
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Polymorph prediction through observed structural isomorphism leading to a new crystalline form of cannabidiol†
Hannah E.
Straker
 *a,
Lynn
McMillan
*a,
Lina
Mardiana
bcd,
Glen R.
Hebberd
b,
Elle
Watson
bc,
Paul G.
Waddell
c,
Michael R.
Probert
*bc and
Michael J.
Hall
*bc
*a,
Lynn
McMillan
*a,
Lina
Mardiana
bcd,
Glen R.
Hebberd
b,
Elle
Watson
bc,
Paul G.
Waddell
c,
Michael R.
Probert
*bc and
Michael J.
Hall
*bc
aJazz Pharmaceuticals, Kent Science Park, Sittingbourne, Kent ME9 8AG, UK. E-mail: hannah.straker@jazzpharma.com; lynn.mcmillan@jazzpharma.com
bIndicatrix Crystallography Ltd, Bedson Building, Newcastle University, Newcastle upon Tyne, Tyne and Wear NE1 7RU, UK. E-mail: michael.probert@newcastle.ac.uk; michael.hall@newcastle.ac.uk
cChemistry – School of Natural and Environmental Sciences, Bedson Building, Newcastle University, Newcastle upon Tyne, Tyne and Wear NE1 7RU, UK
dDepartment of Chemistry, Universitas Indonesia, Depok, Jawa Barat 16424, Indonesia
First published on 29th March 2023
Abstract
Cannabidiol (CBD) is a naturally occurring phytocannabinoid, approved for use in the US and other countries for the treatment of seizures associated with Dravet syndrome, Lennox–Gastaut syndrome and tuberous sclerosis complex, and is commonly isolated and used in its well-known stable crystalline form (form 1). We present a study of the crystal structures of an homologous series of CBD analogues, containing variations in the alkyl sidechain at C-5′, which shows that CBD form 1 is an unusual structural outlier. CBD C-5′ homologues display homologous isomorphism, existing as isomorphic variants in the solid state, whereas CBD form 1 shows notably different crystal packing, suggesting the potential existence of a previously uncharacterized isomorphic CBD polymorph. This predicted isomorphic CBD polymorph (form 2) was subsequently discovered through the use of high throughput crystallisation techniques (ENaCt) in combination with CBD homologue seeding, with single crystal X-ray diffraction analysis demonstrating its fit within the larger isomorphic series. This work represents the first example of directed polymorph discovery using high throughput ENaCt techniques.
1. Introduction
Cannabidiol (CBD) is one of at least eighty-five naturally occurring phytocannabinoids found in cannabis plants and accounts for around 40–50% of the crude plant extract alongside other significant phytocannabinoids including cannabidivarin (CBDV), cannabigerol (CBG) and Δ9-tetrahydrocannabinol (THC).1–3 CBD naturally exists in the plant as cannabidiolic acid (CBDA), which is converted to CBD through thermal decarboxylation upon isolation.4 CBD was discovered and first isolated in 1940 by the groups of both Adams and Todd,5,6 with relative and absolute stereochemistry determined by subsequent work from Mechoulam and Sikemeier.7–9Following botanical extraction and decarboxylation, high purity CBD may be isolated commercially by crystallisation.10,11 CBD is not euphoric and has become increasingly important as an active pharmaceutical ingredient (API). The medicinal product Sativex™, which is used for symptom improvement in patients with moderate to severe spasticity due to multiple sclerosis (MS), contains CBD and THC as the most abundant phytocannabinoids while Epidiolex™, which is used for the treatment of seizures associated with Dravet syndrome, Lennox–Gastaut syndrome and tuberous sclerosis complex, contains highly purified CBD.12,13
The crystal structure of CBD was first reported in 1977 by the two groups of Kennard14 and Rosenqvist15 (CANDOM01, CANDOM10), with Bodensteiner et al. later confirming the absolute stereochemistry via accurate measurement of the anomalous dispersion effects on the Friedel pairs of reflections (CANDOM11).16 CBD crystallises in the monoclinic space group P21, with two independent molecules in the unit cell (Z′ = 2). These molecules are connected through a single hydrogen bond, linking two of the phenolic OH groups. A network of weaker C–H⋯O–H interactions complete the molecular network within the crystal structure. The major difference between the two molecules in the asymmetric unit arises from differences in the torsion angles within the n-pentyl chain, attached at the C-5′ position. In one of the molecules the n-pentyl chain adopts a more linear arrangement, whilst in the other molecule the n-pentyl chain is folded back towards the body of molecule (Fig. 1).
