
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Formation of calcium phosphate nanoparticles in the presence of carboxylate molecules: a time-resolved in situ synchrotron SAXS and WAXS study†
Dritan
Siliqi
 a,
Alessio
Adamiano
b,
Massimo
Ladisa
c,
Cinzia
Giannini
a,
Michele
Iafisco
*b and
Lorenzo
Degli Esposti
*b
a,
Alessio
Adamiano
b,
Massimo
Ladisa
c,
Cinzia
Giannini
a,
Michele
Iafisco
*b and
Lorenzo
Degli Esposti
*b
aInstitute of Crystallography (IC), National Research Council (CNR), Via Amendola 122/O, 70126, Bari, Italy
bInstitute of Science, Technology and Sustainability for Ceramics (ISSMC, former ISTEC), National Research Council (CNR), Via Granarolo 64, 48018, Faenza, Italy. E-mail: michele.iafisco@istec.cnr.it; lorenzo.degliesposti@istec.cnr.it
cIstituto per le Applicazioni del Calcolo “Mauro Picone” (IAC), National Research Council (CNR), Via Amendola 122/I, 70126, Bari, Italy
First published on 5th December 2022
Abstract
In this work we have studied in situ the formation and growth of calcium phosphate (CaP) nanoparticles (NPs) in the presence of three calcium-binding carboxylate molecules having different affinities for Ca2+ ions: citrate (Cit), hydroxycitrate (CitOH), and glutarate (Glr). The formation of CaP NPs at several reaction temperatures ranging from 25 °C to 80 °C was monitored in situ through simultaneous Small and Wide X-ray Scattering (SAXS/WAXS) using synchrotron light. SAXS was used to investigate the first stages of NP formation where a crystalline order is not yet formed. In this regard we have developed a new bivariate mesh data analysis method for identifying the SAXS curves associated with the most relevant timeframes for performing curve modeling. WAXS was used to study the formation of crystalline phases and their evolution over time. The combined SAXS/WAXS data allowed us to track NP nucleation, their size and morphology, and their evolution up to mature hydroxyapatite (HA) nanocrystals. We have assessed that in the first stages of reaction (80 seconds) amorphous, elongated primary NPs nucleate whose size and morphology depend on the temperature and type of carboxylate molecule. The temperature controls the release of Ca2+ ions from carboxylate molecules, and thus induces the formation of a higher amount of amorphous particles and increases their size and aspect ratio. As the reaction time progresses, amorphous particles evolve into crystalline ones, whose kinetics of crystal growth are controlled by temperature and carboxylate ions. Stronger Ca-binding carboxylates (CitOH > Cit > Glr) have a more pronounced inhibiting effect on HA crystallization, retarding the formation and growth of crystalline domains, while a rise of temperature promotes crystallization. This work allowed us to shed more light on the formation of HA in the presence of growth-controlling molecules, as well as present the potential of combined SAXS/WAXS for studying the formation of highly relevant NPs for different applications.
Introduction
Calcium phosphate (CaP) nanoparticles (NPs) are widely studied nanomaterials, as biogenic CaP NPs constitute the mineral component of mammalian hard tissues (i.e. bones and teeth), and synthetic ones are extensively used materials for bone tissue engineering and nanomedicine.1Their formation in vivo as well as in vitro proceeds through a complex sequence of steps which involve reactions between precursor ions, nucleation of ion clusters, and growth processes that lead to mature NPs.2–4 Understanding CaP NP formation and crystallization is of critical importance to comprehend the processes of bone formation and bone pathogenesis, as well as to design tailored materials for nanomedicine, bone regeneration, environment remediation, agriculture, and many other applications.
With the recent advancement in X-ray scattering techniques, in situ analysis during NP formation can be performed, providing important and new insights into the reaction and growth mechanisms. The use of these techniques allows (i) probing the nanomaterials at different size resolutions (i.e. atom scale, particle scale, etc.), (ii) investigation of pre-nucleation clusters and other metastable early products, and (iii) the study of particle properties such as size, morphology, and aggregation, as well as the crystalline order.5
This approach gives the best results in combination with synchrotron light, as the high radiation brilliance allows acquiring data with high temporal resolution as well as low-intensity signals, and thus is perfect for investigating the early stages of NP formation or subtle changes that might occur during crystallization.5
The use of Small Angle X-ray Scattering (SAXS) allows evaluation of particle size, morphology, and their distribution in the population. Therefore it enables monitoring of the formation of nuclei, their growth into particles, and particle maturation, especially in the case of nanomaterials.6 Differently, Wide Angle X-ray Scattering (WAXS) gives information on the atomic ordering of the material, and thus describes the processes of crystallization and crystal growth. Therefore, the combination of Small and Wide X-ray Scattering (SAXS/WAXS) allows full tracking of the growth kinetics of crystalline NPs such as CaP NPs, thus obtaining simultaneous structural and chemical insights ranging from the atomic to nanometric scale with high time resolution.5,7,8 Synchrotron light combined SAXS/WAXS was previously used to study biogenic and synthetic CaP, but only as ex situ measurements.2,9
Recently, we have investigated the formation of CaP NPs and their crystallization into hydroxyapatite (HA, Ca10(PO4)6(OH)2) nanocrystals in the presence of three structurally similar calcium-binding carboxylate molecules in terms of crystal growth, chemical composition, and morphology.10 These molecules were: citrate (Cit), which is a tricarboxylate anion involved in the growth of HA nanocrystals during bone formation, and two carboxylate molecules, namely hydroxycitrate (CitOH) and glutarate (Glr), which share the same structure of Cit but bear different functional groups in the central region.10
These polycarboxylate molecules complex Ca2+ ions in solution, and de-complex them with heating, favoring a gradual CaP particle formation;10,11 in addition, they also attach to specific HA NP surfaces and control their morphology.2,3,12,13
Our ex situ analysis of HA crystallized in the presence of Cit, CitOH or Glr at 80 °C showed that crystallinity, particle dimension and morphology were controlled by the complexation strength of carboxylate molecules with Ca2+, where CitOH complexes calcium ions more strongly than Cit and thus has a higher inhibition effect on HA crystallization, while Glr has a weaker interaction and thus a weaker inhibition. Cit and CitOH seem to induce the same crystallization pathway with the direct conversion from an amorphous precursor into HA forming platy, elongated nanocrystals, while Glr leads to the formation of platelets of octacalcium phosphate (OCP, Ca8H2(PO4)6·5H2O) as the precursor phase that converts into HA nanorods.
Overall, the early steps of HA formation in the presence of these carboxylates remain unclear, as well as their crystallization pathway at temperatures lower than 80 °C. Ex situ characterization techniques (powder X-ray scattering, vibrational spectroscopy, etc.) do not allow the study of HA precursors (i.e. pre-nucleation clusters, metastable early NPs, etc.) nor immature NPs due to their instability.4,14 Therefore, there is a need of a comprehensive, in situ investigation of the early formation of HA nanocrystals to understand the role and mechanism of action of Cit and the other carboxylates during HA nucleation and growth.
Herein, we present a time-resolved in situ study of the formation and growth of HA in the presence of Cit, CitOH, and Glr using simultaneous SAXS/WAXS with synchrotron light to investigate the unstable early stages of particle formation and crystallization, as well as using a new advanced method to verify the ex situ results. SAXS/WAXS scattering curves were collected in real time in situ during HA nucleation and growth in an aqueous solution until particle crystallization was completed. Several reaction temperatures ranging from 25 °C to 80 °C were used to study the influence of temperature on HA nucleation and crystallization, as well as on thermal decomplexation of carboxylate–Ca2+ species.
