
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceJourneys in crystal energy landscapes: actual and virtual structures in polymorphic 5-nitrobenzo[c][1,2,5]thiadiazole†
Francesco Silvio
Gentile
 a,
Emmanuele
Parisi
b and
Roberto
Centore
*c
a,
Emmanuele
Parisi
b and
Roberto
Centore
*c
aDepartment of Pharmacy, University of Salerno, Via Giovanni Paolo II 132, I-84084, Fisciano, Italy
bDepartment of Applied Science and Technology, Politecnico of Turin, Corso Duca degli Abruzzi 24, I-10129, Turin, Italy
cDepartment of Chemical Sciences, University of Naples Federico II, Via Cintia, I-80126, Naples, Italy. E-mail: roberto.centore@unina.it
First published on 2nd January 2023
Abstract
A new polymorph of 5-nitrobenzo[c][1,2,5]thiadiazole, polymorph II, has been discovered. This polymorph is obtained by crystallization from solutions containing Cu(II) ions, which inhibit the formation of the already known polymorph I. The packings of the two polymorphs (actual crystal structures) are compared between each other and also with seven virtual polymorphs generated from the crystal structures of similar compounds retrieved from the CSD and optimized by ab initio calculations with periodic boundary conditions. The comparison is based on the analysis of the synthons present in the different crystal structures, and on calculated lattice energy and density. For the specific case examined, our analysis suggests that among crystal structures containing a given synthon, or a given combination of synthons, only the one with the lowest Ulat is observed; crystal structures with slightly higher Ulat (within 2 kcal mol−1) can be observed, but they must be based on different synthons.
Introduction
In some respects, the formation of crystal polymorphs is still a mystery.1Once crystals have formed, we can accurately calculate their free energy and evaluate the relative stability of different crystal forms of the same compound by using thermodynamic and quantum mechanical methods. Also, the growth of nuclei, after they have formed, can be described in the realm of thermodynamics.Instead, the initial stages before nucleation, when molecules in solution dynamically aggregate (and disaggregate) into clusters, are basically still obscure. Pre-nucleation processes are complex and, moreover, they can be quite different for different molecules.2 Yet, these fuzzy stages are fundamental in relation to the outcome, i.e. the macroscopic crystal form that eventually develops.
Pre-nucleation clusters are formed by a few molecules (from tens to thousands), and they have no definite periodic lattice structure. Recent simulation experiments have begun to provide important information about pre-nucleation phenomena.3–5 They have shown that, in some cases, pre-nucleation clusters do not evolve regularly, by simply adding more bricks to the same wall. During the growth, clusters change their structure while accommodating entering molecules and pass through a series of different structurally related arrangements, which represent local energy minima. This size-dependent and stepwise growth of clusters goes on up to a critical size. Afterwards, the clusters' structure will no longer change, and clusters will grow keeping a definite periodic lattice structure: they have become nuclei. Clusters' structures and growth are driven by several internal and external factors, including optimal intermolecular local pairing between entering molecules. So, intermolecular pairing, molecular recognition and synthon formation are key elements of pre-nucleation steps, which can be inferred, in retrospect, by crystal structure analysis.6
Fused 1,2,5-thia(selena)diazoles have a large spectrum of interesting chemical properties with applications as agrochemicals,7 antitumour,8 and π-conjugated building blocks for organic electronics,9 to name only a few. Here, we report the discovery of a new polymorph of 5-nitrobenzo[c][1,2,5]thiadiazole (Chart 1), hereafter denoted as compound 1, and a detailed analysis of actual and virtual structures of this compound, with the scope of finding a rationale for the two observed crystal structures, out of the many possible ones.
 | ||
| Chart 1 Chemical diagram of 5-nitrobenzo[c][1,2,5]thiadiazole. | ||
Results and discussion
Description of the actual crystal structures of 1
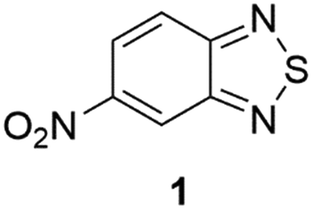
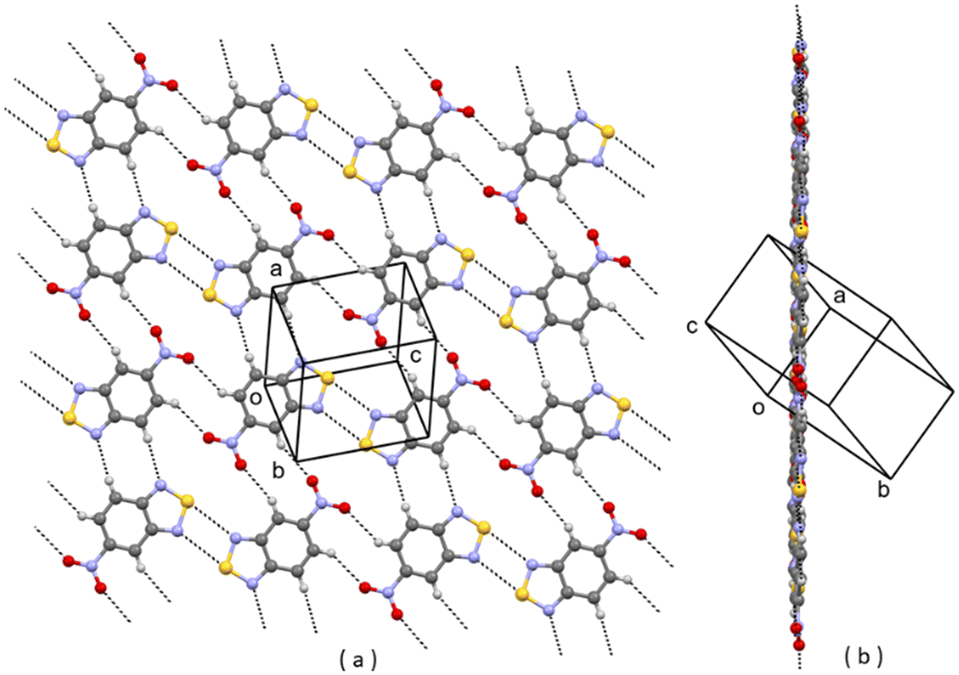
In the CSD10 is described a triclinic polymorph of 1 (refcode GINTII) with the space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) , hereafter denoted as polymorph I, Fig. 1.11 We have isolated a new polymorph II, which is monoclinic, with the space group Pc, Fig. 2. In both polymorphs, the crystal structure is of a layered type, with infinite planar layers, generated by in-plane intermolecular interactions, piled up on each other. In polymorph I, Fig. 1, layers are generated by [N⋯S]2 chalcogen bonded dimers12 and by C–H⋯O and C–H⋯N weak H-bonding interactions, with formation of R22(10) and R22(8) ring patterns respectively. In polymorph II, Fig. 2, O⋯S chalcogen bonds and C–H⋯N and C–H⋯O weak H-bonding interactions hold molecules within the layers.
