
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Microcrystal electron diffraction (MicroED) structure determination of a mechanochemically synthesized co-crystal not affordable from solution crystallization†
Toshiyuki
Sasaki‡
 *a,
Takanori
Nakane‡
b,
Akihiro
Kawamoto
b,
Tomohiro
Nishizawa
c and
Genji
Kurisu
*bde
*a,
Takanori
Nakane‡
b,
Akihiro
Kawamoto
b,
Tomohiro
Nishizawa
c and
Genji
Kurisu
*bde
aDepartment of Materials System Science, Yokohama City University, 22-2 Seto, Kanazawa-ku, Yokohama, Kanagawa 236-0027, Japan. E-mail: tsasaki@yokohama-cu.ac.jp; t.devi.sasaki@gmail.com
bInstitute for Protein Research, Osaka University, 3-2 Yamadaoka, Suita, Osaka 565-0871, Japan. E-mail: gkurisu@protein.osaka-u.ac.jp
cDepartment of Medical Life Science, Yokohama City University, 1-7-29 Suehiro-cho, Tsurumi-ku, Yokohama 230-0045, Japan
dDepartment of Macromolecular Science, Graduate School of Science, Osaka University, Toyonaka 560-0043, Japan
eInstitute for Open and Transdisciplinary Research Initiatives, Osaka University, 2-1 Yamadaoka, Suita, Osaka 565-0871, Japan
First published on 24th December 2022
Abstract
Solid-state grinding can provide “mechano-distinctive” co-crystals that are not accessible from solutions. Herein, we demonstrate the structure determination of a powdered mechano-distinctive co-crystal of 2-aminopyrimidine and succinic acid in a 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 molar ratio using microcrystal electron diffraction.
:1 molar ratio using microcrystal electron diffraction.
Solid-state grinding (SSG) or mechanochemical synthesis has become an increasingly common technique in the preparation of co-crystals.1–7 Co-crystallization by SSG has been applied to enhance dissolution, solubility, and bioavailability of many drugs in co-crystal engineering.8–10 In contrast to solution methods, SSG is ecofriendly and can be used for low-solubility compounds. Further, SSG can yield crystals with specific stoichiometries and crystal structures that are not attainable using solution methods.1,4,6 Consequently, SSG is an attractive and key technique in materials science.
The samples prepared by SSG are in a powder form comprising small crystals with sizes ranging from nano- to micrometers. These are too small for single-crystal X-ray diffraction (XRD), which is the most common technique used for crystal structure analyses. Powder XRD is an alternative technique for analyzing ground samples.11 However, this technique is susceptible to diffraction peak overlap due to coexistence of polymorphs and crystal-size-dependent peak broadening which hinders the analysis of the crystal structure.7,12
Microcrystal electron diffraction (MicroED)13–18 is another technique for determining the structure of nano crystals based on electron microscopy. MicroED was initially developed for protein crystals19–21 but has been used for an increasing variety of small-molecule crystals in recent years.22–27 Very thin crystals show clear electron diffraction patterns owing to the strong interaction between electrons and matter, providing crystal structures directly from the powders.16,17,28 By exploiting this advantage, MicroED was applied to a ground co-crystal, whose structure was reported to be not accessible by the solution method in this study. Fig. 1 shows a representative example, focusing on a 2:1 co-crystal of 2-aminopyrimidine (2AP) and succinic acid (SA), which was first synthesized by Etter et al. in 1990.4