Experimental
Materials
All reagents were purchased from Sigma Aldrich (St. Louis, MO, USA) and were used as received; these include: calcium chloride dihydrate (CaCl2·2H2O, ≥99.0%), glutaric acid (C5H8O4, ≥99.0%, H2(Glr)), potassium hydroxycitrate tribasic monohydrate (K3(C6H5O8)·H2O, ≥95.0%, (K3(CitOH))), sodium citrate tribasic dihydrate (Na3(C6H5O7)·2H2O, ≥99.0%, (Na3(Cit))), sodium hydroxide (NaOH, ≥99.0%), and sodium phosphate dibasic dihydrate (Na2HPO4·2H2O, ≥99.0%).Precursor solutions
Before the experimental data collection, calcium and phosphate precursor solutions were prepared as reported by Degli Esposti et al.10 The calcium precursor solution (hereafter called Ca + carboxylate solution) contained CaCl2 100 mM + X 400 mM (where X was either Na3(Cit), H2(Glr), or K3(CitOH)), and its pH was set to 8.5 with 1 M NaOH. After preparation Ca + carboxylate solutions were stored at 4 °C to avoid the formation of calcium carboxylate salts and immediately before use left to spontaneously warm up to 25 °C. The phosphate precursor solution (hereafter called PO4 solution) contained 120 mM Na2HPO4. After preparation, the solution was heated to 70 °C to completely dissolve Na2HPO4 and subsequently left to cool down to 25 °C before use.Simultaneous synchrotron light SAXS/WAXS data collection of real-time HA formation in situ
Simultaneous Small and Wide Angle X-ray Scattering (SAXS/WAXS) patterns of HA formation in the presence of carboxylate molecules were recorded at the Austrian SAXS beamline of the ELETTRA Synchrotron (Trieste, Italy), operated at 2 GeV (ref. 15) with a custom setup. The setup, which is shown in Fig. 1, allowed investigation of the growth and crystallization of HA NPs in aqueous suspension in real time. In detail, the setup consisted in a liquid flow circuit that comprised: (I) a glass round-bottomed flask containing 12.5 mL of PO4 solution under magnetic stirring and immersed in a thermostatic oil bath, (II) a peristaltic pump that collects the liquid from the flask and sends it to (III) the SAXS/WAXS sample holder, a ϕ 2 mm special quartz capillary (WJM-Glas Müller GmbH, Berlin-Pankow, DE) inserted in a steel frame and placed orthogonally to the X-ray beam. Outside of the circuit, there was (IV) a 25 mL syringe containing 12.5 mL of Ca + carboxylate solution; the syringe was placed in a syringe pump and connected to the reaction flask. All components of the circuit were connected to each other by using ϕ 1.65 mm PVC tubing. The liquid flow was set to 10 mL min−1. The incident beam energy was set at 8 keV and allowed measurement of the SAXS range from a q-value of 0.08 to ca. 6 nm−1, while the WAXS detector allowed measurement of the range from a q-value of 15 to ca. 28 nm−1. The 2D SAXS scattering image was collected with a Pilatus 1 M detector (Dectris, CH), while a Pilatus 100 k detector (Dectris, CH) was used to simultaneously acquire the 2D WAXS pattern. As the first step, the PO4 solution filled the circuit and was left to recirculate in a 10 mL min−1 flow until all the fluid in the circuit reached the temperature set in the reactor, which was either 25 °C, 37 °C, 60 °C, or 80 °C. Before the beginning of the HA precipitation experiment, the X-ray beam was sent to the sample and the SAXS/WAXS 2D scattering images of the sample holder containing a flow of the PO4 solution were collected to be used for background subtraction. Afterwards, while SAXS/WAXS 2D scattering images were collected continuously with a time resolution of 6 seconds (4 s of exposition time and 2 s interval) the Ca–carboxylate solution was injected in the reactor with a flow of 25 mL min−1 monitoring continuously the evolution of SAXS/WAXS curves during particle