 | ||
| Fig. 1 Structure of CBD form 1. Left, a pair of CBD molecules in the unit cell, H-bonds highlighted. Right, overlay of the crystallographically inequivalent CBD molecules in the unit cell showing conformational differences. Anisotropic displacement parameters shown at 50% probability, all carbon bound hydrogen atoms removed for clarity. | ||
CBD is one of a series of naturally occurring homologous phytocannabinoids which differ only in the length of the alkyl chain at the C-5′ position. Due to the potential bioactivity of molecules in this series, and the variations in activity that this alkyl chain may impose, we have investigated the solid forms of a subset of CBD homologues. This subset is formed of the naturally occurring n-propyl substituted cannabidivarin (CBD-3 or CBDV), n-butyl substituted CBD-4, and n-pentyl substituted parent cannabidiol (CBD-5 or CBD) and the synthetic n-hexyl substituted CBD-6 (Fig. 2).
 | ||
| Fig. 2 Molecular structures of investigated cannabinoids, (1) CBD-3 (cannabidivarin; CBDV; n = 2), (2) CBD-4 (n = 3), (3) CBD-5 (cannabidiol; CBD; n = 4) and (4) CBD-6 (n = 5). | ||
Following relatively straightforward crystallisation processes, our analysis of the crystal structures of CBD-3 (CBDV), CBD-4 and CBD-6 revealed a particularly interesting feature. All three of the molecular forms, irrespective of the alkyl side chain, crystallised in the orthorhombic space group P212121, with only one molecule in the asymmetric unit. Furthermore, the structures demonstrated an isomorphic packing arrangement, providing a near unique set of structures where the inclusion of successively longer alkyl chains only increases the molecular separation but does not disrupt the packing motif.
Crystallographic studies on molecular series differing only by alkyl chain length have previously shown odd–even alternation effects, in which odd homologues or even homologues have similar crystal forms, but differ from odd to even.17,18 However only a small number of examples are known in which molecular homologues show isomorphic packing, described by Kitaigorodskii as “homologous isomorphism”.19
This rare case of “homologous isomorphism” within the CBD homologues, prompted our interest, particularly with the knowledge that these structures did not correlate to the parent molecule CBD, having an entirely different solid-state structure. We therefore hypothesized that there may be a hitherto unknown crystalline form of CBD, an isomorphic polymorph to the studied series. Herein, we have validated our prediction with the discovery of a new polymorph of CBD (form 2), through the use of template seeding in combination with high throughput encapsulated nanodroplet crystallisation (ENaCt).20 ENaCt involves the use of high-throughput robotics to generate crystallisation experiments in 96 well format sealed glass plates, in which a nanolitre scale droplet of analyte is encapsulated within an inert oil. The oil encapsulation aids in controlling subsequent crystallisation of the analyte, typically over the course of a few days. ENaCt has found previous application in the crystallisation of a range of small organic molecules, including the discovery of new polymorphs, albeit without the use of seeding.21–23 ENaCt was chosen for this study, over other modern crystallisation methods, as it allowed for the rapid exploration of experimental crystallisation space in organic solvents that would be required to discover new CBD polymorphs.24
2. Experimental
2.1 Material