, hereafter denoted as polymorph I, Fig. 1.11 We have isolated a new polymorph II, which is monoclinic, with the space group Pc, Fig. 2. In both polymorphs, the crystal structure is of a layered type, with infinite planar layers, generated by in-plane intermolecular interactions, piled up on each other. In polymorph I, Fig. 1, layers are generated by [N⋯S]2 chalcogen bonded dimers12 and by C–H⋯O and C–H⋯N weak H-bonding interactions, with formation of R22(10) and R22(8) ring patterns respectively. In polymorph II, Fig. 2, O⋯S chalcogen bonds and C–H⋯N and C–H⋯O weak H-bonding interactions hold molecules within the layers.
 | ||
| Fig. 1 A planar layer of polymorph I of 1; (a) face view; (b) edge view. Intermolecular contacts shorter than the sum of van der Waals radii of the atoms involved are shown by dashed lines. | ||
 | ||
| Fig. 2 A planar layer of polymorph II of 1; (a) face view; (b) edge view. Intermolecular contacts shorter than the sum of van der Waals radii of the atoms involved are shown by dashed lines. | ||
In I, layers are parallel to the lattice planes with Miller indices 12, and the stacking distance of the planes is d12 = 3.19 Å. In II, layers are parallel to the 10![[2 with combining macron]](https://www.rsc.org/images/entities/char_0032_0304.gif) lattice planes with a slightly longer stacking distance d10 = 3.21 Å.
lattice planes with a slightly longer stacking distance d10 = 3.21 Å.
The layered nature of the two polymorphs is also proven by the fact that the 12 and 10 reflections are the most intense in the whole diffraction pattern for, respectively, polymorph I and polymorph II.
The experimental observation is that polymorph II is formed in the presence of Cu(II) ions. In the crystal packings of I and II, as shown in Fig. 1 and 2, is the possible key to rationalizing this. In fact, we can suppose that Cu(II) ions in solution coordinate to molecules of 1 at the N donor sites.13 So, in the presence of Cu(II) ions, the aggregation of molecules in solution through the [N⋯S]2 synthon, typical of form I, is strongly disfavoured, while the formation of clusters with the S⋯O synthon, typical of form II, is allowed. As a matter of fact, Cu(II) ions act as inhibitors for the formation of polymorph I.
Analysis of actual and virtual crystal structures of 1
As shown above, the title compound is polymorphic. Polymorphs I and II of 1 are actual crystal structures (hereafter denoted as A1 and A2 respectively), in the sense that they are experimentally observed. To explore the crystal energy landscape of 1, other packings should be considered. Recent developments in crystal structure prediction (CSP) studies have provided different methods for generating reliable crystal packings of a given compound and exploring the crystal energy landscape: systematic grid search or Monte Carlo search on the multidimensional potential energy surface, genetic algorithms, molecular dynamics exploration of the packing space, small clusters methods and synthon approach methods.14 The approach of actual and virtual structures provides a tool for generating reliable crystal structures of a given compound by using experimental crystal structures of similar compounds.15–18 Following this approach, we have searched the CSD for crystal structures of compounds similar to 1. We have searched for benzo(thia)(selena)(tellura)diazoles with a non-ring substituent at position 5. Seven hits were found and selected, and they are shown in Chart 2. | ||
| Chart 2 Parent crystal structures used to generate virtual crystal structures of 1. | ||
In these seven parent crystal structures, we have just replaced the original molecules with molecules of 1, by simply changing the original 5-substituent (e.g. Br, CH3, NH2, F, CF3, COOEt, COOi-pr) with a nitro group and, in the case of DOLROO and GASFAK, also Se with S. We have then optimized the corresponding seven virtual crystal structures V1–V7 (see Experimental part). We name these as virtual crystal structures because they have not (yet) been observed experimentally for 1. The optimization was successful in all cases, with keeping of the original packings, as shown in Fig. 3–9, and modest variations of lattice parameters, as shown in Table S1 (see the ESI†). So, molecule 1, for which two different packings are found experimentally (i.e. A1 and A2), well fits also the seven packings V1–V7 which are experimentally observed for compounds with different substituents at position 5. Finding a lot of possible packings for a given molecule is not surprising, as this is one general result of modern CSP studies.19 Our result, however, is noteworthy per se, if we consider that virtual structures are generated from experimental packings and if we look at the great variability of the substituent in position 5 in the seven parent structures.