precipitation. The data collection continued until no further evolution of SAXS/WAXS curves was observed, ranging from 1 hour to 14 hours depending on the experimental conditions. In the case of long data collections (>1.5 h), the exposition time was incremented, and the time resolution was decreased to improve the signal-to-noise ratio. Each data collection was named based on the carboxylate used (Cit, CitOH, or Glr) and on the reaction temperature (25 °C, 37 °C, 60 °C, and 80 °C). In the case of CitOH, data collection was performed only at 25 °C and 80 °C due to limited available beamtime and extremely slow crystallization kinetics due to strong growth inhibition exerted by CitOH (see the results reported below).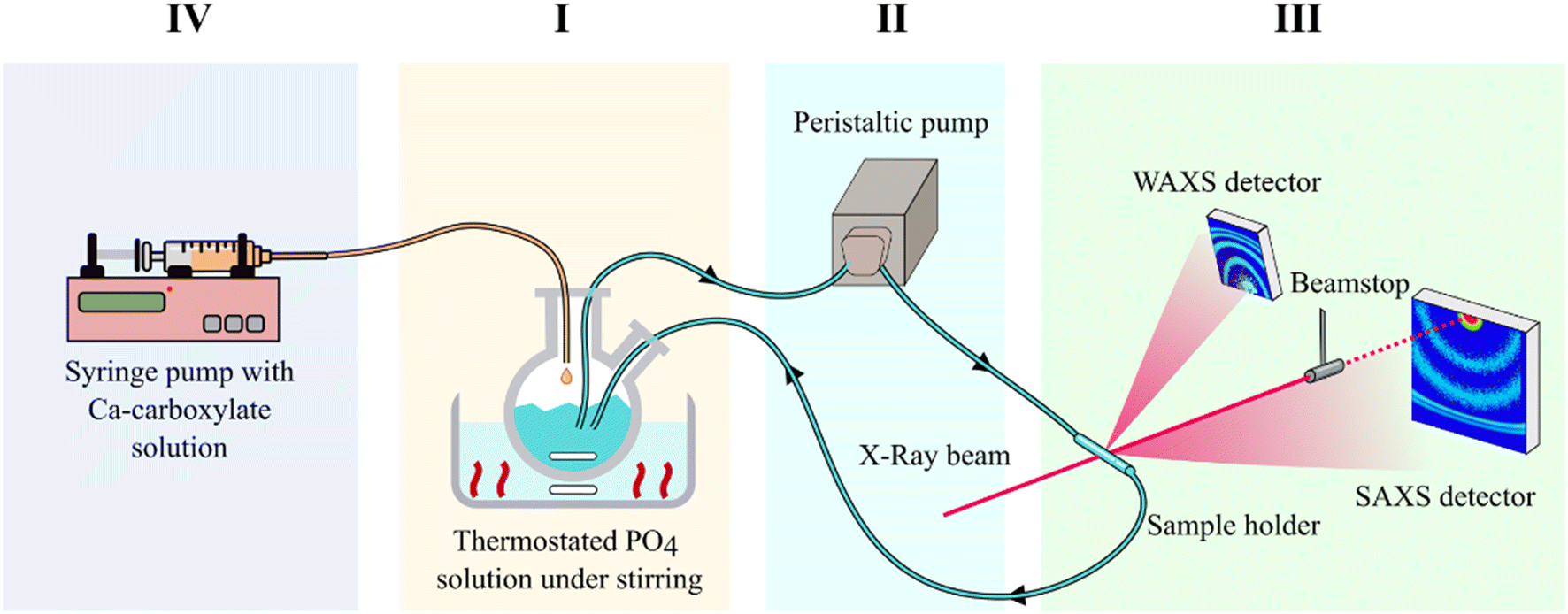 | ||
| Fig. 1 Schematic representation of the setup employed for real-time in situ SAXS/WAXS measurements during the formation of HA nanoparticles. The setup measures SAXS and WAXS simultaneously in the same sample volume. (I) Thermostated reaction vessel with PO4 solution, (II) peristaltic pump, (III) sample holder and SAXS/WAXS detectors, (IV) Ca solution syringe pump injector. | ||
SAXS/WAXS data analysis and modelling
The 2D SAXS images were normalized by transmission values and circularly averaged to be converted into conventional SAXS curves. This step was performed with the software Fit2D available at the beamline.16 Therefore, the background was removed with the software Igor Pro (WaveMetrics, Inc., Lake Oswego, OR, USA). SAXS data analysis and modeling were performed with ATSAS package V3.0.317 and SasView package V5.0.3 (https://www.sasview.org).SAXS data modeling was performed, first by estimating the overall parameters such as the radius of gyration (Rg) and maximum size of NPs (Dmax), and then using an ellipsoid model18 or a custom model as a combination of ellipsoid18 and cylinder19 models. The Schulz distribution was used to estimate the average particle size as well as the polydispersity of samples for the selected structural model.20,21
The 2D WAXS images were normalized and then circularly averaged, producing one-dimensional curves, which underwent background subtraction. In order to improve the signal-to-noise ratio, WAXS curves collected in short timeframes were averaged. Data analysis was performed with the software TOPAS5.22 A full-profile peak broadening analysis was conducted on the (002) peak of HA, calculating the HA c unit cell parameter and the dimensions of crystalline domains along HA (002) directions (D(002)). For all analyses, the baseline of the patterns was calculated through the Chebychev function (16th order polynomial).
Results and discussion
SAXS
SAXS time-resolved curves were collected continuously before (frames 0–10, corresponding to 60 s) and after the introduction of the Ca–carboxylate solution into the PO4 solution, which was set at a defined reaction temperature. Fig. 2 and S1† show an overview of the evolution over time of scattering patterns for the various carboxylate/temperature combinations. Before Ca–carboxylate addition there is no scattering except for the solvent and sample holder, which is removed by background subtraction, generating a flat curve. After injection of calcium precursor solutions, in all cases there is a sudden increase of the curve at q < 0.5 nm−1 that indicates the formation of scattering particles. With time, in all samples there are subtle changes of the curve shape, which suggest variations in particle morphology and aggregation. An example is the slight decrease of the scattering intensity in the range near q ≈ 0.1 nm−1 that is then recovered with time for all samples, but with different kinetics depending on the nature of carboxylate and