All materials were provided by GW Pharma, a Jazz Pharmaceuticals company, with cannabidiol (CBD-5) and cannabidivarin (CBD-3) isolated from botanical materials using proprietary methods and CBD-4 and CBD-6 prepared synthetically following literature methods.252.2 Classical crystallisation of CBD-3, CBD-4, CBD (form 1) and CBD-6
Single crystals suitable for analysis via single crystal X-ray diffraction (SCXRD) were prepared as follows. CBD-3 was crystallised from an n-heptane/toluene solution, CBD-4 was crystallised from an n-heptane solution and CBD-6 crystallised from a neat oil upon prolonged storage at −20 °C. CBD form 1 crystals were grown via slow evaporation of solutions of CBD in n-pentane, n-hexane, n-heptane and dichloromethane (see ESI,† page S4).2.3 CBD form 1 by ENaCt
ENaCt crystallisation of CBD was carried out in 96 well glass plates, using nine hydrocarbon and halogenated solvents in which CBD was highly soluble (pentane, hexane, heptane, chloroform, 1,2-dichloroethane (1,2-DCE), toluene, chlorobenzene, fluorobenzene and hexafluorobenzene (C6F6)), at three different concentrations and in the presence of four oils (FC 40, Fomblin YR, PDMSO, or mineral oil), resulting in a total of 864 individual ENaCt experiments.After 14 days, crystallisation experiments were assessed visually by cross-polarised optical microscopy and outcomes were ranked as: (F) failed; (1) remaining in solution; (2) oil or amorphous solid; (3) microcrystalline material; (4) crystals suitable for SCXRD. 18 of the 864 ENaCt experiments (2.1%) gave single crystals suitable for SCXRD (3 from chloroform, 4 from 1,2-DCE, 6 from toluene, and 5 from fluorobenzene). The majority of single crystals grew from mineral oil at the higher concentration and displayed needle-like morphologies. A representative selection of crystals was examined by SCXRD and gave unit cell parameters that matched the known CBD form 1. A full SCXRD data set was also obtained from a crystal grown from toluene/mineral oil, and the refined structure also matched the known CBD form 1 (see ESI,† pages S5–S7).
2.4 CBD form 2 by ENaCt including template seeding
ENaCt crystallisation of CBD was undertaken seeded with CBD-3 and CBD-4, with control experiments involving non-seeded and seeding with CBD (form 1), using five solvents (chloroform (CHCl3), 1,2-dichloroethane (DCE), 1,1,2,2-tetrachloroethane (TeCE), toluene and 4-fluorotoluene) and four oils (FC 40, Fomblin YR, PDMSO, or mineral oil).Seed crystals were prepared starting from classically recrystallized material or commercially supplied crystalline material and were crushed using a spatula to provide a stock of seed crystals. Seeding of ENaCt plates was completed via use of a Hampton micro-tool. This was used to pick up a small quantity of crushed seed crystals which were then manually placed in each well.
CBD gave crystals across a wide range of experimental conditions, providing needle-like crystal morphologies, with and without seeding with CBD (form 1). Crystals grown of CBD, with CBD-3 and CBD-4 seeding, gave block-like crystal morphologies indicative of a new polymorph (form 2) from all tested solvents.
Unit cell data were collected for a series of crystals including CBD form 1 (seeded w. CBD, DCE, mineral oil) and CBD form 2 (seeded w. CBD-3, DCE, mineral oil), and full SCXRD data sets were also obtained of CBD form 1 grown with CBD seeding (seeded w. CBD, DCE, mineral oil) and CBD form 2 grown with both CBD-3 (DCE, mineral oil) and CBD-4 seeding (4-fluorotoluene, mineral oil) (CCDC 2234293–2234299) (see ESI,† pages S8–S10).
