3-substituted 4,5-dichloroisothiazole derivatives is reported herein. Moreover, the X-ray structures of six new derivatives are described, showing some recurrent motifs like R22(12) synthons and HB/XB 1D supramolecular polymers. The interactions have been characterized using a combination of QTAIM and NCIplot computational tools and rationalized using MEP surface analysis. The latter is useful to reveal the existence of σ-holes opposite to the C–Cl bonds of the 4,5-dichloroisothiazole ring and rationalize the formation of Cl⋯N and Cl⋯O interactions. It is also important to demonstrate the strong H-bond acceptor ability of the sp-hybridized N-atom in compounds 4 and 5, thus explaining the formation of the R22(12) synthons. Finally, the energetic analysis discloses that both H-bonds and π-stacking interactions are equally important, dictating the X-ray packing of the compounds.Author contributions
I. A. K., A. G. P. and V. I. P.: synthesis and spectral analysis; V. V. B. and R. M. G.: investigation and methodology; M. S. G. and A. P. N.: X-ray experiments; A. F. and F. I. Z.: international collaboration, conceptualization, supervision, validation, project administration, writing – original draft, and writing – review & editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This publication has been supported by the Russian Science Foundation (project number 23-43-10024) (FIZ) and the Belarusian Republican Foundation for Fundamental Research (project number X23RNF-051) (VIP). We thank the MICIU/AEI from Spain for financial support (project number PID2020-115637GB-I00, FEDER funds). We also thank the CTI (UIB) for computational facilities. X-ray diffraction experiments were performed at the Center for Shared Use of Physical Methods of Investigation at the Frumkin Institute of Physical Chemistry and Electrochemistry, RAS.Notes and references
- J. Chen, Q. Peng, X. Peng, H. Zhang and H. Zeng, Chem. Rev., 2022, 122, 14594–14678 CrossRef CAS PubMed.
- K. E. Riley and P. Hobza, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2011, 1, 3–17 CAS.
- E. Persch, O. Dumele and F. Diederich, Angew. Chem., Int. Ed., 2015, 54, 3290–3327 CrossRef CAS PubMed.
- A. K. Nangia and G. R. Desiraju, Angew. Chem., Int. Ed., 2019, 58, 4100–4107 CrossRef CAS PubMed.
- G. Desiraju, J. Am. Chem. Soc., 2013, 135, 9952–9967 CrossRef CAS PubMed.
- G. Desiraju, Angew. Chem., Int. Ed., 2007, 46, 8342–8356 CrossRef CAS PubMed.
- (a) I. Alkorta, J. Elguero and A. Frontera, Crystals, 2020, 10, 180 CrossRef CAS; (b) A. Frontera, D. Quiñonero, C. Garau, A. Costa, P. Ballester and P. M. Deyà, J. Phys. Chem. A, 2006, 110, 5144–5148 CrossRef CAS PubMed; (c) G. Mahmoudi, A. Bauzá, M. Amini, E. Molins, J. T. Mague and A. Frontera, Dalton Trans., 2016, 45, 10708–10716 RSC; (d) A. Bauzá, T. J. Mooibroek and A. Frontera, Chem. Commun., 2014, 50, 12626–12629 RSC.
- M. Juanes, R. T. Saragi, W. Caminati and A. Lesarri, Chem. – Eur. J., 2019, 25, 11402–11411 CrossRef CAS PubMed.
- R. F. W. Bader, Chem. Rev., 1991, 91, 893–928 CrossRef CAS.
- E. R. Johnson, S. Keinan, P. Mori-Sanchez, J. Contreras-Garcia, A. J. Cohen and W. Yang, J. Am. Chem. Soc., 2010, 132, 6498–6506 CrossRef CAS PubMed.
- M. Đaković, Crystallogr. Rev., 2020, 26, 69–100 CrossRef.
- (a) M. S.-C. Pedras and M. Suchy, Org. Biomol. Chem., 2005, 3, 2002–2007 RSC; (b) A. V. Kletskov, N. A. Bumagin, F. I. Zubkov, D. G. Grudinin and V. I. Potkin, Synthesis, 2020, 159–188 CAS.