reaction temperature. This variation might suggest the occurrence of particle–particle interactions that give rise to a structure factor contribution to the scattering, whose kinetics is temperature- and carboxylate-dependent. Another example is the change of the slope at 0.2–0.4 < q < 2 nm−1 that occurs for Cit/60 °C, Cit/80 °C, CitOH/80 °C, and Glr (all temperatures), which could be associated with a change in particle morphology and/or the formation of a polydispersion of particle morphologies (see below). To gain insight into SAXS data, we choose to organize the data on a bivariate mesh (Fig. 3), where each pixel represents the integral intensity of the corresponding one-dimensional plot (as in Fig. 2) for a certain time and temperature (respectively x- and y-axes). A pre-processing step to merge the different timescales and to normalize the intensity values was performed.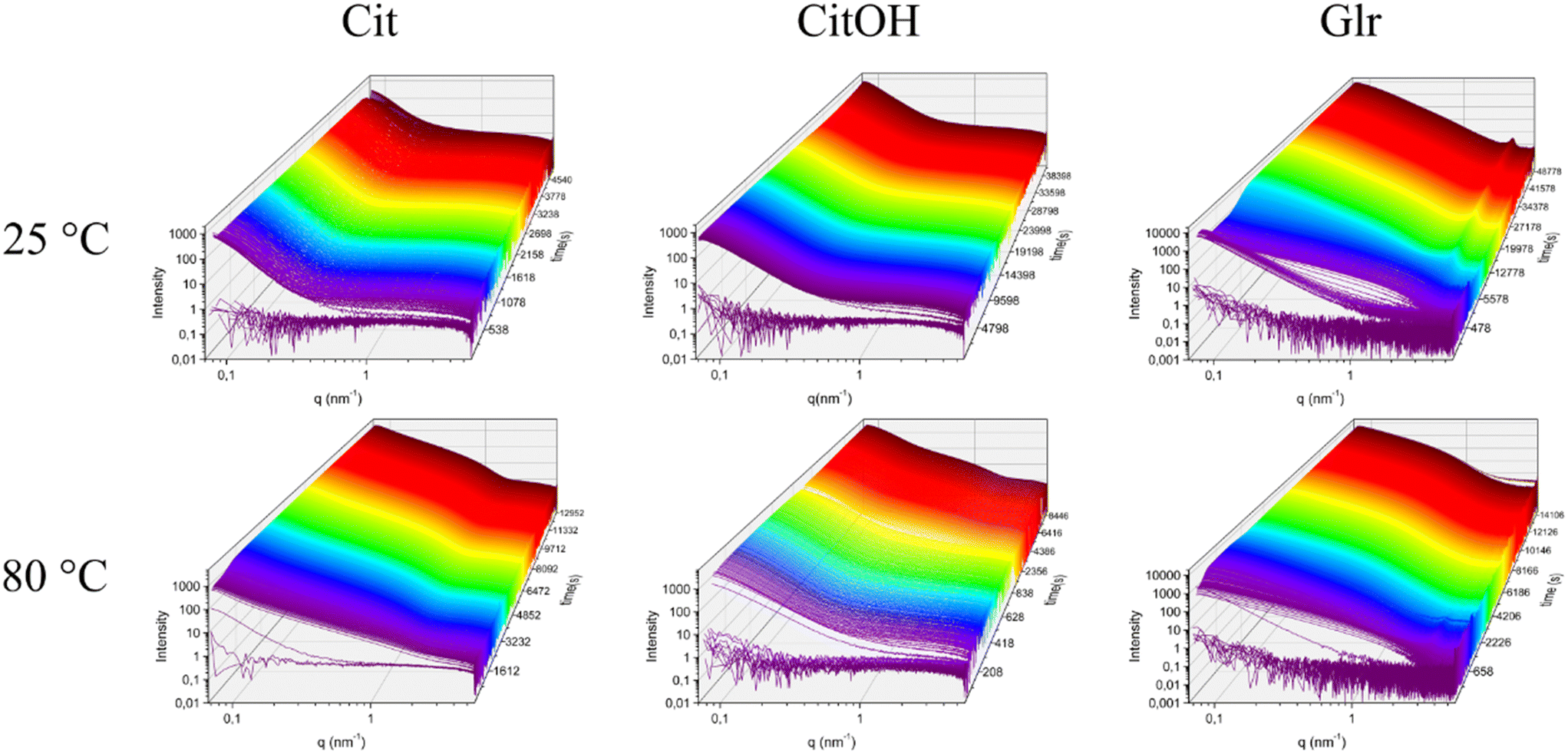 | ||
| Fig. 2 Examples of stack-plots of the measured SAXS curves as a function of time during the formation of the HA nanoparticles in the presence of different carboxylate molecules and reaction temperatures. | ||
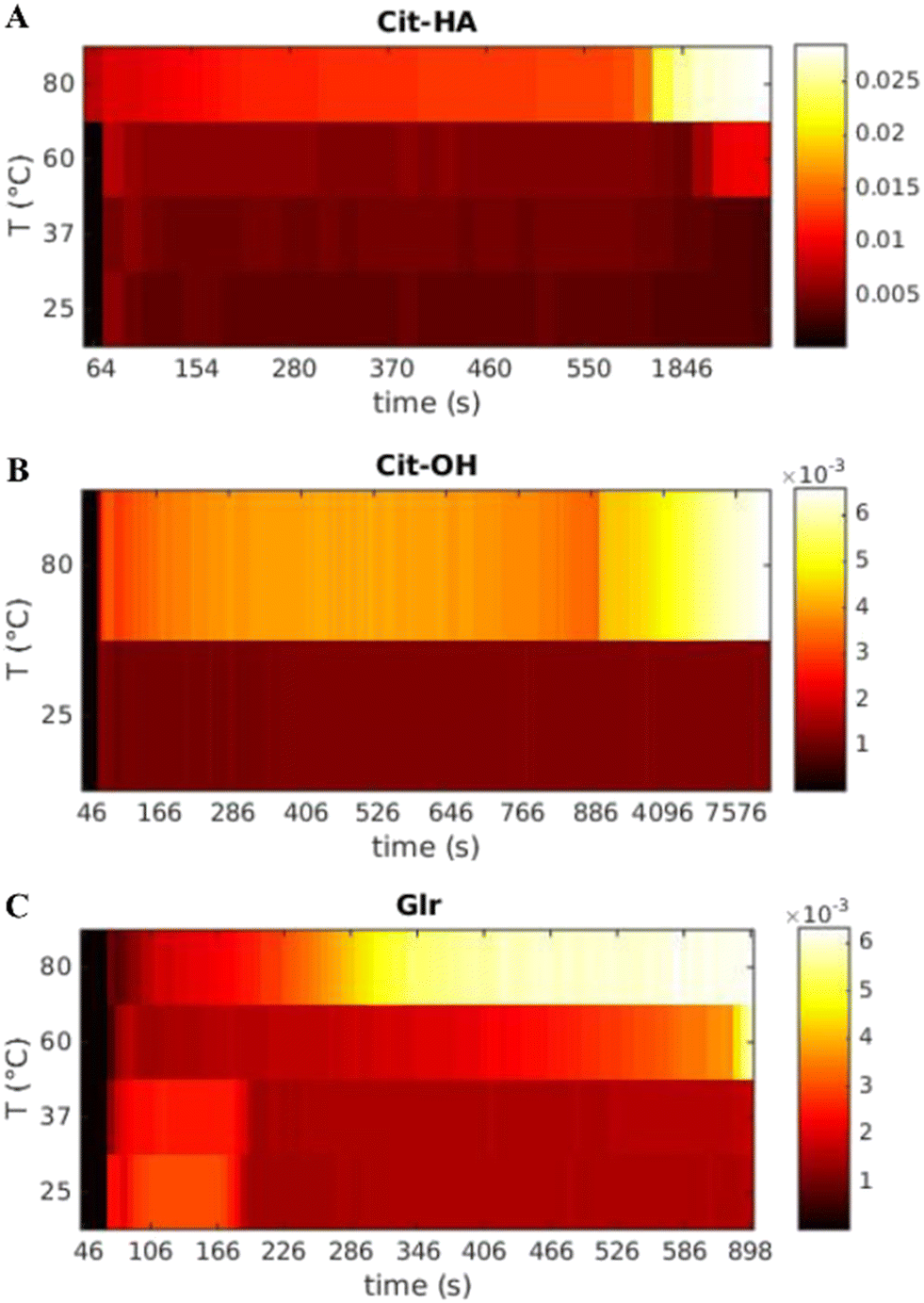 | ||
| Fig. 3 Bivariate mesh of the integrated SAXS intensity as a function of time and temperature for each carboxylate molecule used. (A) Cit, (B) CitOH, and (C) Glr. | ||
In the case of Cit, the first signal occurs within 80 s from the injection of the Ca–carboxylate solution at each temperature (Fig. 3A). The overall intensity at the beginning of particle formation is directly correlated to the increase of reaction temperature. These results suggest that the nucleation of primary CaP particles is controlled by the thermal decomplexing of Cit–Ca species, which gradually increase the concentration of free Ca2+ available for nucleation.11 Cit/25 °C and Cit/37 °C meshes do not show a significant change of the integrated intensity over time, while for both Cit/60 °C and Cit/80 °C there is a strong increase of integrated intensity respectively at 2400–3000 s (40–50 min) and 600–1200 s (10–20 min), respectively.
CitOH has a similar behavior to Cit (Fig. 3B), without significant variations of the integrated intensity over time at 25 °C, while at 80 °C, changes occurring in the range 900–1800 s (15–30 min) clearly show the influence of temperature.
Finally, for Glr at all temperatures the first increase of intensity happens after 80 s similarly to the other carboxylates (Fig. 3C). At high temperatures, there is a gradual increase of intensity that occurs at 900 s (15 min, 60 °C) or 300 s (5 min, 80 °C), while at lower temperatures the intensity is immediately higher but decreases after 180 s (3 min). A similar trend of the intensity can be observed also in Glr/60 °C but occurs in a short timeframe at ca. 90 s (1.5 min) of reaction. This behavior can be attributed to the fast particle formation, in which Glr–HA NPs quickly grow outside the SAXS measuring range and thus give a decrease of integrated intensity (see below for a detailed explanation). This out-growing phenomenon occurs at each reaction temperature, but is faster at higher temperatures, and thus at 80 °C is so fast to be non-detectable with the current time resolution (ca. 10 s).
Based on the bivariate mesh maps, relevant frames corresponding to significant changes in integrated intensity were identified and underwent SAXS curve modeling. The relevant frames were: for Cit, 80 s for all temperatures, 2400 and 3400 s for 60 °C and 600 and 1200 s for 80 °C; for CitOH, 80 s for all temperatures, 900 and 1800 s for 80 °C; for Glr, 80 s for all temperatures, 150 s for 25 °C and 37 °C, 600 s for 60 °C, and 290, 390, and 600 s for 80 °C (Fig. 4).