2.5 Melting point experiments for CBD form 2 by ENaCt
CBD form 2 crystals were grown from four 96-well plates, consisting of a total of 384 experiments (DCE, mineral oil, seeded with CBD-3). 361 wells (94%) showed block-like morphologies, indicative of CBD form 2 crystals, from which crystals were manually selected and packed into melting point tubes for measurements (see ESI,† pages S8–S10).2.6 Crystallographic data
All data are deposited with the CSD and can be accessed through ref codes (2234293–2234299). Where compounds have been previously studied and published data are available, studies were repeated to provide comparable experimental data to allow for more meaningful direct structural comparisons. In these cases, no significant differences were noted from the published structures.3. Results and discussion
Crystals of suitable quality for SCXRD studies were obtained for all compounds including CBD-3, CBD-4, CBD (form 1) and CBD-6 (Fig. 3). | ||
| Fig. 3 Packing diagrams of the structures of CBD homologues, CBD-3, CBD-4, CBD (form 1) and CBD-C6 (from top left to bottom right) as viewed down the crystallographic c-axis. Hydrogen atoms have been removed for clarity. | ||
CBD-3, CBD-4 and CBD-6 all crystallise within the P212121 space group with the same packing, and the structures can be described as isomorphous, an example of homologous isomorphism. The molecules pack into a layered structure, with each layer bound by multiple weak C–H⋯O–H intermolecular interactions. The oxygen atom of the hydroxyl groups, involved in the intra-layer interactions, forms connections with several locally oriented H–C groups (both intermolecular and intramolecular, reducing molecular conformational freedom). These layers then pack together through classically stronger O–H⋯O–H interactions. The differences between the structures across the isomorphous series are imposed by the longer alkyl chain which increases the spacing of the molecules within the layers, resulting in an increase in the corresponding unit cell a-axis lengths. Thus, across the CBD-3, CBD-4 and CBD-6 series the length of the unit cell a-axis increases from 9.5113(4) to 9.7147(5) and finally to 11.1727(13) Å, with alkyl chains aligned closely with the a-axis in the structures. This observed homologous isomorphism across the series of molecules CBD-3, CBD-4 and CBD-6 is highly unusual for systems varying only in alkyl chain length. It should be noted that the unit cell b-axis varies non-linearly across the series, the c-axis cell dimensions show a marginal decrease, and the densities remain similar (Table 1).
| Compound | CBD-3 | CBD-4 | CBD-5 (form 1) | CBD-5 (form 2) | CBD-6 |
|---|---|---|---|---|---|
| Chemical formula | C19H26O2 | C20H28O2 | C21H30O2 | C21H30O2 | C22H32O2 |
| MW | 286.41 | 300.44 | 314.47 | 314.47 | 328.50 |
| Temperature (K) | 150 | 150 | 150 | 150 | 150 |
| Crystal system | Orthorhombic | Orthorhombic | Monoclinic | Orthorhombic | Orthorhombic |
| Space group | P212121 | P212121 | P21 | P212121 | P212121 |
| a (Å) | 9.5113(4) | 9.7147(5) | 10.43482(16) | 10.2114(2) | 11.1727(13) |
| b (Å) | 12.2963(5) | 13.1197(7) | 10.88547(17) | 13.0916(3) | 12.5417(14) |
| c (Å) | 14.3259(5) | 13.9392(8) | 16.7749(3) | 13.6676(3) | 13.6848(16) |
| α (°) | 90 | 90 | 90 | α = 90 | 90 |
| β (°) | 90 | 90 | 95.4518(15) | β = 90 | 90 |
| γ (°) | 90 | 90 | 90 | γ = 90 | 90 |
| V (Å3) | 1675.47(11) | 1776.61(17) | 1896.81(5) | 1827.13(7) | 1917.6(4) |
| ρ calc (g cm−3) | 1.135 | 1.123 | 1.101 | 1.143 | 1.138 |
| Z | 4 | 4 | 4 | 4 | 4 |
| Z′ | 1 | 1 | 2 | 1 | 1 |
| Reflections collected | 11![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 921 921 |
10363 |
26857 |
15895 |
14661 |
| Independent reflections | 2963 [Rint = 0.0166, Rsigma = 0.0136] | 3103 [Rint = 0.0189, Rsigma = 0.0157] | 6671 [Rint = 0.0440, Rsigma = 0.0339] | 3223 [Rint = 0.0184, Rsigma = 0.0125] | 3394 [Rint = 0.0406, Rsigma = 0.0332] |
| Final R indexes [all data] | R 1 = 0.0259, wR2 = 0.0676 | R 1 = 0.0287, wR2 = 0.0768 | R 1 = 0.0425, wR2 = 0.0856 | R 1 = 0.0244, wR2 = 0.0625 | R 1 = 0.0297, wR2 = 0.0776 |
| Flack | 0.07(3) | −0.03(4) | −0.07(9) | 0.00(2) | −0.03(6) |
| CCDC number | 2234293 | 2234294 | 2234295 | 2234299 | 2234296 |
CBD (form 1) is a significant outlier in the series, having an entirely different packing motif within the P21 space group, with Z′ = 2, and with the crystallographically independent molecules having significantly different conformations (as previously shown in Fig. 1).