 | ||
| Fig. 3 (a) Partial packing of JIWVAP; (b) partial packing of V1. Some intermolecular contacts shorter than the sum of van der Waals radii of the atoms involved are shown by dashed lines. | ||
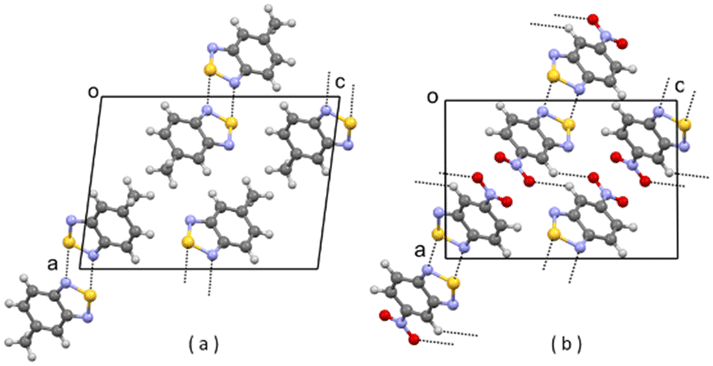 | ||
| Fig. 4 (a) Partial packing of JIWWEU; (b) partial packing of V2. Some intermolecular contacts shorter than the sum of van der Waals radii of the atoms involved are shown by dashed lines. | ||
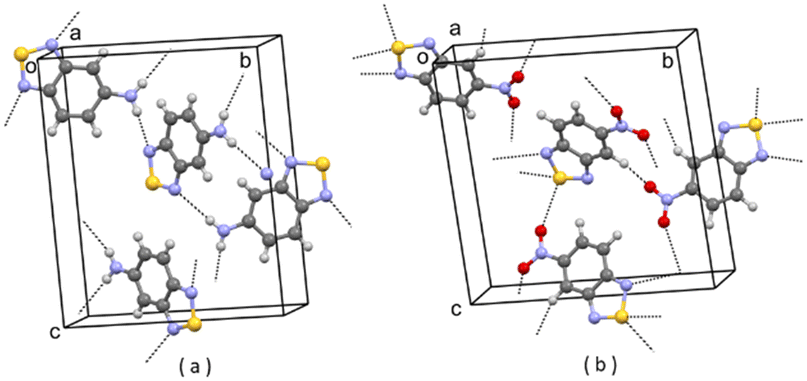 | ||
| Fig. 5 (a) Partial packing of JIWWUK; (b) partial packing of V3. Some intermolecular contacts shorter than the sum of van der Waals radii of the atoms involved are shown by dashed lines. | ||
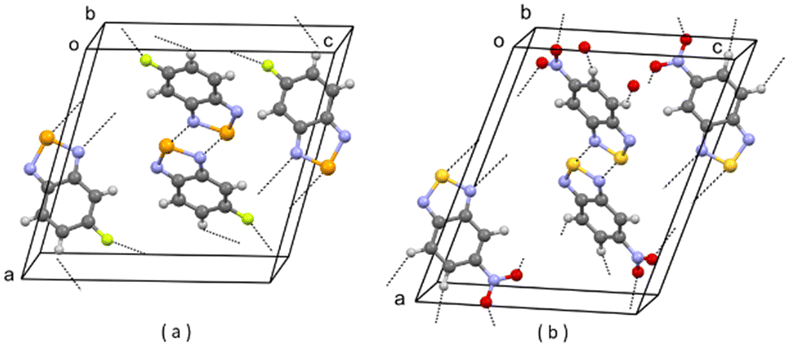 | ||
| Fig. 6 (a) Partial packing of DOLROO; (b) partial packing of V4. Some intermolecular contacts shorter than the sum of van der Waals radii of the atoms involved are shown by dashed lines. | ||
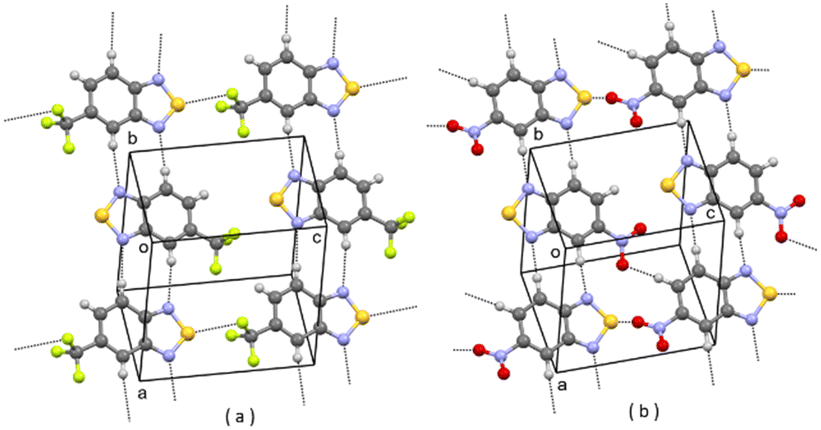 | ||
| Fig. 7 (a) Partial packing of JIXFII, showing a layer of molecules; (b) partial packing of V5 showing a layer of molecules. Some intermolecular contacts shorter than the sum of van der Waals radii of the atoms involved are shown by dashed lines. | ||
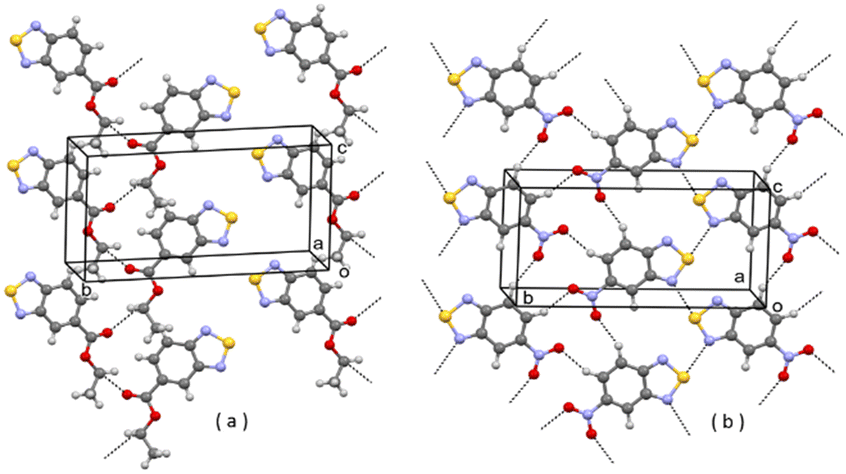 | ||
| Fig. 8 (a) Partial packing of REBHIQ; (b) partial packing of V6. Some intermolecular contacts shorter than the sum of van der Waals radii of the atoms involved are shown by dashed lines. | ||
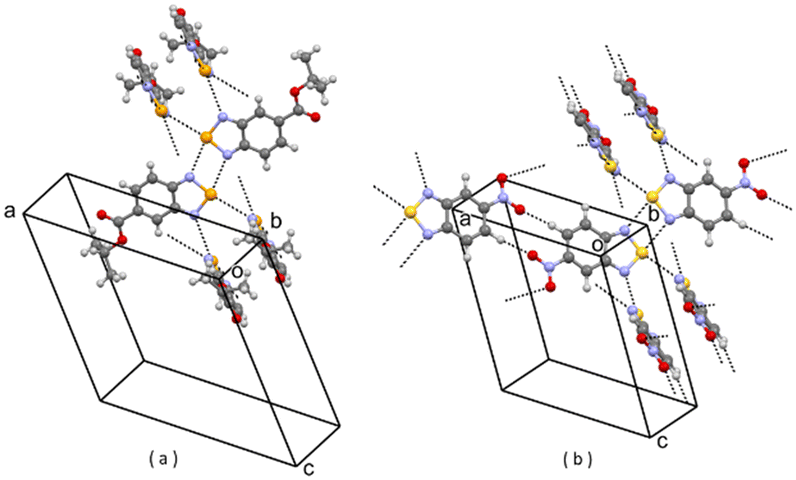 | ||
| Fig. 9 (a) Partial packing of GASFAK; (b) partial packing of V7. Some intermolecular contacts shorter than the sum of van der Waals radii of the atoms involved are shown by dashed lines. | ||