- (a) Q. F. Wu, B. Zhao, Z. J. Fan, J. B. Zhao, X.-F. Guo, D.-Y. Yang, N.-L. Zhang, B. Yu, T. Kalinina and T. Glukhareva, RSC Adv., 2018, 8, 39593–39601 RSC; (b) G.-N. Zong, F.-Y. Li, Z.-J. Fan, W.-T. Mao, H.-B. Song, L. Chen, Y.-J. Zhu, J.-H. Xu, Y.-Q. Song and J.-R. Wang, Chin. J. Struct. Chem., 2015, 34, 871–878 CAS; (c) R. Slack and K. R.-H. Wooldridge, Adv. Heterocycl. Chem., 1965, 4, 107–120 CrossRef CAS PubMed; (d) M. Davis, Adv. Heterocycl. Chem., 1972, 14, 43–98 CrossRef CAS.
- (a) A. Adams, W. A. Freeman, A. Holland, D. Hossack, J. Inglis, J. Parkinson, H. W. Reading, K. Rivett, R. Slack, R. Sutherland and R. Wien, Nature, 1960, 186, 221–222 CrossRef CAS PubMed; (b) Z. Machon, Z. Wieczorek and M. Zimecki, Pol. J. Pharmacol., 2001, 53, 377–383 CAS.
- (a) C. E. Blunt, C. Torcuk, Y. Liu, W. Lewis, D. Siegel, D. Ross and C. J. Moody, Angew. Chem., Int. Ed., 2015, 54, 8740–8745 CrossRef CAS PubMed; (b) K. Stratmann, J. Belli, C. M. Jensen, R. E. Moore and G. M.-L. Patterson, J. Org. Chem., 1994, 59, 6279–6281 CrossRef CAS.
- (a) H. R. Howard, J. A. Lowe III, T. F. Seeger, P. A. Seymour, S. H. Zorn, P. R. Maloney, F. E. Ewing, M. Newman, A. W. Schmidt, J. S. Furman, G. L. Robinson, E. Jackson and C. J. Morrone, J. Med. Chem., 1996, 39, 143–148 CrossRef CAS PubMed; (b) T. F. Seeger, P. A. Seymour, A. W. Schmidt, S. H. Zorn, D. W. Schulz, L. A. Lebel, S. McLean, V. Guanowsky, H. R. Howard, J. A. Lowe III and J. Heym, J. Pharmacol. Exp. Ther., 1995, 275, 101–113 CAS; (c) C. Prakash, A. Kamel and D. Cui, Drug Metab. Dispos., 1997, 25, 897–901 CAS.
- (a) A. R. Rovira, A. Fin and Y. Tor, Chem. Sci., 2017, 8, 2983–2993 RSC; (b) E. Krzyżak, M. Śliwińska and W. Malinka, J. Fluoresc., 2015, 25, 277–282 CrossRef PubMed.
- J. A. Eremina, E. V. Lider, T. S. Sukhikh, L. S. Klyushova, M. L. Perepechaev, D. G. Sheven, A. S. Berezin, A. Y. Grishanov and V. I. Potkin, Inorg. Chim. Acta, 2020, 510, 119778 CrossRef CAS.
- J. A. Eremina, K. S. Smirnova, E. V. Lider, L. S. Klyushova, D. G. Sheven and V. I. Potkin, Transition Met. Chem., 2022, 47, 19–30 CrossRef CAS.
- M. Bihani, P. P. Bora and G. Bez, J. Chem., 2013, 2013, 785930 Search PubMed.
- S. A. Khedkar and P. B. Auti, Mini-Rev. Med. Chem., 2014, 14, 282–290 CrossRef CAS PubMed.
- M. Kamalzare, M. Bayat and A. Maleki, R. Soc. Open Sci., 2020, 7, 200385 CrossRef CAS PubMed.
- J. Safaei-Ghomi, A. Ziarati and S. Zahedi, J. Chem. Sci., 2012, 124, 933–939 CrossRef CAS.
- E. Pourian, S. Javanshir, Z. Dolatkhah, S. Molaei and A. Maleki, ACS Omega, 2018, 3, 5012–5020 CrossRef CAS PubMed.
- M. Rucins, A. Plotniece, E. Bernotiene, W.-B. Tsai and A. Sobolev, Catalysts, 2020, 10, 1019 CrossRef CAS.
- A. V. Kletskov, V. I. Potkin, I. A. Kolesnik, S. K. Petkevich, A. V. Kvachonak, M. O. Dosina, D. O. Loiko, M. V. Larchenko, S. G. Pashkevich and V. A. Kulchitsky, Nat. Prod. Commun., 2018, 13, 1507–1510 CrossRef.