Based on the observed homologous isomorphism of CBD-3, CBD-4 and CBD-6 and the structural outlier that was CBD form 1, we hypothesised that CBD should have a ‘missing polymorph’ which would be isomorphous to the structural series. However, both classical and high throughput ENaCt crystallisation of CBD only resulted in the observation of the needle-like CBD form 1.
Since our hypothesised polymorph of CBD would be isomorphous with the rest of the series, we initiated a further experimental study based around a seeding approach, in which we would attempt to template the growth of a novel CBD polymorph with seeds of the isomorphic crystals of its homologues. ENaCt experiments were therefore undertaken in which we attempted to grow crystals of CBD seeded with CBD-3 or CBD-4, with control experiments including the crystallisation of CBD alone and CBD seeded with CBD, using an experimental space composed of 5 solvent/4 oil combinations. CBD-6 was not used for seeding due to its low melting point making seeding experimentally challenging.
CBD typically crystallised with a needle-like morphology from all solvent/oil combinations, on its own and when seeded with CBD form 1, the morphology matching that typically observed for CBD form 1. Interestingly in the cases when CBD was seeded with CBD-3, a new block-like morphology was consistently observed from all solvent/oil combinations, with 89 crystals from 160 experiments (56%) suitable for SCXRD. Similar results were obtained for seeding with CBD-4, with 85 from 160 experiments (53%) showing block-like crystals suitable for SCXRD. Unit cell measurements were taken for both needle-like crystals (CBD seeded with CBD) and block-like morphologies (CBD seeded with CBD-3 and CBD seeded with CBD-4) showing the presence of two different polymorphs, form 1 and form 2. Interestingly, the newly observed form 2 crystallised in the same P212121 space group as native CBD-3, CBD-4 and CBD-6 (Fig. 4, Table 1).
 | ||
| Fig. 4 Examples of CBD crystals grown via ENaCt, for which full structures were obtained by SCXRD: A) needle-like crystals of CBD form 1 (seeded w. CBD, DCE, mineral oil); B) block-like crystals of CBD form 2 (seeded w. CBD-3, DCE, mineral oil); C) block-like crystals of CBD form 2 (seeded w. CBD-4, 4-fluorotoluene, mineral oil) grown via ENaCt. | ||
Full structural analysis by SCXRD of CBD crystals with the block-like morphology confirmed the presence of a new CBD polymorph (for CBD seeded with CBD-3, see ESI,† page S15). This new polymorphic form of CBD, form 2, was found to match the idealised prediction from the homologous series of CBD variants. Form 2 was found to crystallise in the P212121 space group with Z′ = 1. The structure indeed being found to be isomorphous with respect to the observed crystal structures of the series of CBD homologues CDB-3, CBD-4 and CBD-6 (Fig. 5). Additionally, the a-axis length of this new form was found to fit with the trend of the previously observed structures giving refined values of 10.1971(6) and 10.2114(2) Å respectively for the CBD-3 and CBD-4 seeding experiments, bisecting the values of 9.7147(5) and 11.1727(13) Å from the CBD-4 and CBD-6 variants respectively.
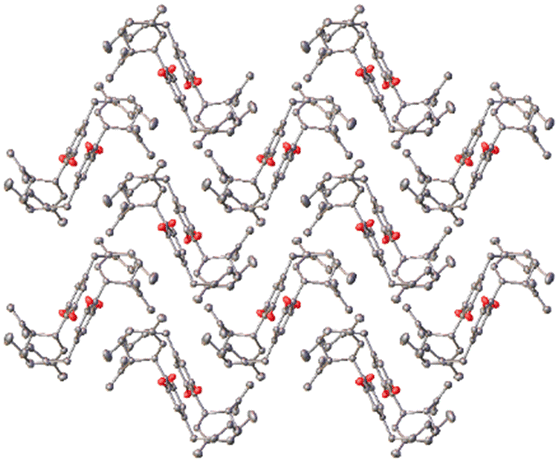 | ||
| Fig. 5 CBD-5 form 2, viewed down the crystallographic c-axis. Hydrogen atoms have been removed for clarity. | ||
Finally, to better understand how CBD form 2 fits within the polymorph landscape of CBD, a series of melting point experiments were undertaken. Firstly, a single crystal of CBD was confirmed as form 2, via unit cell analysis, and was then slowly warmed within the nitrogen gas flow of an Oxford Cryosystems Cryostream 800 in situ on a Bruker D8 Venture diffractometer. Loss of crystallinity, as observed both visually and through a disappearance of a discrete diffraction pattern, occurred at approximately 45 °C.