This interchangeability among different compounds and packings, which we have verified also in other cases,15–17 suggests some general considerations. For instance, a given crystal structure can be considered as a sort of elastic system, in the sense that it can be deformed to fit molecules of different shapes and/or electronic character. Surprisingly, in many cases, this deformation is accomplished at a low cost of lattice energy and density.
The lattice energy and density (at 0 K) of the optimized actual and virtual structures are reported in Table 1.
| U lat (kcal mol−1) | ρ lat (g cm−3) | |
|---|---|---|
| A1 | −50.30 | 1.977 |
| A2 | −48.36 | 1.959 |
| V1 | −47.14 | 1.927 |
| V2 | −46.28 | 1.931 |
| V3 | −45.53 | 1.915 |
| V4 | −44.05 | 1.892 |
| V5 | −45.72 | 1.890 |
| V6 | −45.02 | 1.895 |
| V7 | −46.11 | 1.882 |
The most stable predicted structure, at 0 K, is A1, followed by A2. The lattice energy of A2 is predicted as 1.94 kcal mol−1 higher than that of A1, and this is in line with the observed differences of lattice energy between polymorphs.1 Also, the density of A2 is slightly lower than that of A1. This suggests that A1 is the most stable polymorph of 1, and this is consistent with the measured thermodynamic properties (see Fig. S3 and S4 in the ESI†): for phase I, the melting temperature is 128 °C and the melting enthalpy is 146.4 J g−1; for phase II, the corresponding values are 125 °C and 120.7 J g−1. Calculations predict that at about 545 K (272 °C), A2 has a lower free energy than A1, Fig. 10. The transformation, however, cannot be realized because the melting temperature of both A1 and A2 is lower.