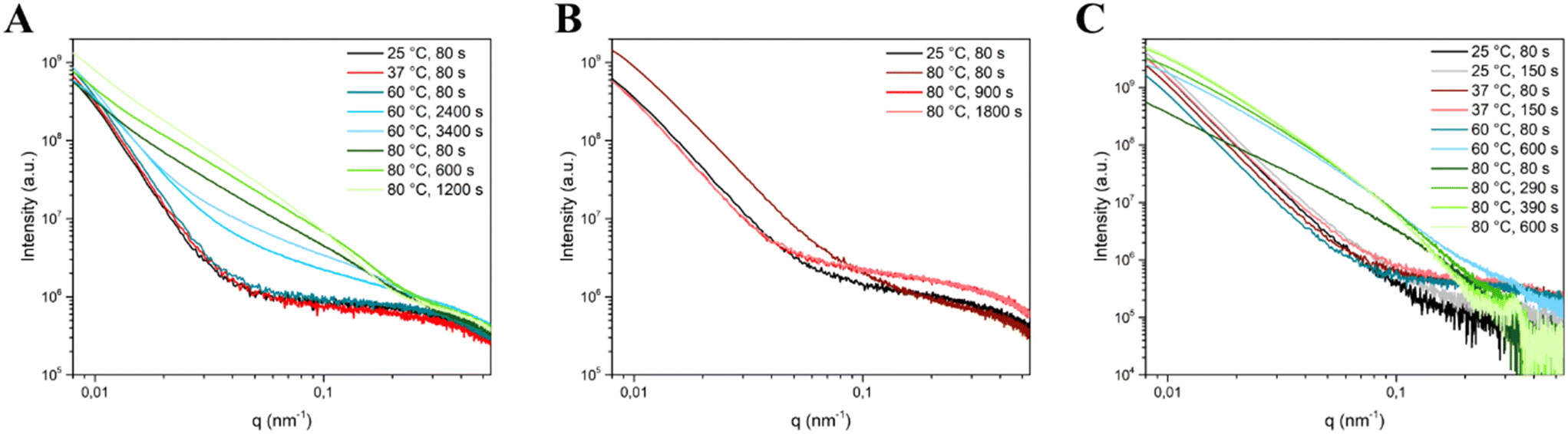 | ||
| Fig. 4 SAXS curves of the selected timeframes corresponding to significant changes in integrated intensity used for SAXS curve modelling for (A) Cit, (B) CitOH, and (C) Glr crystallization. | ||
Interestingly, for Cit/80 °C SAXS curves continue to evolve up to 1200 s (20 min), while for CitOH/80 °C the evolution was longer and concludes at 1800 s (30 min), and for Glr/80 °C it ended at 600 s (10 min). This difference in the duration of evolution is a confirmation of the results of our previous work, in which we have shown that CitOH is a stronger HA growth inhibitor than Cit – and hence particle growth requires more time – while Glr is the weakest.10
For the selected frames reported above, in the first step a preliminary analysis of the NP size was performed by estimating the radius of gyration (Rg), which is calculated from the slope of the Guinier plot of the SAXS curve,23 and the maximum size of the NPs (Dmax) which can be obtained by means of the Pair-Distance Distribution Function [P(r)] by using the Indirect Fourier Transformation as described by Glatter et al.24 (Tables 1–3). Both parameters were evaluated by using the AUTORG25 and GNOM26 programs of the ATSAS package,17 taking into account a monodisperse system. Afterwards, to estimate the NP shape the experimental curves were fitted with an ellipsoid model, refining both model parameters and the associated polydispersity by using the SasView program (Tables 1–3). In this model the parameters are Rpolar that is the radius along the y-coordinate and Requatorial representing the radius along the x-coordinate. Due to the high shape polydispersity of the NPs at the late stages of maturation at high reaction temperature, especially for Glr particles (where the conversion of OCP platelets into HA particles also occurs), the classical ellipsoid model did not allow satisfactory fits to be obtained, therefore we decided to use a custom shape-polydisperse model made by the combination of an ellipsoid model with a cylindrical model, which is defined by its radius (Rcyl) and its length (Hcyl). In this way, we were able to estimate approximately the relative abundance of each shape in the mixture of the particles, which is expressed as their normalized scale factor (expressed as % in Tables 1–3). The polydispersity of the refined parameters, reported in the bracket near the data in Tables 1–3, was evaluated as a ratio between the standard deviation and the mean value.
| Temperature (°C) | Time (s) | R g|Dmax (nm) | Scale (%) | Model parameters | |
|---|---|---|---|---|---|
| Ell.|Cyl. | Ellipsoid model | Cylinder model | |||
| R polar|Requatorial (nm) | R cyl|Hcyl (nm) | ||||
| 25 | 80 | 22.5|65.1 | 100 | 15.9 (0.2)|31.5 (0.0) | — |
| 37 | 80 | 23.4|73.9 | 100 | 17.2 (0.2)|31.7 (0.7) | — |
| 60 | 80 | 23.2|67.9 | 100 | 17.3 (0.2)|31.9 (0.1) | — |
| 2400 | 19.9|65.2 | 67|33 | 2.2 (0.1)|32.3 (0.1) | 4.4 (0.1)|54.4 (0.0) | |
| 3400 | 22.9|71.4 | 64|36 | 4.6 (0.2)|42.2 (0.1) | 22.2 (0.1)|63.9 (0.0) | |
| 80 | 80 | 23.3|77.3 | 100 | 0.2 (0.1)|35.1 (0.0) | — |
| 600 | 21.8|74.7 | 67|33 | 0.9 (0.5)|33.4 (0.1) | 1.5 (0.0)|55.8 (0.4) | |
| 1200 | 21.3|63.5 | 79|21 | 0.9 (0.0)|32.5 (0.0) | 1.4 (0.0)|54.9 (0.0) | |
| Temperature (°C) | Time (s) | R g|Dmax (nm) | Scale (%) | Model parameters | |
|---|---|---|---|---|---|
| Ell.|Cyl. | Ellipsoid model | Cylinder model | |||
| R polar|Requatorial (nm) | R cyl|Hcyl (nm) | ||||
| 25 | 80 | 20.5|67.3 | 64|36 | 9.9 (0.1)|29.7 (0.0) | 2.9 (0.0)|50.9 (0.0) |
| 80 | 80 | 20.1|66.0 | 70|30 | 3.5 (0.1)|26.7 (0.0) | 4.8 (0.0)|30.2 (0.0) |
| 900 | 21.4|65.3 | 42|58 | 10.4 (0.1)|30.5 (0.0) | 1.2 (0.0)|53.1 (0.0) | |
| 1800 | 21.6|65.1 | 32|68 | 12.5 (0.1)|29.1 (0.0) | 3.5 (0.0)|51.9 (0.0) | |
| Temperature (°C) | Time (s) | R g|Dmax (nm) | Scale (%) | Model parameters | |
|---|---|---|---|---|---|
| Ell.|Cyl. | Ellipsoid model | Cylinder model | |||
| R polar|Requatorial (nm) | R cyl|Hcyl (nm) | ||||
| 25 | 80 | 23.1|67.6 | 100 | 14.3 (0.3)|33.4 (0.0) | — |
| 150 | 23.2|68.6 | 100 | 15.4 (0.4)|34.0 (0.0) | — | |
| 37 | 80 | 24.4|74.0 | 100 | 15.6 (0.2)|33.1 (0.2) | — |
| 150 | 22.7|66.1 | 100 | 12.5 (0.5)|32.4 (0.0) | — | |
| 60 | 80 | 19.8|60.6 | 71|29 | 10.2 (0.1)|38.4 (0.1) | 10.4 (0.0)|260.0 (0.0) |
| 600 | 16.2|54.5 | 92|8 | 1.1 (0.0)|24.5 (0.3) | 7.6 (0.0)|62.3 (0.0) | |
| 80 | 80 | 19.1|65.9 | 35|65 | 3.9 (0.1)|28.2 (0.0) | 2.2 (0.0)|18.3 (0.0) |
| 290 | 16.8|56.9 | 91|9 | 2.6 (0.0)|24.6 (0.0) | 8.6 (0.0)|14.3 (0.0) | |
| 390 | 17.2|60.2 | 76|24 | 2.1 (0.0)|24.4 (0.0) | 2.4 (0.0)|89.1 (0.1) | |
| 600 | 17.4|59.4 | 79|21 | 2.4 (0.2)|24.7 (0.0) | 2.4 (0.0)|35.5 (0.0) | |
In the presence of Cit (Table 1), a Rg value of ca. 22 nm for HA NPs is obtained at each maturation time and temperature, suggesting that from the first moments of precipitation the Rg of particles does not change for all the experimental conditions. On the other hand, at higher temperatures the Dmax values become progressively higher, indicating a temperature-controlled particle growth. Together, these two parameters suggest that the NP population is constituted on average by particles comprised between 20 and 70 nm. Regarding the particle morphology, the ellipsoid model for Cit/25 °C, Cit/37 °C, and the early stages of Cit/60 °C SAXS curves (shown in Fig. 4A) describes a particle ca. 34 nm along one axis and 62 nm along the other axis, i.e. an elongated particle with a ratio between the major and minor dimension of ca. 2, in agreement with our previous ex situ electron microscopy data, which described Cit–HA as elongated platy NPs ca. 70 nm in length and 30–40 nm wide, with a thickness of ca. 3–4 nm.10 Unfortunately, it was not possible to model the NP thickness, as in this experimental setup SAXS is an ensemble measurement of free-moving particles in a flowing suspension, and thus all dimensions are averaged together and the small contribution of thickness is negligible. At higher maturation times for Cit/60 °C and for Cit/80 °C there is a progressive evolution in the curve shape (Fig. 4A) that is modeled as a decrease of the Rpolar parameter up to the values of 1–2 nm together with the introduction of shape polydispersity to have a suitable fit. These changes suggest that particles become progressively thinner as a function of time at high reaction temperatures. For these cases, our previous ex situ study proved that NPs grow preferentially along their main axis incrementing their aspect ratio (i.e., length/width ratio). These modifications are reflected in SAXS modeling parameters, as the particle length goes outside the measured SAXS range, i.e. below a q-value of 0.08 nm−1 corresponding to ca. 100 nm. Consequently, the contribution of the particle thickness to scattering becomes more relevant, and thus the particle width and thickness are modeled as ellipsoids and cylinders. On the other hand, the Rg and Dmax values remain unaffected as they depend only on the average measurable particle dimensions. The similarity between ellipsoidal and cylindrical model parameters (Rpolar and Requatorialvs. Rcyl and Hcyl, see Table 1) suggests that in the polydisperse particle population the overall morphology is similar (i.e. elongated particles), but individually the particle size may vary.