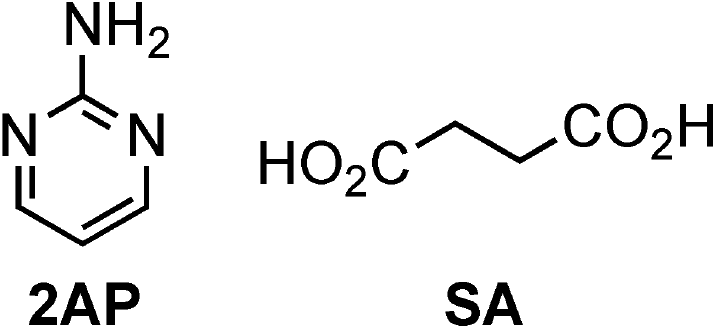 | ||
| Fig. 1 Molecular structures of 2-aminopyrimidine (2AP) and succinic acid (SA). | ||
 | ||
| Fig. 2 Cryo-EM image of the 2:1 co-crystals. | ||
A 2:1 co-crystal in powder form was prepared for MicroED by grinding a 2:1 mixture of 2AP and SA using an agate mortar and pestle for 10 min. This sample was aged at 22–30 °C for nine days (ESI† Fig. S1†). Although the main component of the mixture was the 2:1 co-crystal after the aging, the diffraction peaks indicate existence of small amount of the 1:1 co-crystal (2θ = 11.8°, 13.5°), 2AP (2θ = 18.5°, 28.8°, 29.6°) and SA (2θ = 26.2°). The powdered co-crystals were gently dusted on a copper EM grid (Quantifoil R1.2/1.3 Cu 200 mech) and loaded onto a Talos Arctica microscope (Thermo Fisher Scientific). Data collection was performed using SerialEM29 with a strategy described in the literatures.23,24 Crystals were manually flagged on square atlases (Fig. 2) and diffraction patterns were automatically collected using Verlox controlled by SerialEM via AutoHotKeys. The dataset was collected under parallel illumination conditions at 200 kV, a gun lens 8, a spot size of 11 in the nanoprobe mode and with an electron dose rate of 0.05 e− Å−2 s−1. With a 100 μm condenser aperture and without a selected-area diffraction aperture, the beam diameter was approximately 1.35 μm. During data collection, the temperature of the specimen grid was maintained at 79 K. The diffraction patterns were recorded on a Ceta detector (CMOS 4k × 4k, Thermo Fisher Scientific) running at 1 frame per s, while the crystals were rotated by 60–70° at 1° s−1. Diffraction patterns were indexed, integrated, and scaled using DIALS.30–32 With a sample set in excess of 500 recorded crystals, diffraction patterns from 14 best crystals were merged to provide the final structure factors (ESI† Table S1). The crystal structure was solved using charge flipping and refined kinematically by SHELXL with Olex2 (ESI† Table S2).33–35 Owing to unmodeled dynamical scatterings and accuracy limitations of the virtual camera distance (618 mm), the standard deviations of the refined parameters are considered underestimated.
Both the 2:1 (this work) and 1:1 (refcode: SERMOR)4 co-crystal structures belong to the monoclinic space group P21/c (P21/n), but they exhibit different molecular conformations and hydrogen-bonding networks, as seen in Fig. 3. In the 2:1 co-crystal, SA has a trans conformation and forms a ring-type hydrogen-bonding network with 2AP, which is denoted as R22(8) according to the graph-set analysis shown in Fig. 3a(i).36–39 Another ring-type hydrogen-bonding network of R22(8) is formed between neighboring 2AP. Consequently, the 2:1 co-crystal formed a zigzag (Ψ = 57.1°) supramolecular tape with inversion centers at the centers of SA and a ring-type hydrogen-bonding network of 2AP. The tapes leaning against the b-axis stack along the a-axis to form a 2-dimensional (2D) supramolecular layer, as shown in Fig. 3b(i). The 3-dimensional (3D) crystal structure is an assembly of layers along the c-axis. Fig. 3a(ii) depicts the 1:1 co-crystal forms, which form a zigzag (Ψ = 81.4°) supramolecular tape with no inversion centers by ring-type hydrogen-bonding networks of R22(8) between SA with the gauche conformation and 2AP. 2D supramolecular layers constructed by stacking the tape along the a-axis yield a 3D crystal structure, as shown in Fig. 3b(ii).