Secondly, to allow for a bulk melting point analysis, four 96-well plates were prepared containing CBD seeded with CBD-3, in DCE and mineral oil. After 5 days, large numbers of CBD form 2 crystals were identified by morphology, the crystallisation plates were opened, and crystals removed from multiple wells. These were collected on a glass slide, placed in capillary tubes and melting point experiments were carried out. CBD form 2 crystals melted at around 43–46 °C, lower than the corresponding CBD form 1 (66 °C). Since CBD form 2 has a lower melting point to form 1 by approximately 20 °C, we suggest that form 2 is a kinetically formed ‘via templating’ metastable polymorph, with form 1 as the more thermodynamically stable polymorph. The presence of multiple polymorphs for small organic molecule is not unusual,26,27 for example glycine28 or the much studied ROY,29 however this is the first example of such polymorphism in the cannabidiols.
Conclusions
Based on our observation of homologous isomorphism within a series of CBD C-5′ homologues, we investigated the anomalous parent CBD molecule for the presence of a predicted new polymorphic form. Through the use of template seeding in combination with extensive crystallisation space exploration, by high throughput ENaCt techniques, we have discovered a new polymorph (form 2) of CBD, which we believe to be a metastable form based on melting point analysis. With the discovery of CBD form 2, we now report a rare case of four small molecules CBD-3, CBD-4, CBD-5 and CBD-6 differing only by a single methylene unit, which display homologous isomorphism in the solid state across the series. Furthermore, this is the first report of seed directed polymorph discovery by ENaCt techniques, which further expands on the impact of this high throughput nanoscale crystallisation method.Conflicts of interest
HS and LMcM are employees of Jazz Pharmaceuticals and hold stock and/or stock options in Jazz Pharmaceuticals, plc., GRH, EW and LM are employees of Indicatrix Crystallography, and MRP and MJH are directors and shareholders in Indicatrix Crystallography.Acknowledgements
LM thanks The Indonesian Endowment Fund for Education (LPDP) for PhD funding, MJH and MRP thank Newcastle University for funding support.References
- R. Mechoulam and L. Hanus, Chem. Phys. Lipids, 2002, 121, 35–43 CrossRef CAS PubMed.
- L. D. Schuman, D. Lu, D. A. Kendall, A. C. Howlett and A. H. Lichtman, in Substance Use Disorders. Handbook of Experimental Pharmacology, ed. M. Nader and Y. Hurd, Springer, Cham, 2020, vol. 258, pp. 323–353 Search PubMed.
- A. R. Aguillón, R. A. C. Leão, L. S. M. Miranda and R. O. M. A. de Souza, Chem. – Eur. J., 2021, 27, 5577–5600 CrossRef PubMed.
- B. A. Whittle, C. A. Hill, I. R. Flockhart, D. V. Downs, P. Gibson and G. W. Wheatley, US Pat., US20200306328A1, 2019 Search PubMed.
- R. Adams, M. Hunt and J. H. Clark, J. Am. Chem. Soc., 1940, 62, 196–200 CrossRef CAS.
- A. Jacob and A. R. Todd, J. Chem. Soc., 1940, 649–653 RSC.
- R. Mechoulam and Y. Shvo, Tetrahedron, 1963, 19, 2073–2078 CrossRef CAS PubMed.
- R. Mechoulam and Y. Gaoni, Tetrahedron Lett., 1967, 8, 1109–1111 CrossRef PubMed.
- T. Petrzilka, W. Haefliger and C. Sikemeier, Helv. Chim. Acta, 1969, 52, 1102–1134 CrossRef CAS.