In conclusion, particle morphology modeling proved that at low reaction temperatures the particle shape is in agreement with average dimensions estimated by Rg and Dmax, while at higher temperatures the formation of highly anisotropic thin and elongated particles occurs. This change of morphology is almost concomitant with crystallization (see below), suggesting that the progressive increase of temperature which induced the particle growth is associated with the formation of the crystalline phase.
Crystallization of HA NPs in the presence of CitOH was studied at 25 and 80 °C (Table 2). All Rg and Dmax values are comparable to Cit, indicating a similar particle size distribution on average, while polydisperse shape modeling parameters are different. Indeed, their Requatorial values are independent from time and reaction temperature and are similar to Cit values, while the Rpolar ones are shorter – indicating a diameter of ca. 20 nm – and only the cylindrical sub-population decreases at 80 °C. This difference between CitOH and Cit confirms that for CitOH particles growth is more inhibited, with smaller particles that do not elongate at higher reaction temperatures as also proved by the more limited change in CitOH/80 °C SAXS curves reported in Fig. 4B. It must be considered that all values here reported for CitOH/80 °C are bigger than the ones previously reported by ex situ analysis. This discrepancy might be due to the post-synthesis product work-up necessary for the ex situ characterization.
SAXS modelling parameters of HA crystallization in the presence of Glr are reported in Table 3. For Glr/25 °C and Glr/37 °C both Rg and Dmax as well as Rpolar and Requatorial values are comparable to Cit (as also shown by similar SAXS curves reported in Fig. 4C). At higher reaction temperatures the curve shape changes (Fig. 4C), which leads to a lower Rg and the need to use the polydispersity of shapes in the fitting. Similarly to Cit, also for Glr at 60 and 80 °C the particle length falls outside the probed SAXS range and the particle thickness is modeled with the Rpolar/Rcyl values. This suggests an influence of temperature on particle evolution similar to Cit, where higher reaction temperatures induce the formation of highly anisotropic particles in concomitance with crystallization. However, the ex situ characterization pointed out that at 80 °C and <300 s of maturation the sample is constituted by sub-micrometric OCP platelets, whose dimensions fall outside the SAXS scattering range.15 Therefore, it is likely that values modeled by in situ SAXS curves correspond only to the fraction of smaller particles in the system.
WAXS
Complementary to SAXS data analysis, WAXS patterns were collected to study the crystallization of the particles observed by SAXS (Fig. 5 and S2†). In the case of precipitation in the presence of Glr at any temperature and for Cit/80 °C several diffraction peaks appear with time, which were all indexed to HA (powder diffraction file 00-009-0432). Our previous work evinced that in the presence of Glr also the OCP phase (powder diffraction file 00-026-1056) forms at short crystallization times as a precursor of HA,10 so we hypothesize that the same occurs here but it could not be confirmed due to the low intensity of WAXS peaks and the high superposition between HA and OCP peaks. For Cit/25 °C and CitOH/25 °C there are no signals of crystallization at each time point, in the case of Cit/37 °C, Cit/60 °C, and CitOH/80 °C the formation of a single diffraction peak can be observed at 18.1 nm−1, which was indexed to the (002) peak of HA. This peak commonly has a high relative intensity and is well resolved; its position is associated with the HA crystallographic c-axis and its broadness is directly proportional to the main axis of elongated HA nanocrystals.10 The appearance of this peak suggests that in the presence of a weak inhibitor such as Glr or at higher precipitation temperatures – T ≥ 37 °C (Cit) or T ≥ 80 °C (CitOH) – particles crystallize into HA, while in the presence of strong inhibitors and at low temperatures particles remain amorphous or form nanocrystals that are too small and poorly crystalline to be detected by WAXS analysis. | ||
| Fig. 5 Examples of stack-plots of the measured WAXS curves as a function of time during the formation of the HA nanoparticles in the presence of different carboxylate molecules and reaction temperatures. | ||
From the variations in the broadness and position of the (002) peak at 18.1 nm−1 we have calculated the time evolution of the D(002) crystalline domain as well as the variation of the HA c cell axis for the conditions where HA formation was detected. It must be mentioned that the (002) peak has an anomalous broadness, as it appears to be very narrow and with a peak broadness almost comparable to instrumental peak broadening. It is likely that in the capillary sample holder the high liquid flow rate, and thus shear rate, oriented the anisotropic HA NPs aligning their main axis (c-axis) with the flow direction. The consequence of this effect is that the scattering of the (002) peak is enhanced by preferential orientation; thus, the instrumental response function of the experimental apparatus could not be calculated and the broadness of the (002) peak could not be correlated to a numeric value of the crystalline domain size. Therefore, the D[002] crystalline domain values are expressed as arbitrary units and only their trends over maturation time are discussed.