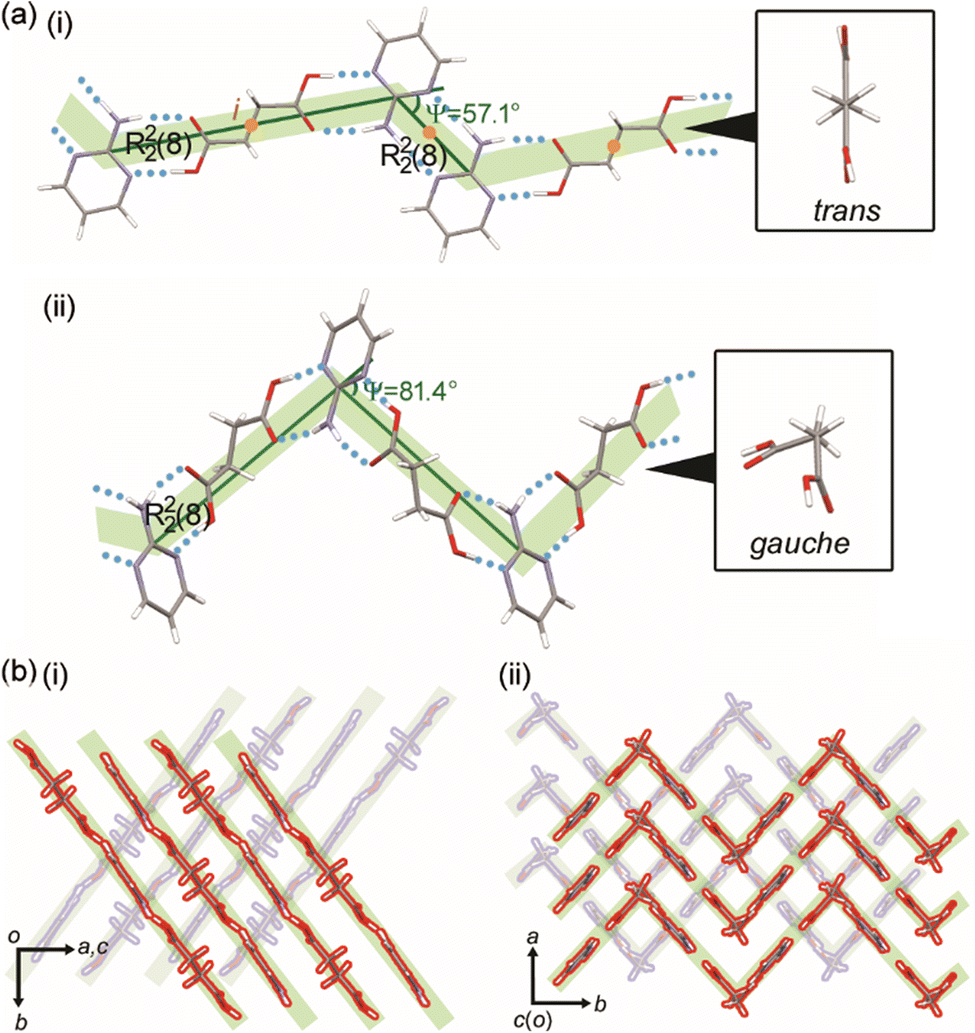 | ||
| Fig. 3 (a) Hydrogen-bonding networks and (b) packing diagrams of the (i) 2:1 (this work) and (ii) 1:1 (refcode: SERMOR)4 co-crystals. | ||
The differences in the crystal structures and intermolecular interactions such as hydrogen-bonding networks can change the stability of the co-crystal as investigated by thermal analyses. The 2:1 co-crystal showed a 32% mass reduction starting at approximately 100 °C owing to sublimation. It melted at 149 °C in thermal measurements performed for thermogravimetric (TG) and differential thermal analysis (DTA), as seen in Fig. 4a and S2a.†1H-NMR spectroscopy showed that the sublimated solid was 2AP (Fig. S3†). However, the 1:1 co-crystal prepared by mixing 2AP and SA in a 1:1 molar ratio in ethanol had no peaks before the melting point of 149.5 °C, similar to the peaks of the 2:1 co-crystal (Fig. S2b†). This indicates that the 2:1 co-crystal transforms into the 1:1 co-crystal by releasing 2AP. Fig. 4b shows that powder XRD confirmed the formation of 1:1 co-crystals (Fig. S1†). Diffraction peaks, e.g. at, 2θ = 17.7°, characteristic of the 1:1 co-crystal appeared when heating the 2:1 co-crystal at 120 °C for 2 min. The co-crystals formed SA in addition to the remaining ca. 7 mol% of the 1:1 co-crystal, as determined by 1H-NMR spectroscopy after heating for further 13 min (Fig. S4†). Moreover, the 2:1 co-crystals gradually transformed into 1:1 co-crystals when kept at room temperature. Fig. 4b and c show that approximately 80% of a batch of 2:1 co-crystals transformed into 1:1 co-crystals after 3 months (Fig. S5†). This shows that the 2:1 co-crystal has a lower stability than that of the 1:1 co-crystal under ambient conditions.