- N. A. Bumagin, V. M. Zelenkovskii, A. V. Kletskov, S. K. Petkevich, E. A. Dikusar and V. I. Potkin, Russ. J. Gen. Chem., 2016, 86, 68–81 CrossRef CAS.
- U. SAINT, V8.40B, Bruker AXS Inc., Madison, WI, 2020 Search PubMed.
- L. Krause, R. Herbst-Irmer, G. M. Sheldrick and D. Stalke, J. Appl. Crystallogr., 2015, 48, 3–10 CrossRef CAS PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Adv., 2015, 71, 3–8 CrossRef PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3–8 Search PubMed.
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, J. Appl. Crystallogr., 2009, 42, 339–341 CrossRef CAS.
- C. R. Groom, I. J. Bruno, M. P. Lightfoot and S. C. Ward, Acta Crystallogr., Sect. B: Struct. Sci., Cryst. Eng. Mater., 2016, 72, 171–179 CrossRef CAS PubMed.
- C. Adamo and V. Barone, J. Chem. Phys., 1999, 110, 6158–6169 CrossRef CAS.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed.
- F. Weigend and R. Ahlrichs, Phys. Chem. Chem. Phys., 2005, 7, 3297–3305 RSC.
- R. Ahlrichs, M. Bar, M. Haser, H. Horn and C. Kolmel, Chem. Phys. Lett., 1989, 162, 165–169 CrossRef CAS.
- S. B. Boys and F. Bernardi, Mol. Phys., 1970, 19, 553–566 CrossRef CAS.
- J. Contreras-García, E. Johnson, S. Keinan, R. Chaudret, J.-P. Piquemal, D. Beratan and W. Yang, J. Chem. Theory Comput., 2011, 7, 625–632 CrossRef PubMed.
- T. Lu and F. Chen, J. Comput. Chem., 2012, 33, 580–592 CrossRef CAS PubMed.
- W. Humphrey, A. Dalke and K. Schulten, J. Mol. Graphics, 1996, 14, 33–38 CrossRef CAS PubMed.
- (a) N. Martin, C. Seoane and J. L. Soto, Tetrahedron, 1988, 44, 5861–5868 CrossRef CAS; (b) L. L. W. Cheung, S. A. Styler and A. P. Dicks, J. Chem. Educ., 2010, 87, 628–630 CrossRef CAS; (c) S. K. Petkevich, T. D. Zvereva, P. S. Shabunya, H. Zhou, E. V. Nikitina, A. A. Ershova, V. P. Zaytsev, V. N. Khrustalev, A. A. Romanycheva, A. A. Shetnev and V. I. Potkin, Chem. Heterocycl. Compd., 2022, 58, 333–341 CrossRef CAS.
- I. Skrastiņa, A. Baran, D. Muceniece and J. Popelis, Chem. Heterocycl. Compd., 2014, 50, 87–102 CrossRef.
- (a) R. W. Newberry and R. T. Raines, Acc. Chem. Res., 2017, 50, 1838–1846 CrossRef CAS PubMed; (b) S. K. Singh and A. Das, Phys. Chem. Chem. Phys., 2015, 17, 9596–9612 RSC; (c) A. P. Novikov, M. A. Volkov, A. V. Safonov and M. S. Grigoriev, Crystals, 2022, 12, 271 CrossRef CAS.
- (a) P. Sharma, A. Gogoi, A. K. Verma, A. Frontera and M. K. Bhattacharyya, New J. Chem., 2020, 44, 5473–5488 RSC; (b) D. Dutta, T. Baishya, R. M. Gomila, A. Frontera, M. Barceló-Oliver, A. K. Verma and M. K. Bhattacharyya, J. Mol. Struct., 2023, 1274, 134568 CrossRef CAS; (c) D. Dutta, P. Sharma, A. Frontera, A. Gogoi, A. K. Verma, D. Dutta, B. Sarma and M. K. Bhattacharyya, New J. Chem., 2020, 44, 20021–20038 RSC; (d) S. M. Nashre-ul-Islam, D. Dutta, A. Frontera and M. K. Bhattacharyya, Inorg. Chim. Acta, 2019, 487, 424–432 CrossRef CAS.
- E. V. Bartashevich and V. G. Tsirelson, Russ. Chem. Rev., 2014, 83, 1181–1203 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: CCDC 2236983–2236986, 2243955 and 2243956. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3ce00112a |
| This journal is © The Royal Society of Chemistry 2023 |