The size of the D(002) crystalline domains of the samples and their evolution over time and temperature are reported in Table 4 and Fig. S3.† The crystalline domain size depends on both the nature of the carboxylic molecule and the temperature. For Cit, (Table 4 and Fig. S3A and D†) the appearance of the crystalline phase occurs at progressively shorter times as a function of temperature, ranging from no crystallization over 170 min at 25 °C to the formation of the HA phase after 3 min of reaction at 80 °C. In addition, the evolution of the crystalline domain size is directly correlated to the reaction temperature. Indeed, at 80 °C there is a burst growth of D(002) in less than 50 min, while at 60 °C the crystalline domain is ca. half smaller than that at 80 °C and has no change over time. At 37 °C the D(002) crystalline domain size is again half in comparison to the one at 60 °C and slowly grows in ca. 100 min. These results are in agreement with SAXS data on the formation of Cit–HA nanocrystals. Indeed, SAXS proved that Cit–HA nucleation is controlled by the thermal decomplexation of Cit–Ca species in solution, while WAXS proved that the reaction temperature accelerates the conversion of the first amorphous NPs into nanocrystals, and the subsequent crystal growth into bigger particles.
| Sample | Temperature (°C) | Appearance of (002) peak (min) | Time evolution of D[002] | Initial c parameter (Å, min) | Final c parameter (Å, min) |
|---|---|---|---|---|---|
| Cit | 25 | — | — | — | — |
| 37 | 115 | 155% increase from 115 to 195 m | 6.930, 150 m | 6.935, 500 m | |
| 60 | 60 | 20% increase from 60 to 130 m | 6.933, 60 m | 6.933, 130 m | |
| 80 | 3 | 100% increase from 3 to 120 m | 6.934, 3 m | 6.945, 120 m | |
| CitOH | 25 | — | — | — | — |
| 80 | 37 | 60% increase from 37 to 150 m | 6.931, 40 m | 6.943, 150 m | |
| Glr | 25 | 4 | 150% increase from 4 to 800 m | 6.918, 4 m | 6.902, 800 m |
| 37 | 4 | 10% increase from 4 to 50 m | 6.921, 4 m | 6.910, 50 m | |
| 60 | 2 | 15% increase from 2 to 50 m | 6.923, 2 m | 6.913, 50 m | |
| 80 | 4 | 100% increase from 4 to 120 m | 6.923, 4 m | 6.944, 200 m |
A similar situation occurs for CitOH, which is a stronger crystallization inhibitor than Cit, as HA is formed only at 80 °C after ca. 40 min of maturation, and the D(002) crystalline domain is ca. two to four-fold smaller than the D(002) of Cit at the same reaction temperature (Table 4 and Fig. S3B and E†). During maturation the CitOH–HA crystalline domain size doubles in ca. 110 min, and after 150 min of reaction no changes occur anymore.
For Glr, which is the weakest crystallization inhibitor, temperature has less influence as the HA/OCP phase was always detected in the first moment of reaction independently from reaction temperature, and always with similar values of the crystalline domain size (Table 4, Fig. S3C and F†). In details, the evolution of the D[002] crystalline domain size at 80 °C is the same for Glr and Cit systems (curves are superimposed), while at 60 °C and 37 °C there is almost no change over time. The independence of HA formation from reaction temperature suggests a lesser relevance of thermal decomplexation for the crystallization in the presence of Glr. The evolution of Glr/25 °C is particularly interesting, as the growth of the crystalline domain progresses in discrete steps, suggesting a different evolution mechanism, probably related to OCP formation and subsequent transformation into HA.
The variations of HA c cell axis over time (Table 4 and Fig. S4†) are less correlated to carboxylic acid and precipitation temperature than the D(002) crystalline domain. For all the carboxylates at 80 °C a cell expansion from ca. 6.92–6.93 Å to 6.945 Å occurs over time. These values are slightly higher than the ones calculated by ex situ crystallization (ca. 6.87 Å), but this difference is not so relevant as a slight expansion of the c cell parameter with maturation in the reaction solution has been reported previously.10,27 At lower reaction temperatures this cell parameter remains constant at ca. 6.93 Å for Cit and CitOH, while in the presence of Glr it decreases to ca. 6.91 Å, and the speed of this decrease is directly correlated with temperature. This suggests that for Glr at temperatures <80 °C a different process occurs in comparison to the other carboxylates. A tentative interpretation for the decrease of the c-axis (i.e., the shift of the (002) peak at higher q) can be due to a progressive formation and evolution of the OCP phase at lower reaction temperature, while at 80 °C OCP quickly transforms into HA and thus the peak shifts in the opposite direction. In fact, the (002) peak situated at 18.2 nm−1 for the Glr system could be composed by the overlapping of the HA (002) peak (theoretical position 18.07 nm−1) and the OCP (002) peak (theoretical position 18.35 nm−1).
Overall, considering our SAXS/WAXS data and the previous studies on the crystallization of HA in the presence of Cit, we hypothesize that HA nanocrystal formation follows a 4-step process: (i) Cit anions bind partially calcium ions, forming Ca–Cit complexes. Then, (ii) when brought into contact with phosphate ions there is an immediate nucleation of non-crystalline CaP clusters/nuclei independently from reaction temperature. These amorphous particles (observed within 1 min after mixing) are anisotropic and are ca. 50–60 nm long. With time (iii) particles grow and elongate, preferentially along their main axis. Finally, (iv) the HA crystalline phase nucleates and grows within the amorphous particles, as described by Delgado-López et al. and Iafisco et al.2,28 Depending on the reaction temperature, steps (iii) and (iv) might be consequential (<60 °C) or simultaneous (80 °C). The temperature influences the nucleation rate, crystal formation rate, and particle/crystal growth speeds. Regarding the influence of carboxylate molecules on the formation of nanoparticles, a stronger complexation (CitOH) does not change the development of early particles, but the process is slowed down, smaller, and less anisotropic particles are formed, and the formation of the crystalline phase is inhibited. The weaker Ca–carboxylate interaction of Glr gives fewer clear results, due to the simultaneous occurrence of several phenomena, i.e. particle precipitation, OCP formation, and OCP conversion into HA.
Conclusions
We have studied in situ the formation and growth of CaP NPs in the presence of calcium-binding dicarboxylate molecules at several reaction temperatures through simultaneous SAXS/WAXS using synchrotron light.SAXS investigation was focused on the early stages of particle formation, proving that both the nature of carboxylate molecules and reaction temperature had a strong influence on the amount, size, and morphology of first-nucleated NPs. The structural features of the molecules controlled the Ca2+–carboxylate complexation and thus ion supersaturation and association with nanoparticles, while the temperature influenced the decomplexation rate.
In particular, SAXS data analysis and modeling showed that: (i) nucleation of amorphous particles is almost immediate independently from reaction temperature and carboxylate molecules; (ii) at low temperatures the first-formed particles have similar morphologies and dimensions (elongated particles ca. 50–60 nm in length and 20–30 nm in width) for Cit and Glr, while the CitOH morphology is similar but the width is smaller; (iii) at high temperatures particles quickly elongate and become polydisperse and with heterogeneous morphology.