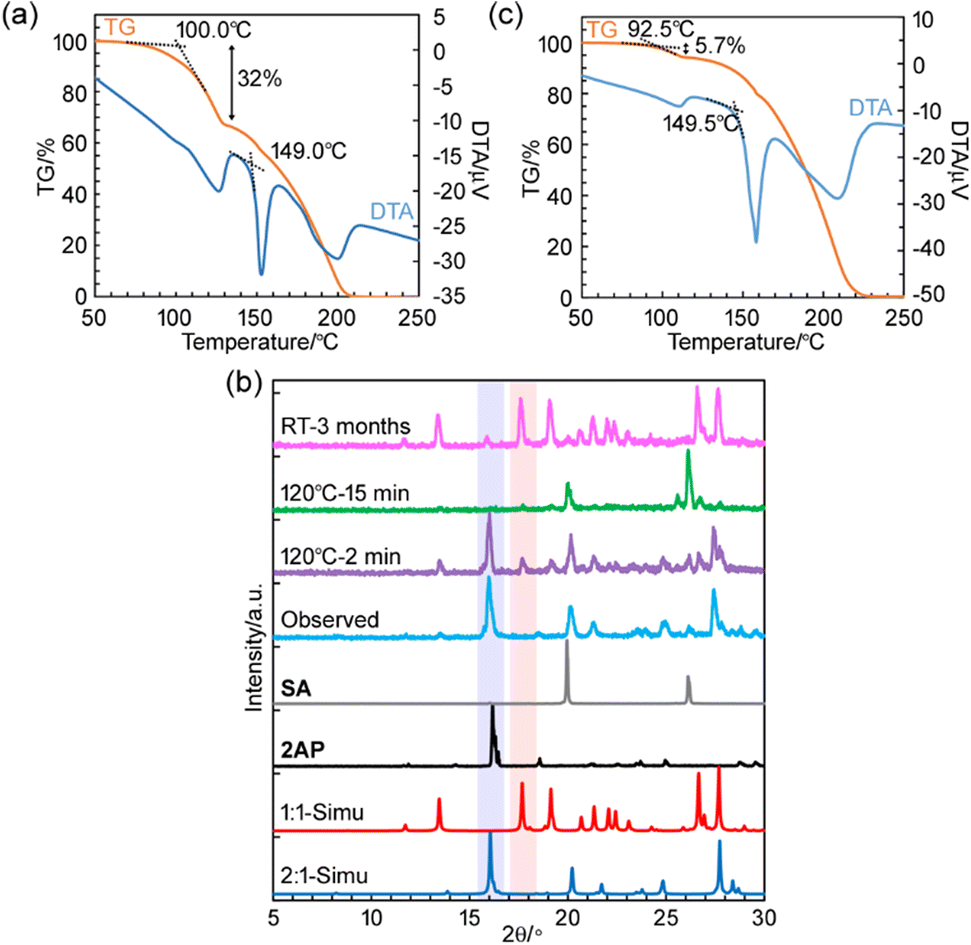 | ||
| Fig. 4 (a) TG/DTA diagram of the 2:1 co-crystal. (b) Powder XRD patterns measured at 24 °C: simulated from the crystal structures of the 2:1 co-crystal (2:1-Simu, blue) and 1:1 co-crystal (1:1-Simu, red); experimental patterns of 2AP (2AP, black), SA (SA, gray), the 2:1 co-crystal before (Observed, light blue) and after heating for 2 (120 °C–2 min, purple) and 15 min (120 °C–15 min, green); and the 2:1 co-crystal after aging for 3 months under ambient conditions (RT-3 months, pink). Representative diffraction peaks of the 2:1 and 1:1 co-crystals are highlighted by blue and red, respectively. (c) TG/DTA diagram of the 2:1 co-crystal after aging for 3 months under ambient conditions. | ||
The stabilities of the co-crystals were further evaluated with regard to the theoretically calculated energies listed in Tables 1 and S3.† The crystal structures, which were solved based on different techniques, i.e. microcrystal electron and power X-ray diffraction,4 were optimized and their lattice energies (Elat) were calculated using CONFLEX9 (MMFF94s).40,41 BSSE-corrected intermolecular interaction energies (Eint) between hydrogen-bonded molecules (2AP⋯2AP and 2AP⋯SA) were calculated at the B3LYP-D3/6-311G** level of theory42–44 using the counterpoise method45 with Gaussian 16 (ref. 46) and Gauss view 6.0.16.47 This was based on the dimers retrieved from the optimized crystal structures. The Elat of the 2:1 co-crystal (−125.47 kcal mol−1) indicates its lower stability than that of the 1:1 co-crystal (−182.97 kcal mol−1). Further, while the supramolecular tape in the 1:1 co-crystal is constructed by a robust hydrogen-bonding network (Eint (2AP⋯SA) = −16.31 kcal mol−1), both this network (Eint (2AP⋯SA) = −16.01 kcal mol−1) and a relatively weak hydrogen-bonding network (Eint (2AP⋯2AP) = −12.36 kcal mol−1) are formed in the 2:1 co-crystal.