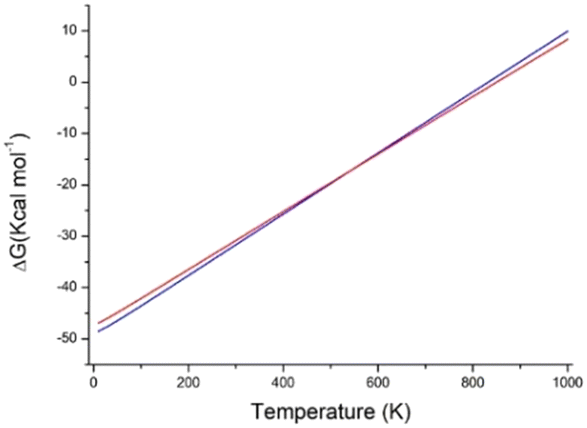 | ||
| Fig. 10 Calculated lattice free energy of A1 (blue line) and A2 (red line) as a function of temperature. | ||
For the virtual structures, lattice energies are predicted higher and lattice densities lower than those of A1 and A2. Altogether, the nine crystal structures in Table 1, which all rely on experimentally observed packings, are all within 6.25 kcal mol−1 lattice energy and within 0.105 g cm−3 lattice density. Taking for instance virtual structure V1, it has lattice energy only 1.22 kcal mol−1 higher than that of A2, so, one could wonder why V1 is not observed and, more generally, what is the physical meaning, if any, of the virtual structures. If we consider that they are generated from experimentally observed crystal structures of similar molecules, and that they correspond to true minima of the lattice energy of 1,20 we should conclude that the virtual structures represent crystal structures that could form. Why, then, are they not observed?21 Of course, we cannot say to have explored all the experimental conditions suitable for their formation, and only static lattice energies have been considered in our analysis, neglecting the vibrational entropy contributions to the lattice free energy.22 Moreover, recent computational studies based on basin hopping and Monte Carlo threshold algorithms have clearly shown that local minima of the crystal energy landscape, as virtual structures are, can collapse to a single minimum as they are separated by small energy barriers.23–25 With all the above caveats, we think that virtual structures, by telling us what could have been and wasn't, can help to shed some light, indirectly, on the initial stages of crystallization.
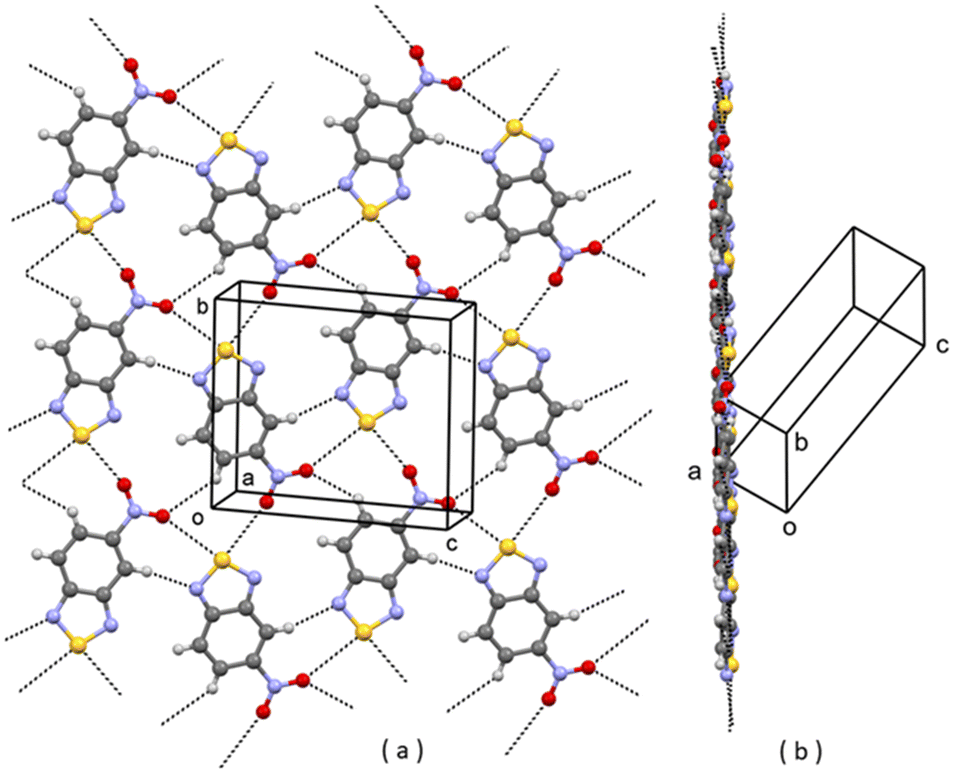
One reason why virtual structures are not observed is that their formation is bypassed because the corresponding clusters, during growth and before reaching the critical size, transform into clusters leading to other structures, such as those experimentally observed (I or II).21 So, virtual structures do not exist simply because they do not form. Taking for instance virtual structures V1, V2, V4 and V7, they all contain the [N⋯S]2 synthon, which is present in A1, and some (e.g. V7) contain the R22(10) ring pattern formed by C–H⋯O weak H-bonding interactions, also present in A1. We can suppose that clusters potentially leading to final structures V1, V2, V4, and V7 do form in solution but, while growing, they evolve into clusters leading to A1, which is the final selected structure containing the [N⋯S]2 synthon. As for V3, the parent structure JIWWUK contains a strong H-bonded N–H⋯N synthon, Fig. 5(a), that is not present in V3, where it is replaced by an O⋯S chalcogen bond, Fig. 5(b). This, however, is also present in lower Ulat actual structure A2. In V5, ring patterns R22(8) through weak C–H⋯N interactions are present, Fig. 7. These, however, are also present in lower Ulat actual structure A1. The only virtual structure featuring different synthons as compared with actual structures is V6, which shows different synthons as compared also with the parent REBHIQ structure, Fig. 8. In fact, C–H⋯O interactions with the formation of R33(12) ring patterns are observed in V6, and N⋯S chalcogen bonds with the formation of C(4) chain patterns. These synthons lead to the formation of infinite planar layers, Fig. 8(b), piled up on each other, as in A1 and A2.