WAXS data gave complementary insights on the transformation of the amorphous particles in the HA nanocrystals, showing that the formation of the crystalline order is temperature- and carboxylate-dependent. In the presence of Cit and CitOH the crystallization to HA occurs between 180 s and 7200 s (120 min) at 80° and 37 °C, respectively, while at 25 °C crystallization does not occur. Moreover, the combination of SAXS and WAXS data shows that crystallization is simultaneous with particle elongation. Finally, the CaP formation in the presence of Glr follows a different mechanism as the OCP phase dominates and grows in a temperature-dependent fashion at reaction temperatures <60 °C, while at 80 °C there is the conversion of OCP into the HA phase.
To the best of our knowledge, the present study is the first report on the use of simultaneous SAXS/WAXS with synchrotron light for studying the formation of CaP NPs. By using custom data analysis tools, we have shed more light on the influence of growth inhibitor molecules on HA nucleation and growth, showing the relations between carboxylate–Ca2+ interaction, temperature, and NP formation. This study proves the potential of SAXS/WAXS as an innovative approach for the detailed characterization of nanomaterials, which could be extended to several types of biomimetic and bio-inspired nanomaterials for advanced applications.
Author contributions
D. Siliqi: investigation, writing – original draft, writing – review & editing. A. Adamiano: investigation, writing – review & editing. M. Ladisa: investigation, visualization. C. Giannini: investigation, writing – original draft, writing – review & editing. M. Iafisco: conceptualization, writing – original draft, writing – review & editing, supervision. L. Degli Esposti: investigation, visualization, writing – original draft, writing – review & editing, funding acquisition.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the CERIC-ERIC Consortium for financial support and for giving access to ELETTRA SAXS beamline (proposal CERIC-ERIC-20175403). The authors acknowledge Dr. Heinz Amenitsch for the scientific and technical support during SAXS data collection.References
- M. Epple, K. Ganesan, R. Heumann, J. Klesing, A. Kovtun, S. Neumann and V. Sokolova, J. Mater. Chem., 2010, 20, 18–23 RSC.
- J. M. Delgado-López, R. Frison, A. Cervellino, J. Gómez-Morales, A. Guagliardi and N. Masciocchi, Adv. Funct. Mater., 2014, 24, 1090–1099 CrossRef.
- K. Chatzipanagis, M. Iafisco, T. Roncal-Herrero, M. Bilton, A. Tampieri, R. Kroger and J. M. Delgado-Lopez, CrystEngComm, 2016, 18, 3170–3173 RSC.
- M. Li, L. Wang, W. Zhang, C. V. Putnis and A. Putnis, Cryst. Growth Des., 2016, 16, 4509–4518 CrossRef CAS.
- S. Wu, M. Li and Y. Sun, Angew. Chem., Int. Ed., 2019, 58, 8987–8995 CrossRef CAS PubMed.
- T. Li, A. J. Senesi and B. Lee, Chem. Rev., 2016, 116, 11128–11180 CrossRef CAS PubMed.
- M. A. Graewert and D. I. Svergun, Curr. Opin. Struct. Biol., 2013, 23, 748–754 CrossRef CAS PubMed.
- J. M. Delgado-López and A. Guagliardi, in New Perspectives on Mineral Nucleation and Growth, Springer, 2017, pp. 211–225 Search PubMed.
- F. Bertolotti, F. J. Carmona, G. Dal Sasso, G. B. Ramírez-Rodríguez, J. M. Delgado-López, J. S. Pedersen, F. Ferri, N. Masciocchi and A. Guagliardi, Acta Biomater., 2021, 120, 167–180 CrossRef CAS PubMed.
- L. Degli Esposti, A. Adamiano, D. Siliqi, C. Giannini and M. Iafisco, Bioact. Mater., 2021, 6, 2360–2371 CrossRef CAS PubMed.
- J. M. Delgado-López, M. Iafisco, I. Rodríguez, A. Tampieri, M. Prat and J. Gómez-Morales, Acta Biomater., 2012, 8, 3491–3499 CrossRef PubMed.
- M. Li, J. Zhang, L. Wang, B. Wang and C. V. Putnis, J. Phys. Chem. B, 2018, 122, 1580–1587 CrossRef CAS PubMed.
- Y. H. Tseng, Y. Mou, P. H. Chen, T. W. Tsai, C. I. Hsieh, C. Y. Mou and J. C. Chan, Magn. Reson. Chem., 2008, 46, 330–334 CrossRef CAS.
- F. Nudelman, K. Pieterse, A. George, P. H. Bomans, H. Friedrich, L. J. Brylka, P. A. Hilbers, G. de With and N. A. Sommerdijk, Nat. Mater., 2010, 9, 1004–1009 CrossRef CAS.
- H. Amenitsch, M. Rappolt, M. Kriechbaum, H. Mio, P. Laggner and S. Bernstorff, J. Synchrotron Radiat., 1998, 5, 506–508 CrossRef CAS.
- A. Hammersley, European Synchrotron Radiation Facility Internal Report ESRF97HA02T, 1997, vol. 68, p. 58 Search PubMed.
- K. Manalastas-Cantos, P. V. Konarev, N. R. Hajizadeh, A. G. Kikhney, M. V. Petoukhov, D. S. Molodenskiy, A. Panjkovich, H. D. Mertens, A. Gruzinov and C. Borges, J. Appl. Crystallogr., 2021, 54, 343–355 CrossRef CAS PubMed.
- J. Finnigan and D. Jacobs, J. Phys. D: Appl. Phys., 1971, 4, 72 CrossRef.
- J. S. Pedersen, Adv. Colloid Interface Sci., 1997, 70, 171–210 CrossRef CAS.
- M. Kotlarchyk and S. H. Chen, J. Chem. Phys., 1983, 79, 2461–2469 CrossRef CAS.
- M. Kotlarchyk, R. B. Stephens and J. S. Huang, J. Phys. Chem., 1988, 92, 1533–1538 CrossRef CAS.
- A. Coelho, Coelho Software, 2012 Search PubMed.
- A. Guinier, Ann. Phys., 1939, 11, 161–237 Search PubMed.
- O. Glatter, J. Appl. Crystallogr., 1977, 10, 415–421 CrossRef.
- M. V. Petoukhov, P. V. Konarev, A. G. Kikhney and D. I. Svergun, Appl. Crystallogr., 2007, 40, s223–s228 CrossRef CAS.
- D. Svergun, J. Appl. Crystallogr., 1992, 25, 495–503 CrossRef CAS.
- F. J. Martínez-Casado, M. Iafisco, J. M. Delgado-López, C. Martínez-Benito, C. Ruiz-Pérez, D. Colangelo, F. Oltolina, M. Prat and J. Gómez-Morales, Cryst. Growth Des., 2015, 16, 145–153 CrossRef.
- M. Iafisco, G. B. Ramirez-Rodriguez, Y. Sakhno, A. Tampieri, G. Martra, J. Gomez-Morales and J. M. Delgado-Lopez, CrystEngComm, 2015, 17, 507–511 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: SAXS and WAXS scattering curves as a function of time during the formation of the HA nanoparticles at 37 °C and 60 °C reaction temperature (PDF). See DOI: https://doi.org/10.1039/d2ce01227h |
| This journal is © The Royal Society of Chemistry 2023 |