| Entry | E lat | E int (2AP⋯2AP) | E int (2AP⋯SA) |
|---|---|---|---|
| 2:1 co-crystal |
−125.47 | −12.36 | −16.01 |
| 1:1 co-crystal |
−182.97 | — | −16.31 |
In conclusion, the co-crystal structure of 2AP and SA in a 2:1 molar ratio synthesized by SSG are not accessible by solution methods. This precludes the preparation of large single crystals suitable for structure determination by single-crystal XRD. However, the crystal structure of the 2:1 cocrystal was determined using MicroED. Although more accurate atomic positions of the 2:1 co-crystal might have been obtained by dynamical refinement, we limited our analysis to kinematical treatment due to technical difficulties. Kinematical refinement affords atomic positions accurate enough to discuss crystal packings. The 2:1 and 1:1 co-crystals of 2AP and SA have different stabilities owing to their different hydrogen-bonding networks and crystal structures, as determined by the thermal analyses and theoretical calculations. This demonstrates that MicroED is suitable to determine the crystal structure of powdered samples including those produced exclusively by SSG, or which are “mechano-distinctive”, and is useful in co-crystal engineering where SSG is a prevalent preparation method.
The refined structure of the 2:1 co-crystal was deposited to CCDC (accession code: 2211429) and COD (accession code: 3000424). The raw MicroED diffraction images were deposited to XRDa (accession code: XRD-109, https://doi.org/10.51093/xrd-00109). Input and output files for computational chemistry calculations and powder X-ray diffraction data were deposited to Zenodo (https://doi.org/10.5281/zenodo.7435859).
Author contributions
G. K. supervised this study. T. S. designed the project, prepared the samples, carried out theoretical calculations, and wrote the manuscript. T. N., A. K., T. N., and G. K. conducted MicroED experiments. All authors contributed to the discussion and manuscript review.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by JSPS KAKENHI Grant Number 22K05054 and Hitachi Metals–Materials Science Foundation for T. S. and Research Support Project for Life Science and Drug Discovery (BINDS) from AMED under Grant Number JP22ama121001. We thank Dr. Yanagisawa and Dr. Yamashita for their advice on semi-automatic MicroED data collection with SerialEM macros. We are also grateful to Dr. Suzuki for pre-evaluation of nanocrystals for MicroED by a fluorescence microscope and to Dr. Irie for measurements and analyses of 1H-NMR spectra.Notes and references
- A. O. Patil, D. Y. Curtin and I. C. Paul, J. Am. Chem. Soc., 1984, 106, 348–353 CrossRef.
- A. O. Patil, D. Y. Curtin and I. C. Paul, J. Am. Chem. Soc., 1984, 106, 4010–4015 CrossRef.
- F. Toda, K. Tanaka and A. Sekikawa, J. Chem. Soc., Chem. Commun., 1987, 279–280 RSC.
- M. C. Etter and D. A. Adsmond, J. Chem. Soc., Chem. Commun., 1990, 589–591 RSC.
- A. V. Trask, D. A. Haynes, W. D. S. Motherwell and W. Jones, Chem. Commun., 2006, 51–53 RSC.