In the specific case we have examined, the analysis of the crystal energy landscape seems to suggest that among the crystal structures containing a given synthon, or a given combination of synthons, only the one with the lowest Ulat is observed; crystal structures with slightly higher Ulat (say within 2 kcal mol−1) can be observed, but they must be based on different synthons. This, however, cannot be considered a general case, and exceptions do exist. For instance, in some cases, crystals are produced by removal of solvent molecules from a solvate; these crystal structures have higher energy but are based on synthons similar to the lower energy solvate structure (pseudopolymorphism).26,27
Conclusion
We have proposed a hypothesis in which the analysis of actual and virtual structures can provide indirect confirmation of results emerging from recent simulation experiments of prenucleation phenomena. At variance with other ways of generating crystal structures of a given compound, in our approach, virtual structures are generated starting from known crystal structures of similar compounds. So, the packings of virtual structures are experimentally observed, but on similar compounds. The fact that a few crystal structures are observed for a given compound, out of the many possible ones, points to a strong selection playing at the level of prenucleation clusters in solution.18Experimental part
Synthesis of 1
As in many cases of fused ring heterocycles,28–30 the synthesis starts from a 1,2-disubstituted benzene. A solution of 4-nitro-o-phenylenediamine (5 g, 32.3 mmol), dimethylformamide (50 mL) and triethylamine (7.7 mL) was poured into a round bottom flask equipped with a condenser and put in an ice–water bath under a nitrogen flux. Thionyl chloride (5.7 mL) was added dropwise, and the reaction was left for 4 h under stirring at 0 °C. The resulting mixture was poured in 400 mL of distilled water and the precipitate was collected by filtration under vacuum, washed with distilled water and dried. The product was extracted with chloroform and evaporated to yield 3.379 g (18.5 mmol) of a red solid. Mp 128 °C, yield: 57.3%. 1H-NMR (400 MHz, CHCl3-d6): δ 8.86 (s, 1H), δ 8.35 (d, 2H), 8.02 (d, 2H).Crystallization procedure for polymorph II of 1
A layering of four different solutions was realized in a slim glass tube with a 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ligand to metal molar ratio. Chloroform (1 mL) and ethanol (1 mL) were layered in sequence on a solution of 1 (30 mg) in chloroform (1 mL). Then, an ethanol solution (1 mL) of copper(II) chloride salt was slowly layered on top. The resulting four-layer system was left undisturbed at RT until complete evaporation occurred. Orange plate crystals (mp 125 °C) were collected after one week.
:1 ligand to metal molar ratio. Chloroform (1 mL) and ethanol (1 mL) were layered in sequence on a solution of 1 (30 mg) in chloroform (1 mL). Then, an ethanol solution (1 mL) of copper(II) chloride salt was slowly layered on top. The resulting four-layer system was left undisturbed at RT until complete evaporation occurred. Orange plate crystals (mp 125 °C) were collected after one week.
X-ray analysis
Data for crystal structure determination were measured on a Bruker-Nonius Kappa CCD diffractometer, using graphite monochromated MoKα radiation (λ = 0.71073 Å). Reduction of data and semiempirical absorption correction were done using the SADABS program.31 The structures were solved by direct methods (SIR97 program32) and refined by the full-matrix least-squares method on F2 using the SHELXL-2016 program33 with the aid of the program WinGX.34 H atoms were placed in calculated positions and refined by the riding model. For all H atoms, Uiso = 1.2 × Ueq of the carrier atom was assumed. The analysis of the crystal packing was performed using the program Mercury.35Polymorph II: C6H3N3O2S, Mr = 181.17, monoclinic, Pc, Z = 2, orange, a = 3.812(3) Å, b = 8.905(4) Å, c = 10.498(5) Å, β = 95.02(4)°, V = 355.0(4) Å3, T = 293 K, 2117 reflections collected, 1288 independent reflections (Rint = 0.0537), R (I > 2σ(I)) = 0.0478, GOF = 1.044. CCDC 2204969.
Computational details
Quantum mechanical ab initio simulations were performed using a development version of CRYSTAL17 package36 adopting a density functional theory (DFT) formalism with 3D periodic boundary conditions. The main ingredients of the computational scheme are the adoption of the linear combination of atomic orbitals (LCAO) and the hybrid functional, with a percentage of Hartree Fock exchange to avoid electron self-interaction. In the LCAO approximation, each crystalline orbital is represented as a sum of Bloch functions centred at the nuclei. Each Bloch function, in turn, is expressed as a linear combination of Gaussian type orbitals (GTO) basis sets. A modified version (specific for the solids) of Ahlrichs double-ζ basis set,37 C_pob_DZVP_rev2,38 was adopted with the B3LYP global hybrid functional,39,40 and the Grimme D3 empirical correction.41 Grimme correction is mandatory to improve the description of the dispersion interactions in molecular crystals to obtain reliable optimized geometries. The DFT exchange–correlation contribution and its gradient were evaluated by numerical integration over the unit cell volume. The generation of the integration grid points was based on an atomic partition method, originally proposed by Becke,42 in which the radial and angular points are obtained from Gauss–Legendre quadrature and Lebedev two-dimensional distributions respectively. In these calculations, a pruned grid with 99 radial and 1454 angular points has been used with a total of 991454 points. The percentage error in the total electron density integration is less than 10−5 electrons per cell.