- B. Whittle, C. A. Hill, I. R. Flockhart, D. V. Downs, P. Gibson and G. W. Wheatley, US Pat., US7344736B2, 2002 Search PubMed.
- I. Flockhart, G. W. Wheatley, S. Dring and L. Archer, EU Pat., EP1542952A1, 2003 Search PubMed.
- G. Guy, S. Wright, A. Mead and O. Devinsky, US Pat., US9949937B2, 2017 Search PubMed.
- G. Guy, S. Wright, A. Mead and O. Devinsky, US Pat., US10111840B2, 2015 Search PubMed.
- P. G. Jones, L. Falvello, O. Kennard, G. M. Sheldrick and R. Mechoulam, Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem., 1977, 33, 3211–3214 CrossRef.
- T. Ottersen, E. Rosenqvist, C. E. Turner and F. S. El-Feraly, Acta Chem. Scand., 1977, 31, 807–812 CrossRef CAS.
- T. Mayr, T. Grassl, N. Korber, V. Christoffel and M. Bodensteiner, IUCrData, 2017, 2, x170276 CrossRef CAS.
- P. Zugenmaier and A. Heiske, Liq. Cryst., 1993, 15, 835–849 CrossRef CAS.
- Y. Yang, H. Ikedo, H. Huang, I. Yoshikawa and H. Houjou, Cryst. Growth Des., 2021, 21, 4121–4132 CrossRef CAS.
- A. I. Kitaigorodsky, Molecular Crystals and Molecules, Academic Press, New York, 1973 Search PubMed.
- A. R. Tyler, R. Ragbirsingh, C. J. McMonagle, P. G. Waddell, S. E. Heaps, J. W. Steed, P. Thaw, M. J. Hall and M. R. Probert, Chem, 2020, 6, 1755–1765 CAS.
- J. Zhu, I. Moreno, P. Quinn, D. S. Yufit, L. Song, C. M. Young, Z. Duan, A. R. Tyler, P. G. Waddell, M. J. Hall, M. R. Probert, A. D. Smith and A. C. O'Donoghue, J. Org. Chem., 2022, 87, 4241–4253 CrossRef CAS PubMed.
- Z. Y. AlSubeh, A. Waldbusser, H. A. Raja, C. J. Pearce, K. L. Ho, M. J. Hall, M. R. Probert, N. H. Oberlies and S. Hematian, J. Org. Chem., 2022, 87, 2697–2710 CrossRef CAS PubMed.
- M. S. Cooper, L. Zhang, M. Ibrahim, K. Zhang, X. Sun, J. Röske, M. Göhl, M. Brönstrup, J. K. Cowell, L. Sauerhering, S. Becker, L. Vangeel, D. Jochmans, J. Neyts, K. Rox, G. P. Marsh, H. J. Maple and R. Hilgenfeld, J. Med. Chem., 2022, 65, 13328–13342 CrossRef CAS PubMed.
- J. P. Metherall, R. C. Carroll, S. J. Coles, M. J. Hall and M. R. Probert, Chem. Soc. Rev., 2023, 52, 1995–2010 RSC.
- G. Guy, V. Knappertz, B. Whalley, R. Gray, J. Cilia, A. Patel and H. Straker, UK Pat., GB2579179A, 2018 Search PubMed.
- D. Mangin, F. Puel and S. Veesler, Org. Process Res. Dev., 2009, 13, 1241–1253 CrossRef CAS.
- B. A. Nogueira, C. Castiglioni and R. Fausto, Commun. Chem., 2020, 3, 34 CrossRef PubMed.
- N. F. Xavier, A. M. da Silva and G. F. Bauerfeldt, Cryst. Growth Des., 2020, 20(7), 4695–4706 CrossRef CAS.
- G. J. O. Beran, I. J. Sugden, C. Greenwell, D. H. Bowskill, C. C. Pantelides and C. S. Adjiman, Chem. Sci., 2022, 13, 1288–1297 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details, images of crystal forms. CCDC 2234293–2234299. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3ce00041a |
| This journal is © The Royal Society of Chemistry 2023 |