- A. V. Trask and W. Jones, Top. Curr. Chem., 2005, 254, 41–70 CrossRef.
- T. Friščić and W. Jones, Cryst. Growth Des., 2009, 9, 1621–1637 CrossRef.
- M. R. Caira, L. R. Nassimbeni and A. F. Wildervanck, J. Chem. Soc., Perkin Trans. 2, 1995, 2213–2216 RSC.
- J. F. Remenar, S. L. Morissette, M. L. Peterson, B. Moulton, J. M. MacPhee, H. R. Guzmán and Ö. Almarsson, J. Am. Chem. Soc., 2003, 125, 8456–8457 CrossRef PubMed.
- R. Thakuria, A. Delori, W. Jones, M. P. Lipert, L. Roy and N. Rodríguez-Hornedo, Int. J. Pharm., 2013, 453, 101–125 CrossRef PubMed.
- E. Y. Cheung, S. J. Kitchin, K. D. M. Harris, Y. Imai, N. Tajima and R. Kuroda, J. Am. Chem. Soc., 2003, 125, 14658–14659 CrossRef PubMed.
- J. A. Kaduk, S. J. L. Billinge, R. E. Dinnebier, N. Henderson, I. Madsen, R. Černý, M. Leoni, L. Lutterotti, S. Thakral and D. Chateigner, Nat. Rev. Methods Primers, 2021, 1, 77 CrossRef.
- B. L. Nannenga and T. Gonen, Nat. Methods, 2019, 16, 369–379 CrossRef PubMed.
- M. J. de la Cruz, J. Hattne, D. Shi, P. Seidler, J. Rodriguez, F. E. Reyes, M. R. Sawaya, D. Cascio, S. C. Weiss, S. K. Kim, C. S. Hinck, A. P. Hinck, G. Calero, D. Eisenberg and T. Gonen, Nat. Methods, 2017, 14, 399–402 CrossRef PubMed.
- C. G. Jones, M. W. Martynowycz, J. Hattne, T. J. Fulton, B. M. Stoltz, J. A. Rodriguez, H. M. Nelson and T. Gonen, ACS Cent. Sci., 2018, 4, 1587–1592 CrossRef PubMed.
- M. Gemmi, E. Mugnaioli, T. E. Gorelik, U. Kolb, L. Palatinus, P. Boullay, S. Hovmöller and J. P. Abrahams, ACS Cent. Sci., 2019, 5, 1315–1329 CrossRef PubMed.
- X. Mu, C. Gillman, C. Nguyen and T. Gonen, Annu. Rev. Biochem., 2021, 90, 431–450 CrossRef PubMed.
- Z. Huang, E. S. Grape, J. Li, A. K. Inge and X. Zou, Coord. Chem. Rev., 2021, 427, 213583 CrossRef.
- B. L. Nannenga, D. Shi, A. G. W. Leslie and T. Gonen, Nat. Methods, 2014, 11, 927–930 CrossRef PubMed.
- K. Yonekura, K. Kato, M. Ogasawara, M. Tomita and C. Toyoshima, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 3368–3373 CrossRef PubMed.
- D. Shi, B. L. Nannenga, M. G. Iadanza and T. Gonen, eLife, 2013, 2, e01345 CrossRef PubMed.
- T. Gruene, J. T. C. Wennmacher, C. Zaubitzer, J. J. Holstein, J. Heidler, A. Fecteau-Lefebvre, S. De Carlo, E. Müller, K. N. Goldie, I. Regeni, T. Li, G. Santiso-Quinones, G. Steinfeld, S. Handschin, E. van Genderen, J. A. van Bokhoven, G. H. Clever and R. Pantelic, Angew. Chem., Int. Ed., 2018, 57, 16313–16317 CrossRef PubMed.
- H. Hamada, T. Nakamuro, K. Yamashita, H. Yanagisawa, O. Nureki, M. Kikkawa, K. Harano, R. Shang and E. Nakamura, Bull. Chem. Soc. Jpn., 2020, 93, 776–782 CrossRef.
- H. Lu, T. Nakamuro, K. Yamashita, H. Yanagisawa, O. Nureki, M. Kikkawa, H. Gao, J. Tian, R. Shang and E. Nakamura, J. Am. Chem. Soc., 2020, 142, 18990–18996 CrossRef PubMed.