Reciprocal space sampling was based on a regular Pack–Monkhorst43 sub-lattice grid centred at the Γ-point, i.e. at the centre of the first Brillouin zone, leading to 4 sample points along each of the reciprocal lattice vectors in the irreducible part of the Brillouin zone (the total number of k-points is symmetry dependent, and is different in each case).
The crystal structures were optimized with respect to both the atomic fractional coordinates and the lattice parameters, keeping the experimental space group symmetry. The vibrational problem of the lattice was handled, within the harmonic approximation, from the one-point numerical derivatives of the analytical gradient of the Born–Oppenheimer potential energy surface. The optical phonons were calculated only at the Γ point of the reciprocal space. The nature of true minima of the optimized structures was confirmed by the positive values of all the vibrational eigenvalues.
The lattice energy (Ulat) at 0 K was calculated as the difference between the DFT molar energy of the optimized crystal at 0 K and the DFT molar energy of the free molecule. Lattice energies were corrected for basis set superposition error (BSSE) using the counterpoise method.44 The curves in Fig. 10 were obtained by calculating the difference between the molar Gibbs free energy of the optimized crystal and the molar free energy of the free molecule in the temperature range of 0–1000 K.
Author contributions
The manuscript was written through contributions of all the authors, and all the authors have given approval to the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support from the University of Naples Federico II within the FRA-MEREMOC project is gratefully acknowledged. We also thank the CRdC NTAP of Regione Campania (Italy) for the X-ray facility.Notes and references
- A. J. Cruz-Cabeza, S. M. Reutzel-Edens and J. Bernstein, Chem. Soc. Rev., 2015, 44, 8619–8635 RSC.
- M. W. Anderson, M. Bennett, R. Cedeno, H. Cölfen, S. J. Cox, A. J. Cruz-Cabeza, J. J. De Yoreo, R. Drummond-Brydson, M. K. Dudek, K. A. Fichthorn, A. R. Finney, I. Ford, J. M. Galloway, D. Gebauer, R. Grossier, J. H. Harding, A. Hare, D. Horváth, L. Hunter, J. Kim, Y. Kimura, C. E. A. Kirschhock, A. A. Kiselev, W. Kras, C. Kuttner, A. Y. Lee, Z. Liao, L. Maini, S. O. Nilsson Lill, N. Pellens, S. L. Price, I. B. Rietveld, J. D. Rimer, K. J. Roberts, J. Rogal, M. Salvalaglio, I. Sandei, G. Schuszter, J. Sefcik, W. Sun, J. H. ter Horst, M. Ukrainczyk, A. E. S. Van Driessche, S. Veesler, P. G. Vekilov, V. Verma, T. Whale, H. P. Wheatcroft and J. Zeglinski, Faraday Discuss., 2022, 235, 219–272 RSC.
- J. Anwar and D. Zahn, Angew. Chem., Int. Ed., 2011, 50, 1996–2013 CrossRef CAS PubMed.
- D. Zahn, ChemPhysChem, 2015, 16, 2069–2075 CrossRef CAS PubMed.
- F. Zhang, J. A. Gavira, G. W. Lee and D. Zahn, Crystals, 2021, 11, 174 CrossRef CAS.
- R. J. Davey, K. Allen, N. Blagden, W. I. Cross, H. F. Lieberman, M. J. Quayle, S. Righini, L. Seton and G. J. T. Tiddy, CrystEngComm, 2002, 4, 257–264 RSC.
- M. Shimono, S. Sugano, A. Nakayama, C.-J. Jiang, K. Ono, S. Toki and H. Takatsujia, Plant Cell, 2007, 19, 2064–2076 CrossRef CAS PubMed.
- S. Deng, D. Zeng, Y. Luo, J. Zhao, X. Li, Z. Zhao and T. Chen, RSC Adv., 2017, 7, 16721–16729 RSC.
- P. Herguth, X. Jiang, M. S. Liu and Alex K.-Y. Jen, Macromolecules, 2002, 35, 6094–6100 CrossRef CAS.
- C. R. Groom, I. J. Bruno, M. P. Lightfoot and S. C. Ward, Acta Crystallogr., Sect. B: Struct. Sci., Cryst. Eng. Mater., 2016, 72, 171–179 CrossRef CAS PubMed.
- L. S. Konstantinova, E. A. Knyazeva, N. V. Obruchnikova, Y. V. Gatilov, A. V. Zibarev and O. A. Rakitin, Tetrahedron Lett., 2013, 54, 3075–3078 CrossRef CAS.
- R. Centore, F. Borbone, A. Carella, M. Causà, S. Fusco, F. S. Gentile and E. Parisi, Cryst. Growth Des., 2020, 20, 1229–1236 CrossRef CAS.
- E. Parisi, A. Carella, F. Borbone, F. Chiarella, F. S. Gentile and R. Centore, CrystEngComm, 2022, 24, 2884–2890 RSC.
- G. M. Day, Crystallogr. Rev., 2011, 17, 3–52 CrossRef.
- R. Centore, M. Causà, F. Cerciello, F. Capone and S. Fusco, CrystEngComm, 2014, 16, 9168–9175 RSC.