- M. Ueda, T. Aoki, T. Akiyama, T. Nakamuro, K. Yamashita, H. Yanagisawa, O. Nureki, M. Kikkawa, E. Nakamura, T. Aida and Y. Itoh, J. Am. Chem. Soc., 2021, 143, 5121–5126 CrossRef PubMed.
- C. Guzmán-Afonso, Y.-L. Hong, H. Colaux, H. Iijima, A. Saitow, T. Fukumura, Y. Aoyama, S. Motoki, T. Oikawa, T. Yamazaki, K. Yonekura and Y. Nishiyama, Nat. Commun., 2019, 10, 3537–3546 CrossRef PubMed.
- E. T. Broadhurst, H. Xu, S. Parsons and F. Nudelman, IUCrJ, 2021, 8, 860–866 CrossRef PubMed.
- D. Marchetti, F. Guagnini, A. E. Lanza, A. Pedrini, L. Righi, E. Dalcanale, M. Gemmi and C. Massera, Cryst. Growth Des., 2021, 21, 6660–6664 CrossRef.
- D. N. Mastronarde, J. Struct. Biol., 2005, 152, 36–51 CrossRef PubMed.
- G. Winter, D. G. Waterman, J. M. Parkhurst, A. S. Brewster, R. J. Gildea, M. Gerstel, L. Fuentes-Montero, M. Vollmar, T. Michels-Clark, I. D. Young, N. K. Sauter and G. Evans, Acta Crystallogr., Sect. D: Struct. Biol., 2018, 74, 85–97 CrossRef CAS PubMed.
- M. T. B. Clabbers, T. Gruene, J. M. Parkhurst, J. P. Abrahams and D. G. Waterman, Acta Crystallogr., Sect. D: Struct. Biol., 2018, 74, 506–518 CrossRef CAS PubMed.
- J. Beilsten-Edmands, G. Winter, R. Gildea, J. Parkhurst, D. Waterman and G. Evans, Acta Crystallogr., Sect. D: Struct. Biol., 2020, 76, 385–399 CrossRef CAS PubMed.
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, J. Appl. Crystallogr., 2009, 42, 339–341 CrossRef CAS.
- L. J. Bourhis, O. V. Dolomanov, R. J. Gildea, J. A. K. Howard and H. Puschmann, Acta Crystallogr., Sect. A: Found. Adv., 2015, 71, 59–75 CrossRef CAS PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3–8 Search PubMed.
- M. C. Etter, J. C. MacDonald and J. Bernstein, Acta Crystallogr., Sect. B: Struct. Sci., Cryst. Eng. Mater., 1990, 46, 256–262 CrossRef PubMed.
- M. C. Etter, Acc. Chem. Res., 1990, 23, 120–126 CrossRef.
- J. Bernstein, R. E. Davis, L. Shimoni and N.-L. Chang, Angew. Chem., Int. Ed. Engl., 1995, 34, 1555–1573 CrossRef.
- T. Sasaki, Y. Ida, I. Hisaki, T. Yuge, Y. Uchida, N. Tohnai and M. Miyata, Chem. – Eur. J., 2014, 20, 2478–2487 CrossRef PubMed.
- H. Goto, S. Obata, N. Nakayama and K. Ohta, CONFLEX 8, CONFLEX Corporation, Tokyo, Japan, 2017 Search PubMed.
- T. A. Halgren, J. Comput. Chem., 1996, 17, 490–519 CrossRef.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef.
- C. Lee, W. Yang and R. G. Parr, Phys. Rev. B, 1988, 37, 785–789 CrossRef CAS PubMed.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed.
- S. F. Boys and F. Bernardi, Mol. Phys., 1970, 19, 553–566 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery, Jr., J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 16, Revision C.01, Gaussian, Inc., Wallingford, CT, 2019 Search PubMed.
- R. D. Dennington II, T. A. Keith and J. M. Millam, GaussView 6.0.16, Semichem, Inc., 2000–2016 Search PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: CCDC 2211429. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d2ce01522f |
| ‡ Contributed equally as co-first author. |
| This journal is © The Royal Society of Chemistry 2023 |