- R. Centore, S. Fusco, F. Capone and M. Causà, Cryst. Growth Des., 2016, 16, 2260–2265 CrossRef CAS.
- J. C. Cole, C. R. Groom, M. G. Read, I. Giangreco, P. McCabe, A. M. Reilly and G. P. Shields, Acta Crystallogr., Sect. B: Struct. Sci., Cryst. Eng. Mater., 2016, 72, 530–541 CrossRef CAS PubMed.
- M. Causà and R. Centore, CrystEngComm, 2017, 19, 1320–1327 RSC.
- R. Nikhar and K. Szalewicz, Nat. Commun., 2022, 13, 3095 CrossRef CAS PubMed.
- Actually, the optimized structure V4 does not correspond to a true minimum of Ulat at 0 K, because one negative frequency is observed. This is very likely related to a specific feature of the parent structure DOLROO. In fact, this crystal structure is disordered, with two orientations of the fluorine atom, resulting in a (statistical) C2 symmetry of the molecule. Only one of the two positions has been selected for building and optimizing virtual structure V4. Evidently, the disordered crystal structure DOLROO does not correspond to a minimum of lattice energy at 0 K, but to a minimum of free energy, i.e. including the residual entropy contribution due to the disordered fluorine substituent, and this translates also to V4. We think that this could be a general feature of crystal structures with positional disorder.
- S. L. Price, Acta Crystallogr., Sect. B: Struct. Sci., Cryst. Eng. Mater., 2013, 69, 313–328 CrossRef CAS PubMed.
- J. Nyman and G. M. Day, CrystEngComm, 2015, 17, 5154–5165 RSC.
- E. C. Dybeck, D. P. McMahon, G. M. Day and M. R. Shirts, Cryst. Growth Des., 2019, 19, 5568–5580 CrossRef CAS.
- S. Yang and G. M. Day, J. Chem. Theory Comput., 2021, 17, 1988–1999 CrossRef CAS PubMed.
- S. Yang and G. M. Day, Commun. Chem., 2022, 5, 86 CrossRef.
- V. Suitchmezian, I. Jess and C. Näther, Cryst. Growth Des., 2009, 9, 774–782 CrossRef CAS.
- M. Nowak, A. J. Dyba, J. Janczak, A. Morritt, L. Fábián, B. Karolewicz, Y. Z. Khimyak, D. E. Braun and K. P. Nartowski, Mol. Pharmaceutics, 2022, 19, 456–471 CrossRef CAS PubMed.
- U. Caruso, R. Centore, A. Roviello and A. Sirigu, Macromolecules, 1992, 25, 2290–2293 CrossRef CAS.
- P. Ambrosanio, R. Centore, S. Concilio, B. Panunzi, A. Sirigu and N. Tirelli, Polymer, 1999, 40, 4923–4928 CrossRef CAS.
- A. Castaldo, R. Centore, A. Peluso, A. Sirigu and A. Tuzi, Struct. Chem., 2002, 13, 27–36 CrossRef CAS.
- SADABS, Bruker-Nonius, Delft, The Netherlands, 2002 Search PubMed.
- A. Altomare, M. C. Burla, M. Camalli, G. L. Cascarano, C. Giacovazzo, A. Guagliardi, G. G. Moliterni, G. Polidori and R. Spagna, J. Appl. Crystallogr., 1999, 32, 115–119 CrossRef CAS.
- G. M. Sheldrick, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3–8 Search PubMed.
- L. J. Farrugia, J. Appl. Crystallogr., 2012, 45, 849–854 CrossRef CAS.
- C. F. Macrae, I. J. Bruno, J. A. Chisholm, P. R. Edgington, P. McCabe, E. Pidcock, L. Rodriguez-Monge, R. Taylor, J. van de Streek and P. A. Wood, J. Appl. Crystallogr., 2008, 41, 466–470 CrossRef CAS.
- R. Dovesi, V. R. Saunders, C. Roetti, R. Orlando, C. M. Zicovich Wilson, F. Pascale, B. Civalleri, K. Doll, N. M. Harrison, I. J. Bush, P. D'Arco, M. Llunel, Y. Noël, L. Maschio, M. Causà, A. Erba, M. Rérat and S. Casassa, CRYSTAL17 user's manual, 2018, https://www.crystal.unito.it/Manuals/crystal17.pdf Search PubMed.
- F. Weigend and R. Ahlrichs, Phys. Chem. Chem. Phys., 2005, 7, 3297–3305 RSC.
- D. Vilela Oliveira, M. F. Peintinger, J. Laun and T. Bredow, J. Comput. Chem., 2019, 40, 2364–2376 CrossRef CAS PubMed.
- A. D. Becke, J. Chem. Phys., 1992, 96, 2155–2160 CrossRef CAS.
- C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785–789 CrossRef CAS PubMed.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed.
- A. D. Becke, J. Chem. Phys., 1988, 88, 2547–2553 CrossRef CAS.
- H. J. Monkhorst and J. D. Pack, Phys. Rev. B: Solid State, 1976, 13, 5188–5192 CrossRef.
- R. M. Balabin, J. Chem. Phys., 2008, 129, 164101 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: 1H-NMR spectrum of 1, DSC and PXRD analysis of polymorphs, and atomic coordinates of optimized actual and virtual structures in CIF format. CCDC 2204969. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d2ce01619b |
| This journal is © The Royal Society of Chemistry 2023 |